

- Laboratory of Neuroendocrinology, Department of Neurobiology, Brain Research Institute, David Geffen School of Medicine at University of California Los Angeles, Los Angeles, CA, USA
Estradiol has profound actions on the structure and function of the nervous system. In addition to nuclear actions that directly modulate gene expression, the idea that estradiol can rapidly activate cell signaling by binding to membrane estrogen receptors (mERs) has emerged. Even the regulation of sexual receptivity, an action previously thought to be completely regulated by nuclear ERs, has been shown to have a membrane-initiated estradiol signaling (MIES) component. This highlighted the question of the nature of mERs. Several candidates have been proposed, ERα, ERβ, ER-X, GPR30 (G protein coupled estrogen receptor), and a receptor activated by a diphenylacrylamide compound, STX. Although each of these receptors has been shown to be active in specific assays, we present evidence for and against their participation in sexual receptivity by acting in the lordosis-regulating circuit. The initial MIES that activates the circuit is in the arcuate nucleus of the hypothalamus (ARH). Using both activation of μ-opioid receptors (MOR) in the medial preoptic nucleus and lordosis behavior, we document that both ERα and the STX-receptor participate in the required MIES. ERα and the STX-receptor activation of cell signaling are dependent on the transactivation of type 1 metabotropic glutamate receptors (mGluR1a) that augment progesterone synthesis in astrocytes and protein kinase C (PKC) in ARH neurons. While estradiol-induced sexual receptivity does not depend on neuroprogesterone, proceptive behaviors do. Moreover, the ERα and the STX-receptor activation of medial preoptic MORs and augmentation of lordosis were sensitive to mGluR1a blockade. These observations suggest a common mechanism through which mERs are coupled to intracellular signaling cascades, not just in regulating reproduction, but in actions throughout the neuraxis including the cortex, hippocampus, striatum, and dorsal root ganglias.
Introduction
Estrogens have broad and profound effects on the structure and function of the CNS. In the hippocampus, estradiol enhances cognition, and memory and learning (Yildirim et al., 2008; Mamiya et al., 2009), effects which are in large part due to spinogenesis that increase synapses (Woolley and McEwen, 1992; Cooke and Woolley, 2005; Sasahara et al., 2007). In the cortex and substantia nigra, estradiol has been shown to be neuroprotective (Quesada and Micevych, 2004; Wise et al., 2005; Dubal et al., 2006; Quesada et al., 2008), whereas in sensory systems, estradiol modulates nociception (Bradshaw and Berkley, 2003; Cason et al., 2003; Chaban et al., 2003, 2004; Chaban and Micevych, 2005; Berkley et al., 2007; Loyd and Murphy, 2008; Murphy et al., 2009). In the hypothalamus, estradiol from the ovaries induces the synthesis of neuroprogesterone (neuroP) that is required for estrogen positive feedback inducing the luteinizing hormone (LH) surge (reviewed in Micevych et al., 2009). Recent results from a number of laboratories, including ours, have demonstrated that in addition to classical nuclear-initiated actions, estrogens act on membrane receptors to affect cell signaling that underlie many of these actions. This paper will review studies including those in our laboratory that demonstrate that membrane-initiated estradiol signaling (MIES) is an important component of female sexual receptivity measured by lordosis – a behavior traditionally thought to be dominated by nuclear estradiol action.
Lordosis Behavior
Sexual behavior in female rats can be distinctly categorized into proceptive and receptive behaviors. Proceptive behaviors in estrus female rats include behaviors that primarily attract and solicit mounting by male rats such as abrupt hopping and darting sequences usually accompanied by a crouching posture, and ear-wiggling (reviewed in Erskine, 1989). Another aspect of female proceptive behavior is pacing, which is defined as the intermittent approach and withdrawal of the female from the male (McClintock and Adler, 1978). This component of proceptive behavior allows the female to control the rate of coital stimulation received and is a vital factor as it can have positive reinforcing components on sexual behavior (Bermant and Westbrook, 1966) and induce a reward state (Erskine, 1985; Paredes and Alonso, 1997). When experimentally testing sexual behaviors under traditional laboratory conditions such as in small encased testing arenas, hopping, and darting as well as ear-wiggling are commonly observed. However when rats are in their natural habitat, pacing seems to be the more commonly observed event.
In contrast to proceptive behaviors, receptive behaviors are often equally observed in either environment since these behaviors are characterized by a consummatory act. When the male rat has successfully pursued the female rat and subsequently mounted her from the back, a lordosis reflex is elicited. This reflex is caused by appropriate hormonal priming and palpation of the female flanks and perineum by the male. As a result, the female exhibits a distinct spinal dorsiflexion of the tail, arching of the back, extension of the neck, and elevation of the hindquarters and rump to allow for male intromission (Beach, 1948). To quantify sexual receptivity, a lordosis quotient, which is defined as the number of lordotic postures displayed by the female divided by the number of mounts × 100, has been commonly used (Beach, 1948; Beach and LeBoeuf, 1967).
Sexual receptivity is regulated by a well-defined circuit that spans the limbic system and hypothalamus to include the posterodorsal medial amygdala (MeApd), bed nucleus of the stria terminalis (BNST), medial preoptic nucleus (MPN), ventromedial nucleus of the hypothalamus (VMH), and the arcuate nucleus (ARH). Together these brain regions integrate hormonal information, sensory input from the accessory olfactory system (AOS), and tactile stimulation from the perineum and flanks to elicit the lordosis reflex. The AOS plays a necessary role in the detection of relevant olfactory cues during sexual receptivity as its sensory neurons project to the BNST by way of the MeApd. Together with somatic sensory cues into the periaqueductal gray (PAG), reticular formation, and vestibular nuclei, these inputs will integrate and converge to provide descending projections to the spinal cord to activate medial lying dorsal horn motoneurons to produce the stereotypic lordotic posture.
