 Mauricio P. Pinto1*
Mauricio P. Pinto1* Britta M. Jacobsen1 and Kathryn B. Horwitz1,2
Britta M. Jacobsen1 and Kathryn B. Horwitz1,2
- 1 Division of Endocrinology, Metabolism and Diabetes, Department of Medicine, University of Colorado Anschutz Medical Campus, Aurora, CO, USA
- 2 Department of Pathology, University of Colorado Anschutz Medical Campus, Aurora, CO, USA
The development of in vitro three-dimensional cell culture matrices offers physiologically relevant alternatives to traditional culture on plastic surfaces. However methods to analyze cell subpopulations therein are poor. Here we present a simple and inexpensive method to analyze cell subpopulations in mixed-cell colonies using standard immunohistochemical (IHC) techniques. Briefly, Matrigel™ blocks are sandwiched between two layers of HistoGel™, hardened by rapid cooling then processed for routine fixation, paraffin embedding, and IHC. We demonstrate the assay using mono- and co-cultured normal human breast, human breast cancer, and transformed mouse stromal cells along with hormone treated breast cancer cells. Judicious selection of specific antibodies allows different cell types within heterotypic colonies to be identified. A brief pulse of bromodeoxyuridine in living colonies allows proliferation of cell subpopulations to be quantified. This simple assay is useful for multiple cell types, species, and conditions.
Introduction
For decades, the ex vivo culture of cell lines has been fundamental to understanding the mechanisms involved in cancer development and progression. In most cases these studies are performed by culturing cells on two-dimensional (2D) plastic surfaces. Under these conditions non-malignant cells often lose differentiation, and the state of malignant cells differs from that of their solid tumor counterparts (Birgersdotter et al., 2007). This is explained at least in part by the lack of extracellular matrix signals. Indeed, some morphological and functional properties can be restored by growing cells in reconstituted basement membranes (Barcellos-Hoff et al., 1989; Streuli and Bissell, 1990; Schmidhauser et al., 1992). Since its discovery in 1983, laminin-rich extracellular matrix [also known as Matrigel, Cultrex, or Engelbreth-Holm Swarm (EHS) matrix; Kleinman and Martin, 2005] has been used to culture cells in 3 dimensions (3D) in conditions that allow cells to replicate some of the features present in tissues and tumors (Debnath and Brugge, 2005). Malignant cells show characteristic morphologies when cultured in 3D that are not observed in 2D (Kenny et al., 2007). Additionally, 3D culture generally preserves the proliferation rate (dit Faute et al., 2002), gene expression profile (Li et al., 2006), and drug sensitivity of cells (dit Faute et al., 2002; Weaver et al., 2002). Recent studies demonstrate that the stromal layer surrounding the epithelium plays a pivotal role in a wide variety of biological processes including malignancies (Bissell and Radisky, 2001; Wiseman and Werb, 2002). Therefore the study of cell interactions in heterotypic cultures, for example mixed epithelial and stromal cultures, has become particularly interesting (Yamada and Cukierman, 2007; Weigelt and Bissell, 2008). Such studies are preferentially carried out in a 3D environment because of the intrinsic limitations of mixed-cell 2D culture. Indeed, the study of cells in 3D has become a valid alternative to the use of animal models (Yamada and Cukierman, 2007) providing a reproducible, controlled microenvironment that resembles the behavior of cancer cells in solid tumors (Jacks and Weinberg, 2002; Birgersdotter et al., 2007).
The lack of reliable immunostaining techniques with cell-specific antibodies is a major limitation of 3D cultures. Currently two techniques are available: The first involves permeabilizing cells and visualizing them using expensive equipment such as confocal microscopy. The second involves whole-culture immunostaining (Lee et al., 2007) performed in the same chambers in which cells are grown, which also eventually requires confocal microscopy for optimal results. In both cases concerns are raised about access of antibodies into the cells at deeper layers. Here, we present an inexpensive and reliable alternative method to immunostain cells and cell subpopulations grown in 3D culture. This method is applicable to any species, cell type, and antigens for which appropriate antibody combinations are available.
Materials and Methods
Cells Lines
MCF10A cells were obtained from the University of Colorado Cancer Center Tissue Culture Core. MCF7 cells were from the Michigan Cancer Foundation. T47D cells were from Iafa Keydar (Israel). BT-474 cells were from the American Type Culture Collection (ATCC, Manassas, VA, USA). All cell lines were authenticated by Single Tandem Repeat analysis at the University of Colorado Cancer Center Sequencing Core. The BJ3Z mouse mammary gland malignant stromal cell line was generated in our laboratory (Jacobsen et al., 2006). MCF10A cells were routinely passaged in MEGM medium (Lonza, Walkersville, MD, USA). All other cell lines were passaged in MEM (Invitrogen, Carlsbad, CA, USA) containing 5% fetal bovine serum (FBS, HyClone, Logan, UT, USA).
