 Eric Kenneth Parkinson*†
Eric Kenneth Parkinson*† Stephen Stewart Prime*†
Stephen Stewart Prime*†- Centre for Immunobiology and Regenerative Medicine, Barts and the London School of Medicine and Dentistry, Blizard Institute, London, United Kingdom
Cellular senescence is an irreversible cell cycle arrest occurring following multiple rounds of cell division (replicative senescence) or in response to cellular stresses such as ionizing radiation, signaling imbalances and oxidative damage (stress-induced premature senescence). Even very small numbers of senescent cells can be deleterious and there is evidence that senescent cells are instrumental in a number of oral pathologies including cancer, oral sub mucous fibrosis and the side effects of cancer therapy. In addition, senescent cells are present and possibly important in periodontal disease and other chronic inflammatory conditions of the oral cavity. However, senescence is a double-edged sword because although it operates as a suppressor of malignancy in pre-malignant epithelia, senescent cells in the neoplastic environment promote tumor growth and progression. Many of the effects of senescent cells are dependent on the secretion of an array of diverse therapeutically targetable proteins known as the senescence-associated secretory phenotype. However, as senescence may have beneficial roles in wound repair, preventing fibrosis and stem cell activation the clinical exploitation of senescent cells is not straightforward. Here, we discuss biological mechanisms of senescence and we review the current approaches to target senescent cells therapeutically, including senostatics and senolytics which are entering clinical trials.
Introduction
There have been several reviews on the subject of senescent cells in human disease (1, 2). Classical cellular senescence is defined as an irreversible cell cycle arrest that is distinct from quiescence, terminal differentiation and apoptosis. This form of senescence occurs following multiple rounds of cell division [replicative senescence (3)] or following a wide range of cellular stresses such as ionizing radiation, signaling imbalances or oxidative damage [stress-induced premature senesce or SIPS; (4)]. More recently, the definition of senescence has been broadened to include developmental senescence, oncogene-induced senescence (OIS) and cancer therapy-induced senescence where an irreversible cell cycle arrest is either unstable (4) or may not occur at all (5). OIS does not occur in young healthy cells (6–8) with low levels of p16INK4A and not all oncogenes induce senescence (9), so cells need to be damaged before OIS operates and even then, only when expressed at a sufficient level (10). Senescence is a double-edged sword in that it operates as a suppressor in the early stages of malignancy (11) but also, has the capacity to promote tumor development (12) and tumor progression (11, 13) once the tumor has developed via molecules of the senescence-associated secretory phenotype (SASP) such as interleukins, matrix metalloproteinases (MMPs), vascular endothelial growth factor (13) and certain metabolites (14) (summarized in Figure 1). Even small numbers of senescent cells can be deleterious (15) and there is evidence that senescent cells are instrumental in age-related pathologies (16), in response to cancer therapy (17) and in chronic inflammatory conditions (18).
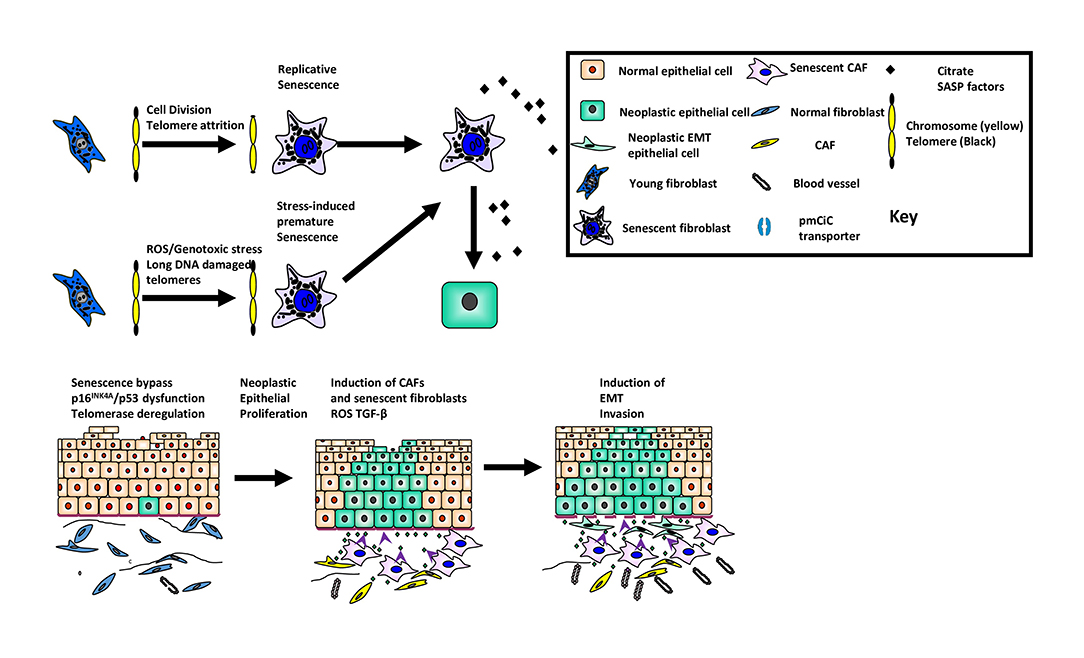
Figure 1. The role of cellular senescence in oral cancer development and progression. The figure summarizes the role of senescence as a suppressor of malignancy in keratinocytes and its paradoxical role as a promoter of tumor progression in the developing tumor environment.
The SASP and the Extracellular Senescence Metabolome (ESM)
During the establishment of senescence, an array of proteins (13) and metabolites (19) accumulate in the extracellular milieu and collectively are referred to as the senescence-associated secretory phenotype [SASP; (13)] or the extracellular senescence metabolome [ESM; (19)], respectively.
More specifically, the SASP consists of a large number of cytokines, chemokines and immunomodulatory molecules, growth factors, shed surface molecules and survival factors, together with promoters of angiogenesis, fibrosis and tissue re-modeling (13). The mechanisms of senescence and the constituents of the SASP vary between cell types which makes the SASP cell type- and mechanism-specific. More recent data indicate that the SASP is highly heterogeneous and varies temporally even within populations of the same cell type (20). Some of the cytokines of the SASP increase to detectable levels in the plasma of older humans and have been proposed to be biomarkers of age-related conditions such as frailty (21). This sterile inflammation is known as inflammaging (22) and is thought to be due to the accumulation of senescent cells which display inappropriate levels of cytoplasmic chromatin; it is mediated by the cyclic GMP–AMP synthase (cGAS)–stimulator of the interferon gene (STING) (cGAS-STING) pathway that induces inflammatory cytokines (23).