Hormonal Priming of Sexual Behavior
In gonadally intact animals, female rodent sexual receptivity is induced by the sequential actions of peak estradiol on proestrus followed by progesterone on the limbic-hypothalamic circuit. This robust behavior is easily quantifiable and exquisitely sensitive to exogenous estradiol and progesterone, as seen in studies conducted with ovariectomized (OVX) animals. In such rats, both estradiol-only and estradiol + progesterone paradigms have been used to study the inhibitory and facilitative circuits involved in sexual behavior and each has been used to garner important information about the steroid responsiveness of the CNS circuits regulating lordosis behavior. Since each steroid priming paradigm has inherent advantages, the choice of which to use has been dictated by the question to be addressed. It is clear from half a century of investigation that although the behavioral output is similar, estradiol and estradiol + progesterone priming either activates different circuits or activates the same circuits in a temporally distinct way. In animals primed with just estradiol, the dose required is much higher than the dose needed when priming animals with estradiol + progesterone to obtain sexual receptivity (Pfaff, 1970). Moreover, when using estradiol-only, repetitive estradiol treatments are required every 4 days to ramp animals to a constant level of receptivity compared to one big bolus treatment of estradiol followed by progesterone (Sodersten and Eneroth, 1981; Bloch et al., 1987). Following estradiol treatment, sexual receptivity cannot be facilitated by progesterone for approximately 20 h (Sinchak and Micevych, 2001).
The interactions between estradiol and progesterone are important in the understanding of how estradiol regulates the cell signaling that underlies sexual behavior. It is becoming apparent that the interaction between steroids of peripheral- and central-origin is important for this regulation. Endogenous ovarian or exogenous estradiol dramatically increases neuroP synthesis in the hypothalamus (Micevych et al., 2003, 2007). NeuroP synthesis is regulated by estradiol through a membrane associated estrogen receptor (mER) that increases free cytoplasmic calcium concentration ([Ca2+]i). This de novo synthesis of neuroP in the hypothalamus is critical for regulating the LH surge – the central event of reproduction. Blocking neuroP synthesis prevents the E2-induced LH surge and blocking hypothalamic steroid synthesis in cycling rats disrupts the estrous cycle. While all this has been shown to be important for the regulation of estrogen positive feedback of the LH surge (reviewed in Micevych and Sinchak, 2007), the synthesis of neuroP or activation of classical progesterone receptors does not appear to affect estradiol-only induced lordosis behavior (Micevych and Sinchak, 2007). On the other hand, estradiol-only induced proceptive behavior is blocked by trilostane (TRI) or aminoglutethimide (AGT), antagonists of progesterone synthesis, indicating a role for neuroP in mediating proceptive behaviors (Figure 1; Micevych et al., 2008). These results are congruent with the long standing idea that sexual receptivity is primarily driven by estradiol and proceptivity needs progesterone (reviewed in Barfield et al., 1984).

Figure 1. Effects of blocking the synthesis of progesterone and progesterone receptors in the CNS on sexual behavior. Blocking either progesterone synthesis (A,B) with aminoglutethimide (AGT) or trilostane (TRI) or activation of progesterone receptors (C) with RU486 reduces expression of proceptive behaviors, but has no effect on expression of lordosis in OVX/ADX rats treated every 4 days with 10 μg 17β-estradiol benzoate (EB) and then 1 h before the test with EB and drug. Animals were tested 53–56 h after the initial EB injection for sexual receptivity, as measured by lordosis quotient (LQ; Beach, 1948; Beach and LeBoeuf, 1967) and expression of proceptive behaviors (proceptivity scale; Tennent et al., 1980). Treatments to block progesterone synthesis or progesterone receptors were delivered subcutaneously and started just before the EB treatment and on the following mornings before testing. AGT (10 mg per treatment) blocks P450 side chain cleavage that converts cholesterol to pregnenolone (A); TRI blocks the enzyme 3β-hydroxysteroid dehydrogenase (16.5 mg per treatment; 3β-HSD) which converts pregnenolone to progesterone (B). RU486 (5 mg per treatment) is a progesterone receptor antagonist (C). Data are means ± SEM of 12 animals. * Represents significantly less than control treatment within behavior group as determined by Mann–Whitney, where p > 0.05 (from Micevych et al., 2008).
We have used both steroid treatments, estradiol-only and estradiol + progesterone, to study steroid activation of CNS circuits and our data point to the ARH as the site at which estradiol initially activates the lordosis-regulating limbic-hypothalamic circuit (Mills et al., 2004; Dewing et al., 2007). Here, MIES stimulates a microcircuit involving the neuropeptide Y (NPY) innervation of the pro-opiomelanocortin (POMC) neurons (expressing β-endorphin) in the ARH. Activation of NPY-Y1 receptors on neurons that project to the MPN increases the release of β-endorphin. As a result, μ-opioid receptor (MOR) neurons in the MPN that project to the VMH are inhibited, producing a transient inhibition necessary for full lordosis behavior (Torii et al., 1996, 1999; Sinchak and Micevych, 2001; Mills et al., 2004). Subsequently, estradiol induces necessary gene transcription and translation for the expression of lordosis behavior (reviewed in Micevych and Sinchak, 2007). The integrated response of the entire limbic-hypothalamic circuit is funneled through the VMH to descending projections that activate spinal motoneurons innervating muscles responsible for lordosis behavior (Calizo and Flanagan-Cato, 2003; Sinchak et al., 2010; reviewed in Pfaff et al., 2008).
Membrane-Initiated Signaling: Estrogen Receptors
The current situation with respect to membrane estrogen receptors (ERs) remains unsettled. A number of proteins have been suggested as ERs, including the classical nuclear receptors ERα and ERβ, ER-X, STX-activated membrane ER (mER), and GRP30, also known as G protein coupled estrogen receptor (GPER). Lordosis behavior is dependent on estradiol priming. To date, a preponderance of evidence points to ERα as the primary ER mediating reproduction (Rissman et al., 1997; Micevych et al., 2003; Wintermantel et al., 2006). However, other putative receptors, such as ERβ, STX-receptor, and GRP30, have been suggested to participate in regulating reproduction in different models (Carmeci et al., 1997; Qiu et al., 2003, 2006; Revankar et al., 2005; Kuo et al., 2009). These have been recently reviewed (Micevych and Mermelstein, 2008; Micevych and Dominguez, 2009) and this review will focus on their ability to influence lordosis behavior.
ER-X is a novel membrane ER that is expressed primarily in the cortex during development (post-natal days 7–10) or after trauma, uterus, and lung plasma membrane microdomains associated with caveolin proteins (Pappas et al., 1995; Toran-Allerand et al., 2002; Toran-Allerand, 2005). Unlike other ERs, it is activated by estradiol stereoisomers, 17α-estradiol and 17β-estradiol, but preferentially binds the former, which is inactive at ERα or ERβ. The ER antagonist ICI 182,780, which is considered a “universal” ER antagonist, paradoxically activates ER-X. In terms of female sexual receptivity, ICI 182,780 prevented the estradiol-induced activation/internalization of MOR. This result suggests that ER-X is not involved since ICI 182,780 is an agonist at the putative ER-X and would have facilitated the estradiol action. Ergo, its developmental profile and its promiscuous binding of both estradiol stereoisomers make it unlikely that ER-X is the ER responsible for mediating sexual receptivity.
