 Maud Kamal1,2,3
Maud Kamal1,2,3 Ralf Jockers1,2,3*
Ralf Jockers1,2,3*
- 1 Department of Endocrinology, Metabolism and Cancer, INSERM U06, Institut Cochin, Paris, France
- 2 CNRS UMR 8104, Paris, France
- 3 University Paris Descartes, Paris, France
Clustering of proteins in higher order complexes is a common theme in biology and profoundly influences protein function. The idea that seven-transmembrane spanning G protein-coupled receptors (GPCRs) might form dimers or higher order oligomeric complexes has been formulated more than 20 years ago. Since then, this phenomenon has been investigated with many different biochemical and biophysical techniques. The more recent notion of GPCR heteromerization describes the specific association of two different GPCRs. GPCR heteromerization may be of primary importance in neuroendocrinology, as this may explain at least some of the functional crosstalks described between different hormonal systems. Importantly, many GPCR heteromers have distinct functional properties compared to their corresponding homomers. Heteromer-specific pharmacological profiles might be exploited for drug design and open new therapeutic options. GPCR heteromerization has been first studied in heterologous expression systems. Today, increasing evidence for the existence of GPCR heteromers in endogenous systems is emerging providing crucial evidence for the physiological function of GPCR heteromerization.
Introduction
The seven-transmembrane spanning G protein-coupled receptor (GPCR) family represent in humans, with more than 800 members, the largest family of cell surface proteins corresponding to about 3% of all genes. Although generally expressed at relatively low levels, every cell typically expresses at least one and in most cases several GPCRs at their cell surface. Not surprisingly, GPCRs are involved in all major biological processes ranging form cell migration, proliferation, inflammation, immunity, etc… GPCRs respond to a remarkable diversity of ligands ranging from photons, to ions, metabolites, amino acids, lipids, peptides, and proteins. Over the last 20 years, GPCRs have been among the most tractable targets for the development of therapeutic small molecule drugs.
The canonical signaling pathway of GPCRs involves monomeric receptors, heterotrimeric G proteins, and various effector systems. Subsequently, several features have been added to this basic concept. Notably, addition of GPCR interacting proteins other than G proteins opened the possibility of fine-tuning of signaling in a receptor subtype- and tissue-specific manner. Moreover, alternative, G protein-independent, i.e., β-arrestin-dependent, signaling pathways have been described. Further diversity has been introduced by the notion that GPCRs might not only exist as monomeric peptides but can also cluster in dimeric or oligomeric structures. The current review gives a short overview of the proposed functions of GPCR oligomers and discusses the biological relevance and participation of such complexes in functional crosstalk. In the second part, examples of GPCR oligomers in the neuro-endocrine system will be critically assessed.
GPCR Oligomers, what are they Good for?
The concept of GPCR di(oligo)merization has raised considerable interest over the last 20 years. Supporting evidence comes form numerous reports using biochemical, biophysical, and functional assays (see Bouvier, 2001; Angers et al., 2002 for review). Whereas the first pioneering studies considered the clustering of one specific GPCR into homomers, it become rapidly evident that different types of GPCRs might also assemble into heteromers, thus broadening the potential impact of GPCR oligomerization considerably (see Prinster et al., 2005; Milligan, 2009 for review). Having accepted that GPCR dimers exist, the question of the functional significance of these dimers arises.
In the case of obligatory dimers, the functional significance of GPCR dimers is obvious. Indeed, early studies showed that co-expression of GABAB1 and GABAB2 subunits is necessary for the functional expression of the GABAB receptor (Jones et al., 1998; White et al., 1998). Subsequently, it was shown that the GABAB1 subunit binds the natural ligand whereas the GABAB2 subunit is necessary for G protein coupling and cell surface expression of the heteromeric complex. Similar observations have been made for sweet and umami taste receptors (Nelson et al., 2001, 2002). The issue is however more complex for non-obligatory dimers. The major difficulty resides in preparing pure monomeric and dimeric GPCR samples rather than measuring their biological function. An elegant way to determine the function of monomeric GPCRs was achieved by the reconstitution of purified β2AR in high-density lipoprotein phospholipid bilayer particles. Importantly, the geometry of these particles allows only integration of monomeric receptor protomers. Addition of purified heterotrimeric G proteins showed that G protein activation was as efficient as in intact cells (Whorton et al., 2007). Similar conclusions were drawn by two further studies using rhodopsin as model receptor (Bayburt et al., 2007; Ernst et al., 2007). These studies show that monomeric GPCRs are sufficient for G protein activation leaving the question of the function of GPCR dimers unanswered.
The role of GPCR dimer formation is clear in non-obligatory heteromers with distinct functional properties (Prinster et al., 2005; Milligan, 2009). GPCR heteromerization between orphan GPCRs for which no natural ligand is known and non-orphan GPCRs with known ligand is very interesting in the sense that it might define the function of orphan GPCRs. The existence of ligand-independent functions of some of the more than 100 still orphan GPCRs has indeed been determined in several heteromers containing non-orphan GPCRs (Levoye et al., 2006).
A number of studies have suggested that GPCR oligomerization may be important in cell surface delivery implying that oligomerization occurs early in the biosynthetic pathway (Bulenger et al., 2005). According to this model, successful oligomerization is part of the quality control process. A prominent example is the GABAB receptors, where the GABAB2 subunit controls the proper cell surface expression of GABAB1.
Formation of functional dimers offers the possibility for allosteric regulation of one protomer by the other. Several reports confirmed indeed the existence of allosteric interactions between the two orthosteric binding sites within a dimer. Negative and positive allosteric effects have been reported (Durroux, 2005).
Allosteric interactions between the protomers of GPCR dimers imply asymmetry within the GPCR cluster. The concept of asymmetric dimers on the level of the ligand binding site can be further extended toward G protein coupling or binding of GPCR interacting proteins (GIPs) in general. Assymetric binding of G proteins to only one of the two protomers was shown for several GPCRs. In the case of GABAB receptors, ligand binding to the GB1 subunit trans-activates the GB2 subunit that binds to the G proteins (Galvez et al., 2001). In contrast, in the leukotriene B4 BLT1 receptor the same protomer binds the ligand and activates the G protein in a cis manner, therefore questioning the function of the second protomer (Baneres and Parello, 2003). Recent studies on the melatonin MT1 receptor complex with Gi proteins and the regulator of G protein (RGS) 20 suggest that one protomer binds to the G protein whereas the second protomer binds to other GIPs, RGS20 in the present case. These results provide an additional justification for dimer formations and for the fine-tuning of GPCR signaling through GIPs.
Functional Crosstalk between GPCRs with or without Dimerization
Signal integration between different hormones is a common feature of the endocrine system. Indeed, each cell expresses typically several GPCR subtypes that integrate different hormonal signals in time and space to produce an adequate cellular response. The ability of one hormone, acting on a specific GPCR, to modulate the function of a second GPCR does not inherently imply heteromerization between these two GPCRs. Indeed receptor heteromerization represent only one out of several ways of functional crosstalk at the cellular level (Figure 1). Heterologous sensitization and desensitization, i.e., through activation of protein kinase A or C, are often involved in the integration of cellular signals (Jockers et al., 1998). Crosstalk may also occur at receptor-proximal levels through competition for a common and limited G protein pool as indicated in a recent study on the virally encoded BILF1 receptor (Nijmeijer et al., 2010). Clustering of this Gi-coupled constitutively active receptor with either the chemokine CXCR4 or the histamine H4 receptor appears to sequester Gi proteins away from the two latter receptors impairing their Gi-dependent signal transduction. Furthermore, potentiation of cellular signals at the level of Gβγ subunits has been reported in a number of studies (see Prezeau et al., 2010 for review). Stimulation of Gi-coupled GABAB receptor is known to potentiate Ca2+ signaling of the Gq-coupled mGlu1a receptor, in cortical neurons co-expressing both receptors (Hirono et al., 2001). Studies in transfected HEK293 cells confirmed this functional crosstalk and indicated that Gβγ subunits, liberated upon activation of GABAB receptor, are responsible for the Ca2+ potentiation in the absence of any evidence of receptor heteromerization (Rives et al., 2009). Importantly, similar potentiating effects can be seen between other Gi- and Gq-coupled GPCRs suggesting a general mechanism of signal integration.
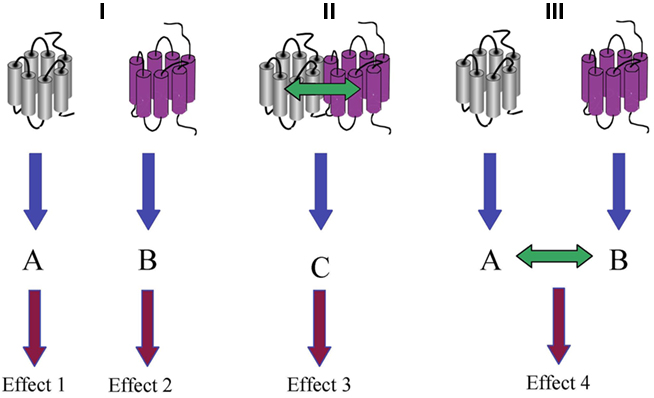
Figure 1. Functional versus physical interaction of GPCRs. (I) In the absence of any physical or functional interaction, the activation of two different GPCRs induces two independent signaling pathways A and B, which consequently result in two independent effects 1 and 2. (II) Physical interaction of two different GPCRs results in the activation of the heteromer-specific signaling pathway C that will be responsible of a downstream effect 3. (III) In the absence of physical interaction, a functional interaction can be observed when signaling pathways A and B crosstalk to produce effect 4.
A further mechanism of cellular crosstalk was recently suggested for the corticotropin-releasing factor receptor 1 (CRF1) and serotonin 5-HT2 receptors (Magalhaes et al., 2010) Cellular 5-HT2 responses and serotonin-dependent anxiety were sensitized by CRF stimulation. Sensitization of 5-HT2 responses correlated with increased cell surface expression of 5-HT2 receptors, which are known to constitutively internalize. Interestingly this effect was dependent on the presence of functional PDZ domain binding motifs of the CRF1 and 5-HT2 receptors and intact endocytotic and recycling pathways suggesting a putative scaffolding function of multi-PDZ domain-containing proteins to facilitate recycling of 5-HT2 and CRF1 receptors.
Crosstalk at the level of GPCR oligomers has been demonstrated in several cases. For example, discovery of heteromer-specific signaling pathways (Rashid et al., 2007) and heteromer-selective ligands strongly suggest the importance of opioid receptor (OR) heteromers in physiology (Waldhoer et al., 2005). Unique heteromer-specific cellular responses have been also associated with the 5-HT2A/mGlu2 heteromer, which has been implicated in psychosis (Gonzalez-Maeso et al., 2008). By constructing chimeric receptors between mGlu2, which interacts with 5-HT2A receptors and mGlu3, which does not interact with 5-HT2A, the authors convincingly showed that the cellular responses were exclusively dependent on the formation of 5-HT2A/mGlu2 heteromers.
Collectively, these data show that functional crosstalk may occur at different levels of cellular signaling and trafficking that may or may not depend on GPCR heteromerization. The examples presented in the chapter “Examples in the neuro-endocrine system” will be analyzed in respect of the molecular level of crosstalk.