3D Matrigel Culture
Cells were seeded into eight-well plastic-chambered glass microscope slides (BD Falcon, Cat No.354118) containing growth-factor reduced Matrigel™ or phenol red-free growth-factor reduced Matrigel™ (BD Biosciences, Bedford, MA, USA, Cat Nos. 354230 or 356231 respectively; hereafter called Matrigel) following a protocol described by Lee et al. (2007) with minor modifications. Briefly, Matrigel (0.5 ml aliquots) was thawed on ice for 3–4 h. Individual wells were coated with 50 μl of Matrigel using a p200 micropipette and a 1-ml syringe plunger to spread it evenly. Coated chambers were incubated at 37°C while cells were trypsinized and counted. Cell numbers used were: MCF10A and MCF7 10,000 cells/well or BT-474 and BJ3Z 50,000 cells/well. Cells were seeded on the surface of Matrigel-coated wells in a total volume of 200 μl MEM supplemented with 5% twice dextran-coated charcoal (DCC)-stripped FBS, except for MCF10A for which MEGM supplemented with 2% donor horse serum (DHS; GemCell, Gemini, West Sacramento, CA, USA) was used. Cells were maintained 7 days in an incubator at 37°C, 5% CO2 with fresh medium added every 2 days.
Phase Contrast Microscopy
Live cells in eight-well chambers were photographed on day 6 using a phase contrast filter in a Nikon Eclipse microscope model Ti, coupled to a DS-Qi1Mc camera (Nikon Corp., Japan). Images were acquired using NIS-Elements Advance Research software version 3.1 (Nikon).
Isolation of 3D Cultured Cells
Matrigel/cell blocks were processed on day 7 using the protocol depicted in Figure 1: a tube of HistoGel™ (Thermo Scientific Richard-Allan Scientific, Kalamazoo, MI, USA; Cat. HG-4000-012) specimen processing gel was thawed in a water-bath at 65–70°C for at least 2 h or until the gel was completely liquefied, then kept in the water-bath at all times. In Step 1, 5–10 min. before harvesting cells, biopsy cryomolds (Disposable vinyl specimen molds 10 mm × 10 mm × 5mm, Tissue-Tek, Sakura Finetek, Torrance, CA, USA; Cat No. 4565) were coated with 100–150 μl of warmed Histogel, which was spread evenly to completely cover the surface of the mold. In Step 2, the surface medium of Matrigel/cell wells was removed with a pipette tip and aspirator without disturbing the cells. The plastic chamber was removed from the glass slide using the tool provided by the manufacturer, leaving the Matrigel/cell blocks. These were scraped off the slide with a clean razor blade (0.009′ single edge, Smith Brand). In Step 3 the Matrigel/cell blocks were transferred onto the Histogel pre-coated mold using a sterile scalpel (Swann-Morton, Sheffield England; Cat No.DSS-20). A single Matrigel block (out of eight) can be transferred, but optimal results were obtained when four blocks were combined in a single mold. In Step 4 another 100–150 μl of warm Histogel was added on top of the Matrigel to form a “sandwich.” In Step 5 the mold was immediately transferred to ice and allowed to solidify for 10 min. Matrigel–Histogel “sandwiches” were then easily transferred by pressing the back of the molds, popping them out into processing/embedding blue pathology cassettes (Lab Storage, Cat #6054), which were closed, fixed ~16 h in 4% paraformaldehyde, transferred into 70% ethanol, and processed as usual before embedding in paraffin.