Recently, estradiol has been shown to stimulate a seven transmembrane integral protein, GPR30, expressed throughout the brain and periphery (Revankar et al., 2005; Prossnitz et al., 2008). Studies have shown that estradiol stimulation of cells transfected with GPR30 can be blocked by the ER antagonist ICI 182,780. However, other results have shown the converse (Thomas et al., 2007). In our hands, stimulation of GPR30 with the selective agonist, G-1, did not induce internalization of MOR in the MPN or affect lordosis behavior suggesting that GPR30 does not activate the ARH – MPN circuit with regards to lordosis behavior (Dewing et al., 2007). These results are consistent with an inability to detect ER-X or GPR30 on the cell membrane after surface biotinylation on primary neuronal cultures.
Interestingly, STX acting on a yet uncharacterized receptor mimics the actions of estradiol. STX, a diphenylacrylamide compound that structurally resembles 4-OH tamoxifen, is a ligand for a proposed novel mER (Qiu et al., 2003). This synthetic estrogen receptor modulator has been shown to induce activation of a G protein signaling cascade through phospholipase C/inositol triphosphate and have estrogenic effects in a system absent of ERα and ERβ (Roepke et al., 2011). The STX-binding protein binds stereospecifically to estradiol and is blocked by the classical ER antagonist ICI 182,780. When microinjected into the ARH of OVX rats, STX activates the MPN–ARH circuit by inducing internalization of MOR, a similar effect is seen with estradiol (Figure 2). STX also facilitates sexual receptivity, as measured by a lordosis quotient, in estrogen primed females compared with estrogen alone females (Christensen et al., 2011). Further evidence for STX action was obtained by using immortalized ARH neurons, N-38 cells, in which STX increased [Ca2+]i through the release of intracellular stores (Kuo, personal communication). How these data align with various studies that demonstrate the necessity of ERα for reproductive behavior, including MOR internalization, is not readily apparent and will require further experimentation.
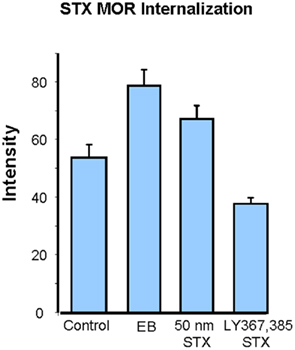
Figure 2. Comparison of estradiol with STX-induced μ-opioid receptor (MOR) in the medial preoptic nucleus (MPN). Estradiol, STX, LY 367,385, and control (aCSF) were injected into the arcuate nucleus of the hypothalamus of OVX rats and then transcardially perfused 30 min later with chilled 0.9% saline followed by 4% paraformaldehyde in Sorenson’s buffer. Sections from the arcuate nucleus (ARH) were processed for MOR internalization using rabbit primary antibodies directed against MOR (1:24,000; Neuromics). Histogram illustrates the ability of STX to induce MOR internalization similar to EB, as measured by immunofluorescence staining intensity in the MPN. Blocking mGluR1a with the antagonist LY 367,385 attenuates STX-induced MOR internalization. * Represents p < 0.05 compared to control as determined by one-way ANOVA.
While the dependence of sexual receptivity on ERα appears clear, how MIES fits into this model is now beginning to emerge. We and others have identified full length ERα in membrane fractions (Chaban et al., 2004; Gorosito et al., 2008; Bondar et al., 2009; Dominguez and Micevych, 2010). In native and immortalized hypothalamic neurons, the major membrane ERα-immunoreactive protein is coded for by an alternative splicing of the ERα gene (Bondar et al., 2009; Dewing and Micevych, unpublished observations). In addition to the full length ERα (66 kDa), a prominent 52 kDa ERα immunoreactive protein was revealed in native hypothalamic and N-38 neurons. PCR experiments demonstrated that the 52-kDa ERα variant was coded for by an ERα mRNA with a deletion of the fourth exon, ERαΔ4. An interesting observation had been that MIES-mediated MOR activation/internalization was temporally constrained in vivo (Dewing et al., 2007). In vitro, estradiol modulated membrane levels of ERα, providing a mechanism to explain the observation of temporal constraint of MIES in our lordosis assay (Bondar et al., 2009; Dominguez and Micevych, 2010). Under estrogen-starved conditions, estradiol activated ER-mediated events, suggesting a population of mER on the cell membrane (Eckersell et al., 1998; Chaban et al., 2004). Surface biotinylation confirmed ERα and ERαΔ4 on the cell plasma membrane (Gorosito et al., 2008; Bondar et al., 2009; Dominguez and Micevych, 2010) that were transiently increased with estradiol treatment. This trafficking of ERα and ERαΔ4 to the membrane is dependent on activation of protein kinase C θ (PKCθ) in neurons. Consequently, this same novel PKC was activated by MIES in vivo.
Estradiol also regulates the internalization of mER. Internalization of agonist bound receptors is a well-described characteristic of many membrane receptors and is an assay for receptor activation (reviewed in Sinchak and Micevych, 2003). In hypothalamic neurons, the appearance of pits in the plasma membrane suggests endocytic processes (Olmos et al., 1987; Garcia-Segura et al., 1988). The mechanism of internalization involves phosphorylation by G protein coupled receptor (GPCR) kinases (GIRKs), binding of β-arrestin and adaptor/scaffolding proteins, and sequestration into early endosomes (Dominguez et al., 2009; Micevych and Dominguez, 2009). As illustrated in Figure 3, once the receptor has released its ligand, the endosomes can either recycle back to the plasma membrane or fuse with a lysosome leading to degradation of the receptor or down-regulation (reviewed in Ritter and Hall, 2009). Thus, internalization is a relatively quick process and does not diminish the number of receptors. On the other hand, degradation/down-regulation is a slower process that, as its name suggests, produces a down-regulation in receptor number. The two events are related but receptor internalization is not necessarily coupled to down-regulation. For example, estradiol-induced MOR internalization did not result in a change in the total number of receptors (Eckersell et al., 1998).