Biological Significance of GPCR Heteromerization – Criteria
Evidence for the formation of GPCR heteromers is mostly based on observations made in transfected cell lines raising the question of the biological significance of these complexes. Nevertheless, an increasing number of articles provide evidence of GPCR heteromerization in endogenous tissues, some of which will be discussed in the next chapter. To provide some guidance in defining the physiological relevance of GPCR heterodimers, the International Union of Basic and Clinical Pharmacology (IUPHAR) released some recommendations in 2007 (Pin et al., 2007). The first criterion concerned the evidence for physical interaction (or proximity) of both receptors in native tissues or primary cells. This may be achieved by performing co-immunoprecipitation experiments with selective antibodies or energy transfer techniques between labeled ligands or antibodies. The second criterion requests evidence for heteromer-specific properties such as heteromer-specific signaling properties, the identification of heteromer-selective ligands and the presence of allosteric ligand binding properties. The third criterion recommends supporting evidence from knockout animals or RNAi technologies to further validate the possible functional role of heteromers in vivo. Obviously, none of these criteria alone provides conclusive evidence for the existence and functional significance of GPCR heteromers. The comity therefore proposes that at least two of the three criteria have to be fulfilled for the acceptance of GPCR heteromers.
Examples in the Neuro-Endocrine System
G protein-coupled receptor oligomerization has become an extensively studied field. The current chapter will focus on the description of GPCR heteromers relevant in neuroendocrinology. A more extensive list of GPCR heteromers is provided in Table 1. Complementary information on GPCR heteromerization may be also found in other recent expert reviews (Birdsall, 2010; Ferre et al., 2010; Rozenfeld and Devi, 2010).
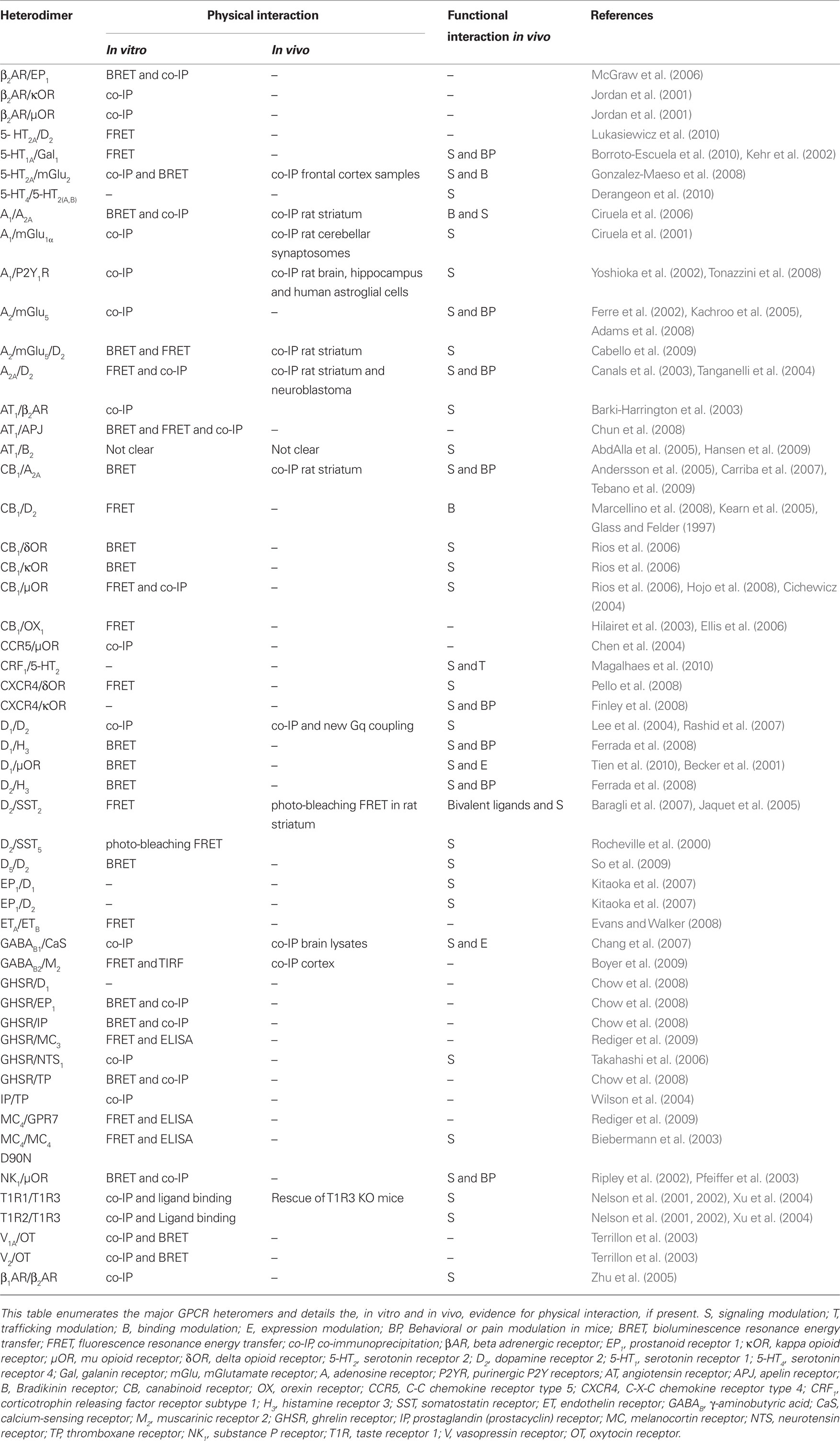
Table 1. Non-exhaustive list of GPCR heteromers in the neuro-endocrine system.
GPCR Heteromers in the Central Nervous System
G protein-coupled receptors are widely distributed in the nervous system, and mediate key physiological processes including cognition, mood, appetite, pain, and synaptic transmission. The important role of GPCRs in central nervous system (CNS) disorders is outlined by the great quest of pharmaceutical companies for drug discovery. Several GPCRs, in particular the metabotropic glutamate, adenosine, dopamine as well as serotonin receptor, and cannabinoid receptors, were shown to be implicated in CNS disorders such as Parkinson, Alzheimer, schizophrenia, depression, and psychosis. Despite the lack of direct evidence for the implication of GPCR heteromers in neuropathologies, several examples in the literature show the importance of heteromerization in receptor functions suggesting that alterations in GPCR crosstalk can directly impact health.
Opioid and Cannabinoid receptor heteromers
Opioid receptors bind endogenous peptides with effects resembling those of opiate drugs and are very well known for their roles in pain, analgesia and reward. Heteromerization between the different OR subtypes was extensively studied over the last 20 years (see van Rijn et al., 2010 for review) with some evidence in vivo. For example, the δ/κ OR selective ligand, the 6′-guanidinonaltrindole (6′-GNTI), is an analgesic and has the unique property of preferentially activating OR heterodimers over the corresponding homomers, indicating that OR heteromers are functionally relevant in vivo (Waldhoer et al., 2005). In the current review we will address only examples of OR heteromerization with other GPCRs.
Opiates and exogenous cannabinoids are analgesics used for the treatment of patients with neuropathic pain. Considerable behavioral, anatomical, and biochemical evidence describe similarities between the OR and the cannabinoid receptor systems. The first study to suggest direct physical interactions between the cannabinoid CB1 receptor and κOR and δOR (CB1/κOR and CB1/δOR) was conducted in transfected cells by Rios et al. (2006). The authors identified a reciprocal antagonistic relationship between the two receptors in transfected cells and in native tissues exposed to increasing concentrations of an agonist for one receptor along with non-activating concentrations of an agonist for the other receptor. Another study showed, in 2008, a direct interaction between CB1 and μOR using FRET and co-immunoprecipitation experiments in CHO cells exogenously expressing both receptors (Hojo et al., 2008). In vivo, functional interaction of these receptors is very well exemplified by 9-tetrahydrocannabinol, the major psychoactive constituent of marijuana, which enhances the potency of opioids such as morphine in animal models (Cichewicz, 2004). Moreover, anatomical studies confirmed the co-localization of μOR and CB1 in common somatodendritic compartments of catecholaminergic neurons in the locus coeruleus, the center of opiate addiction and withdrawal, and also revealed CB1-positive axon terminals forming synaptic contact with μOR-containing dendrites (Scavone et al., 2010).
Recently, ORs were shown to interact functionally with chemokine receptors (Salanga et al., 2009). CXCR4 is able to cross-desensitize the chemotaxis and calcium mobilization response as well as analgesia associated with κOR. Activation of CXCR4 with CXCL12 induces desensitization of κOR and this leads to an impairment of analgesic activity in vivo (Finley et al., 2008). Using FRET, CXCR4 was shown to form heteromeric complexes with δOR. Intriguingly, simultaneous application of δOR and CXCR4 agonists to human monocytes (MM-1 cells) expressing both receptors endogenously, abolished functional effects observed upon treatment of these cells with either agonist alone (Pello et al., 2008).
CCR5 was reported to interact physically, in vitro, with μOR using co-immunoprecipitation assays inducing cross desensitization of the receptor without modulation of receptor internalization. DAMGO (μOR agonist) and RANTES induced chemotaxis in CHO cells co-expressing both receptors. Preincubation with either DAMGO or RANTES inhibited chemotaxis caused by the other ligand. Moreover, DAMGO pretreatment enhanced phosphorylation of CCR5 and reduced RANTES-promoted GTPγS binding suggesting heterologous desensitization (Chen et al., 2004).
Several different GPCRs are implicated in the regulation of striatal functions. Notably, in vivo functional crosstalks have been reported between dopamine and ORs. As an example, methamphetamine-induced zif268 (transcription factor also known as Egr1, Early Growth Response Protein 1) expression was abolished in μOR-knockout mice in which dopamine receptors were blocked by haloperidol. However, the expression of zif268 mRNA was not altered in μOR knockout mice without blockade of dopamine receptors or in wild-type mice with blockade of dopamine receptors (Tien et al., 2010). These results suggest a crosstalk between the μ-opioid system and the dopamine system in the modulation of the expression of zif268 induced by methamphetamine. D1 knockout mice were shown to have a reduced μOR expression in striatal patches without alterations in κOR and δOR expression patterns (Becker et al., 2001). Co-localization of these receptors was shown in rat striatal and cortical neurons, however direct interactions were only reported in transfected cells using the BRET assay (Juhasz et al., 2008).
Another example is the μOR and the substance P (NK1) receptor, which is highly expressed in brain regions implicated in depression, anxiety, and stress. They are also present in the nucleus accumbens, which mediates the motivational properties of drugs of abuse including opioids. The rewarding effects of morphine are absent in mice lacking NK1 suggesting that these receptors are critical for the reinforcing properties of morphine, and for adaptive responses elicited by repeated opiate administration (Ripley et al., 2002). Direct interaction between NK1 and μOR was reported in vitro and functional characterization of cells expressing both receptors revealed cross-modulation of receptor internalization and desensitization (Pfeiffer et al., 2003).
Cannabinoid receptors family remain one of the most important GPCR drug discovery targets due to the intense interest in CB1 receptor antagonists for treating obesity and the metabolic syndrome. Heteromerization of CB1 in the context of metabolism will be discussed later. The literature reports many examples of functional interaction between cannabinoid and dopamine receptors. Functional antagonism was shown in rat brain where the CB1 agonist CP55,940 reduced the affinity of D2 agonist binding sites in the dorsal and ventral striatum. Similarly, in vivo administration of CP55,940 inhibited D2-promoted locomotor activity (Marcellino et al., 2008). In vitro, heteromer formation was demonstrated using FRET (Marcellino et al., 2008). Co-expression of both receptors in HEK293 cells showed that co-stimulation of D2 and CB1 results in a switch of G protein coupling preference of these Gi/o-coupled receptors toward the pertussis toxin-insensitive Gαs protein (Kearn et al., 2005). Similar observations have been reported in primary striatal neuron cultures (Glass and Felder, 1997) supporting the physiological relevance of D2/CB1 heteromer formation.
