 Estelle Tallet1,2
Estelle Tallet1,2 Isabelle Fernandez1,2 Chi Zhang1,2 Marion Salsac1,2 Nathalie Gregor3
Isabelle Fernandez1,2 Chi Zhang1,2 Marion Salsac1,2 Nathalie Gregor3 Mohammed Akli Ayoub4,5,6 Jean Philippe Pin4,5,6 Eric Trinquet3
Mohammed Akli Ayoub4,5,6 Jean Philippe Pin4,5,6 Eric Trinquet3 Vincent Goffin1,2*
Vincent Goffin1,2*- 1 INSERM, U845, Centre de Recherche “Croissance et Signalisation”, Equipe “Physiopathologie des hormones PRL/GH”, Paris, France
- 2 Université Paris Descartes, Sorbonne Paris Cité, Faculté de Médecine, Paris, France
- 3 Cisbio Bioassays, Codolet, France
- 4 Centre National de la Recherche Scientifique, Unité Mixte de Recherche 5203, Institut de Génomique Fonctionnelle, Montpellier, France
- 5 INSERM, U661, Montpellier, France
- 6 Université Montpellier 1 and 2, Montpellier, France
The prolactin receptor (PRLR) is emerging as a therapeutic target in oncology. Knowledge-based drug design led to the development of a pure PRLR antagonist (Del1-9-G129R-hPRL) that was recently shown to prevent PRL-induced mouse prostate tumorogenesis. In humans, the first gain-of-function mutation of the PRLR (PRLRI146L) was recently identified in breast tumor patients. At the molecular level, the actual mechanism of action of these two novel players in the PRL system remains elusive. In this study, we addressed whether constitutive PRLR activation (PRLRI146L) or PRLR blockade (antagonist) involved alteration of receptor oligomerization and/or of inter-chain distances compared to unstimulated and PRL-stimulated PRLR. Using a combination of various biochemical and spectroscopic approaches (co-IP, blue native electrophoresis, BRET1), we demonstrated that preformed PRLR homodimers are altered neither by PRL- or I146L-induced receptor triggering, nor by antagonist-mediated blockade. These findings were confirmed using a novel time-resolved fluorescence resonance energy transfer (TR-FRET) technology that allows monitoring distance changes between cell surface tagged receptors. This technology revealed that PRLR blockade or activation did not involve detectable distance changes between extracellular domains of receptor chains within the dimer. This study merges with our previous structural investigations suggesting that the mechanism of PRLR activation solely involves intermolecular contact adaptations leading to subtle intramolecular rearrangements.
Introduction
The prolactin receptor (PRLR) is emerging as a novel therapeutic target in human diseases (Goffin et al., 2006; Rouet et al., 2010). The main indications include prostate and breast cancer in which locally produced prolactin (PRL) probably more than the circulating hormone has been proposed to promote tumor growth (Clevenger et al., 2003; Li et al., 2004; Dagvadorj et al., 2007; McHale et al., 2008). In addition, we recently identified the first gain-of-function mutation of this receptor (so-called PRLRI146L) in patients presenting with breast tumors, further supporting the involvement of PRLR signaling in human tumorogenesis (Bogorad et al., 2008). Current anti-PRL drugs are dopamine agonists, which efficiently target pituitary PRL production (Molitch, 2005). However, such compounds are per se inappropriate in targeting extrapituitary-produced PRL (whose expression is believed to be dopamine-independent) or PRLRI146L (whose constitutive activity is PRL-independent). Strategies targeting the receptor itself are thus necessary. Engineered ligands exhibiting antagonistic properties are currently viewed as a promising approach (Tallet et al., 2008). Accordingly, our group has recently developed pure competitive antagonists, the prototype of which was named Del1-9-G129R-hPRL (Bernichtein et al., 2003b). The latter efficiently down-regulates PRLR signaling triggered by autocrine PRL (Dagvadorj et al., 2007; Rouet et al., 2010) as well as by PRLRI146L (Bogorad et al., 2008). The development of novel therapeutic compounds, either engineered from the PRL core or chemically synthesized, requires a better understanding of the molecular/atomic changes underlying PRLR activation and pharmacological blockade. Within the past few years, our group has provided structural insight into the PRL family by determining three-dimensional structures of free agonist/antagonist ligands (PRLWT, Del1-9-G129R-hPRL) and PRL-receptor complexes (Teilum et al., 2005; Jomain et al., 2007; Broutin et al., 2010; Van Agthoven et al., 2010). Although these structures provided very helpful atomic level characterization of protein–protein interaction sites, the comparison of free and bound structures allowed only limited speculation on the dynamic properties of membrane-anchored receptors, especially regarding the recently discovered PRLRI146L variant.