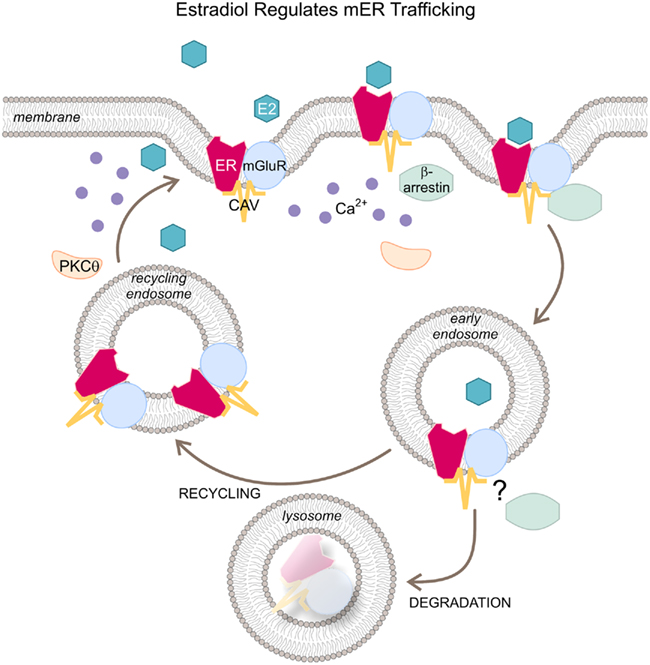
Figure 3. Schematic representation of estradiol’s regulation of mER trafficking. Upon activation by a ligand (E2), membrane ER is internalized and the agonist-receptor complex is phosphorylated by G protein coupled receptor kinases (GIRKs). β-arrestins are attached to the receptor along with adaptor/scaffolding proteins (e.g., caveolin) and rapidly internalized into early endosomes. At this point, the receptor is either dissociated from its agonist and recycled back to the plasma membrane or degraded by fusing with lysosomes. Abbreviations: E2, estradiol; ER, estrogen receptor; CAV, caveolin; mGluR1, metabotropic glutamate receptor 1; PKCθ, protein kinase Cθ.
In primary hypothalamic neuronal cultures, the rate of mER internalization increased in parallel with its insertion into the membrane, suggesting a coupling between receptor activation and trafficking. Both the number of receptors in the membrane and their activation/internalization peaked at 30 min while continuous estradiol treatment reduced the levels of mERα to basal. These data directly imply that estradiol can significantly increase membrane levels of the full length and variants of ERα. They also illustrate another important feature of MIES – that estradiol can temporally regulate ERα trafficking into and out of the plasma membrane, recapitulating our in vivo observations (Bondar et al., 2009). Two hours of estradiol treatment was no longer sufficient to induce the same levels of trafficking and internalization. By 24 h of estradiol treatment, internalized (and surface membrane) levels of mERα were below basal suggesting ER degradation, i.e., down-regulation (Bondar et al., 2009; Dominguez and Micevych, 2010). Thus, by regulating the level of ER on the membrane, estradiol controls its own level of signaling.
Membrane-Initiated Signaling: Estrogen Receptor has Partners for Signaling
Over the years, a common mechanism of MIES has been proposed: membrane estradiol actions involve interactions of ERs with traditional cell surface receptors. Two kinds of interactions have been demonstrated. The first is ER interactions with growth factor receptors such as insulin-like growth factor 1 (IGF-1) receptors (Toran-Allerand et al., 1988; Fernandez-Galaz et al., 1999; Quesada and Etgen, 2002; Quesada and Micevych, 2004) to stimulate signal transduction. In the hypothalamus, ERα has been reported to regulate lordosis behavior through an interaction with the IGF-1 receptor (Quesada and Etgen, 2002). Blocking IGF-1 receptors abates the neuroprotective effects of estradiol, suggesting that the neuroprotection actions of estrogen are through the IGF-1 system. The mechanism by which ER and IGF-1 receptors transduce signaling is still unclear, although there is evidence that ERα, ERβ, and IGF-1 receptor colocalize in both neurons and glial cells throughout adult rat brain (Cardona-Gomez et al., 2000). In addition, ERβ interacts with IGF-1 receptors in the midbrain to provide neuroprotection for nigrostriatal neurons (Quesada and Micevych, 2004; Quesada et al., 2007, 2008) and ER co-immunoprecipitates with IGF-1 and their activation subsequently phosphorylates phosphatidylinositol 3-kinase (IP3K; Mendez et al., 2003; Quesada et al., 2008). Thus emerging data has demonstrated crosstalk between ER and IGF-1 receptors resulting in stimulation of rapid signaling cascades.
More recently, another mER association has been discovered that explains how MIES can be stimulatory or inhibitory by interacting with different metabotropic glutamate receptors (mGluR). Acting through type I mGluRs, ER can activate MAPK dependent signaling, CREB or release intracellular Ca2+ stores. Acting through type II mGluRs, ERs can inhibit adenylyl cyclase leading to reduced influx through L-type voltage gated calcium channels (VGCC; Boulware et al., 2005; Dewing et al., 2007; Chaban et al., 2011; reviewed in Mermelstein and Micevych, 2008; Micevych and Mermelstein, 2008; Micevych and Dominguez, 2009). Such mER–mGluR interactions have been discovered throughout the neuraxis, from primary sensory neurons of the dorsal root ganglia (DRG) to the hippocampus, striatum, and hypothalamus in neurons and astrocytes. Estradiol regulation of sexual receptivity requires the interaction of ERα and mGluR1a, as demonstrated by a lack of MOR internalization and lordosis behavior following the blockade of mGluR1a in the ARH of estradiol primed rats (Dewing et al., 2007). In vivo, ERα was colocalized in mGluR1a expressing neurons in the ARH and co-immunoprecipitation studies demonstrated the potential physical interactions of these receptors in membranes collected from the ARH (Dewing et al., 2008). Further, we established that MIES involving ER–mGluR1a was responsible for estradiol facilitation of neuroP synthesis in hypothalamic astrocytes (Kuo et al., 2009). Finally, in DRG neurons, MIES attenuated ATP-induced Ca2+ influx through VGCC, a cellular event associated with nociceptive signaling (Chaban et al., 2003; Chaban and Micevych, 2005). In DRG neurons, ERα transactivates mGluR2/3, which inhibits adenylyl cyclase and PKA blocking the VGCC (Chaban et al., 2004, 2011; reviewed in Micevych and Mermelstein, 2008). Together these results demonstrate that by interacting with different mGluRs, ERα can mediate inhibitory or excitatory actions of estradiol, which speaks to the global importance of ER–mGluR interactions mediating MIES.