In the last few years, it has been suggested that A2A receptors could exert a permissive role on CB1 function in a way that a basal adenosinergic tone would be important for cannabinoid-mediated effects. Specifically, it has been demonstrated that the ability of a CB1 agonist to inhibit motor activity requires the presence and the activation of A2A (Andersson et al., 2005; Carriba et al., 2007). The activation state of A2A seems also to regulate synaptic effects of CB1, which is partly dependent on mGlu5 (Tebano et al., 2009). Heteromer formation between CB1 and A2A was shown in vitro, in cells exogenously expressing both receptors using BRET and, in vivo, by immunoprecipitation assays in rat striatum suggesting that heteromer formation may participate in the functional crosstalk between these two receptors (Carriba et al., 2007).
Adenosine, Dopamine, and Glutamate receptor heteromers
Adenosine activates four GPCRs, A1, A2A, A2B, and A3, which are widespread throughout the body. They are involved in a variety of physiological processes and pathologies including neurological, cardiovascular, inflammatory diseases, and cancer (see Trincavelli et al., 2010 for review).
Heteromerization between the different adenosine receptor subtypes is exemplified by the A1/A2A heteromer, which was reported in vitro and in vivo in 2006 by Ciruela et al. (2006). Immunogold detection and co-immunoprecipitation experiments confirmed the heteromer formation in rat striatal glutamatergic nerve terminals. The authors showed that A2A activation reduces the affinity of A1 for agonists, which might explain the stimulatory effect of adenosine on glutamatergic neurotransmission at higher ligand concentrations. Moreover, the authors speculate about a possible role of A1/A2A heteromers in the tolerance to the psychostimulant effects of caffeine (Ciruela et al., 2006).
Using co-immunoprecipitation assays Ciruela et al. (2001) showed a subtype-specific interaction between mGlu1α and A1 receptors in both rat cerebellar synaptosomes and co-transfected HEK293 cells. This heteromer was reported to be implicated in neuroprotection suggesting a potential role in neurodegenerative diseases such as Alzheimer. Importantly, the timing of mGlu1α and A1 receptor activation is very important to achieve a maximal effect in adenosine- and glutamate-mediated neuroprotection/neurodegeneration (Ciruela et al., 2001).
Purinergic P2Y receptors are involved in neuromodulation and neuron–glia interactions and respond to a wider range of agonists, including di- or tri-phosphates of nucleosides, and uridine diphosphate-glucose. The P2Y1 subtype has been reported to form heteromers with the A1 receptor in vivo as determined by co-localization and co-immunoprecipitation experiments in rat brain (Yoshioka et al., 2002), rat hippocampus (Tonazzini et al., 2007), and in human astroglial cells (Tonazzini et al., 2008). P2Y1 stimulation impaired the potency of A1 coupling to G proteins, whereas the stimulation of A1 increased the functional responsiveness of P2Y1, indicating a functional interaction between these receptors in rat hippocampus (Tonazzini et al., 2007).
Several reports highlight the physical and functional interaction between A2 and D2 receptors. BRET and FRET assays as well as co-immunoprecipitation in neuroblastoma cells and striatum indicate formation of A2/D2 heteromers (Canals et al., 2003). It was also reported that A2A receptor antagonists increase locomotion in reserpinized (dopamine-depleted) mice and produce contralateral rotational behavior in rats only after the treatment with sub-threshold doses of the D2-like receptor agonist quinpirole (Tanganelli et al., 2004).
A physical interaction between A2 and mGlu5 was shown in vitro by co-immunoprecipitation. Simultaneous stimulation of cells co-expressing both receptors led to a synergistic increase in ERK phosphorylation (Ferre et al., 2002). In vivo, the anti-Parkinsonian effects of mGlu5 antagonists, known to increase motor control, were potentiated by an A2A antagonist confirming a functional interaction between these two receptors (Kachroo et al., 2005). A combination of a sub-threshold dose of SCH 58261 (A2A antagonist) with a sub-threshold dose of MTEP (mGlu5 antagonist) reduced alcohol self-administration (Adams et al., 2008). In addition, A2A and mGlu5 seem to form higher order oligomeric complexes with D2. Cabello et al. (2009) were able to show in vitro using BRET and FRET assays as well as co-immunoprecipitation the presence of the latter three receptors in the same complex. Similar results were obtained in rat striatum homogenates (Cabello et al., 2009).
Dopamine is a neurotransmitter in many brain regions, and regarding Parkinson disease – the most important is the striatum. In addition, relevant studies regarding receptor oligomerization are described in nucleus accumbens, i.e., a nucleus of the striatum. Dopamine binds and activates five types of receptors, D1, D2, D3, D4, and D5. The literature reports many examples of dopamine subtype heteromerization and postulate their implication in neuropathologies such as Parkinson and dyskinesia (Missale et al., 2010). D1/D2 heteromers are convincingly demonstrated in vivo by co-immunoprecipitation (Lee et al., 2004). Whereas D1 and D2 homomers are coupled to Gs and Gi proteins, respectively, D1/D2 heteromers are coupled to Gq/11 when both protomers are activated. Activation of this heteromer-specific Gq/11 pathway increases levels of calcium/calmodulin-dependent protein kinase IIα in the nucleus accumbens, unlike activation of the Gs-coupled D1 receptors, indicating a mechanism by which D1/D2 may contribute to synaptic plasticity (Rashid et al., 2007).
Recently, D1 and D2 were reported to form heteromeric complexes with histamine H3 (but not H4) receptors in vitro using BRET. Functional interaction in mice is suggested in light of the important role of postsynaptic H3 receptors in the modulation of dopaminergic transmission by means of a negative modulation of D2 receptor function. Indeed, selective H3 receptor agonists inhibit D1- and D2-promoted locomotor activity and the H3 receptor antagonist thioperamide has a potentiating effect. Moreover, radioligand binding experiments in striatal membrane preparations showed that activation of H3 decreased the binding affinity of D2 (Ferrada et al., 2008).
Pituitary tumors express both somatostatin and dopamine receptors (Colao et al., 2007). D2 and somatostatin SST2 receptors are co-localized in three different brain regions including the cerebral cortex, striatum, and substantia nigra. In vivo, D2/SST2 heteromer formation was confirmed in rat striatal neurons using photo-bleaching FRET (Baragli et al., 2007). Another evidence supporting D2/SST2 formation comes from the high efficacy of bivalent ligands, designed to target somatostatin and dopamine receptors, on the inhibition of growth hormone (GH) and prolactin secretion from GH-secreting pituitary adenomas, which are partially responsive to somatostatin analog therapy (Jaquet et al., 2005).
The literature reports several studies pinpointing a functional crosstalk between the prostanoid receptor EP1 as well as D1 and D2 receptors without evidences for physical interactions. EP1 is expressed in more than one-half of medium spiny neurons in the striatum, and its ligand, prostaglandin E2 (PGE2) is produced in the striatum in response to stimulation with a D1 or D2 agonist. The PGE2–EP1 pathway facilitates both D1 and D2 signaling as indicated by the Thr34 phosphorylation of the dopamine- and cAMP-Regulated PhosphoProtein 32 (DARPP-32) in vitro in striatal slices. EP1-knock out mice exhibit a significant suppression of hyperlocomotion induced by cocaine or SKF81297, a D1 agonist, and significant attenuation of catalepsy induced by raclopride, a D2 antagonist (Kitaoka et al., 2007).
GABA receptor heteromers
GABAB are critical receptors in the CNS, where their function is mainly the suppression of neuronal activity. The GABAB1/GABAB2 heteromer, is one of the best characterized examples of GPCR heteromerization as detailed above. There is also some evidence for the formation of heteromers between GABAB1 and the calcium-sensing receptor (CaS) in vitro and in hippocampal neurons. CaS receptors control systemic calcium balance via the regulation of parathyroid hormone secretion and renal calcium excretion. Heteromer formation seems to modulate the expression of CaS as expression of the CaS is increased in brain lysates of GABAB1 knockout mice and in cultured hippocampal neurons (Chang et al., 2007).
GABAB2 was recently shown to interact functionally and physically with muscarinic M2 receptors. In vitro, a combination of FRET and TIRF microscopy detected the heteromers at the cell surface. GABAB2 and M2 are both localized in the somatosensory cortex and are shown to interact in vivo by co-immunoprecipitation. In PC12 cells, GABAB2 expression rescues M2 surface expression, M2-induced activation of GIRKs, and inhibition of cAMP production (Boyer et al., 2009).
Serotonin receptor heteromers
Serotonin (5-HT) receptors represent one of the largest subfamilies of GPCRs. There are 14 different 5-HT receptor subtypes widely distributed throughout the peripheral and CNS, representing therapeutic targets for drugs used to treat anxiety, depression, schizophrenia, obesity, and other disorders (Millan et al., 2008).
Alterations in either serotonin or dopamine neurotransmission seems to be implicated in many human neurological and psychiatric disorders, including depression, anxiety, and schizophrenia (de Almeida et al., 2008). Recently, formation of 5-HT2A/D2 heteromers was reported in vitro using FRET but the functional properties of this complex are not known. Nevertheless, 5-HT2A and D2 receptors are co-localized in the medial prefrontal cortex as well as in the pars reticulate of the substantia nigra in rat, suggesting, at least, the possibility of functional crosstalk between these two receptors in vivo (Lukasiewicz et al., 2010).
The 5-HT2A/mGlu2 heteromer plays a role in anxiety and constitutes one of the most interesting examples of GPCR heteromerization reported so far. Compelling pharmacological and biochemical evidence in vivo confirmed the physical interaction between 5-HT2A and mGlu2. Moreover, the metabotropic glutamate agonist, LY379268 increases the affinity of different hallucinogens for the 5-HT2A receptor-binding site but also, a selective 5-HT2A receptor agonist decreases the affinity of different agonists for the mGlu2 receptor-binding site (Gonzalez-Maeso et al., 2008).
Galanin is co-expressed with and modulates serotonin and noradrenaline release, two neurotransmitters implicated in depression. Stimulation of galanin GAL1 and/or GAL3 receptors results in a depression-like phenotype, while activation of the GAL2 receptor attenuates depression-like behavior (Kuteeva et al., 2008). 5-HT1A and GAL1 interact with each other in vitro as demonstrated by FRET. After simultaneous activation of both receptors, Gi and MAPK signaling did not show any additive or synergistic effects indicating that activation of each receptor alone is sufficient to reach maximal signaling capacity for these specific pathways (Borroto-Escuela et al., 2010). However, an antagonistic effect of GAL1 activation on 5-HT1A-induced hippocampal serotonin release and hypothermia and locomotor activity were shown in the rat brain (Kehr et al., 2002).
Another example of functional crosstalk with no evidence, at least for now, of physical interaction has been described for CRF1 and 5HT2 receptors involved in anxiety as detailed above (Magalhaes et al., 2010).
GPCR Heteromers in the Cardiovascular–Renal System
The most commonly studied and clinically targeted cardiac GPCRs include adrenergic, angiotensin, endothelin, and adenosine receptors. Drugs, targeting adrenergic, and angiotensin receptor signaling pathways alone, account for the majority of prescriptions for cardiovascular diseases (Salazar et al., 2007).
Angiotensin II (AngII) causes vasoconstriction both by a direct action on smooth muscle cells and, indirectly, through the facilitation of noradrenaline release from postganglionic sympathetic neurons. AngII binds to two main receptors, AT1 and AT2. Via stimulation of AT1 receptors, AngII causes virtually all of its physiological actions, namely the cardiovascular, neuronal, renal, endocrine, and hepatic effect see (Guimaraes and Pinheiro, 2005) for review. Bradykinin (BK), an endogenous vasoactive peptide mainly produced from plasma with a physiological role in inflammation and nociception, binds two main receptors, B1 and B2 (Fincham et al., 2009). AbdAlla et al. (2000) reported functional and physical interaction between AT1 and B2 receptors with major implications in pre-eclampsia and hypertensions. In the cortical thick ascending limb in the kidney, BK exerts negative modulatory effects on AngII-induced calcium responses dependent on tyrosine kinase and MAPK pathways. In the presence of BK AngII-induced sodium transport is suppressed (Hus-Citharel et al., 2010). The AT1/B2 heteromer has also been suggested to contribute to AngII hyper-responsiveness of mesangial cells in experimental hypertension (AbdAlla et al., 2005). However, the existence of AT1/B2 heteromers has been questioned by several groups suggesting that the documented in vivo crosstalk between these two hormones is most likely based on a functional rather than physical interactions (Hansen et al., 2009).