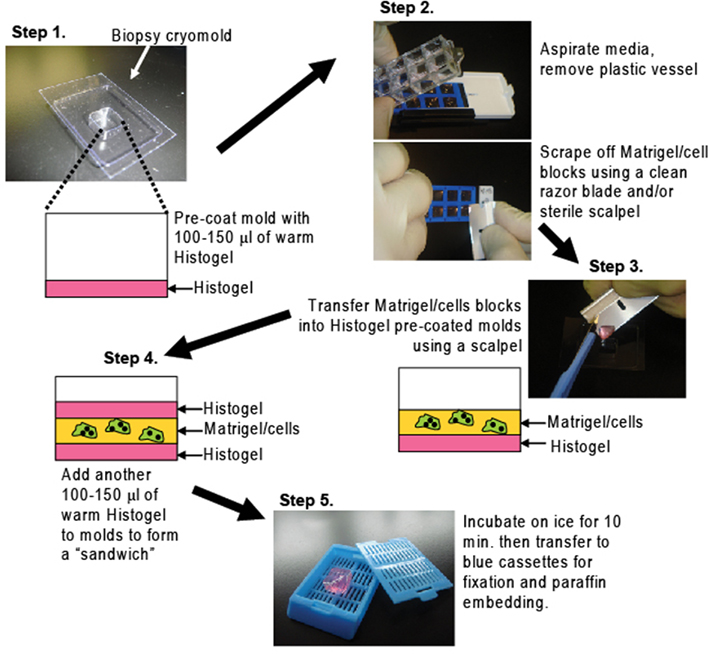
Figure 1. A method to process cells grown in Matrigel for paraffin embedding. Matrigel/cell blocks are encapsulated in a Histogel “sandwich” in biopsy cryomolds then transferred to blue pathology cassettes for paraffin embedding and routine H&E or IHC analyses (details in Materials and Methods)
Fluorescent Immunostaining
Paraffin blocks containing cells in Matrigel were sectioned using a standard rotary microtome (Leica Biosystems, Nussloch, Germany; RM2235). Four or 5 μm sections were floated in a 42–45°C water-bath then transferred onto pre-cleaned microscope glass slides (Mercedes Medical, Sarasota, FL, USA). Dried slides were baked at 65°C for 1 h and used for hematoxylin and eosin (H&E) staining or IHC. For IHC, sections were deparaffinized and antigen retrieval was performed in a pressure cooker (Biocare Medical) at 20 psi for 5 min in citrate buffer (10 mM sodium citrate, 0.05% Tween-20, pH 6.0). Sections were blocked 30 min with 10% normal goat serum and primary antibodies were applied against: Cytokeratin 5 (CK5, 1:200, rabbit monoclonal, Epitomics #2290-1) Pan-cytokeratin (pan-CK, 1:100, mouse monoclonal, BioLegend #628601); fibroblast activation protein (FAP, 1:100, rabbit polyclonal, abcam, Cambridge, MA, USA; #ab53066,). Fluorescent secondary antibodies were: Alexa fluor 555 (red) goat anti-rabbit IgG (1:300) and Alexa fluor 488 (green) goat anti-mouse IgG (1:400; both Invitrogen). Cell nuclei were counterstained with 4′-6-Diamidino-2-phenylindole (DAPI). Fluorescent images were obtained using a Nikon Eclipse E600 fluorescent microscope coupled to a RGB-MS-C micro color camera, and Image Pro Plus software version 4.5 (MediaCybernetics, Silver Spring, MD, USA).
Dual Fluorescence IHC
For MCF10A/BJ3Z co-cultures, a mouse monoclonal antibody against CK5 (1:100, Leica, NCL-L-CK5) and a rabbit polyclonal against alpha-smooth muscle actin (SMA; 1:400, Epitomics, Burlingame, CA, USA; #1184-1) were used with the above mentioned secondary antibodies. Fluorescent images were obtained as described.
BrdU Incorporation
To assess proliferation, live cells in Matrigel were incubated with BrdU (0.25 mg/ml final concentration) 1 h prior to harvesting then processed as described (Figure 1). Paraffin sections were used for dual fluorescence IHC with two additional steps: after antigen retrieval, slides were washed in water and, incubated in 2 N HCl for 1 h and in 0.1 M sodium borate decahydrate (pH 10.5) for 5 min before blocking. MCF10A cells were tagged with anti-CK14 (1:200, Thermo Scientific, Fremont, CA; RB9020-P1), MCF7 and BT-474 cells were tagged with anti-CK18 (1:400, Calbiochem #AP1021), and proliferating cells were tagged with anti-BrdU (1:50, Becton Dickinson, San Jose, CA, USA; 347580). For T47D cells in Figure 4 a CK5 rabbit monoclonal was used (Epitomics). Primary antibodies were detected with the above secondary antibodies, nuclei were counterstained with DAPI, and fluorescent microscope images were obtained.
Hormonal Treatments
For hormone treatment experiments T47D cells were plated as described in phenol red-free Matrigel and treated with ethanol (1:1,000 v/v) or estrogen (10 nM) plus progesterone (100 nM) for 2 weeks, adding fresh hormone or ethanol every 2 or 3 days. At day 14 cells were incubated in BrdU for 1 h, harvested and stained for CK5 and BrdU as described.
Results
Cells were plated in Matrigel as described by Lee et al. (2007) with minor modifications. Matrigel/cell blocks were then processed as summarized in Figure 1. Briefly, Matrigel/cell blocks are transferred to HistoGel pre-coated biopsy cryomolds then covered with a second HistoGel layer to stabilize the Matrigel block, and hardened. The HistoGel/Matrigel sandwich is fixed, paraffin embedded, and cut into serial sections by standard methods. These can be stained with (H&E) to assess structure or processed for multi-color immunohistochemistries (IHC) to identify specific cell subpopulations or measure proliferation indices.