Recently, the oral keratinocyte SASP has been examined in more detail as part of a comprehensive survey of SIPS (24). Many of the transcripts and proteins of the SASP are consistent between fibroblasts and mortal pre-neoplastic keratinocytes. However, senescence-specific exosomes induce the interferon pathway in neighboring monocytes via the cGAS-STING pathway (25) and numerous SASP factors (particular prostaglandins) and metabolites are dependent on the keratinocyte senescence program (26, 27). Although there is no evidence for senescence in human pre-malignant lesions in vivo, it has been described in other human pre-malignant conditions (28) and p16INK4A is upregulated in high-grade pre-malignant lesions (29). Current thinking indicates that the SASP may be released in two waves which are dependent on different pathways and which modulate the immune system in different ways with the common goal being the promotion of neoplasia (30).
The ESM metabolites (citrate, C-mannosyl tryptophan, urate, and eicosapentaenoate), are amongst the 22 metabolites most significantly elevated with chronological age and aging traits [loss of lung function and bone mineral density (31)]; the function of most of these metabolites in age-related diseases, however, is unclear. A role for C-mannosyl tryptophan in apoptosis has been predicted based on bioinformatics and, more recently, a role for citrate has been suggested in the pathobiology of type 2 diabetes (32), memory (33), heart rate (34), blood pressure (34), and cancer (35, 36).
ORAL Keratinocyte Snescence
Whilst the senescence regulators telomerase, p53 and pRB/p16INK4A are common to both keratinocytes and fibroblasts, their exact roles are not quite the same. Mechanistically, following telomere dysfunction, disabling p53 in both fibroblasts (37) and keratinocytes (38) extends the proliferative lifespan of fibroblasts but not keratinocytes, whereas knockdown of p16INK4A has no effect on its own. However, the combined knockdown of p53 and p16INK4A extends the proliferative lifespan of fibroblasts further and induces a phenomenon resembling crisis in both cell types (37, 38). In serum-free culture systems, p16INK4A, but not p14ARF, accumulates following proliferative exhaustion in the absence of telomere attrition (39). p16INK4A accumulation in keratinocytes is also associated with the expression of laminin gamma 5 delta 2, a hyper-motile phenotype seen in both carcinoma-in-situ, and in experimental wounding in vitro (29). This form of senescence is bypassed by plating keratinocytes on collagen type 1, disabling p16INK4A or p53 and is delayed by inhibition of the transforming growth factor beta (TGF-β) pathway (40). Epidermal keratinocytes do undergo a phenotype resembling senescence in aging humans (41) and this appears to be mediated by a paracrine mechanism associated with senescent melanocytes that induce telomere-associated DNA damage foci (TAFs) in neighboring suprabasal keratinocytes. However, there is no evidence of p16INK4A accumulation in aged keratinocytes in situ and it is not clear whether TAFs persist in the basal layer of squamous epithelia because telomerase should gradually resolve the TAFs (4, 42).
Senescence as a Suppressor of Oral Squamous Cell Carcinoma Development
RAS and RAF oncogenes induce senescence when over-expressed (10). Senescence is observed in pre-malignant lesions of several cancer types (43, 44) and the elimination of senescent cells in mice precipitates tumor progression (11). Whilst RAS and RAF mutations are rare in oral cancer (45, 46), p16INK4A accumulates in oral carcinoma in situ (29) which is suggestive of senescence. Both SIPS and classical senescence are mediated by the pRB/p16INK4A and p14ARF/p53 pathways (1) and upon their dysfunction in oral cancer, telomere crisis and genetic instability ensue (38, 47) followed by telomerase deregulation and cellular immortality (38, 47–49). The dysfunction of p16INK4A, p53, and telomerase in both human papillomavirus (HPV) positive and negative head and neck squamous cell carcinomas (HNSCC) is nearly ubiquitous (50). Further, deregulation of telomerase by activating mutations of the promoter of the catalytic component of the enzyme TERT is a common event in HPV negative SCCs (51), but this is not enough to account for the increase in telomerase in 90% of HNSCC (52) and further genetic alterations are required (53).
Fibroblast Senescence in the Oral Cancer Environment
The tumor microenvironment consists of neoplastic epithelial cells and non-neoplastic stromal cells. The predominant stromal cell-type are fibroblasts and when these cells become “activated,” the term cancer associated fibroblast (CAF) is used. Fibroblast activation occurs de novo by reactive oxygen species (ROS) derived from the epithelial cancer cells (Figure 1) and by constituents of the SASP [IL-1β, IL-6, osteopontin (54)]. CAFs are heterogeneous and express a variety of different proteins, the significance of which is unclear. Current thinking indicates that fibroblast activation is context-dependent, plastic and likely to fall along a continuum rather than into discrete subsets (55). These observations may account for recent observations where CAFs with pro-tumorigenic and anti-tumorigenic phenotypes have been described. Stromal features are predictive of mortality (56) and ECM deposition and organization correlate with poor prognosis (57) in oral squamous cell carcinoma (OSCC).
There is a plethora of data showing that CAFs possess the ability to influence the hallmarks of cancer (54). In OSCC, for example, CAFs promote invasion and epithelial dis-cohesion, induce epithelial-mesenchymal transition and cause resistance to apoptosis (30, 58, 59). CAFs regulate fibroblast activation in an autocrine manner, remodel the ECM in a paracrine fashion which leads to a pro-fibrogenic phenotype and also, maintain persistent chronic inflammation. Further, CAFs show up-regulation of glycolysis and down-regulation of oxidative phosphorylation [reverse Warburg effect (60)] which creates a favorable hypoxic environment for epithelial tumor development and progression.
CAFs undergo senescence through secondary senescence induction from other senescent cells although at present, it is unclear whether this leads to functional heterogeneity. Whilst senescent fibroblasts are likely to contribute to the tumor-promoting environment, they probably do so in concert with non-senescent CAFs in a complementary manner. It is unknown whether stromal cells help keratinocytes to escape cellular senescence and this warrants further investigation.
The role of senescence in fibrosis is complex. In the short term, senescent cells can promote wound repair (61) and can also ameliorate fibrosis of the skin in vivo (62). Further, senescent cells are targeted by both the innate and the adaptive immune systems (11) and disabling either senescence or the immune system promotes fibrosis (63). Paradoxically, persistent inflammation, a property of many oral diseases, can induce senescence (64) and if senescent cells evade the immune system and persist, they can increase fibrosis. Damaged senescent cells avoid immune recognition through matrix metalloproteinase-dependent shedding of NKG2D ligands reinforced via paracrine suppression of NKG2D receptor-mediated immunosurveillance (65).