AT1/β2AR heteromers were described in mouse cardiac myocytes transfected with the corresponding cDNAs. The main functional consequence of this interaction is the enhancement of agonist-induced β2AR signaling and the silencing of the spontaneous activation of β2AR, suggesting that heteromerization mutually stabilizes both receptor subtypes in their respective inactive conformations in the absence of agonist. In vivo, selective blockade of β2AR in mouse cardiomyocytes inhibits angiotensin-induced contractility and administration of the AT1 antagonist valsartan reduces the maximal response to catecholamine-induced elevation of heart rate (Barki-Harrington et al., 2003), further supporting the physiological relevance of heteromerization.
Another heteromeric partner described for AT1 is the apelin receptor (APJ). Apelin seems to be an important factor in the regulation of vascular tone and cardiovascular function (Kalea and Batlle, 2010). AT1 was shown to physically interact with APJ in vitro by BRET, FRET, and co-immunoprecipitation assays. Whereas in vivo evidence for AT1/APJ heteromer formation is still lacking, many examples support an in vivo functional crosstalk. In apelin knockout mice, exogenous Ang II induces atherosclerosis and abdominal aortic aneurysm formation while co-infusion of apelin abrogated these effects. Moreover, in a vein graft mode, apelin treatment rescued AngII-mediated increase in neointimal formation and vascular remodeling (Chun et al., 2008).
Adrenergic receptors are responsible for translating chemical messages from the sympathetic nervous system into cardiovascular responses. Three β-adrenergic receptor subtypes are found in the mammalian heart β1, β2, and β3-AR (Salazar et al., 2007). Heteromerization between the different subtypes has been described and is implicated in many cardiovascular functions. For example, alterations in receptor pharmacology and function were seen in intact adult mouse cardiomyocytes lacking β1AR and β2AR, where heteromerization reduced spontaneous receptor activity and enhanced responsiveness to catecholamines (Zhu et al., 2005).
Another key player in the regulation of the cardiovascular systems are endothelin receptors. Endothelins are composed of a family of 21 amino acid peptides (ET-1, ET-2, and ET-3). ET-1 is the predominant endothelin in the cardiovascular system binding to endothelin A (ETA) and B (ETB) receptors (Sakurai et al., 1990). ETA and ETB have been reported to form heteromers, which induce sustained calcium signaling as compared to the corresponding homomers, known to induce a more transient calcium release in transfected HEK293 cells (Evans and Walker, 2008).
Serotonin was found in multiple cardiovascular tissues, including heart, blood vessels, brain, platelets, adrenal gland, and kidney. 5-HT4 (mainly 5-HT4b), 5-HT2A, and 5-HT2B receptors coexist in auricular myocytes of newborn rats. 5-HT4 and 5-HT2 receptors appear to regulate intercellular communication at rat cardiac myocyte gap junctions. Unexpectedly, 5-HT4 activation reduces cAMP, L-type calcium currents, and intercellular coupling while 5-HT2A or 5-HT2B activation enhances gap junctional intercellular communication (Derangeon et al., 2010). These results suggest receptor heteromerization, which are still to be assessed.
Prostacyclin (PGI2) and Thromboxane A2 (TxA2) are biological opposites: PGI2 is a vasodilator and inhibitor of platelet aggregation, which limits the deleterious actions of TxA2, a vasoconstrictor, and platelet activator. The molecular mechanisms involved in the counter-regulation of PGI2 and TxA2 promoted signaling are unclear. Physical and functional interaction between the PGI2 receptor (IP1) and TxA2 receptor (TP) were reported in vitro in HEK293 cells. IP1 seems to facilitate TP-mediated generation of cAMP in a manner independent of IP1-induced cAMP formation (Wilson et al., 2004).
Opioid receptors and β2ARs have been shown to coexist and functionally interact in cardiac myocyte sarcolemma. Low doses of selective opioids are known to inhibit norepinephrine-mediated functions (Pepe et al., 1997). Physical interaction was reported by co-immunoprecipitation studies in CHO cells exogenously expressing both receptors. In vitro, heteromerization results in an alteration of trafficking properties with no significant alterations in ligand binding. Moreover, opioid-selective agonists alter β2AR internalization, thus providing a possible molecular mechanism for the lack of β2AR agonist-mediated effects after OR activation (Jordan et al., 2001).
Prostanoids EP1 receptors and β2AR form heteromers in vitro (BRET, co-immunoprecipitation). EP1 and β2AR colocalize in airway smooth muscle cells, where activation of EP1 was reported to reduce β2AR-stimulated cAMP without affecting β2AR phosphorylation or trafficking. EP1/β2AR heteromers might be implicated in asthma (McGraw et al., 2006).
P2Y receptors play various physiological roles, including mediation of vasodilatation in the endothelium, vasoconstriction of smooth muscles, and control of mitogenic actions and secretory processes. A2A/P2Y1 and A2A/P2Y12 heteromers were detected in human platelet membranes by co-immunoprecipitation techniques, and seem to have a crucial role in the physiology of platelet aggregation (Nakata et al., 2010).
GPCR Heteromers in Taste
Taste, or gustation, is mediated by a specialized anatomically and physiologically defined chemosensory gustatory system. The gustatory system in mammals includes taste receptor cells (TRC) organized in taste buds located within the gustatory papillae, located mainly, but not exclusively in the tongue. Reception of taste qualities that humans describe as sweet, umami, and bitter involves GPCRs from the T1R and T2R subfamilies (Bachmanov and Beauchamp, 2007). Whereas T1R are typical class C GPCRs with a large N-terminal “Venus fly trap” (VFT) domain, T2R belong to rhodopsin-like class A GPCRs with small N-terminal domains. The T1R family is composed of three subtypes T1R1, T1R2, and T1R3 that have been shown to form obligatory T1R1/T1R3 and T1R2/T1R3 heteromers (Nelson et al., 2001, 2002). Binding of sweet stimuli such as sucrose or aspartame to the VFT of T1R2 and umami taste stimuli such as l-glutamate to the orthosteric binding site of the VFT of T1R1 activate T1R2/T1R3 and T1R1/T1R3 heterodimers, respectively (Nelson et al., 2001, 2002; Xu et al., 2004). Several molecules binding to allosteric binding site within the VFT domain or the TM domain of taste receptors have been characterized.
For example, IMP and GMP bind near the opening of the orthosteric glutamate binding site of T1R1 thus further stabilize the closed conformation of the VFT (Zhang et al., 2008). Newly developed enhancers of sweet taste bind to a similar site in the T1R2 stabilize the sucrose-promoted closure of the VFT (Zhang et al., 2010). Such compounds could help reduce the caloric content in food and beverages. Other compounds such as lactisole and cyclamate have been proposed to bind to allosteric binding site located in the TM domain of T1R3 and thus regulate orthosteric ligand binding to the VFT of T1R1 and T1R2 in the respective heterodimers.
Much less is known about the T2R subfamily that comprises approximately 40 members in humans (Bachmanov and Beauchamp, 2007). T1R and T2R are expressed in distinct subsets of TRC excluding any functional crosstalk at the cellular level. However, nearly all T2R members are typically co-expressed within individual TRCs suggesting signal integration at the cellular level. The existence and potential functional importance of T2R heteromers remains to be studied.
GPCR Heteromers in Reproduction
Different GPCRs participate in reproductive functions at the level of the hypothalamic–pituitary axes and reproductive organs. Oxytocin is a hypothalamic hormone stored in the posterior pituitary, which has uterine-contracting and milk-releasing actions. The receptor for oxytocin, OT, has been shown to cluster into homomers but also into heteromers with closely related vasopressin V1A and V2 receptors in transfected HEK293 cells (Terrillon et al., 2003). The existence of OT homomers has been recently confirmed in mammary gland tissue using classical radioligand binding assays and a newly developed time-resolved FRET assay between two fluorescently labeled OT ligands (Albizu et al., 2010). In contrast to previous energy transfer assays that require expression of modified receptors fused to energy donors and acceptors, this novel assay can be applied to unmodified wild-type receptors and is thus likely to boost research on GPCR homo- and heteromerization in native tissues (Figure 2).
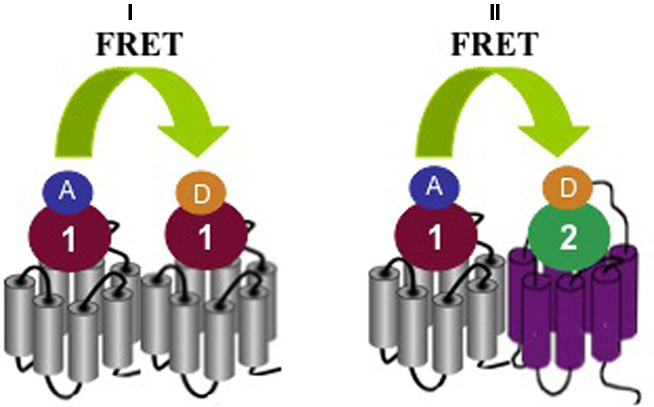
Figure 2. Detection of homo/heteromer formation by FRET using fluorescent ligands. (I) In the case of homomer formation a FRET signal can be detected between a selective donor (D) ligand I that binds the first protomer and a selective acceptor (A) ligand 1 that binds the second protomer The donor and acceptor ligands can be the same molecule but labeled differently. (II) In the case of heteromer formation, a FRET signal can be detected between a selective donor ligand 1 that binds the first protomer and a selective acceptor ligand 2 that binds the second protomer. In this case, donor and acceptor ligands are different molecules.
The luteinizing hormone (LH) is produced by the anterior pituitary gland. Whereas LH stimulates testosterone production in Leydig cells in males, it triggers ovulation in females. LH constitutes together with the follicle-stimulating hormone (FSH) and the thyroid-stimulating hormone (TSH) the glycoprotein hormone family. Their respective receptors LHR belong to a distinct subgroup of GPCRs that are characterized by a large N-terminal ligand binding domain-containing leucine-rich repeats and the typical heptahelical transmembrane domain. Biophysical and pharmacological assays were employed to show the formation of LHR homomers that display negative cooperativity between the ligand binding sites of receptor protomers (Urizar et al., 2005). More recently, the existence and functional relevance of LHR homomers was shown in vivo by using an elegant trans-complementation assay (Rivero-Muller et al., 2010). By generating transgenic mice co-expressing signaling deficient and ligand binding deficient forms of the LHR with targeted deletion of the wild-type LHR, the authors succeeded in completely restoring normal gonadal and genital function. This study provides compelling in vivo evidence for GPCR oligomerization by intermolecular functional complementation of two mutant receptors (Figure 3).