Membrane-Initiated Signaling: Cellular Events
Lordosis behavior depends on an ERα-dependent β-endorphin projection from the ARH that acts on MOR in the MPN (Micevych et al., 2003). Interestingly, blocking MIES in the ARH with a mGluR1a antagonist prevented MOR internalization and completely abrogated lordosis behavior (Dewing et al., 2007). Furthermore, when mGluR1 was activated in animals primed with a sub-behavioral dose of estradiol, lordosis was greatly facilitated. Thus, MIES is involved in both rapid signaling and may participate in a membrane to nuclear signaling that is important for the expression of proteins needed for lordosis behavior.
Estradiol activated a number of intracellular pathways in the ARH, including phosphorylated PKCθ and protein kinase A (PKA; Dewing et al., 2008). Site-specific blockade of PKC in ARH with a general PKC inhibitor, bisindolylmaleimide (BIS), significantly attenuated estradiol-induced MOR internalization in the MPN (Figure 4). Furthermore, disruption of PKC signaling within the ARH at the time of estradiol treatment significantly diminished the lordosis reflex. Even when mGluR1 was activated with an agonist, (S)-3,5-dihydroxyphenylglycine (DHPG), blocking PKC prevented MOR internalization. Conversely, activating PKC with phorbol 12, 13-dibutyrate (PDBu) with and without estradiol-induced MOR internalization to levels equivalent to estradiol-only control animals. These data indicate that PKC activation is downstream of mGluR1a and PKC is a critical step for membrane ERα-initiated mGluR1a-mediated cell signaling. PDBu significantly facilitated lordosis compared with rats primed with a sub-behavioral dose of estradiol. Together with ERα–mGluR1a co-immunoprecipitation, these findings indicate that ERα transactivation of mGluR1a is necessary for phosphorylation of the novel Ca2+-independent PKCθ in the ARH (Dewing et al., 2008). This rapid activation of cell signaling in vivo initiates the pathway leading to MOR internalization and modulation of lordosis behavior (Dewing et al., 2007, 2008; reviewed in Micevych and Dominguez, 2009). Interestingly, STX initiates MIES by activating similar cell signaling cascades to those activated by ERα-induced MIES. Both activate novel PKCs, PKCδ by STX and PKCθ by estradiol (Qiu et al., 2003). Additionally, STX-induced [Ca2+]i is blocked by the antagonism of mGluR1a, suggesting a convergence of signaling by ERα and STX-activated receptors.
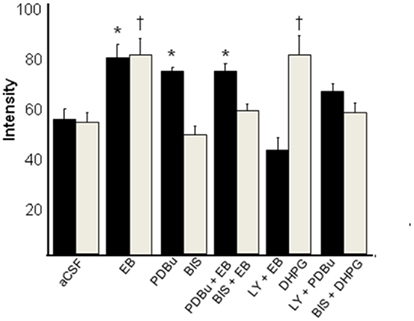
Figure 4. Estradiol-induced μ-opioid receptor (MOR) internalization in the medial preoptic nucleus (MPN) is dependent on protein kinase C θ (PKCθ) activation in the arcuate nucleus of the hypothalamus (ARH). This graph represents two experiments: animals infused with a PKC inhibitor, bisindolylmaleimide (BIS; 50 nmol, gray bars), and animals infused with a PKC activator, phorbol 12,13-dibutyrate (PDBu; 25 nmol, black bars). Animals sacrificed 30 min after EB injections displayed a significant increase in MOR internalization compared to oil injected animals. Antagonizing with BIS attenuated this EB-induced MOR internalization in the MPN. PDBu did not increase the level of internalization above that seen in EB-only treated animals. However the PKC activator did induced MOR internalization in the absence of EB treatment suggesting that estradiol and PDBu act through the same signaling pathway. The mGluR1 antagonist (LY367385) and agonist (DHPG) were also infused in combination with BIS or PDBu to determine whether PKC activation was downstream of ER/mGluR1 signaling. These data correlate with behavioral data that show that rats treated with BIS before EB were less receptive than aCSF-treated, EB-primed rats, and that PDBu did not induce lordosis behavior in animals that were not treated with a sub-behavioral dose of EB (2 μg; Dewing et al., 2008; Yildirim et al., 2008). * = statistical significance at the p < 0.05 level compared with aCSF + oil group as determined by two-way ANOVA and post hoc analysis (from Dewing et al., 2008).
Conclusion
Recent observations have begun painting an interesting picture of steroid signaling in the brain. The idea of MIES has gained widespread support from experiments that demonstrated a rapid estradiol activation of cell signaling. This was bolstered with observations that membrane-impermeable constructs, in spite of theoretical concerns, would also activate a cornucopia of kinases and mobilize intracellular stores of calcium. Estradiol acting through membrane receptors has even shown to activate sexual receptive behavior – a phenomenon long thought to be driven by direct nuclear actions of estradiol. Membrane ERs appear to behave like GPCRs since they rapidly activate cellular signaling systems. These actions require interacting directly or indirectly with different classes of membrane receptors such as the IGF-1 receptor, the oxytocin receptor, and the mGluRs. Transactivation of mGluRs provide a mechanism through which estradiol can produce both facilitation and inhibition of cellular events. The direction of these events was determined ultimately by which mGluR was coupled with the ER. Regulation of sexual receptivity necessitated transactivation of the mGluR1a. Significantly, estradiol controlled its membrane signaling by regulating ER trafficking and internalization of ERα (and ERαΔ4). In addition to these “post-synaptic” events, in recent years, the idea that the brain synthesizes neurosteroids has been revived. Thus, neurons and glial cells have been shown to synthesize sex steroids that like other fourth generation (4-G) neurotransmitters (i.e., nitric oxide, endocannabinoids, and carbon monoxide) are regulated at the point of synthesis (reviewed in Micevych and Sinchak, 2008). Neuroprogesterone is an important component of the CNS control of female proceptive behaviors. Although it remains puzzling that MIES-activated neuroP synthesis is sufficient to trigger the LH surge and augment estradiol initiated proceptive behaviors, we have not been able to demonstrate any actions of neuroP on sexual receptive behaviors. Regardless, it is clear that the brain can no longer be considered a passive recipient of steroid information from the ovaries. Rather, there is a complex interaction of peripheral steroids and neurosteroids that regulate cell signaling and transcription to cause significant biological consequences in brain function and behavior.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Ms. Amy Christensen for her assistance with this manuscript. Work from our laboratory presented here was supported by the National Institutes of Health Grants HD042635 and DA013185.