Figure 2 (left panels) show characteristic phase contrast 3D colony morphologies: MCF10A cells form round, apparently hollow colonies as described previously (Debnath et al., 2003); MCF7 and BT-474 cells form solid clusters classified as “masses” (Kenny et al., 2007); BJ3Z cells form irregular solid masses confirming their malignant state. Paraffin-embedded colonies were serial sectioned and one section was stained by H&E (Figure 2, center). The method we describe efficiently preserves colony architecture, confirming for example, the hollow nature of the MCF10A spheroids. Other sections were used for IHC with primary antibodies selected for the desired cell type, fluorescent secondary antibodies best suited to the primary, plus counterstaining with DAPI (Figure 2, right). They were: for MCF10A cells, anti-Cytokeratin 5 (CK5; red); for MCF7 and BT-474 cells, pan-cytokeratin (pan-CK; green) and for BJ3Z cells, FAP-alpha (red).
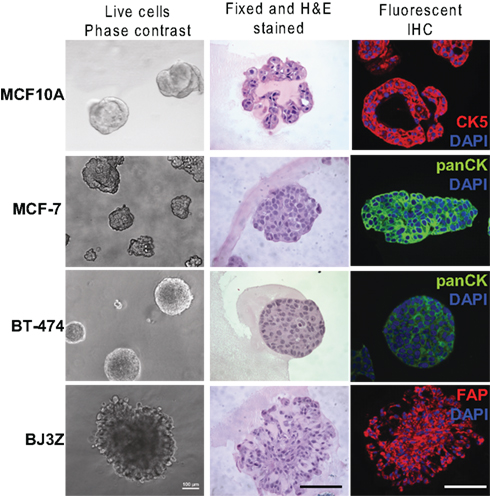
Figure 2. Colonies grown in Matrigel were analyzed by phase contrast, and by H&E and IHC of paraffin-embedded serial sections. MCF10A normal human breast, MCF7, and BT-474 human breast cancer, or BJ3Z activated mouse stromal cells were grown in Matrigel for 7 days. Live cells were imaged on day 6 by phase contrast (left). Matrigel/cell blocks were processed on day 7 as described and 5 μm paraffin sections were stained with H&E (center). For IHC (right), CK5 detected MCF10A cells (red); pan-CK detected the human breast cancer cells (green); fibroblast activation protein (FAP, red) detected the mouse stromal cells. Appropriate red or green fluorescent secondary antibodies were used and sections were counterstained with DAPI (blue). Scale: 100 μm
The stroma and its cellular components influence a wide variety of biological processes (Woodward et al., 1998; Bissell and Radisky, 2001; Weaver and Gilbert, 2004). Therefore study of heterotypic cultures consisting of two or more different cell types in 3D is of particular interest to understand cell-cell interactions in a controlled environment. For example, Figure 3A shows the 3D structures that form in a co-culture of human mammary epithelial MCF10A cells with mouse stromal BJ3Z cells, processed by our method. Cell clusters were stained against human-specific CK5, a marker of MCF10A cells (green) and against SMA, a marker of stromal cells (red). Note that the stromal BJ3Z cells are at the center, with the MCF10A cells growing as clusters on the stromal surface.