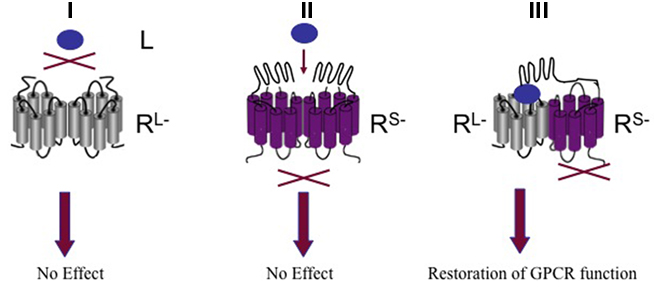
Figure 3. G protein-coupled receptors oligomerization by intermolecular functional complementation of mo mutant receptors. (I) Oligomerization of ligand binding deficient receptor mutant (RL−) does not trigger GPCR function. (II) Oligomerization of signaling deficient receptor mutant (Rs−) does not trigger GPCR function. (III) Co-expression of RL− and Rs− restores GPCR function. L; ligand.
Follicle-stimulating hormone, another hormone produced by the anterior pituitary gland, acts synergistically with LH to regulate pubertal maturation and reproductive processes. Based on the recent resolution of the crystal structure of FSH together with ectodomain of its receptor (FSHR) suggested formation of a tetrameric complex composed of a FSH dimer that bridges two FSHR ectodomains (Fan and Hendrickson, 2005). Such complexes were observed in the crystals and in solution, at high concentrations (μM range), suggesting the possible stabilization of FSHR oligomers in the presence of FSH. Subsequent studies did not confirm a major role of FSH binding and/or the ectodomain in oligomerization of the FSHR but pointed rather to the central role of the TM domain in stabilizing constitutive oligomers (Guan et al., 2010).
The existence of FSHR dimers inspired chemists to synthesize FSHR antagonist composed of two pharmacophores connected through ethylene glycol spacers (Bonger et al., 2009). These antagonists were indeed more potent than dimeric compounds in which one of the pharmacophores was replaced by an inactive confomer.
The gonadotropin-releasing hormone (GnRH) is produced in the hypothalamus and responsible for the release of FSH and LH from the anterior pituitary. Oligomerization of human GnRH receptors has been documented in transfected cells but remain still controversial in vivo (Pfleger et al., 2004). Convincing evidence for GnRH receptor heteromerization has been gathered in the protochordate Ciona intestinalis, which expresses six GnRH isoforms (tGnRH-3 to -8) and four GnRH receptor subtypes (R1-4). Heteromerization between R1, which binds to tGnRH-6, and the orphan R4 has been demonstrated in Ciona ovaries in co-immunoprecipitation experiments (Sakai et al., 2009). Importantly, tGnRH-6-induced ERK1/2 activation of R1 was robustly potentiated in the presence of R4 most likely originating from R1/R4 heteromerization.
GPCR Heteromers in Metabolism
The increasing prevalence of obesity is responsible for the dramatic increase in the number of scientific and clinical studies on the control of energy homeostasis. Regulation of energy balance relies on a well-orchestrated communication between the CNS and peripheral organs. Many GPCRs, including cannabinoid, ghrelin, and melanocortin (MC) receptors, play a central role in the regulation of lipid metabolism and are therefore of major interest for drug development. In the current chapter, we will discuss some examples of GPCR heteromerization, which might be implicated in energy metabolism and related diseases as type 2 diabetes and obesity.
The endocannabinoid system comprises the enzymatic machinery for endocannabinoid synthesis and degradation, and cannabinoid CB1 and CB2 receptors. The endocannabinoid system controls energy balance and lipid metabolism centrally, in the hypothalamus and mesolimbic pathways, and peripherally, in adipocytes, liver, skeletal muscle, and pancreatic islet cells, acting through numerous anorexigenic and orexigenic pathways. Obese people display an increased endocannabinoid tone, driving CB1 in a feed-forward dysfunction. Several CB1 antagonists/inverse agonists were developed for the treatment of obesity. However, beside their efficiency in reducing food intake, these drugs were not devoid of side effects like psychiatric disorders and suicide (Bermudez-Silva et al., 2010). Orexins are orexigenic mediators selectively expressed in the hypothalamus, within neurons of the lateral hypothalamic area. Orexin A is involved in food intake in satiated rats but also in the regulation of drinking behavior. Co-expression of CB1 and the orexin OX1 receptor in CHO cells enhances the potency of orexin A to stimulate ERK kinase phosphorylation, and this effect was blocked by addition of SR-141716A, a CB1 antagonist (Hilairet et al., 2003). FRET assays indicate that CB1 and OX1 can form heteromers in vitro in CHO cells exogenously expressing these receptors. Heteromer formation was shown to modulate receptor distribution in vitro (Ellis et al., 2006).
Ghrelin plays an important role in energy homeostasis (Castaneda et al., 2010). It is synthesized by the stomach and is believed to exert the majority of its actions through the ghrelin receptor (GHSR), which is present in high density in both the hypothalamus and the pituitary gland. Neurotensin is a 13-amino acid neurohormone, located in the synaptic vesicles and released from the neuronal terminals in a calcium-dependent manner. It plays an important role in the physiology of pain-induction, central blood pressure control, and inflammation. There are three known neurotensin receptors, NTS1, NTS2, NTS3 but only NTS1 and NTS2 belong to the GPCR family (Stolakis et al., 2010). GHSR and NTS1 are shown to physically interact in vitro and neuromedin U was described as ligand for this heteromer. GHSR/NTS1 heteromers have been suggested to play a role for an autocrine growth-promoting pathway in non–small cell lung cancers by modulating the transcription of downstream target genes including the Forkhead box protein M1 (FOXM1; Takahashi et al., 2006).
Jiang et al. (2006) identified neurons co-expressing GHSR and D1 in GHSR-IRES-tauGFP mice, suggesting at least a functional interaction between these two receptors. In vitro, the activation of GHSR by ghrelin amplifies dopamine/D1-induced cAMP accumulation (Jiang et al., 2006). In vivo, ghrelin and dopamine crosstalk was suggested based on the observation in rats that ghrelin administration increased dopamine-related hyperactivity (Wellman et al., 2005). Till date, there is no direct evidence in vitro or in vivo confirming GHSR/D1 heteromerization. GHSR can constitutively hetero-oligomerize with members of the prostanoid receptor family. GHSR is reported to heteromerize with EP3-I, IP1, and TPα receptors in vitro using BRET and co-immmunoprecipitation. The constitutive activation of phospholipase C by GHSR was significantly decreased in HEK 293 cells co-transfected with EP3-I, IP, or TPα. Moreover, cell surface expression of GHSR was reduced in presence of the prostanoids receptors (Chow et al., 2008). Despite lack of direct evidence, the expression of GHSR and prostanoid receptors in atherosclerotic plaques suggests heteromer implication in this pathology (Gomez-Hernandez et al., 2006).
Melanocortin receptors have a diverse range of physiological functions; MC1 controls skin pigmentation, MC2, the receptor for the adrenocorticotropic hormone (ACTH), plays a critical role in the hypothalamic–pituitary–adrenal axis, whereas MC3 and MC4 have essential roles in energy homeostasis and MC5 is believed to be involved in exocrine function (Cone, 2006). Mutations in the MC4R gene are the most frequent monogenic cause of severe obesity. Biebermann et al. present an interesting mechanistic explanation why a particular heterozygous inactivating MC4 mutation leads to the development of extreme obesity by a dominant-negative effect. This dominant-negative effect of the D90N mutation is caused by a functionally altered wild-type MC4/D90N receptor heteromer. MC4 can actually form heteromers, at least in vitro, with a mutant MC4 D90N resulting in functional alterations as exemplified by the complete loss of activation of the Gs/adenylyl cyclase pathways (Biebermann et al., 2003). In addition, MC4 can physically interact, in vitro, with GPR7. MC3 was shown to heteromerize with GHSR resulting in the modulation of MC3 trafficking (Rediger et al., 2009).
Conclusions and Perspectives
G protein-coupled receptor oligomerization has been an intensely studied field of research over the last 20 years. There is a broad consensus that GPCR oligomerization is not necessary for efficient activation of heterotrimeric G proteins as monomeric receptors are sufficient to ascertain this basic function. In contrast, GPCR oligomerization appears to be important for allosteric regulation and fine-tuning of signaling and receptor trafficking. It is furthermore admitted that most if not all GPCRs can form oligomers in intact cells. The detailed knowledge of the dimerization interface of some receptors (Guo et al., 2003; Wu et al., 2010) will undoubtedly guide the development of such tools. The lack of adequate tools (specific high-affinity antibodies, specific ligands, etc.) to study GPCR oligomerization in vivo or tissues expressing endogenous receptors has hampered faster progress for a long time. However, by employing original methods and developing new pharmacological tools more and more convincing evidence for the existence of GPCR oligomers is now increasingly gathered. GPCR oligomers, in particular heteromers provide new opportunities for specific pharmacological intervention. Based on the restricted expression profile of heteromers compared to the corresponding homomers, heteromer-specific drugs are expected to be very specific and to have fewer side effects. However, the great potential of heteromers is only beginning to be exploited as only very few heteromer-selective compounds are known today. Identification of such compounds will not only be important for future drug development but will also be indispensable tools for the identification and characterization of these heteromers in vivo. GPCR oligomers do not exist as isolated functional entities. It is now well established that GPCRs are integral part of protein networks composed of GPCRs and GPCR-associated protein complexes (Daulat et al., 2007, 2009; Bockaert et al., 2010). Learning more about the dynamics and tissue-specific composition of these clusters will undoubtedly provide new insights into the function of GPCR involved in neuroendocrinology.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from SERVIER, the Fondation Recherche Médicale (“Equipe FRM”), Institut National de la Santé et de la Recherche Médicale (INSERM), Centre National de la Recherche Scientifique (CNRS). We thank Jean-Luc Guillaume and Dr. Nathalie Clement for critical reading of the manuscript.
References
AbdAlla, S., Abdel-Baset, A., Lother, H., el Massiery, A., and Quitterer, U. (2005). Mesangial AT1/B2 receptor heterodimers contribute to angiotensin II hyperresponsiveness in experimental hypertension. J. Mol. Neurosci. 26, 185–192.
AbdAlla, S., Lother, H., and Quitterer, U. (2000). AT(1)-receptor heterodimers show enhanced G-protein activation and altered receptor sequestration. Nature 407, 94–98.
Adams, C. L., Cowen, M. S., Short, J. L., and Lawrence, A. J. (2008). Combined antagonism of glutamate mGlu5 and adenosine A2A receptors interact to regulate alcohol-seeking in rats. Int. J. Neuropsychopharmacol. 11, 229–241.
Albizu, L., Cottet, M., Kralikova, M., Stoev, S., Seyer, R., Brabet, I., Roux, T., Bazin, H., Bourrier, E., Lamarque, L., Breton, C., Rives, M. L., Newman, A., Javitch, J., Trinquet, E., Manning, M., Pin, J. P., Mouillac, B., and Durroux, T. (2010). Time-resolved FRET between GPCR ligands reveals oligomers in native tissues. Nat. Chem. Biol. 6, 587–594.
Andersson, M., Usiello, A., Borgkvist, A., Pozzi, L., Dominguez, C., Fienberg, A. A., Svenningsson, P., Fredholm, B. B., Borrelli, E., Greengard, P., and Fisone, G. (2005). Cannabinoid action depends on phosphorylation of dopamine- and cAMP-regulated phosphoprotein of 32 kDa at the protein kinase A site in striatal projection neurons. J. Neurosci. 25, 8432–8438.
Angers, S., Salahpour, A., and Bouvier, M. (2002). Dimerization: an emerging concept for G protein-coupled receptor ontogeny and function. Annu. Rev. Pharmacol. Toxicol. 42, 409–435.
Baneres, J. L., and Parello, J. (2003). Structure-based analysis of GPCR function: evidence for a novel pentameric assembly between the dimeric leukotriene B4 receptor BLT1 and the G-protein. J. Mol. Biol. 329, 815–829.