Senescence in Cancer Therapy and the Poor Health of Cancer Survivors
Senescent cells accumulate in mouse tissues following DNA damage (66) and chemotherapy (17) and their removal by genetic or pharmacological deletion ameliorates the side effects of such therapies in mice, including hypo salivation (67), fibrosis (68), and ulceration (69). Further, the tissues of cancer survivors have short telomeres and increased numbers of senescent cells (70–73) and oral mucositis, a major side effect of cancer therapy, has been linked to keratinocyte senescence (74).
ORAL Sub Mucous Fibrosis (OSMF)
OSMF is a debilitating and pre-cancerous condition caused by the chewing of “paan” which contains areca nut and sometimes tobacco (10, 16). In early OSMF, there is epithelial atrophy, juxta-epithelial inflammation and the sub epithelial connective tissues become avascular and thickened which leads to collagen accumulation. In advanced disease, the fibrosis extends into the deeper tissues and is associated with an increased inflammatory infiltrate (17). In OSMF, biologically active alkaloids and flavonoids in areca nut stimulate fibroblasts to increase collagen synthesis (18, 19); concurrently, there is reduced collagen degradation due to increased stability of the collagen structure (20, 21) and reduced collagenase activity (22). These events are mediated by changes in the regulation of several SASP factors such as TGF-β1, plasminogen activator-1 and matrix metalloproteinases (MMPs) and their inhibitors. The result is progressive hyalinization and fibrosis of the oral submucosa.
The role of senescence in OSMF has been comprehensively reviewed recently (75). It has been attributed to areca nut alkaloids (76, 77) and occurs in keratinocytes, fibroblasts and endothelial cells; in fibroblasts, the mechanism is associated with ROS, irreparable DNA damage and an increase in mitochondrial mass and membrane potential (76). Depleting the fibroblast population of senescent cells in OSMF drastically reduces the levels of MMP-1 and MMP-2 in conditioned medium suggesting that, as with CAFs, there are both non-fibrogenic and pro-fibrogenic fibroblasts (76). Senescence in keratinocytes, however, leads to epithelial atrophy and down-regulation of basal stemness, whilst senescence in endothelial cells accounts for a decrease in vascularity which leads to the development of a hypoxic state. The consequence of cellular senescence is the generation of a SASP, ROS generation and the induction of DNA double strand breaks in keratinocytes (78). Only when the keratinocytes escape from senescence does malignant transformation ensue but the myofibroblasts persist because they evade immune clearance.
The Role of Senescence in Other Oral Diseases
The oral cavity is the site of trauma, is susceptible to toxin and drug exposure, responds to the impact of systemic disease and aging and is exposed to microbial infection. Whilst senescence has been implicated in the pathobiology of odontogenesis (79–81), changes in the supporting structures of the teeth (82, 83), the pathology of salivary glands (84) and disruption of homeostasis in the oral mucosa, it is thought to play a fundamental role in the most prevalent human disorder, namely periodontal disease (82). It has been proposed that Gram-negative bacterial infection leads to a DNA damage response in both gingival keratinocytes and fibroblasts, chronic inflammation results in ROS-mediated oxidative DNA damage and keratinocyte senescence leads to breakdown of the cervical epithelial barrier (62). Further, bacterial toxins induce local immunosuppression resulting in a failure to remove the senescent cells and the perpetuation of the disorder.
Targeting Senescent Cells in Disease
Inhibiting SASP Production/Function (Senostatics)
From an early stage, proteins of the SASP have been considered as therapeutic targets in the treatment of cancer (Figure 2). Recent data, however, has shown that the SASP is cell-type specific, thus making this approach less practical. A recent review has highlighted some potential drug targets, particularly those associated with inhibition of the inflammasome and plasminogen activator inhibitor-1 (85). Such targets include p38 mitogen-activated kinase (MAPK14; (86), p38 MAPKAPK2/3 (87), Janus-activated kinase 1 (JAK1; (88) and steroids (89); these drugs have been shown to ameliorate the effects of frailty (88) and type 2 diabetes in mice (90). The MAPK14 inhibitors and steroids have been reported to inhibit the secretion of a diverse array of SASP proteins (86, 89), but JAK1 inhibitors have only been reported to target a subset of the inflammasome (88). Regulators of alternative splicing such as PTPB1 have been shown to inhibit the SASP and may also be attractive targets for therapy (91). In addition, rapamycin, a promoter of autophagy has been shown to ameliorate the effects of oral mucositis linked to senescence (74) in mice, but not as yet in humans. A major problem with senostatics, however, is that they are likely to require continuous administration, thereby increasing the cost per patient (92).
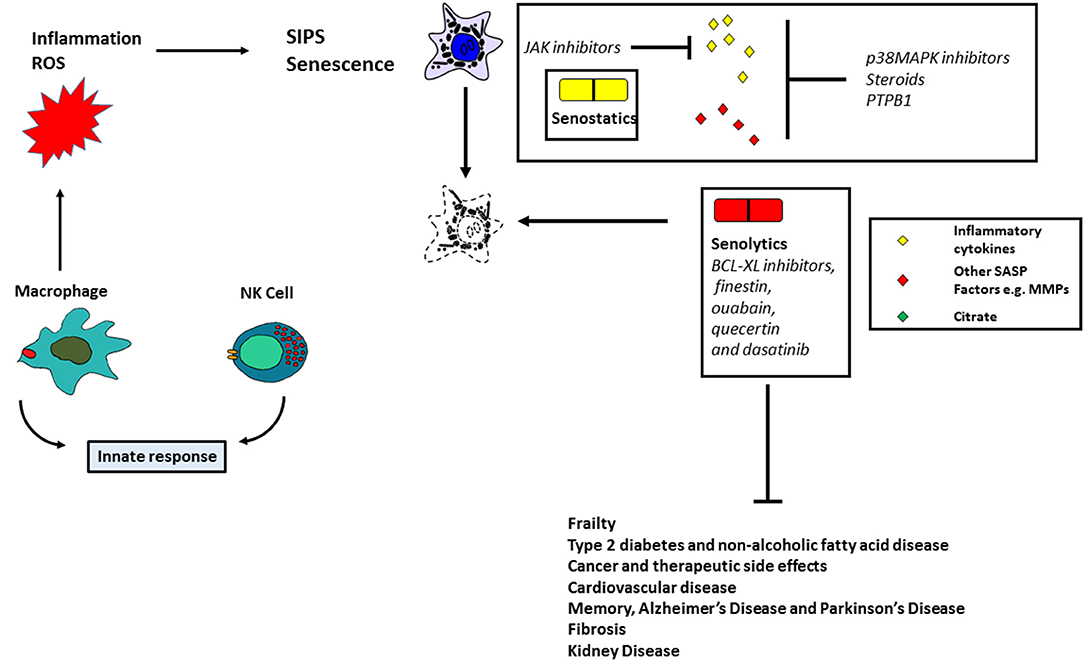
Figure 2. Targeting senescent cells therapeutically. The figure summarizes the instrumental role of senescent cells in human pathologies and the potential role of antioxidant, senostatic, and senolytic therapies.