The PRLR is a pioneering member of the class I hematopoietic cytokine receptor family (Kelly et al., 1991). This non-tyrosine kinase, single-pass transmembrane receptor family comprises nearly 50 members that exhibit wide heterogeneity regarding the stoichiometry of receptor chain assembly (Boulay et al., 2003). Together with the receptors for growth hormone (GHR), leptin (OBR), erythropoietin (EPOR), thrombopoietin (TPOR), and granulocyte colony stimulating factor (G-CSFR), the PRLR defines a subclass of cytokine receptors adopting the simplest model of receptor assembly, since functional receptors involve only a single type of chain which is assumed to homodimerize. Mutational and structural studies of PRL have identified two binding sites, each able to interact with one receptor chain (Goffin et al., 1996b; Broutin et al., 2010). The functional importance of both sites was demonstrated by the fact that mutations of hot spot residues at site 1 prevented receptor binding in cell-based assays (Goffin et al., 1992; Kinet et al., 1996). On the contrary, steric mutations introduced within PRL binding site 2 did not prevent receptor binding but resulted in competitive receptor antagonists unable to trigger signaling (Bernichtein et al., 2003b; Jomain et al., 2007). Surface plasmon resonance (SPR) using immobilized and oriented PRLR extracellular domain (ECD) was used to monitor sequential interaction of two ECDs with PRL binding site 1 then 2. These studies revealed that the affinity of site 1 for the PRLR-ECD was in the nanomolar range (which is similar to the affinity for membrane-anchored PRLR), while that of site 2 was much lower (micromolar). While PRLR antagonists displayed unchanged site 1 affinity, no interaction involving site 2 was detectable (Jomain et al., 2007). Although these findings suggested that the antagonistic properties of site 2 mutants resulted from their inability to interact with a second receptor moiety – which was in good agreement with the original model of sequential receptor dimerization (Fuh et al., 1993; Goffin et al., 1994) – extrapolation of SPR data to membrane-anchored receptors must remain very cautious.
Indeed, recent reports have suggested that the PRLR, as many cytokine receptors (if not all), is present in a pre-assembled form at the plasma membrane. Using BRET1 (fluorescent/bioluminescent tags added to the C-terminus of receptors) and co-immunoprecipitation (co-IP) approaches, Qazi et al. (2006) suggested that the membrane PRLR was constitutively homodimerized (or heterodimerized when long and short isoforms were co-expressed in the same cell). These findings were in agreement with another report involving co-IP, which also concluded to the existence of ligand-independent homodimers of human PRLR isoforms, and proposed a significant role for the transmembrane domain in stabilizing the dimer (Gadd and Clevenger, 2006). These two reports further agreed on the fact that any qualitative or quantitative alteration of PRLR dimerization induced by the ligand was beyond the detection limits of the techniques used. Otherwise, BRET2 technology applied to various C-terminal tagged PRLR isoforms revealed an effect of ligand binding on fluorescence signals, that discriminated receptor agonists (hPRLWT and S179D-hPRL) from a partial agonist/antagonist (G129R-hPRL; Tan et al., 2005). Although at that time the authors could not speculate whether these findings reflected the ability of agonists to recruit monomers into dimers or to induce conformational changes in the cytoplasmic tails of pre-existing dimers, the subsequent demonstration that non-stimulated PRLR exists as preformed dimers (Tan et al., 2005; Qazi et al., 2006) clearly argues for the second hypothesis.