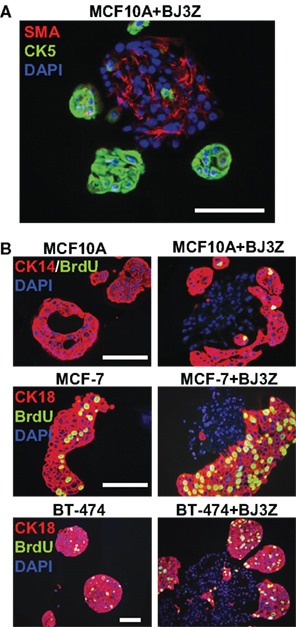
Figure 3. Three-dimensional cell subpopulations and proliferation rates. (A) MCF10A/BJ3Z co-cultures were stained against SMA (red) to tag the mouse stromal cells and anti-human CK5 (green) to tag the MCF10A cells. (B) Proliferation as measured by BrdU incorporation in different cell subpopulations. Human MCF10A, MCF7, or BT-474 cells were grown in 3D mono-culture (left) or in co-culture with BJ3Z mouse stromal cells (right). Anti-CK14 (red) was used for MCF10A cells; anti-CK18 (red) was used for MCF7 and BT-474 cells. All sections were dual stained for BrdU (green) and counterstained with DAPI (blue). Note enhanced proliferation of BJ3Z co-cultured breast cells and lack of stromal cell proliferation. When cells of different species are co-cultured, judicious choice of primary antibody pairs and fluorescent second antibodies is required. Scale: 100 μm
Another major limitation of current 3D culture methods is the lack of reliable protocols to measure proliferation of total cells or cell subpopulations. Most proliferation assays require extraction of the cells from Matrigel by enzymatic digestion to obtain single-cell suspensions, which are then counted with a hemacytometer (Shekhar et al., 2001) or by flow cytometry (Seidl et al., 2002). Not only could these assays alter cell behavior during processing; they also preclude independent analysis of cell subpopulations. Here we used BrdU incorporation to assess the proliferation rates of cell subpopulations in mixed 3D cell cultures by incorporating tandem cell type-specific markers into the assay. Normal or malignant human breast cells were mono-cultured (Figure 3B, left) or co-cultured with BJ3Z mouse stromal cells (Figure 3B, right) in Matrigel. Prior to harvesting, 3D cultures were incubated 1 h with BrdU, which is incorporated into DNA at S-phase and detected immunologically. Figure 3B (top panels) shows MCF10A normal human breast cells in mono-culture (left) or in co-culture with BJ3Z mouse stromal cells (right). MCF10A cells are identified by CK14 expression (red) and BrdU is identified by green fluorescence. In mono-culture, few if any MCF10A cells incorporate BrdU; co-culture with BJ3Z increases MCF10A proliferation. Note that the central DAPI-positive (blue) BJ3Z cells are CK14-negative and non-proliferative.
Figure 3B (middle) shows MCF7 human breast cancer cells, and Figure 3B (bottom) shows BT-474 human breast cancer cells in mono-culture or co-culture with BJ3Z mouse stromal cells. CK18 expression (red) was used to distinguish the breast epithelial cells from the stromal cells that they surround. Proliferation of the breast cancer cells (left) is increased by co-culture with stromal cells (right). The methods shown here can be used for any mono or heterotypic 3D cultured cells to assess their proliferation rate or combined with any other marker of interest provided specific antibodies are available.
Finally, previous studies have shown that ovarian hormones, namely estrogens (E) and progesterone (P) play a key role in breast tumor growth and differentiation (Clemons and Goss, 2001; Hankinson et al., 2004; Harrell et al., 2006) and that markers like CK5 identify cancer cell subpopulations with stem-like properties (Horwitz et al., 2008). Paradoxically, these cell subsets are characterized by low proliferation rates along with their enhanced ability to initiate tumors (Horwitz et al., 2008; Kabos et al., 2010). Recent studies suggest that ovarian hormones promote expansion of a stem-cell pool in the mammary glands of mice (Asselin-Labat et al., 2010; Joshi et al., 2010). Figure 4 shows T47D human breast cancer cells in 3D phenol red-free Matrigel colony cultures treated with either ethanol (Control, left) or a combination of E + P (right). Previous studies have shown that T47D cells express both estrogen (ER) and the progesterone (PR) receptor when cultured under these conditions (not shown). Note that in the absence of hormones (E or P) cells form relatively small colonies that lack CK5. Treatment of cells with E + P enlarges the colony clusters and triggers expression of CK5 (red) in a subset of cells. Pre-incubation of cells with BrdU allows quantification of proliferation in these cell subsets. Importantly, while proliferating cells (green nuclei) are present in both the control and E + P treated CK5-negative subpopulations, the CK5-positive stem-like cells do not take up BrdU and appear to be relatively quiescent.
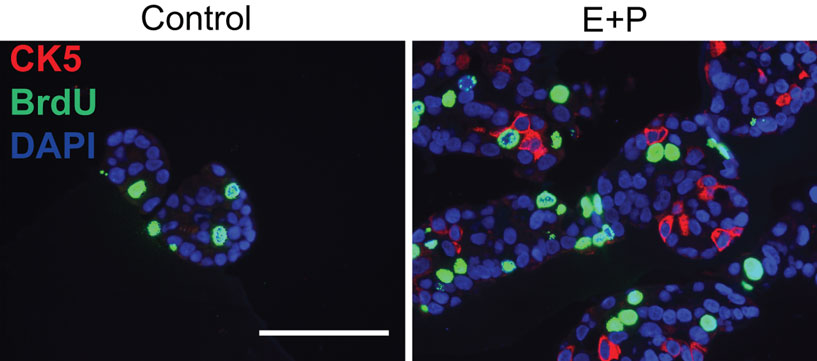
Figure 4. Hormone treatment induces basal-like Cytokeratin 5 (CK5) expression in a subset of luminal breast cancer cells. ER + PR + T47D cells were incubated with ethanol (Control) or a combination of estrogen and progesterone (E + P). Cells were also incubated in BrdU (green) to assess proliferation. Following E + P treatment, a subset of cells expressing CK5 (red) is expanded, that has little or no proliferative activity. Scale: 100 μm.