Senolytics
The above approaches are likely to be specific for certain senescence-derived molecules and whilst this will reduce the likelihood of side effects, an alternative approach is to use drugs which selectively target senescent cells. Senolytics (Figure 2) include BCL-XL inhibitors, finestin, ouabain, quecertin and dasatinib (92). In addition, FOXO4 can bind with the p53 protein to induce cellular senescence and a peptide competing with FOXO4 can act as a senolytic by excluding p53 from the nucleus (93). These drugs ameliorate the effects of many age-related diseases in mice and clinical trials have begun to test their effects in human disease. Whilst senolytics can only be administered for a short time due to known side effects (e.g., thrombocytopenia), there is some evidence that they can ameliorate idiopathic pulmonary fibrosis (94) and can transiently clear senescent cells in the adipose and skin tissue of diabetic kidney disease patients (95). Another approach is to use chimeric antigen receptor (CAR) T cells to target senescent cells (96). Senescent cells become hyper-inflammatory, in response to pathogens and repress anti-viral gene expression in non-senescent cells by a paracrine mechanism (97); senolytics improve the survival of old mice infected with pathogens such as SARS-CoV-2. Apart from the known side effects of senolytics (98), it is cautionary to note that they do not always work (92) suggesting some level of cell-type/mechanism specificity. New drugs, therefore, will need to be identified. Further, SIPS is mediated in part by p16INK4A (see above) which is silenced in subsets of mammary cells in middle-aged females and is thought to be a precursor of breast cancer (85). Therefore, although removal of p16INK4A-positive cells in mice might improve age-related pathologies without a concomitant cancer risk, this might not be the case in humans.
Preventing the Accumulation of Senescent Cells
The two approaches described above are currently being heavily pursued but suffer from the fact that both require long term administration if employed alone and approaches that reduce the production of senescent cells will need to be considered in parallel. Such approaches might include stimulating the immune system, antioxidants such as mitochondrial catalase inhibitors (99–101), activators of DNA repair (102) and inducers of telomerase activity (103, 104) all of which would delay senescence in different ways (Figure 2).
Targeting Senescent Cells in Oral Diseases
As yet, no clinical trials have been published that target senescent cells in oral disease. However, there are many mouse models which show that senolytics might be effective in ameliorating OSMF; steroids block many components of the SASP (89) and are one of the treatments known to be beneficial in OSMF (105). Inhibitors of senescence have not been used in the treatment of OSMF, but it has been proposed that they may be of value for the alleviation of both radiation-induced fibrosis (106) and ulcers (83), and for the treatment of pre-cancerous lesions (107).
Detecting Senescent Cells
Several clinical trials have now assessed the potential of senolytics in human disease (108) but one of the problems is providing evidence that they are actually working. Even in severely ill patients, senescent cells are infrequent and SASP factors are either undetectable (94) or require the screening of several factors to verify senescence (95, 109); even when a few SASP factors are detectable, they are not consistent from patient to patient (95).
Several metabolites do accumulate in the extracellular milieu of senescent fibroblasts in vitro (19) and, indeed, a metabolite (15-deoxy-delta-12,14-prostaglandin J2) indicative of senolytic activity has been identified (110). However, no metabolite has been tested for its utility in senolytic- or senescence-related therapies in humans.
Discussion
Whilst it is clear that senescent cells are instrumental in a wide variety of age-related pathologies in mouse models, translating these findings to humans remains a significant challenge. Senescent cells occur at very low frequencies in diseased tissues and most of the SASP factors overlap considerably with other processes, most notably oxidative damage and inflammation. Deleting senescent cells in humans may also have an associated cancer risk. The levels of SASP factors in the plasma/sera in published studies to date are very low and inconsistent, thereby making proof-of principle of many approaches very difficult. Senolytic therapies hold some promise, but safer and cell type-specific versions are likely to be required. It is clear that much further research will be required to identify and eliminate senescent cells in human disease including those of the oral cavity.
Author Contributions
EP produced the first draft of the manuscript and figures. EP and SP performed the literature searches and modified and edited the manuscript. Both authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors wish to thank Lindsay Parkinson for assistance with graphics.
References
1. Campisi J, Kapahi P, Lithgow GJ, Melov S, Newman JC, Verdin E. From discoveries in ageing research to therapeutics for healthy ageing. Nature. (2019) 571:183–92. doi: 10.1038/s41586-019-1365-2
2. Kirkland JL. Translating the science of aging into therapeutic interventions. Cold Spring Harb Perspect Med. (2016) 6:a025908. doi: 10.1101/cshperspect.a025908
3. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. (1961) 25:585–621. doi: 10.1016/0014-4827(61)90192-6
4. Patel PL, Suram A, Mirani N, Bischof O, Herbig U. Derepression of hTERT gene expression promotes escape from oncogene-induced cellular senescence. Proc Natl Acad Sci USA. (2016) 113:E5024–33. doi: 10.1073/pnas.1602379113
5. Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, et al. Cellular senescence: defining a path forward. Cell. (2019) 179:813–27. doi: 10.1016/j.cell.2019.10.005
6. Benanti JA, Galloway DA. Normal human fibroblasts are resistant to RAS-induced senescence. Mol Cell Biol. (2004) 24:2842–52. doi: 10.1128/MCB.24.7.2842-2852.2004
7. Henrard DR, Thornley AT, Brown ML, Rheinwald JG. Specific effects of ras oncogene expression on the growth and histogenesis of human epidermal keratinocytes. Oncogene. (1990) 5:475–81.