Baragli, A., Alturaihi, H., Watt, H. L., Abdallah, A., and Kumar, U. (2007). Heterooligomerization of human dopamine receptor 2 and somatostatin receptor 2 Co-immunoprecipitation and fluorescence resonance energy transfer analysis. Cell. Signal. 19, 2304–2316.
Barki-Harrington, L., Luttrell, L. M., and Rockman, H. A. (2003). Dual inhibition of beta-adrenergic and angiotensin II receptors by a single antagonist: a functional role for receptor–receptor interaction in vivo. Circulation 108, 1611–1618.
Bayburt, T. H., Leitz, A. J., Xie, G., Oprian, D. D., and Sligar, S. G. (2007). Transducin activation by nanoscale lipid bilayers containing one and two rhodopsins. J. Biol. Chem. 282, 14875–14881.
Becker, A., Grecksch, G., Kraus, J., Peters, B., Schroeder, H., Schulz, S., and Hollt, V. (2001). Loss of locomotor sensitisation in response to morphine in D1 receptor deficient mice. Naunyn Schmiedebergs Arch. Pharmacol. 363, 562–568.
Bermudez-Silva, F. J., Viveros, M. P., McPartland, J. M., and Rodriguez de Fonseca, F. (2010). The endocannabinoid system, eating behavior and energy homeostasis: the end or a new beginning? Pharmacol. Biochem. Behav. 95, 375–382.
Biebermann, H., Krude, H., Elsner, A., Chubanov, V., Gudermann, T., and Gruters, A. (2003). Autosomal-dominant mode of inheritance of a melanocortin-4 receptor mutation in a patient with severe early-onset obesity is due to a dominant-negative effect caused by receptor dimerization. Diabetes 52, 2984–2988.
Birdsall, N. J. (2010). Class A GPCR heterodimers: evidence from binding studies. Trends Pharmacol. Sci. 31, 499–508.
Bockaert, J., Perroy, J., Becamel, C., Marin, P., and Fagni, L. (2010). GPCR interacting proteins (GIPs) in the nervous system: roles in physiology and pathologies. Annu. Rev. Pharmacol. Toxicol. 50, 89–109.
Bonger, K. M., Hoogendoorn, S., van Koppen, C. J., Timmers, C. M., Overkleeft, H. S., and van der Marel, G. A. (2009). Synthesis and pharmacological evaluation of dimeric follicle-stimulating hormone receptor antagonists. ChemMedChem 4, 2098–2102.
Borroto-Escuela, D. O., Narvaez, M., Marcellino, D., Parrado, C., Narvaez, J. A., Tarakanov, A. O., Agnati, L. F., Diaz-Cabiale, Z., and Fuxe, K. (2010). Galanin receptor-1 modulates 5-hydroxtryptamine-1A signaling via heterodimerization. Biochem. Biophys. Res. Commun. 393, 767–772.
Bouvier, M. (2001). Oligomerization of G-protein-coupled transmitter receptors. Nat. Rev. Neurosci. 2, 274–286.
Boyer, S. B., Clancy, S. M., Terunuma, M., Revilla-Sanchez, R., Thomas, S. M., Moss, S. J., and Slesinger, P. A. (2009). Direct interaction of GABAB receptors with M2 muscarinic receptors enhances muscarinic signaling. J. Neurosci. 29, 15796–15809.
Bulenger, S., Marullo, S., and Bouvier, M. (2005). Emerging role of homo- and heterodimerization in G-protein-coupled receptor biosynthesis and maturation. Trends Pharmacol. Sci. 26, 131–137.
Cabello, N., Gandia, J., Bertarelli, D. C., Watanabe, M., Lluis, C., Franco, R., Ferre, S., Lujan, R., and Ciruela, F. (2009). Metabotropic glutamate type 5, dopamine D2 and adenosine A2a receptors form higher-order oligomers in living cells. J. Neurochem. 109, 1497–1507.
Canals, M., Marcellino, D., Fanelli, F., Ciruela, F., de Benedetti, P., Goldberg, S. R., Neve, K., Fuxe, K., Agnati, L. F., Woods, A. S., Ferre, S., Lluis, C., Bouvier, M., and Franco, R. (2003). Adenosine A2A-dopamine D2 receptor–receptor heteromerization: qualitative and quantitative assessment by fluorescence and bioluminescence energy transfer. J. Biol. Chem. 278, 46741–46749.
Carriba, P., Ortiz, O., Patkar, K., Justinova, Z., Stroik, J., Themann, A., Muller, C., Woods, A. S., Hope, B. T., Ciruela, F., Casado, V., Canela, E. I., Lluis, C., Goldberg, S. R., Moratalla, R., Franco, R., and Ferre, S. (2007). Striatal adenosine A2A and cannabinoid CB1 receptors form functional heteromeric complexes that mediate the motor effects of cannabinoids. Neuropsychopharmacology 32, 2249–2259.
Castaneda, T. R., Tong, J., Datta, R., Culler, M., and Tschop, M. H. (2010). Ghrelin in the regulation of body weight and metabolism. Front. Neuroendocrinol. 31:1. doi: 10.1016/j.yfrne.2009.10.008
Chang, W., Tu, C., Cheng, Z., Rodriguez, L., Chen, T. H., Gassmann, M., Bettler, B., Margeta, M., Jan, L. Y., and Shoback, D. (2007). Complex formation with the Type B gamma-aminobutyric acid receptor affects the expression and signal transduction of the extracellular calcium-sensing receptor. Studies with HEK-293 cells and neurons. J. Biol. Chem. 282, 25030–25040.
Chen, C., Li, J., Bot, G., Szabo, I., Rogers, T. J., and Liu-Chen, L. Y. (2004). Heterodimerization and cross-desensitization between the mu-opioid receptor and the chemokine CCR5 receptor. Eur. J. Pharmacol. 483, 175–186.
Chow, K. B., Leung, P. K., Cheng, C. H., Cheung, W. T., and Wise, H. (2008). The constitutive activity of ghrelin receptors is decreased by co-expression with vasoactive prostanoid receptors when over-expressed in human embryonic kidney 293 cells. Int. J. Biochem. Cell Biol. 40, 2627–2637.
Chun, H. J., Ali, Z. A., Kojima, Y., Kundu, R. K., Sheikh, A. Y., Agrawal, R., Zheng, L., Leeper, N. J., Pearl, N. E., Patterson, A. J., Anderson, J. P., Tsao, P. S., Lenardo, M. J., Ashley, E. A., and Quertermous, T. (2008). Apelin signaling antagonizes Ang II effects in mouse models of atherosclerosis. J. Clin. Invest. 118, 3343–3354.
Cichewicz, D. L. (2004). Synergistic interactions between cannabinoid and opioid analgesics. Life Sci. 74, 1317–1324.
Ciruela, F., Casado, V., Rodrigues, R. J., Lujan, R., Burgueno, J., Canals, M., Borycz, J., Rebola, N., Goldberg, S. R., Mallol, J., Cortes, A., Canela, E. I., Lopez-Gimenez, J. F., Milligan, G., Lluis, C., Cunha, R. A., Ferre, S., and Franco, R. (2006). Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1-A2A receptor heteromers. J. Neurosci. 26, 2080–2087.
Ciruela, F., Escriche, M., Burgueno, J., Angulo, E., Casado, V., Soloviev, M. M., Canela, E. I., Mallol, J., Chan, W. Y., Lluis, C., McIlhinney, R. A., and Franco, R. (2001). Metabotropic glutamate 1alpha and adenosine A1 receptors assemble into functionally interacting complexes. J. Biol. Chem. 276, 18345–18351.
Colao, A., Filippella, M., Pivonello, R., Di Somma, C., Faggiano, A., and Lombardi, G. (2007). Combined therapy of somatostatin analogues and dopamine agonists in the treatment of pituitary tumours. Eur. J. Endocrinol. 156(Suppl. 1), S57–S63.
Cone, R. D. (2006). Studies on the physiological functions of the melanocortin system. Endocr. Rev. 27, 736–749.
Daulat, A. M., Maurice, P., Froment, C., Guillaume, J. L., Broussard, C., Monsarrat, B., Delagrange, P., and Jockers, R. (2007). Purification and identification of G protein-coupled receptor protein complexes under native conditions. Mol. Cell Proteomics 6, 835–844.
Daulat, A. M., Maurice, P., and Jockers, R. (2009). Recent methodological advances in the discovery of GPCR-associated protein complexes. Trends Pharmacol. Sci. 30, 72–78.
de Almeida, J., Palacios, J. M., and Mengod, G. (2008). Distribution of 5-HT and DA receptors in primate prefrontal cortex: implications for pathophysiology and treatment. Prog. Brain Res. 172, 101–115.
Derangeon, M., Bozon, V., Defamie, N., Peineau, N., Bourmeyster, N., Sarrouilhe, D., Argibay, J. A., and Herve, J. C. (2010). 5-HT4 and 5-HT2 receptors antagonistically influence gap junctional coupling between rat auricular myocytes. J. Mol. Cell. Cardiol. 48, 220–229.
Durroux, T. (2005). Principles: a model for the allosteric interactions between ligand binding sites within a dimeric GPCR. Trends Pharmacol. Sci. 26, 376–384.
Ellis, J., Pediani, J. D., Canals, M., Milasta, S., and Milligan, G. (2006). Orexin-1 receptor-cannabinoid CB1 receptor heterodimerization results in both ligand-dependent and -independent coordinated alterations of receptor localization and function. J. Biol. Chem. 281, 38812–38824.
Ernst, O. P., Gramse, V., Kolbe, M., Hofmann, K. P., and Heck, M. (2007). Monomeric G protein-coupled receptor rhodopsin in solution activates its G protein transducin at the diffusion limit. Proc. Natl. Acad. Sci. U.S.A. 104, 10859–10864.
Evans, N. J., and Walker, J. W. (2008). Endothelin receptor dimers evaluated by FRET, ligand binding, and calcium mobilization. Biophys. J. 95, 483–492.
Fan, Q. R., and Hendrickson, W. A. (2005). Structure of human follicle-stimulating hormone in complex with its receptor. Nature 433, 269–277.
Ferrada, C., Ferre, S., Casado, V., Cortes, A., Justinova, Z., Barnes, C., Canela, E. I., Goldberg, S. R., Leurs, R., Lluis, C., and Franco, R. (2008). Interactions between histamine H3 and dopamine D2 receptors and the implications for striatal function. Neuropharmacology 55, 190–197.
Ferre, S., Karcz-Kubicha, M., Hope, B. T., Popoli, P., Burgueno, J., Gutierrez, M. A., Casado, V., Fuxe, K., Goldberg, S. R., Lluis, C., Franco, R., and Ciruela, F. (2002). Synergistic interaction between adenosine A2A and glutamate mGlu5 receptors: implications for striatal neuronal function. Proc. Natl. Acad. Sci. U.S.A. 99, 11940–11945.
Ferre, S., Navarro, G., Casado, V., Cortes, A., Mallol, J., Canela, E. I., Lluis, C., and Franco, R. (2010). G protein-coupled receptor heteromers as new targets for drug development. Prog. Mol. Biol. Transl. Sci. 91, 41–52.
Fincham, C. I., Bressan, A., Paris, M., Rossi, C., and Fattori, D. (2009). Bradykinin receptor antagonists – a review of the patent literature 2005–2008. Expert Opin. Ther. Pat. 19, 919–941.
Finley, M. J., Chen, X., Bardi, G., Davey, P., Geller, E. B., Zhang, L., Adler, M. W., and Rogers, T. J. (2008). Bi-directional heterologous desensitization between the major HIV-1 co-receptor CXCR4 and the kappa-opioid receptor. J. Neuroimmunol. 197, 114–123.