Discussion
During the last decade the development of 3D culture models have offered physiologically relevant alternatives to traditional 2D culture on plastic surfaces (Jacks and Weinberg, 2002; Kim et al., 2004; Debnath and Brugge, 2005; Kim, 2005; Weigelt and Bissell, 2008). However, study of cells in 3D cultures has been hampered by lack of simple methods to analyze them. Current techniques are time-consuming, require expensive equipment such as confocal microscopy for optimal results, and are poorly adapted to study cell subpopulations. Here we present a reliable, easy, and inexpensive method to transfer 3D cultured cells into paraffin blocks. Embedded cells can be sectioned in a microtome and used for routine H&E staining and standard IHC assays that target different cell types. A previous study used cells grown in 3D to generate sections for H&E stains (Kunz-Schughart et al., 2001). This study used cells grown on agarose which lacks the basement membrane components of Matrigel that influence cell behavior and morphology (Streuli and Bissell, 1990; Schmidhauser et al., 1992). A second recent study used human plasma and thrombin to generate a solid clot when added to Matrigel (Graham et al., 2009), allowing the transfer of cells for paraffin embedding. Results of this technique are similar to ours by allowing IHC and preserving the 3D morphology of cell colonies. However, processing to obtain plasma is time-consuming, requires regular access to human blood for plasma extraction including human research approval, and addition of plasma and thrombin to cells may affect their physiological properties.
In vivo, cells are influenced not only by the extracellular matrix but also by neighboring cells. Therefore methods to study heterotypic 3D cultures are essential (Kim et al., 2004; Weigelt and Bissell, 2008). A few heterotypic 3D models have been recently described: Shekhar et al. (2001) reported ductal–alveolar morphogenesis when MCF10A cells were co-cultured with tumor-derived breast fibroblasts. This effect was enhanced by the presence of human umbilical cord vascular endothelial cells. In this study, the investigators transferred the 3D cultured cells into paraffin blocks for H&E and colorimetric IHC analyses, but no technical details of their methods were provided. To determine cell proliferation, the cells were counted in a hemacytometer following enzymatic digestion of the Matrigel. Another report (Ronnov-Jessen et al., 1995) used cryo-sections, which are subject to freezing artifacts, changes in morphology and other limitations, to analyze cells in 3D collagen gels. Triple immunostaining showed that primary tumor cells converted normal SMA negative fibroblasts into SMA positive myofibroblasts. A recent report (Sasser et al., 2007) showed that proliferation of breast cancer cell lines in 3D was enhanced by co-culture with human bone marrow stromal cells. While this method efficiently quantified cell numbers in 3D, it required use of fluorescently pre-tagged cells.
The method presented here (Figure 1) preserves the 3D architecture of any cell type in mono- or co-culture with permanent paraffin sections and does not require fluorescent pre-tagged cells. BrdU incorporation allows quantification of proliferation in any desired cell subpopulation provided cell-specific antibodies are available (Figure 3). Of interest is the fact that by employing traditional IHC techniques the method can be used to optimize IHC conditions when tumors or other valuable samples are scarce.
The study of heterotypic 3D cultures is critical for understanding cell-cell interactions and the signal transduction pathways involved in the behavior of normal and cancer cells in an environment that resembles in vivo tissue or tumor conditions (Debnath and Brugge, 2005; Yamada and Cukierman, 2007). The putative role of ovarian hormones in expanding a breast cancer stem-like cell subpopulation (Figure 4) is a case in point. The development of simple IHC techniques that can be applied to 3D models offers excellent alternatives to costly animal models and their intrinsic variability. 3D models allow high throughput mechanistic studies of cell behavior, proliferation, apoptosis and migration, and lend themselves to high throughput drug screening (Friedrich et al., 2009) of anti-cancer agents while targeting cells in appropriate microenvironments (Bissell and Radisky, 2001). The assay described here adds to the power of 3D models.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported by Susan G. Komen Foundation Postdoctoral Fellowship (PDF0706748) to Mauricio P. Pinto Support also provided by the NIH RO1-CA026869, the Breast Cancer Research Foundation and Play for Pink, the National Foundation for Cancer Research, and the Avon Foundation for Women (Kathryn B. Horwitz).
References
Asselin-Labat, M. L., Vaillant, F., Sheridan, J. M., Pal, B., Wu, D., Simpson, E. R., Yasuda, H., Smyth, G. K., Martin, T. J., Lindeman, G. J., and Visvader, J. E. (2010). Control of mammary stem cell function by steroid hormone signalling. Nature 465, 798–802.