8. Scott LA, Vass JK, Parkinson EK, Gillespie DA, Winnie JN, Ozanne BW. Invasion of normal human fibroblasts induced by v-Fos is independent of proliferation, immortalization, and the tumor suppressors p16INK4a and p53. Mol Cell Biol. (2004) 24:1540–59. doi: 10.1128/MCB.24.4.1540-1559.2004
9. Kennedy AL, Morton JP, Manoharan I, Nelson DM, Jamieson NB, Pawlikowski JS, et al. Activation of the PIK3CA/AKT pathway suppresses senescence induced by an activated RAS oncogene to promote tumorigenesis. Mol Cell. (2011) 42:36–49. doi: 10.1016/j.molcel.2011.02.020
10. Sarkisian CJ, Keister BA, Stairs DB, Boxer RB, Moody SE, Chodosh LA. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat Cell Biol. (2007) 9:493–505. doi: 10.1038/ncb1567
11. Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. (2011) 479:547–51. doi: 10.1038/nature10599
12. Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci USA. (2001) 98:12072–7. doi: 10.1073/pnas.211053698
13. Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. (2008) 6:2853–68. doi: 10.1371/journal.pbio.0060301
14. Haferkamp S, Drexler K, Federlin M, Schlitt HJ, Berneburg M, Adamski J, et al. Extracellular citrate fuels cancer cell metabolism and growth. Front Cell Dev Biol. (2020) 8:602476. doi: 10.3389/fcell.2020.602476
15. Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, et al. Senolytics improve physical function and increase lifespan in old age. Nat Med. (2018) 24:1246–56. doi: 10.1038/s41591-018-0092-9
16. Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. (2016) 530:184–9. doi: 10.1038/nature16932
17. Demaria M, O'Leary MN, Chang J, Shao L, Liu S, Alimirah F, et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. (2017) 7:165–76. doi: 10.1158/2159-8290.CD-16-0241
18. Ogrodnik M, Evans SA, Fielder E, Victorelli S, Kruger P, Salmonowicz H, et al. Whole-body senescent cell clearance alleviates age-related brain inflammation and cognitive impairment in mice. Aging Cell. (2021) 20:e13296. doi: 10.1111/acel.13296
19. James EL, Michalek RD, Pitiyage GN, de Castro AM, Vignola KS, Jones J, et al. Senescent human fibroblasts show increased glycolysis and redox homeostasis with extracellular metabolomes that overlap with those of irreparable DNA damage, aging, and disease. J Proteome Res. (2015) 14:1854–71. doi: 10.1021/pr501221g
20. Wiley CD, Flynn JM, Morrissey C, Lebofsky R, Shuga J, Dong X, et al. Analysis of individual cells identifies cell-to-cell variability following induction of cellular senescence. Aging Cell. (2017) 16:1043–50. doi: 10.1111/acel.12632
21. Yousefzadeh MJ, Schafer MJ, Noren Hooten N, Atkinson EJ, Evans MK, Baker DJ, et al. Circulating levels of monocyte chemoattractant protein-1 as a potential measure of biological age in mice and frailty in humans. Aging Cell. (2018) 17:e12706. doi: 10.1111/acel.12706
22. Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. (2014) 69(Suppl.1):S4–9. doi: 10.1093/gerona/glu057
23. Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. (2017) 550:402–6. doi: 10.1038/nature24050
24. Schwartz RE, Shokhirev MN, Andrade LR, Gutkind JS, Iglesias-Bartolome R, Shadel GS. Insights into epithelial cell senescence from transcriptome and secretome analysis of human oral keratinocytes. Aging. (2021) 13:4747–77. doi: 10.18632/aging.202658
25. Niklander SE, Crane HL, Darda L, Lambert DW, Hunter KD. The role of icIL-1RA in keratinocyte senescence and development of the senescence-associated secretory phenotype. J Cell Sci. (2021) 134:jcs252080. doi: 10.1242/jcs.252080
26. Karen-Ng LP, James EL, Stephen A, Bennett MH, Mycielska ME, Parkinson EK. The extracellular metabolome stratifies low and high risk potentially premalignant oral keratinocytes and identifies citrate as a potential non-invasive marker of tumour progression. Cancers. (2021) 13:4212. doi: 10.3390/cancers13164212
27. Niklander S, Bandaru D, Lambert DW, Hunter KD. ROCK inhibition modulates the senescence-associated secretory phenotype (SASP) in oral keratinocytes. FEBS Open Bio. (2020) 10:2740–9. doi: 10.1002/2211-5463.13012
28. Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. (2005) 434:864–70. doi: 10.1038/nature03482
29. Natarajan E, Saeb M, Crum CP, Woo SB, McKee PH, Rheinwald JG. Co-expression of p16(INK4A) and laminin 5 gamma2 by microinvasive and superficial squamous cell carcinomas in vivo and by migrating wound and senescent keratinocytes in culture. Am J Pathol. (2003) 163:477–91. doi: 10.1016/S0002-9440(10)63677-2
30. Lee S, Schmitt CA. The dynamic nature of senescence in cancer. Nat Cell Biol. (2019) 21:94–101. doi: 10.1038/s41556-018-0249-2
31. Menni C, Kastenmuller G, Petersen AK, Bell JT, Psatha M, Tsai PC, et al. Metabolomic markers reveal novel pathways of ageing and early development in human populations. Int J Epidemiol. (2013) 42:1111–9. doi: 10.1093/ije/dyt094
32. Birkenfeld AL, Lee HY, Guebre-Egziabher F, Alves TC, Jurczak MJ, Jornayvaz FR, et al. Deletion of the mammalian INDY homolog mimics aspects of dietary restriction and protects against adiposity and insulin resistance in mice. Cell Metab. (2011) 14:184–95. doi: 10.1016/j.cmet.2011.06.009
33. Fan SZ, Sung CW, Tsai YH, Yeh SR, Lin WS, Wang PY. Nervous system deletion of mammalian INDY in mice mimics dietary restriction-induced memory enhancement. J Gerontol A Biol Sci Med Sci. (2021) 76:50–6. doi: 10.1093/gerona/glaa203
34. Willmes DM, Daniels M, Kurzbach A, Lieske S, Bechmann N, Schumann T, et al. The longevity gene mIndy (I'm Not Dead, Yet) affects blood pressure through sympathoadrenal mechanisms. JCI Insight. (2021) 6:e136083. doi: 10.1172/jci.insight.136083
35. Drexler K, Schmidt KM, Jordan K, Federlin M, Milenkovic VM, Liebisch G, et al. Cancer-associated cells release citrate to support tumour metastatic progression. Life Sci Alliance. (2021) 4:e202000903. doi: 10.26508/lsa.202000903
36. Mycielska ME, Dettmer K, Rummele P, Schmidt K, Prehn C, Milenkovic VM, et al. Extracellular citrate affects critical elements of cancer cell metabolism and supports cancer development in vivo. Cancer Res. (2018) 78:2513–23. doi: 10.1158/0008-5472.CAN-17-2959
37. Jacobs JJ, de Lange T. Significant role for p16INK4a in p53-independent telomere-directed senescence. Curr Biol. (2004) 14:2302–8. doi: 10.1016/j.cub.2004.12.025
38. Rheinwald JG, Hahn WC, Ramsey MR, Wu JY, Guo Z, Tsao H, et al. A two-stage, p16(INK4A)- and p53-dependent keratinocyte senescence mechanism that limits replicative potential independent of telomere status. Mol Cell Biol. (2002) 22:5157–72. doi: 10.1128/MCB.22.19.6930.2002-a
39. Munro J, Stott FJ, Vousden KH, Peters G, Parkinson EK. Role of the alternative INK4A proteins in human keratinocyte senescence: evidence for the specific inactivation of p16INK4A upon immortalization. Cancer Res. (1999) 59:2516–21.