Galvez, T., Duthey, B., Kniazeff, J., Blahos, J., Rovelli, G., Bettler, B., Prezeau, L., and Pin, J. P. (2001). Allosteric interactions between GB1 and GB2 subunits are required for optimal GABA(B) receptor function. EMBO J. 20, 2152–2159.
Glass, M., and Felder, C. C. (1997). Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: evidence for a Gs linkage to the CB1 receptor. J. Neurosci. 17, 5327–5333.
Gomez-Hernandez, A., Martin-Ventura, J. L., Sanchez-Galan, E., Vidal, C., Ortego, M., Blanco-Colio, L. M., Ortega, L., Tunon, J., and Egido, J. (2006). Overexpression of COX-2, Prostaglandin E synthase-1 and prostaglandin E receptors in blood mononuclear cells and plaque of patients with carotid atherosclerosis: regulation by nuclear factor-kappaB. Atherosclerosis 187, 139–149.
Gonzalez-Maeso, J., Ang, R. L., Yuen, T., Chan, P., Weisstaub, N. V., Lopez-Gimenez, J. F., Zhou, M., Okawa, Y., Callado, L. F., Milligan, G., Gingrich, J. A., Filizola, M., Meana, J. J., and Sealfon, S. C. (2008). Identification of a serotonin/glutamate receptor complex implicated in psychosis. Nature 452, 93–97.
Guan, R., Wu, X., Feng, X., Zhang, M., Hebert, T. E., and Segaloff, D. L. (2010). Structural determinants underlying constitutive dimerization of unoccupied human follitropin receptors. Cell. Signal. 22, 247–256.
Guimaraes, S., and Pinheiro, H. (2005). Functional evidence that in the cardiovascular system AT1 angiotensin II receptors are AT1B prejunctionally and AT1A postjunctionally. Cardiovasc. Res. 67, 208–215.
Guo, W., Shi, L., and Javitch, J. A. (2003). The fourth transmembrane segment forms the interface of the dopamine D2 receptor homodimer. J. Biol. Chem. 278, 4385–4388.
Hansen, J. L., Hansen, J. T., Speerschneider, T., Lyngso, C., Erikstrup, N., Burstein, E. S., Weiner, D. M., Walther, T., Makita, N., Iiri, T., Merten, N., Kostenis, E., and Sheikh, S. P. (2009). Lack of evidence for AT1R/B2R heterodimerization in COS-7, HEK293, and NIH3T3 cells: how common is the AT1R/B2R heterodimer? J. Biol. Chem. 284, 1831–1839.
Hilairet, S., Bouaboula, M., Carriere, D., Le Fur, G., and Casellas, P. (2003). Hypersensitization of the Orexin 1 receptor by the CB1 receptor: evidence for cross-talk blocked by the specific CB1 antagonist, SR141716. J. Biol. Chem. 278, 23731–23737.
Hirono, M., Yoshioka, T., and Konishi, S. (2001). GABA(B) receptor activation enhances mGluR-mediated responses at cerebellar excitatory synapses. Nat. Neurosci. 4, 1207–1216.
Hojo, M., Sudo, Y., Ando, Y., Minami, K., Takada, M., Matsubara, T., Kanaide, M., Taniyama, K., Sumikawa, K., and Uezono, Y. (2008). mu-Opioid receptor forms a functional heterodimer with cannabinoid CB1 receptor: electrophysiological and FRET assay analysis. J. Pharmacol. Sci. 108, 308–319.
Hus-Citharel, A., Bouby, N., Iturrioz, X., and Llorens-Cortes, C. (2010). Multiple cross talk between angiotensin II, bradykinin, and insulin signaling in the cortical thick ascending limb of rat kidney. Endocrinology 151, 3181–3194.
Jaquet, P., Gunz, G., Saveanu, A., Dufour, H., Taylor, J., Dong, J., Kim, S., Moreau, J. P., Enjalbert, A., and Culler, M. D. (2005). Efficacy of chimeric molecules directed towards multiple somatostatin and dopamine receptors on inhibition of GH and prolactin secretion from GH-secreting pituitary adenomas classified as partially responsive to somatostatin analog therapy. Eur. J. Endocrinol. 153, 135–141.
Jiang, H., Betancourt, L., and Smith, R. G. (2006). Ghrelin amplifies dopamine signaling by cross talk involving formation of growth hormone secretagogue receptor/dopamine receptor subtype 1 heterodimers. Mol. Endocrinol. 20, 1772–1785.
Jockers, R., Issad, T., Zilberfarb, V., de Coppet, P., Marullo, S., and Strosberg, A. D. (1998). Desensitization of the beta-adrenergic response in human brown adipocytes. Endocrinology 139, 2676–2684.
Jones, K. A., Borowsky, B., Tamm, J. A., and Gerald, C. (1998). GABAb receptors function as a heterotrimeric assembly of the subunits GABAbR1 and GAGbR2. Nature 396, 674–679.
Jordan, B. A., Trapaidze, N., Gomes, I., Nivarthi, R., and Devi, L. A. (2001). Oligomerization of opioid receptors with beta 2-adrenergic receptors: a role in trafficking and mitogen-activated protein kinase activation. Proc. Natl. Acad. Sci. U.S.A. 98, 343–348.
Juhasz, J. R., Hasbi, A., Rashid, A. J., So, C. H., George, S. R., and O’Dowd, B. F. (2008). Mu-opioid receptor heterooligomer formation with the dopamine D1 receptor as directly visualized in living cells. Eur. J. Pharmacol. 581, 235–243.
Kachroo, A., Orlando, L. R., Grandy, D. K., Chen, J. F., Young, A. B., and Schwarzschild, M. A. (2005). Interactions between metabotropic glutamate 5 and adenosine A2A receptors in normal and parkinsonian mice. J. Neurosci. 25, 10414–10419.
Kalea, A. Z., and Batlle, D. (2010). Apelin and ACE2 in cardiovascular disease. Curr. Opin. Investig. Drugs 11, 273–282.
Kearn, C. S., Blake-Palmer, K., Daniel, E., Mackie, K., and Glass, M. (2005). Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors enhances heterodimer formation: a mechanism for receptor cross-talk? Mol. Pharmacol. 67, 1697–1704.
Kehr, J., Yoshitake, T., Wang, F. H., Razani, H., Gimenez-Llort, L., Jansson, A., Yamaguchi, M., and Ogren, S. O. (2002). Galanin is a potent in vivo modulator of mesencephalic serotonergic neurotransmission. Neuropsychopharmacology 27, 341–356.
Kitaoka, S., Furuyashiki, T., Nishi, A., Shuto, T., Koyasu, S., Matsuoka, T., Miyasaka, M., Greengard, P., and Narumiya, S. (2007). Prostaglandin E2 acts on EP1 receptor and amplifies both dopamine D1 and D2 receptor signaling in the striatum. J. Neurosci. 27, 12900–12907.
Kuteeva, E., Hokfelt, T., Wardi, T., and Ogren, S. O. (2008). Galanin, galanin receptor subtypes and depression-like behaviour. Cell. Mol. Life Sci. 65, 1854–1863.
Lee, S. P., So, C. H., Rashid, A. J., Varghese, G., Cheng, R., Lanca, A. J., O’Dowd, B. F., and George, S. R. (2004). Dopamine D1 and D2 receptor Co-activation generates a novel phospholipase C-mediated calcium signal. J. Biol. Chem. 279, 35671–35678.
Levoye, A., Dam, J., Ayoub, M. A., Guillaume, J. L., and Jockers, R. (2006). Do orphan G-protein-coupled receptors have ligand-independent functions? New insights from receptor heterodimers. EMBO Rep. 7, 1094–1098.
Lukasiewicz, S., Polit, A., Kedracka-Krok, S., Wedzony, K., Mackowiak, M., and Dziedzicka-Wasylewska, M. (2010). Hetero-dimerization of serotonin 5-HT(2A) and dopamine D(2) receptors. Biochim. Biophys. Acta 1803, 1347–1358.
Magalhaes, A. C., Holmes, K. D., Dale, L. B., Comps-Agrar, L., Lee, D., Yadav, P. N., Drysdale, L., Poulter, M. O., Roth, B. L., Pin, J. P., Anisman, H., and Ferguson, S. S. (2010). CRF receptor 1 regulates anxiety behavior via sensitization of 5-HT2 receptor signaling. Nat. Neurosci. 13, 622–629.
Marcellino, D., Carriba, P., Filip, M., Borgkvist, A., Frankowska, M., Bellido, I., Tanganelli, S., Muller, C. E., Fisone, G., Lluis, C., Agnati, L. F., Franco, R., and Fuxe, K. (2008). Antagonistic cannabinoid CB1/dopamine D2 receptor interactions in striatal CB1/D2 heteromers. A combined neurochemical and behavioral analysis. Neuropharmacology 54, 815–823.
McGraw, D. W., Mihlbachler, K. A., Schwarb, M. R., Rahman, F. F., Small, K. M., Almoosa, K. F., and Liggett, S. B. (2006). Airway smooth muscle prostaglandin-EP1 receptors directly modulate beta2-adrenergic receptors within a unique heterodimeric complex. J. Clin. Invest. 116, 1400–1409.
Millan, M. J., Marin, P., Bockaert, J., and Mannoury la Cour, C. (2008). Signaling at G-protein-coupled serotonin receptors: recent advances and future research directions. Trends Pharmacol. Sci. 29, 454–464.
Milligan, G. (2009). G protein-coupled receptor hetero-dimerization: contribution to pharmacology and function. Br. J. Pharmacol. 158, 5–14.
Missale, C., Fiorentini, C., Collo, G., and Spano, P. (2010). The neurobiology of dopamine receptors: evolution from the dual concept to heterodimer complexes. J. Recept. Signal Transduct. Res. 30, 347–354.
Nakata, H., Suzuki, T., Namba, K., and Oyanagi, K. (2010). Dimerization of G protein-coupled purinergic receptors: increasing the diversity of purinergic receptor signal responses and receptor functions. J. Recept. Signal Transduct. Res. 30, 337–346.
Nelson, G., Chandrashekar, J., Hoon, M. A., Feng, L., Zhao, G., Ryba, N. J., and Zuker, C. S. (2002). An amino-acid taste receptor. Nature 416, 199–202.
Nelson, G., Hoon, M. A., Chandrashekar, J., Zhang, Y., Ryba, N. J., and Zuker, C. S. (2001). Mammalian sweet taste receptors. Cell 106, 381–390.
Nijmeijer, S., Leurs, R., Smit, M. J., and Vischer, H. F. (2010). The Epstein–Barr virus-encoded G protein-coupled receptor BILF1 hetero-oligomerizes with human CXCR4, scavenges Galphai proteins, and constitutively impairs CXCR4 functioning. J. Biol. Chem. 285, 29632–29641.
Pello, O. M., Martinez-Munoz, L., Parrillas, V., Serrano, A., Rodriguez-Frade, J. M., Toro, M. J., Lucas, P., Monterrubio, M., Martinez, A. C., and Mellado, M. (2008). Ligand stabilization of CXCR4/delta-opioid receptor heterodimers reveals a mechanism for immune response regulation. Eur. J. Immunol. 38, 537–549.
Pepe, S., Xiao, R. P., Hohl, C., Altschuld, R., and Lakatta, E. G. (1997). “Cross talk” between opioid peptide and adrenergic receptor signaling in isolated rat heart. Circulation 95, 2122–2129.
Pfeiffer, M., Kirscht, S., Stumm, R., Koch, T., Wu, D., Laugsch, M., Schroder, H., Hollt, V., and Schulz, S. (2003). Heterodimerization of substance P and mu-opioid receptors regulates receptor trafficking and resensitization. J. Biol. Chem. 278, 51630–51637.
Pfleger, K. D., Kroeger, K. M., and Eidne, K. A. (2004). Receptors for hypothalamic releasing hormones TRH and GnRH: oligomerization and interactions with intracellular proteins. Semin. Cell Dev. Biol. 15, 269–280.