References
Barfield, R. J., Glaser, J. H., Rubin, B. S., and Etgen, A. M. (1984). Behavioral effects of progestin in the brain. Psychoneuroendocrinology 9, 217–231.
Beach, F. A., and LeBoeuf, B. J. (1967). Coital behaviour in dogs. I. Preferential mating in the bitch. Anim. Behav. 15, 546–558.
Berkley, K. J., McAllister, S. L., Accius, B. E., and Winnard, K. P. (2007). Endometriosis-induced vaginal hyperalgesia in the rat: effect of estropause, ovariectomy, and estradiol replacement. Pain 132(Suppl. 1), S150–S159.
Bermant, G., and Westbrook, W. H. (1966). Peripheral factors in the regulation of sexual contact by female rats. J. Comp. Physiol. Psychol. 61, 244–250.
Bloch, G. J., Babcock, A. M., Gorski, R. A., and Micevych, P. E. (1987). Cholecystokinin stimulates and inhibits lordosis behavior in female rats. Physiol. Behav. 39, 217–224.
Bondar, G., Kuo, J., Hamid, N., and Micevych, P. (2009). Estradiol-induced estrogen receptor-alpha trafficking. J. Neurosci. 29, 15323–15330.
Boulware, M. I., Weick, J. P., Becklund, B. R., Kuo, S. P., Groth, R. D., and Mermelstein, P. G. (2005). Estradiol activates group I and II metabotropic glutamate receptor signaling, leading to opposing influences on cAMP response element-binding protein. J. Neurosci. 25, 5066–5078.
Bradshaw, H. B., and Berkley, K. J. (2003). The influence of ovariectomy with or without estrogen replacement on responses of rat gracile nucleus neurons to stimulation of hindquarter skin and pelvic viscera. Brain Res. 986, 82–90.
Calizo, L. H., and Flanagan-Cato, L. M. (2003). Hormonal-neural integration in the female rat ventromedial hypothalamus: triple labeling for estrogen receptor-alpha, retrograde tract tracing from the periaqueductal gray, and mating-induced Fos expression. Endocrinology 144, 5430–5440.
Cardona-Gomez, G. P., DonCarlos, L., and Garcia-Segura, L. M. (2000). Insulin-like growth factor I receptors and estrogen receptors colocalize in female rat brain. Neuroscience 99, 751–760.
Carmeci, C., Thompson, D. A., Ring, H. Z., Francke, U., and Weigel, R. J. (1997). Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics 45, 607–617.
Cason, A. M., Samuelsen, C. L., and Berkley, K. J. (2003). Estrous changes in vaginal nociception in a rat model of endometriosis. Horm. Behav. 44, 123–131.
Chaban, V., Li, J., McDonald, J. S., Rapkin, A., and Micevych, P. (2011). Estradiol attenuates the adenosine triphosphate-induced increase of intracellular calcium through group ii metabotropic glutamate receptors in rat dorsal root ganglion neurons. J. Neurosci. Res. [Epub ahead of print].
Chaban, V. V., Lakhter, A. J., and Micevych, P. (2004). A membrane estrogen receptor mediates intracellular calcium release in astrocytes. Endocrinology 145, 3788–3795.
Chaban, V. V., Mayer, E. A., Ennes, H. S., and Micevych, P. E. (2003). Estradiol inhibits atp-induced intracellular calcium concentration increase in dorsal root ganglia neurons. Neuroscience 118, 941–948.
Chaban, V. V., and Micevych, P. E. (2005). Estrogen receptor-alpha mediates estradiol attenuation of ATP-induced Ca2+ signaling in mouse dorsal root ganglion neurons. J. Neurosci. Res. 81, 31–37.
Christensen, A., Dewing, P., and Micevych, P. (2011). “The STX-activated membrane estrogen receptor regulates female sexual receptivity through interaction with the metabotropic glutamate receptor,” in Society for Neuroscience Annual Meeting, Washington, DC.
Cooke, B. M., and Woolley, C. S. (2005). Gonadal hormone modulation of dendrites in the mammalian CNS. J. Neurobiol. 64, 34–46.
Dewing, P., Boulware, M. I., Sinchak, K., Christensen, A., Mermelstein, P. G., and Micevych, P. (2007). Membrane estrogen receptor-alpha interactions with metabotropic glutamate receptor 1a modulate female sexual receptivity in rats. J. Neurosci. 27, 9294–9300.
Dewing, P., Christensen, A., Bondar, G., and Micevych, P. (2008). Protein kinase C signaling in the hypothalamic arcuate nucleus regulates sexual receptivity in female rats. Endocrinology 149, 5934–5942.
Dominguez, R., Hu, E., Zhou, M., and Baudry, M. (2009). 17beta-estradiol-mediated neuroprotection and ERK activation require a pertussis toxin-sensitive mechanism involving GRK2 and beta-arrestin-1. J. Neurosci. 29, 4228–4238.
Dominguez, R., and Micevych, P. (2010). Estradiol rapidly regulates membrane estrogen receptor alpha levels in hypothalamic neurons. J. Neurosci. 30, 12589–12596.
Dubal, D. B., Rau, S. W., Shughrue, P. J., Zhu, H., Yu, J., Cashion, A. B., Suzuki, S., Gerhold, L. M., Bottner, M. B., Dubal, S. B., Merchanthaler, I., Kindy, M. S., and Wise, P. M. (2006). Differential modulation of estrogen receptors (ERs) in ischemic brain injury: a role for ERalpha in estradiol-mediated protection against delayed cell death. Endocrinology 147, 3076–3084.
Eckersell, C. B., Popper, P., and Micevych, P. E. (1998). Estrogen-induced alteration of mu-opioid receptor immunoreactivity in the medial preoptic nucleus and medial amygdala. J. Neurosci. 18, 3967–3976.
Erskine, M. S. (1985). Effects of paced coital stimulation on estrus duration in intact cycling rats and ovariectomized and ovariectomized-adrenalectomized hormone-primed rats. Behav. Neurosci. 99, 151–161.
Erskine, M. S. (1989). Solicitation behavior in the estrous female rat: a review. Horm. Behav. 23, 473–502.
Fernandez-Galaz, M. C., Naftolin, F., and Garcia-Segura, L. M. (1999). Phasic synaptic remodeling of the rat arcuate nucleus during the estrous cycle depends on insulin-like growth factor-I receptor activation. J. Neurosci. Res. 55, 286–292.