40. Natarajan E, Omobono JD 2nd, Guo Z, Hopkinson S, Lazar AJ, Brenn T, et al. A keratinocyte hypermotility/growth-arrest response involving laminin 5 and p16INK4A activated in wound healing and senescence. Am J Pathol. (2006) 168:1821–37. doi: 10.2353/ajpath.2006.051027
41. Barrandon Y, Green H. Three clonal types of keratinocyte with different capacities for multiplication. Proc Natl Acad Sci USA. (1987) 84:2302–6. doi: 10.1073/pnas.84.8.2302
42. Suram A, Kaplunov J, Patel PL, Ruan H, Cerutti A, Boccardi V, et al. Oncogene-induced telomere dysfunction enforces cellular senescence in human cancer precursor lesions. EMBO J. (2012) 31:2839–51. doi: 10.1038/emboj.2012.132
43. Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. (2006) 444:633–7. doi: 10.1038/nature05268
44. Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. (2005) 436:720–4. doi: 10.1038/nature03890
45. Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. (2015) 517:576–82. doi: 10.1038/nature14129
46. Clark LJ, Edington K, Swan IR, McLay KA, Newlands WJ, Wills LC, et al. The absence of Harvey ras mutations during development and progression of squamous cell carcinomas of the head and neck. Br J Cancer. (1993) 68:617–20. doi: 10.1038/bjc.1993.396
47. Gordon KE, Ireland H, Roberts M, Steeghs K, McCaul JA, MacDonald DG, et al. High levels of telomere dysfunction bestow a selective disadvantage during the progression of human oral squamous cell carcinoma. Cancer Res. (2003) 63:458–67.
48. Dickson MA, Hahn WC, Ino Y, Ronfard V, Wu JY, Weinberg RA, et al. Human keratinocytes that express hTERT and also bypass a p16(INK4a)-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol Cell Biol. (2000) 20:1436–47. doi: 10.1128/MCB.20.4.1436-1447.2000
49. Muntoni A, Fleming J, Gordon KE, Hunter K, McGregor F, Parkinson EK, et al. Senescing oral dysplasias are not immortalized by ectopic expression of hTERT alone without other molecular changes, such as loss of INK4A and/or retinoic acid receptor-beta: but p53 mutations are not necessarily required. Oncogene. (2003) 22:7804–8. doi: 10.1038/sj.onc.1207085
50. Parkinson EK. Senescence as a modulator of oral squamous cell carcinoma development. Oral Oncol. (2010) 46:840–53. doi: 10.1016/j.oraloncology.2009.09.009
51. Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA Jr, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci USA. (2013) 110:6021–6. doi: 10.1073/pnas.1303607110
52. Mao L, El-Naggar AK, Fan YH, Lee JS, Lippman SM, Kayser S, et al. Telomerase activity in head and neck squamous cell carcinoma and adjacent tissues. Cancer Res. (1996) 56:5600–4.
53. Chiba K, Lorbeer FK, Shain AH, McSwiggen DT, Schruf E, Oh A, et al. Mutations in the promoter of the telomerase gene TERT contribute to tumorigenesis by a two-step mechanism. Science. (2017) 357:1416–20. doi: 10.1126/science.aao0535
54. Yoshida GJ, Azuma A, Miura Y, Orimo A. Activated fibroblast program orchestrates tumor initiation and progression; molecular mechanisms and the associated therapeutic strategies. Int J Mol Sci. (2019) 20:2256. doi: 10.3390/ijms20092256
55. Barrett R, Pure E. Cancer-associated fibroblasts: key determinants of tumor immunity and immunotherapy. Curr Opin Immunol. (2020) 64:80–7. doi: 10.1016/j.coi.2020.03.004
56. Marsh D, Suchak K, Moutasim KA, Vallath S, Hopper C, Jerjes W, et al. Stromal features are predictive of disease mortality in oral cancer patients. J Pathol. (2011) 223:470–81. doi: 10.1002/path.2830
57. Mellone M, Hanley CJ, Thirdborough S, Mellows T, Garcia E, Woo J, et al. Induction of fibroblast senescence generates a non-fibrogenic myofibroblast phenotype that differentially impacts on cancer prognosis. Aging. (2016) 9:114–32. doi: 10.18632/aging.101127
58. Cirillo N, Hassona Y, Celentano A, Lim KP, Manchella S, Parkinson EK, et al. Cancer-associated fibroblasts regulate keratinocyte cell-cell adhesion via TGF-beta-dependent pathways in genotype-specific oral cancer. Carcinogenesis. (2017) 38:76–85. doi: 10.1093/carcin/bgw113
59. Hassona Y, Cirillo N, Lim KP, Herman A, Mellone M, Thomas GJ, et al. Progression of genotype-specific oral cancer leads to senescence of cancer-associated fibroblasts and is mediated by oxidative stress and TGF-beta. Carcinogenesis. (2013) 34:1286–95. doi: 10.1093/carcin/bgt035
60. Martinez-Outschoorn UE, Lin Z, Trimmer C, Flomenberg N, Wang C, Pavlides S, et al. Cancer cells metabolically “fertilize” the tumor microenvironment with hydrogen peroxide, driving the Warburg effect: implications for PET imaging of human tumors. Cell Cycle. (2011) 10:2504–20. doi: 10.4161/cc.10.15.16585
61. Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell. (2014) 31:722–33. doi: 10.1016/j.devcel.2014.11.012
62. Jun JI, Lau LF. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol. (2010) 12:676–85. doi: 10.1038/ncb2070
63. Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. (2008) 134:657–67. doi: 10.1016/j.cell.2008.09.015