Pin, J. P., Neubig, R., Bouvier, M., Devi, L., Filizola, M., Javitch, J. A., Lohse, M. J., Milligan, G., Palczewski, K., Parmentier, M., and Spedding, M. (2007). International union of basic and clinical pharmacology. LXVII. recommendations for the recognition and nomenclature of G protein-coupled receptor heteromultimers. Pharmacol. Rev. 59, 5–13.
Prezeau, L., Rives, M. L., Comps-Agrar, L., Maurel, D., Kniazeff, J., and Pin, J. P. (2010). Functional crosstalk between GPCRs: with or without oligomerization. Curr. Opin. Pharmacol. 10, 6–13.
Prinster, S. C., Hague, C., and Hall, R. A. (2005). Heterodimerization of g protein-coupled receptors: specificity and functional significance. Pharmacol. Rev. 57, 289–298.
Rashid, A. J., So, C. H., Kong, M. M., Furtak, T., El-Ghundi, M., Cheng, R., O’Dowd, B. F., and George, S. R. (2007). D1–D2 dopamine receptor heterooligomers with unique pharmacology are coupled to rapid activation of Gq/11 in the striatum. Proc. Natl. Acad. Sci. U.S.A. 104, 654–659.
Rediger, A., Tarnow, P., Bickenbach, A., Schaefer, M., Krude, H., Gruters, A., and Biebermann, H. (2009). Heterodimerization of hypothalamic G-protein-coupled receptors involved in weight regulation. Obes. Facts 2, 80–86.
Rios, C., Gomes, I., and Devi, L. A. (2006). mu opioid and CB1 cannabinoid receptor interactions: reciprocal inhibition of receptor signaling and neuritogenesis. Br. J. Pharmacol. 148, 387–395.
Ripley, T. L., Gadd, C. A., De Felipe, C., Hunt, S. P., and Stephens, D. N. (2002). Lack of self-administration and behavioural sensitisation to morphine, but not cocaine, in mice lacking NK1 receptors. Neuropharmacology 43, 1258–1268.
Rivero-Muller, A., Chou, Y. Y., Ji, I., Lajic, S., Hanyaloglu, A. C., Jonas, K., Rahman, N., Ji, T. H., and Huhtaniemi, I. (2010). Rescue of defective G protein-coupled receptor function in vivo by intermolecular cooperation. Proc. Natl. Acad. Sci. U.S.A. 107, 2319–2324.
Rives, M. L., Vol, C., Fukazawa, Y., Tinel, N., Trinquet, E., Ayoub, M. A., Shigemoto, R., Pin, J. P., and Prezeau, L. (2009). Crosstalk between GABAB and mGlu1a receptors reveals new insight into GPCR signal integration. EMBO J. 28, 2195–2208.
Rocheville, M., Lange, D. C., Kumar, U., Patel, S. C., Patel, R. C., and Patel, Y. C. (2000). Receptors for dopamine and somatostatin: formation of hetero-oligomers with enhanced functional activity. Science 288, 154–157.
Rozenfeld, R., and Devi, L. A. (2010). Receptor heteromerization and drug discovery. Trends Pharmacol. Sci. 31, 124–130.
Sakai, T., Aoyama, M., Kusakabe, T., Tsuda, M., and Satake, H. (2009). Functional diversity of signaling pathways through G protein-coupled receptor heterodimerization with a species-specific orphan receptor subtype. Mol. Biol. Evol. 27, 1097–1106.
Sakurai, T., Yanagisawa, M., Takuwa, Y., Miyazaki, H., Kimura, S., Goto, K., and Masaki, T. (1990). Cloning of a cDNA encoding a non-isopeptide-selective subtype of the endothelin receptor. Nature 348, 732–735.
Salanga, C. L., O’Hayre, M., and Handel, T. (2009). Modulation of chemokine receptor activity through dimerization and crosstalk. Cell. Mol. Life Sci. 66, 1370–1386.
Salazar, N. C., Chen, J., and Rockman, H. A. (2007). Cardiac GPCRs: GPCR signaling in healthy and failing hearts. Biochim. Biophys. Acta 1768, 1006–1018.
Scavone, J. L., Mackie, K., and Van Bockstaele, E. J. (2010). Characterization of cannabinoid-1 receptors in the locus coeruleus: relationship with mu-opioid receptors. Brain Res. 1312, 18–31.
So, C. H., Verma, V., Alijaniaram, M., Cheng, R., Rashid, A. J., O’Dowd, B. F., and George, S. R. (2009). Calcium signaling by dopamine D5 receptor and D5-D2 receptor hetero-oligomers occurs by a mechanism distinct from that for dopamine D1-D2 receptor hetero-oligomers. Mol. Pharmacol. 75, 843–854.
Stolakis, V., Kalafatakis, K., Botis, J., Zarros, A., and Liapi, C. (2010). The regulatory role of neurotensin on the hypothalamic-anterior pituitary axons: emphasis on the control of thyroid-related functions. Neuropeptides 44, 1–7.
Takahashi, K., Furukawa, C., Takano, A., Ishikawa, N., Kato, T., Hayama, S., Suzuki, C., Yasui, W., Inai, K., Sone, S., Ito, T., Nishimura, H., Tsuchiya, E., Nakamura, Y., and Daigo, Y. (2006). The neuromedin U-growth hormone secretagogue receptor 1b/neurotensin receptor 1 oncogenic signaling pathway as a therapeutic target for lung cancer. Cancer Res. 66, 9408–9419.
Tanganelli, S., Sandager Nielsen, K., Ferraro, L., Antonelli, T., Kehr, J., Franco, R., Ferre, S., Agnati, L. F., Fuxe, K., and Scheel-Kruger, J. (2004). Striatal plasticity at the network level. Focus on adenosine A2A and D2 interactions in models of Parkinson’s disease. Parkinsonism Relat. Disord. 10, 273–280.
Tebano, M. T., Martire, A., Chiodi, V., Pepponi, R., Ferrante, A., Domenici, M. R., Frank, C., Chen, J. F., Ledent, C., and Popoli, P. (2009). Adenosine A2A receptors enable the synaptic effects of cannabinoid CB1 receptors in the rodent striatum. J. Neurochem. 110, 1921–1930.
Terrillon, S., Durroux, T., Mouillac, B., Breit, A., Ayoub, M. A., Taulan, M., Jockers, R., Barberis, C., and Bouvier, M. (2003). Oxytocin and vasopressin V1a and V2 receptors form constitutive homo- and heterodimers during biosynthesis. Mol. Endocrinol. 17, 677–691.
Tien, L. T., Ho, I. K., and Ma, T. (2010). Methamphetamine-induced expression of zif268 mRNA is prevented by haloperidol in mice lacking mu-opioid receptor. Neurotoxicology 31, 326–330.
Tonazzini, I., Trincavelli, M. L., Montali, M., and Martini, C. (2008). Regulation of A1 adenosine receptor functioning induced by P2Y1 purinergic receptor activation in human astroglial cells. J. Neurosci. Res. 86, 2857–2866.
Tonazzini, I., Trincavelli, M. L., Storm-Mathisen, J., Martini, C., and Bergersen, L. H. (2007). Co-localization and functional cross-talk between A1 and P2Y1 purine receptors in rat hippocampus. Eur. J. Neurosci. 26, 890–902.
Trincavelli, M. L., Daniele, S., and Martini, C. (2010). Adenosine receptors: what we know and what we are learning. Curr. Top. Med. Chem. 10, 860–877.
Urizar, E., Montanelli, L., Loy, T., Bonomi, M., Swillens, S., Gales, C., Bouvier, M., Smits, G., Vassart, G., and Costagliola, S. (2005). Glycoprotein hormone receptors: link between receptor homodimerization and negative cooperativity. EMBO J. 24, 1954–1964.
van Rijn, R. M., Whistler, J. L., and Waldhoer, M. (2010). Opioid-receptor-heteromer-specific trafficking and pharmacology. Curr. Opin. Pharmacol. 10, 73–79.
Waldhoer, M., Fong, J., Jones, R. M., Lunzer, M. M., Sharma, S. K., Kostenis, E., Portoghese, P. S., and Whistler, J. L. (2005). A heterodimer-selective agonist shows in vivo relevance of G protein-coupled receptor dimers. Proc. Natl. Acad. Sci. U.S.A. 102, 9050–9055.
Wellman, P. J., Davis, K. W., and Nation, J. R. (2005). Augmentation of cocaine hyperactivity in rats by systemic ghrelin. Regul. Pept. 125, 151–154.
White, J. H., Wise, A., Main, M. J., Green, A., Fraser, N. J., Disney, G. H., Barnes, A. A., Emson, P., Foord, S. M., and Marshall, F. H. (1998). Heterodimerization is required for the formation of a functional GABA(B) receptor. Nature 396, 679–682.
Whorton, M. R., Bokoch, M. P., Rasmussen, S. G., Huang, B., Zare, R. N., Kobilka, B., and Sunahara, R. K. (2007). A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc. Natl. Acad. Sci. U.S.A. 104, 7682–7687.
Wilson, S. J., Roche, A. M., Kostetskaia, E., and Smyth, E. M. (2004). Dimerization of the human receptors for prostacyclin and thromboxane facilitates thromboxane receptor-mediated cAMP generation. J. Biol. Chem. 279, 53036–53047.
Wu, B., Chien, E. Y., Mol, C. D., Fenalti, G., Liu, W., Katritch, V., Abagyan, R., Brooun, A., Wells, P., Bi, F. C., Hamel, D. J., Kuhn, P., Handel, T. M., Cherezov, V., and Stevens, R. C. (2010). Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 330, 1066–1071.
Xu, H., Staszewski, L., Tang, H., Adler, E., Zoller, M., and Li, X. (2004). Different functional roles of T1R subunits in the heteromeric taste receptors. Proc. Natl. Acad. Sci. U.S.A. 101, 14258–14263.
Yoshioka, K., Hosoda, R., Kuroda, Y., and Nakata, H. (2002). Hetero-oligomerization of adenosine A1 receptors with P2Y1 receptors in rat brains. FEBS Lett. 531, 299–303.
Zhang, F., Klebansky, B., Fine, R. M., Liu, H., Xu, H., Servant, G., Zoller, M., Tachdjian, C., and Li, X. (2010). Molecular mechanism of the sweet taste enhancers. Proc. Natl. Acad. Sci. U.S.A. 107, 4752–4757.
Zhang, F., Klebansky, B., Fine, R. M., Xu, H., Pronin, A., Liu, H., Tachdjian, C., and Li, X. (2008). Molecular mechanism for the umami taste synergism. Proc. Natl. Acad. Sci. U.S.A. 105, 20930–20934.
Keywords: GPCR, heterodimers, direct physical interaction, functional crosstalk, endocrinology
Citation: Kamal M and Jockers R (2011) Biological significance of GPCR heteromerization in the neuro-endocrine system. Front. Endocrin. 2:2. doi: 10.3389/fendo.2011.00002
Received: 27 November 2010;
Paper pending published: 19 December 2010;
Accepted: 13 January 2011;
Published online: 01 February 2011.
Edited by:
Regina Pekelmann Markus, University of São Paulo, BrazilReviewed by:
Regina Pekelmann Markus, University of São Paulo, BrazilKrystyna Skwarlo-Sonta, University of Warsaw, Poland
Sandra Helena Poliselli Farsky, University of São Paulo, Brazil
Copyright: © 2011 Kamal and Jockers. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Ralf Jockers, Institut Cochin, 22 rue Méchain, 75014 Paris, France. e-mail:cmFsZi5qb2NrZXJzQGluc2VybS5mcg==