Garcia-Segura, L. M., Hernandez, P., Olmos, G., Tranque, P. A., and Naftolin, F. (1988). Neuronal membrane remodelling during the oestrus cycle: a freeze-fracture study in the arcuate nucleus of the rat hypothalamus. J. Neurocytol. 17, 377–383.
Gorosito, S. V., Lorenzo, A. G., and Cambiasso, M. J. (2008). Estrogen receptor alpha is expressed on the cell-surface of embryonic hypothalamic neurons. Neuroscience 154, 1173–1177.
Kuo, J., Hariri, O. R., Bondar, G., Ogi, J., and Micevych, P. (2009). Membrane estrogen receptor-alpha interacts with metabotropic glutamate receptor type 1a to mobilize intracellular calcium in hypothalamic astrocytes. Endocrinology 150, 1369–1376.
Loyd, D. R., and Murphy, A. Z. (2008). Androgen and estrogen (alpha) receptor localization on periaqueductal gray neurons projecting to the rostral ventromedial medulla in the male and female rat. J. Chem. Neuroanat. 36, 216–226.
Mamiya, N., Fukushima, H., Suzuki, A., Matsuyama, Z., Homma, S., Frankland, P. W., and Kida, S. (2009). Brain region-specific gene expression activation required for reconsolidation and extinction of contextual fear memory. J. Neurosci. 29, 402–413.
McClintock, M. K., and Adler, N. T. (1978). The role of the female during copulation in wild and domestic Norway rats (Rattus norvegicus). Behaviour 67, 67–96.
Mendez, P., Azcoitia, I., and Garcia-Segura, L. M. (2003). Estrogen receptor alpha forms estrogen-dependent multimolecular complexes with insulin-like growth factor receptor and phosphatidylinositol 3-kinase in the adult rat brain. Brain Res. Mol. Brain Res. 112, 170–176.
Mermelstein, P. G., and Micevych, P. E. (2008). Nervous system physiology regulated by membrane estrogen receptors. Rev. Neurosci. 19, 413–424.
Micevych, P., and Dominguez, R. (2009). Membrane estradiol signaling in the brain. Front. Neuroendocrinol. 30, 315–327.
Micevych, P., Kuo, J., and Christensen, A. (2009). Physiology of membrane oestrogen receptor signalling in reproduction. J. Neuroendocrinol. 21, 249–256.
Micevych, P., and Sinchak, K. (2007). “The neurochemistry of limbic-hypothalamic circuits regulating sexual receptivity,” in Handbook of Neurochemistry and Molecular Neurobiology: Behavioral Neurochemistry, Neuroendocrinology and Molecular Neurobiology, eds A. Lajtha and J. Blaustein (New York: Springer), 154–193.
Micevych, P., and Sinchak, K. (2008). Estradiol regulation of progesterone synthesis in the brain. Mol. Cell. Endocrinol. 290, 44–50.
Micevych, P., Soma, K. K., and Sinchak, K. (2008). Neuroprogesterone: key to estrogen positive feedback? Brain Res. Rev. 57, 470–480.
Micevych, P. E., Chaban, V., Ogi, J., Dewing, P., Lu, J. K., and Sinchak, K. (2007). Estradiol stimulates progesterone synthesis in hypothalamic astrocyte cultures. Endocrinology 148, 782–789.
Micevych, P. E., and Mermelstein, P. G. (2008). Membrane estrogen receptors acting through metabotropic glutamate receptors: an emerging mechanism of estrogen action in brain. Mol. Neurobiol. 38, 66–77.
Micevych, P. E., Rissman, E. F., Gustafsson, J. A., and Sinchak, K. (2003). Estrogen receptor-alpha is required for estrogen-induced mu-opioid receptor internalization. J. Neurosci. Res. 71, 802–810.
Mills, R. H., Sohn, R. K., and Micevych, P. E. (2004). Estrogen-induced mu-opioid receptor internalization in the medial preoptic nucleus is mediated via neuropeptide Y-Y1 receptor activation in the arcuate nucleus of female rats. J. Neurosci. 24, 947–955.
Murphy, A. Z., Suckow, S. K., Johns, M., and Traub, R. J. (2009). Sex differences in the activation of the spinoparabrachial circuit by visceral pain. Physiol. Behav. 97, 205–212.
Olmos, G., Aguilera, P., Tranque, P., Naftolin, F., and Garcia-Segura, L. M. (1987). Estrogen-induced synaptic remodelling in adult rat brain is accompanied by the reorganization of neuronal membranes. Brain Res. 425, 57–64.
Pappas, T. C., Gametchu, B., and Watson, C. S. (1995). Membrane estrogen receptors identified by multiple antibody labeling and impeded-ligand binding. FASEB J. 9, 404–410.
Paredes, R. G., and Alonso, A. (1997). Sexual behavior regulated (paced) by the female induces conditioned place preference. Behav. Neurosci. 111, 123–128.
Pfaff, D. (1970). Nature of sex hormone effects on rat sex behavior: specificity of effects and individual patterns of response. J. Comp. Physiol. Psychol. 73, 349–358.
Pfaff, D. W., Kow, L. M., Loose, M. D., and Flanagan-Cato, L. M. (2008). Reverse engineering the lordosis behavior circuit. Horm. Behav. 54, 347–354.
Prossnitz, E. R., Oprea, T. I., Sklar, L. A., and Arterburn, J. B. (2008). The ins and outs of GPR30: a transmembrane estrogen receptor. J. Steroid Biochem. Mol. Biol. 109, 350–353.
Qiu, J., Bosch, M. A., Tobias, S. C., Grandy, D. K., Scanlan, T. S., Ronnekleiv, O. K., and Kelly, M. J. (2003). Rapid signaling of estrogen in hypothalamic neurons involves a novel G-protein-coupled estrogen receptor that activates protein kinase C. J. Neurosci. 23, 9529–9540.
Qiu, J., Bosch, M. A., Tobias, S. C., Krust, A., Graham, S. M., Murphy, S. J., Korach, K. S., Chambon, P., Scanlan, T. S., Ronnekleiv, O. K., and Kelly, M. J. (2006). A G-protein-coupled estrogen receptor is involved in hypothalamic control of energy homeostasis. J. Neurosci. 26, 5649–5655.
Quesada, A., and Etgen, A. M. (2002). Functional interactions between estrogen and insulin-like growth factor-I in the regulation of alpha 1B-adrenoceptors and female reproductive function. J. Neurosci. 22, 2401–2408.