64. Jurk D, Wilson C, Passos JF, Oakley F, Correia-Melo C, Greaves L, et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat Commun. (2014) 2:4172. doi: 10.1038/ncomms5172
65. Munoz DP, Yannone SM, Daemen A, Sun Y, Vakar-Lopez F, Kawahara M, et al. Targetable mechanisms driving immunoevasion of persistent senescent cells link chemotherapy-resistant cancer to aging. JCI Insight. (2019) 5:e124716. doi: 10.1172/jci.insight.124716
66. Kumar S, Suman S, Fornace AJ, Datta K. Intestinal stem cells acquire premature senescence and senescence associated secretory phenotype concurrent with persistent DNA damage after heavy ion radiation in mice. Aging. (2019) 11:4145–58. doi: 10.18632/aging.102043
67. Peng X, Wu Y, Brouwer U, van Vliet T, Wang B, Demaria M, et al. Cellular senescence contributes to radiation-induced hyposalivation by affecting the stem/progenitor cell niche. Cell Death Dis. (2020) 11:854. doi: 10.1038/s41419-020-03074-9
68. He Y, Thummuri D, Zheng G, Okunieff P, Citrin DE, Vujaskovic Z, et al. Cellular senescence and radiation-induced pulmonary fibrosis. Transl Res. (2019) 209:14–21. doi: 10.1016/j.trsl.2019.03.006
69. Wang H, Wang Z, Huang Y, Zhou Y, Sheng X, Jiang Q, et al. Senolytics (DQ) mitigates radiation ulcers by removing senescent cells. Front Oncol. (2019) 9:1576. doi: 10.3389/fonc.2019.01576
70. Ariffin H, Azanan MS, Abd Ghafar SS, Oh L, Lau KH, Thirunavakarasu T, et al. Young adult survivors of childhood acute lymphoblastic leukemia show evidence of chronic inflammation and cellular aging. Cancer. (2017) 123:4207–14. doi: 10.1002/cncr.30857
71. Ness KK, Armstrong GT, Kundu M, Wilson CL, Tchkonia T, Kirkland JL. Frailty in childhood cancer survivors. Cancer. (2015) 121:1540–7. doi: 10.1002/cncr.29211
72. Ness KK, Kirkland JL, Gramatges MM, Wang Z, Kundu M, McCastlain K, et al. Premature physiologic aging as a paradigm for understanding increased risk of adverse health across the lifespan of survivors of childhood cancer. J Clin Oncol. (2018) 36:2206–15. doi: 10.1200/JCO.2017.76.7467
73. Rossi F, Di Paola A, Pota E, Argenziano M, Di Pinto D, Marrapodi MM, et al. Biological aspects of inflamm-aging in childhood cancer survivors. Cancers. (2021) 13:4933. doi: 10.3390/cancers13194933
74. Iglesias-Bartolome R, Patel V, Cotrim A, Leelahavanichkul K, Molinolo AA, Mitchell JB, et al. mTOR inhibition prevents epithelial stem cell senescence and protects from radiation-induced mucositis. Cell Stem Cell. (2012) 11:401–14. doi: 10.1016/j.stem.2012.06.007
75. Sharma M, Fonseca FP, Hunter KD, Radhakrishnan R. Loss of oral mucosal stem cell markers in oral submucous fibrosis and their reactivation in malignant transformation. Int J Oral Sci. (2020) 12:23. doi: 10.1038/s41368-020-00090-5
76. Pitiyage GN, Slijepcevic P, Gabrani A, Chianea YG, Lim KP, Prime SS, et al. Senescent mesenchymal cells accumulate in human fibrosis by a telomere-independent mechanism and ameliorate fibrosis through matrix metalloproteinases. J Pathol. (2011) 223:604–17. doi: 10.1002/path.2839
77. Rehman A, Ali S, Lone MA, Atif M, Hassona Y, Prime SS, et al. Areca nut alkaloids induce irreparable DNA damage and senescence in fibroblasts and may create a favourable environment for tumour progression. J Oral Pathol Med. (2016) 45:365–72. doi: 10.1111/jop.12370
78. Illeperuma RP, Kim DK, Park YJ, Son HK, Kim JY, Kim J, et al. Areca nut exposure increases secretion of tumor-promoting cytokines in gingival fibroblasts that trigger DNA damage in oral keratinocytes. Int J Cancer. (2015) 137:2545–57. doi: 10.1002/ijc.29636
79. Choi H, Kim TH, Jeong JK, Strandgren C, Eriksson M, Cho ES. Expression of the Hutchinson-Gilford Progeria mutation leads to aberrant dentin formation. Sci Rep. (2018) 8:15368. doi: 10.1038/s41598-018-33764-6
80. Ok CY, Park S, Jang HO, Takata T, Bae MK, Kim YD, et al. Visfatin induces senescence of human dental pulp cells. Cells. (2020) 9:193. doi: 10.3390/cells9010193
81. Yang RL, Huang HM, Han CS, Cui SJ, Zhou YK, Zhou YH. Serine metabolism controls dental pulp stem cell aging by regulating the DNA methylation of p16. J Dent Res. (2021) 100:90–7. doi: 10.1177/0022034520958374
82. Aquino-Martinez R, Khosla S, Farr JN, Monroe DG. Periodontal disease and senescent cells: new players for an old oral health problem? Int J Mol Sci. (2020) 21:7441. doi: 10.3390/ijms21207441
83. Wang H, Hu Z, Wu J, Mei Y, Zhang Q, Zhang H, et al. Sirt1 promotes osteogenic differentiation and increases alveolar bone mass via Bmi1 activation in mice. J Bone Miner Res. (2019) 34:1169–81. doi: 10.1002/jbmr.3677
84. Pringle S, Wang X, Verstappen G, Terpstra JH, Zhang CK, He A, et al. Salivary gland stem cells age prematurely in primary Sjogren's syndrome. Arthritis Rheumatol. (2019) 71:133–42. doi: 10.1002/art.40659
85. Holst CR, Nuovo GJ, Esteller M, Chew K, Baylin SB, Herman JG, et al. Methylation of p16(INK4a) promoters occurs in vivo in histologically normal human mammary epithelia. Cancer Res. (2003) 63:1596–601.
86. Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. (2011) 30:1536–48. doi: 10.1038/emboj.2011.69
87. Alimbetov D, Davis T, Brook AJ, Cox LS, Faragher RG, Nurgozhin T, et al. Suppression of the senescence-associated secretory phenotype (SASP) in human fibroblasts using small molecule inhibitors of p38 MAP kinase and MK2. Biogerontology. (2016) 17:305–15. doi: 10.1007/s10522-015-9610-z
88. Xu M, Tchkonia T, Ding H, Ogrodnik M, Lubbers ER, Pirtskhalava T, et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc Natl Acad Sci USA. (2015) 112:E6301–10. doi: 10.1073/pnas.1515386112
89. Laberge RM, Zhou L, Sarantos MR, Rodier F, Freund A, de Keizer PL, et al. Glucocorticoids suppress selected components of the senescence-associated secretory phenotype. Aging Cell. (2012) 11:569–78. doi: 10.1111/j.1474-9726.2012.00818.x
90. Ozcan L, Xu X, Deng SX, Ghorpade DS, Thomas T, Cremers S, et al. Treatment of obese insulin-resistant mice with an allosteric MAPKAPK2/3 inhibitor lowers blood glucose and improves insulin sensitivity. Diabetes. (2015) 64:3396–405. doi: 10.2337/db14-1945
91. Georgilis A, Klotz S, Hanley CJ, Herranz N, Weirich B, Morancho B, et al. PTBP1-mediated alternative splicing regulates the inflammatory secretome and the pro-tumorigenic effects of senescent cells. Cancer Cell. (2018) 34:85–102 e9. doi: 10.1016/j.ccell.2018.06.007
92. Song S, Tchkonia T, Jiang J, Kirkland JL, Sun Y. Targeting senescent cells for a healthier aging: challenges and opportunities. Adv Sci. (2020) 7:2002611. doi: 10.1002/advs.202002611
93. Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM, et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell. (2017) 169:132–47 e16. doi: 10.1016/j.cell.2017.02.031
94. Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashmi SK, et al. Senolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study. EBioMedicine. (2019) 40:554–63. doi: 10.1016/j.ebiom.2018.12.052
95. Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, et al. Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine. (2019) 47:446–56. doi: 10.1016/j.ebiom.2019.08.069
96. Amor C, Feucht J, Leibold J, Ho YJ, Zhu C, Alonso-Curbelo D, et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature. (2020) 583:127–32. doi: 10.1038/s41586-020-2403-9
97. Camell CD, Yousefzadeh MJ, Zhu Y, Prata L, Huggins MA, Pierson M, et al. Senolytics reduce coronavirus-related mortality in old mice. Science. (2021) 373:eabe4832. doi: 10.1126/science.abe4832
98. Carpenter VJ, Saleh T, Gewirtz DA. Senolytics for cancer therapy: is all that glitters really gold? Cancers. (2021) 13:723. doi: 10.3390/cancers13040723
99. Dai DF, Santana LF, Vermulst M, Tomazela DM, Emond MJ, MacCoss MJ, et al. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation. (2009) 119:2789–97. doi: 10.1161/CIRCULATIONAHA.108.822403
100. Lee HY, Lee JS, Alves T, Ladiges W, Rabinovitch PS, Jurczak MJ, et al. Mitochondrial-targeted catalase protects against high-fat diet-induced muscle insulin resistance by decreasing intramuscular lipid accumulation. Diabetes. (2017) 66:2072–81. doi: 10.2337/db16-1334
101. Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. (2005) 308:1909–11. doi: 10.1126/science.1106653
102. Tian X, Firsanov D, Zhang Z, Cheng Y, Luo L, Tombline G, et al. SIRT6 is responsible for more efficient DNA double-strand break repair in long-lived species. Cell. (2019) 177:622–38 e22. doi: 10.1016/j.cell.2019.03.043
103. Bernardes de Jesus B, Schneeberger K, Vera E, Tejera A, Harley CB, Blasco MA. The telomerase activator TA-65 elongates short telomeres and increases health span of adult/old mice without increasing cancer incidence. Aging Cell. (2011) 10:604–21. doi: 10.1111/j.1474-9726.2011.00700.x
104. Bernardes de Jesus B, Vera E, Schneeberger K, Tejera AM, Ayuso E, Bosch F, et al. Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer. EMBO Mol Med. (2012) 4:691–704. doi: 10.1002/emmm.201200245
105. Chole RH, Gondivkar SM, Gadbail AR, Balsaraf S, Chaudhary S, Dhore SV, et al. Review of drug treatment of oral submucous fibrosis. Oral Oncol. (2012) 48:393–8. doi: 10.1016/j.oraloncology.2011.11.021
106. Citrin DE, Prasanna PGS, Walker AJ, Freeman ML, Eke I, Barcellos-Hoff MH, et al. Radiation-induced fibrosis: mechanisms and opportunities to mitigate. Report of an NCI workshop, September 19, 2016. Radiat Res. (2017) 188:1–20. doi: 10.1667/RR14784.1
107. Saleh T, Carpenter VJ. Potential use of senolytics for pharmacological targeting of precancerous lesions. Mol Pharmacol. (2021) 100:580–7. doi: 10.1124/molpharm.121.000361
108. Wissler Gerdes EO, Misra A, Netto JME, Tchkonia T, Kirkland JL. Strategies for late phase preclinical and early clinical trials of senolytics. Mech Ageing Dev. (2021) 200:111591. doi: 10.1016/j.mad.2021.111591
109. Suvakov S, Ghamrawi R, Cubro H, Tu H, White WM, Tobah YSB, et al. Epigenetic and senescence markers indicate an accelerated ageing-like state in women with preeclamptic pregnancies. EBioMedicine. (2021) 70:103536. doi: 10.1016/j.ebiom.2021.103536
Keywords: senescence, oral, cancer, fibrosis, telomerase, inflammation, SASP, metabolism
Citation: Parkinson EK and Prime SS (2022) Oral Senescence: From Molecular Biology to Clinical Research. Front. Dent. Med. 3:822397. doi: 10.3389/fdmed.2022.822397
Received: 25 November 2021; Accepted: 18 January 2022;
Published: 01 April 2022.
Edited by:
Monica Caceres, Universidad de Chile, ChileReviewed by:
Mauricio Garrido, University of Chile, ChileCopyright © 2022 Parkinson and Prime. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eric Kenneth Parkinson, ZS5rLnBhcmtpbnNvbkBxbXVsLmFjLnVr; Stephen Stewart Prime, c3RlcGhlbnNwcmltZUBnbWFpbC5jb20=
†These authors have contributed equally to this work