Barcellos-Hoff, M. H., Aggeler, J., Ram, T. G., and Bissell, M. J. (1989). Functional differentiation and alveolar morphogenesis of primary mammary cultures on reconstituted basement membrane. Development 105, 223–235.
Birgersdotter, A., Baumforth, K. R., Porwit, A., Sundblad, A., Falk, K. I., Wei, W., Sjoberg, J., Murray, P. G., Bjorkholm, M., and Ernberg, I. (2007). Three-dimensional culturing of the Hodgkin lymphoma cell-line L1236 induces a HL tissue-like gene expression pattern. Leuk. Lymphoma 48, 2042–2053.
Clemons, M., and Goss, P. (2001). Estrogen and the risk of breast cancer. N. Engl. J. Med. 344, 276–285.
Debnath, J., and Brugge, J. S. (2005). Modelling glandular epithelial cancers in three-dimensional cultures. Nat. Rev. Cancer 5, 675–688.
Debnath, J., Muthuswamy, S. K., and Brugge, J. S. (2003). Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 30, 256–268.
dit Faute, M. A., Laurent, L., Ploton, D., Poupon, M. F., Jardillier, J. C., and Bobichon, H. (2002). Distinctive alterations of invasiveness, drug resistance and cell-cell organization in 3D-cultures of MCF-7, a human breast cancer cell line, and its multidrug resistant variant. Clin. Exp. Metastasis 19, 161–168.
Friedrich, J., Seidel, C., Ebner, R., and Kunz-Schughart, L. A. (2009). Spheroid-based drug screen: considerations and practical approach. Nat. Protoc. 4, 309–324.
Graham, J. D., Mote, P. A., Salagame, U., Balleine, R. L., Huschtscha, L. I., and Clarke, C. L. (2009). Hormone-responsive model of primary human breast epithelium. J. Mammary Gland Biol. Neoplasia 14, 367–379.
Hankinson, S. E., Colditz, G. A., and Willett, W. C. (2004). Towards an integrated model for breast cancer etiology: the lifelong interplay of genes, lifestyle, and hormones. Breast Cancer Res. 6, 213–218.
Harrell, J. C., Dye, W. W., Allred, D. C., Jedlicka, P., Spoelstra, N. S., Sartorius, C. A., and Horwitz, K. B. (2006). Estrogen receptor positive breast cancer metastasis: altered hormonal sensitivity and tumor aggressiveness in lymphatic vessels and lymph nodes. Cancer Res. 66, 9308–9315.
Horwitz, K. B., Dye, W. W., Harrell, J. C., Kabos, P., and Sartorius, C. A. (2008). Rare steroid receptor-negative basal-like tumorigenic cells in luminal subtype human breast cancer xenografts. Proc. Natl. Acad. Sci. U.S.A. 105, 5774–5779.
Jacks, T., and Weinberg, R. A. (2002). Taking the study of cancer cell survival to a new dimension. Cell 111, 923–925.
Jacobsen, B. M., Harrell, J. C., Jedlicka, P., Borges, V. F., Varella-Garcia, M., and Horwitz, K. B. (2006). Spontaneous fusion with, and transformation of mouse stroma by, malignant human breast cancer epithelium. Cancer Res. 66, 8274–8279.
Joshi, P. A., Jackson, H. W., Beristain, A. G., Di Grappa, M. A., Mote, P. A., Clarke, C. L., Stingl, J., Waterhouse, P. D., and Khokha, R. (2010). Progesterone induces adult mammary stem cell expansion. Nature 465, 803–807.
Kabos, P., Haughian, J. M., Wang, X., Dye, W. W., Finlayson, C., Elias, A., Horwitz, K. B., and Sartorius, C. A. (2010). Cytokeratin 5 positive cells represent a steroid receptor negative and therapy resistant subpopulation in luminal breast cancers. Breast Cancer Res. Treat. DOI: 10.1007/s10549-010-1078-6. [Epub ahead of print].
Kenny, P. A., Lee, G. Y., Myers, C. A., Neve, R. M., Semeiks, J. R., Spellman, P. T., Lorenz, K., Lee, E. H., Barcellos-Hoff, M. H., Petersen, O. W., Gray, J. W., and Bissell, M. J. (2007). The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Mol. Oncol. 1, 84–96.
Kim, J. B. (2005). Three-dimensional tissue culture models in cancer biology. Semin. Cancer Biol. 15, 365–377.
Kim, J. B., Stein, R., and O’hare, M. J. (2004). Three-dimensional in vitro tissue culture models of breast cancer – a review. Breast Cancer Res. Treat. 85, 281–291.
Kleinman, H. K., and Martin, G. R. (2005). Matrigel: basement membrane matrix with biological activity. Semin. Cancer Biol. 15, 378–386.