Quesada, A., Lee, B. Y., and Micevych, P. E. (2008). PI3 kinase/Akt activation mediates estrogen and IGF-1 nigral DA neuronal neuroprotection against a unilateral rat model of Parkinson’s disease. Dev. Neurobiol. 68, 632–644.
Quesada, A., and Micevych, P. E. (2004). Estrogen interacts with the IGF-1 system to protect nigrostriatal dopamine and maintain motoric behavior after 6-hydroxdopamine lesions. J. Neurosci. Res. 75, 107–116.
Quesada, A., Romeo, H. E., and Micevych, P. (2007). Distribution and localization patterns of estrogen receptor-beta and insulin-like growth factor-1 receptors in neurons and glial cells of the female rat substantia nigra: localization of ERbeta and IGF-1R in substantia nigra. J. Comp. Neurol. 503, 198–208.
Revankar, C. M., Cimino, D. F., Sklar, L. A., Arterburn, J. B., and Prossnitz, E. R. (2005). A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 307, 1625–1630.
Rissman, E. F., Early, A. H., Taylor, J. A., Korach, K. S., and Lubahn, D. B. (1997). Estrogen receptors are essential for female sexual receptivity. Endocrinology 138, 507–510.
Ritter, S. L., and Hall, R. A. (2009). Fine-tuning of GPCR activity by receptor-interacting proteins. Nat. Rev. Mol. Cell Biol. 10, 819–830.
Roepke, T. A., Ronnekleiv, O. K., and Kelly, M. J. (2011). Physiological consequences of membrane-initiated estrogen signaling in the brain. Front. Biosci. 16, 1560–1573.
Sasahara, K., Shikimi, H., Haraguchi, S., Sakamoto, H., Honda, S., Harada, N., and Tsutsui, K. (2007). Mode of action and functional significance of estrogen-inducing dendritic growth, spinogenesis, and synaptogenesis in the developing Purkinje cell. J. Neurosci. 27, 7408–7417.
Sinchak, K., Garcia, B. L., Bowlby, R., Charukulvanich, P., Garcia, M. P., and Sanathara, N. (2010). “Mu-opioid receptor neurons and opioid receptor-like receptor neurons in the medial preoptic nucleus project to the region of the ventromedial nucleus of the hypothalamus,” in Society for Neuroscience Annual Meeting, San Diego, CA.
Sinchak, K., and Micevych, P. (2003). Visualizing activation of opioid circuits by internalization of G protein-coupled receptors. Mol. Neurobiol. 27, 197–222.
Sinchak, K., and Micevych, P. E. (2001). Progesterone blockade of estrogen activation of mu-opioid receptors regulates reproductive behavior. J. Neurosci. 21, 5723–5729.
Sodersten, P., and Eneroth, P. (1981). Serum levels of oestradiol-17 beta and progesterone in relation to sexual receptivity in intact and ovariectomized rats. J. Endocrinol. 89, 45–54.
Tennent, B. J., Smith, E. R., and Davidson, J. M. (1980). The effects of estrogen and progesterone on female rat proceptive behavior. Horm. Behav. 14, 65–75.
Thomas, P., Pang, Y., Dong, J., Groenen, P., Kelder, J., de Vlieg, J., Zhu, Y., and Tubbs, C. (2007). Steroid and G protein binding characteristics of the seatrout and human progestin membrane receptor alpha subtypes and their evolutionary origins. Endocrinology 148, 705–718.
Toran-Allerand, C. D. (2005). Estrogen and the brain: beyond ER-alpha, ER-beta, and 17beta-estradiol. Ann. N. Y. Acad. Sci. 1052, 136–144.
Toran-Allerand, C. D., Ellis, L., and Pfenninger, K. H. (1988). Estrogen and insulin synergism in neurite growth enhancement in vitro: mediation of steroid effects by interactions with growth factors? Brain Res. 469, 87–100.
Toran-Allerand, C. D., Guan, X., MacLusky, N. J., Horvath, T. L., Diano, S., Singh, M., Connolly, E. S. Jr., Nethrapalli, I. S., and Tinnikov, A. A. (2002). ER-X: a novel, plasma membrane-associated, putative estrogen receptor that is regulated during development and after ischemic brain injury. J. Neurosci. 22, 8391–8401.
Torii, M., Kubo, K., and Sasaki, T. (1996). Influence of opioid peptides on the priming action of estrogen on lordosis in ovariectomized rats. Neurosci. Lett. 212, 68–70.
Torii, M., Kubo, K., and Sasaki, T. (1999). Facilitatory and inhibitory effects of beta-endorphin on lordosis in female rats: relation to time of administration. Horm. Behav. 35, 271–278.
Wintermantel, T. M., Campbell, R. E., Porteous, R., Bock, D., Grone, H. J., Todman, M. G., Korach, K. S., Greiner, E., Perez, C. A., Schutz, G., and Herbison, A. E. (2006). Definition of estrogen receptor pathway critical for estrogen positive feedback to gonadotropin-releasing hormone neurons and fertility. Neuron 52, 271–280.
Wise, P. M., Dubal, D. B., Rau, S. W., Brown, C. M., and Suzuki, S. (2005). Are estrogens protective or risk factors in brain injury and neurodegeneration? Reevaluation after the women’s health initiative. Endocr. Rev. 26, 308–312.
Woolley, C. S., and McEwen, B. S. (1992). Estradiol mediates fluctuation in hippocampal synapse density during the estrous cycle in the adult rat. J. Neurosci. 12, 2549–2554.
Keywords: lordosis behavior, estrogen receptor, STX, cell signaling, neuroprogesterone, neurosteroids
Citation: Micevych PE and Dewing P (2011) Membrane-initiated estradiol signaling regulating sexual receptivity. Front. Endocrin. 2:26. doi: 10.3389/fendo.2011.00026
Received: 20 July 2011;
Paper pending published: 02 August 2011;
Accepted: 16 August 2011;
Published online: 07 September 2011.
Edited by:
Hubert Vaudry, University of Rouen, FranceReviewed by:
James A. Carr, Texas Tech University, USALuis Miguel Garcia-Segura, Consejo Superior de Investigaciones Científicas, Spain
Copyright: © 2011 Micevych and Dewing. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Paul E Micevych, Department of Neurobiology, David Geffen School of Medicine at University of California Los Angeles, Los Angeles, CA 90095-1763, USA. e-mail:cG1pY2V2eWNoQG1lZG5ldC51Y2xhLmVkdQ==