Kunz-Schughart, L. A., Heyder, P., Schroeder, J., and Knuechel, R. (2001). A heterologous 3-D coculture model of breast tumor cells and fibroblasts to study tumor-associated fibroblast differentiation. Exp. Cell Res. 266, 74–86.
Lee, G. Y., Kenny, P. A., Lee, E. H., and Bissell, M. J. (2007). Three-dimensional culture models of normal and malignant breast epithelial cells. Nat. Methods 4, 359–365.
Li, C., Kato, M., Shiue, L., Shively, J. E., Ares, M. Jr., and Lin, R. J. (2006). Cell type and culture condition-dependent alternative splicing in human breast cancer cells revealed by splicing-sensitive microarrays. Cancer Res. 66, 1990–1999.
Ronnov-Jessen, L., Petersen, O. W., Koteliansky, V. E., and Bissell, M. J. (1995). The origin of the myofibroblasts in breast cancer. Recapitulation of tumor environment in culture unravels diversity and implicates converted fibroblasts and recruited smooth muscle cells. J. Clin. Invest. 95, 859–873.
Sasser, A. K., Mundy, B. L., Smith, K. M., Studebaker, A. W., Axel, A. E., Haidet, A. M., Fernandez, S. A., and Hall, B. M. (2007). Human bone marrow stromal cells enhance breast cancer cell growth rates in a cell line-dependent manner when evaluated in 3D tumor environments. Cancer Lett. 254, 255–264.
Schmidhauser, C., Casperson, G. F., Myers, C. A., Sanzo, K. T., Bolten, S., and Bissell, M. J. (1992). A novel transcriptional enhancer is involved in the prolactin- and extracellular matrix-dependent regulation of beta-casein gene expression. Mol. Biol. Cell 3, 699–709.
Seidl, P., Huettinger, R., Knuechel, R., and Kunz-Schughart, L. A. (2002). Three-dimensional fibroblast-tumor cell interaction causes downregulation of RACK1 mRNA expression in breast cancer cells in vitro. Int. J. Cancer 102, 129–136.
Shekhar, M. P., Werdell, J., Santner, S. J., Pauley, R. J., and Tait, L. (2001). Breast stroma plays a dominant regulatory role in breast epithelial growth and differentiation: implications for tumor development and progression. Cancer Res. 61, 1320–1326.
Streuli, C. H., and Bissell, M. J. (1990). Expression of extracellular matrix components is regulated by substratum. J. Cell Biol. 110, 1405–1415.
Weaver, V. M., and Gilbert, P. (2004). Watch thy neighbor: cancer is a communal affair. J. Cell. Sci. 117, 1287–1290.
Weaver, V. M., Lelievre, S., Lakins, J. N., Chrenek, M. A., Jones, J. C., Giancotti, F., Werb, Z., and Bissell, M. J. (2002). beta4 integrin-dependent formation of polarized three-dimensional architecture confers resistance to apoptosis in normal and malignant mammary epithelium. Cancer Cell 2, 205–216.
Weigelt, B., and Bissell, M. J. (2008). Unraveling the microenvironmental influences on the normal mammary gland and breast cancer. Semin. Cancer Biol. 18, 311–321.
Wiseman, B. S., and Werb, Z. (2002). Stromal effects on mammary gland development and breast cancer. Science 296, 1046–1049.
Woodward, T. L., Xie, J. W., and Haslam, S. Z. (1998). The role of mammary stroma in modulating the proliferative response to ovarian hormones in the normal mammary gland. J. Mammary Gland Biol. Neoplasia 3, 117–131.
Keywords: three-dimensional culture, immunohistochemistry, breast cancer, Matrigel, proliferation
Citation: Pinto MP, Jacobsen BM and Horwitz KB (2011) An immunohistochemical method to study breast cancer cell subpopulations and their growth regulation by hormones in three-dimensional cultures. Front. Endocrin. 2:15. doi: 10.3389/fendo.2011.00015
Received: 16 May 2011;
Paper pending published: 27 May 2011;
Accepted: 09 June 2011;
Published online: 21 June 2011.
Edited by:
Antonino Belfiore, University Magna Græ cia of Catanzaro, ItalyReviewed by:
Marcello Maggiolini, University of Calabria, ItalyDidier Picard, University of Geneva, Switzerland
Copyright: © 2011 Pinto, Jacobsen and Horwitz. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Mauricio P. Pinto, Department of Medicine, University of Colorado Anschutz Medical Campus, Mail Stop 8106, 12801 East 17th Avenue, Aurora, CO 80045, USA. e-mail:bWF1cmljaW8ucGludG9AdWNkZW52ZXIuZWR1