These findings have pushed one step forward several unanswered questions regarding the actual activation mechanism of this receptor. These include: do agonists trigger receptor signaling by inducing important conformational changes of the ECDs, that next reverberate onto the cytoplasmic domain? Does PRLRI146L intrinsically adopt a conformation similar to that of PRL-activated PRLRWT? Does Del1-9-G129R-hPRL prevent these conformational changes or, even disrupt the preformed dimer? Does activation of the latter involve significant distance changes between the two ECDs, or only subtle intramolecular conformational changes within the ECD(s)? Are the interacting regions of PRL and PRLR the best targets to design inhibitors, or could the understanding of PRLR activation identify alternative/more efficient regions for future drug design? Although resonance energy transfer technologies (BRET, FRET) are currently the most powerful experimental approaches to address such issues, several drawbacks are encountered using these approaches. First, the low signal-to-noise ratio due to high background and/or to signal generated by fluorescent/bioluminescent proteins present in the intracellular compartment can hamper the detection of specific signal induced by extracellular ligands. This is especially true for the PRLR which mainly resides inside the cell. Another important limit of classical FRET/BRET involves the interpretation of the results. Indeed, the absence of signal does not necessarily reflect the absence of physical protein–protein interactions, since unsuitable relative orientation of fluorophore dipoles can prevent energy transfer between interacting proteins (Pfleger and Eidne, 2006; Vogel et al., 2006). Conversely, as stated above, it is virtually impossible to translate changes of signal intensities (e.g., induced by ligands) into changes of distance, of orientation or both.
To address this issue, we have used the newly developed time-resolved fluorescence resonance energy transfer (TR-FRET) technology, which circumvents such drawbacks (Degorce et al., 2009). It involves the use of terbium cryptate (Lumi4 Tb) as a donor, and red or green fluorophores as acceptors (see Experimental Procedures). Such fluorophore combinations allow much higher signal-to-noise ratio compared to classical FRET. A second advantage of TR-FRET is that signals only depend on the distances between interacting proteins, as energy transfer efficiency is mostly independent of the relative dipole orientation of the donor and acceptor molecules. Third, when fluorophores are specifically targeted to the plasma membrane thanks to the use of receptor-tag chimera labeled with non-permeant synthetic substrates, one can selectively analyze the proximity between cell surface receptors without being hampered by the pool of intracellular tagged receptors. Finally, as tags are linked to the N-terminal end of the receptors, TR-FRET is suitable to monitor distance changes (movements) between ECDs. This feature was particularly appropriate for our purpose since: (i) I146L mutation is located within the S2 subdomain of the ECD (Bogorad et al., 2008), and (ii) the pure antagonistic properties of Del1-9-G129R-hPRL are obviously mediated though the interaction of the latter with the PRLR-ECD. In the present work, we thus applied TR-FRET technology to study various activation states of the PRLR, which involved comparisons between PRLRWT and PRLRI146L, either unbound or bound to receptor agonist (PRL) or pure antagonist (Del1-9-G129R-hPRL). As this was the first time TR-FRET technology was applied to a member of the cytokine family, it was combined to more classical approaches (BRET, co-IP, native electrophoresis) in order to allow direct comparison with previously published data in the field.
Materials and Methods
Hormones and Antibodies
Recombinant, chromatography purified hPRL and Del1-9-G129R-hPRL antagonist were prepared and characterized as previously described (Bernichtein et al., 2003b). Purity of hormone preparations used in this study was >95% as judged from SDS-PAGE analysis. For ligand binding FRET assays, both recombinant hormones were labeled with Red fluorophore using the d2 labeling kit (Ref. 62D2DPEA, Cisbio Bioassays, Codolet, France). Various batches of labeled protein were obtained with final molar ratio of 0.8–3. The bioactivity of labeled proteins was assessed using the lactogenic hormone response element (LHRE)-luciferase reporter gene assay previously described (Bernichtein et al., 2003a). Labeled prolactin exhibited bioactivity similar to unlabeled hormone, although a slight shift to the right of dose–response curves was observed for the batches carrying the highest level of fluorophores (data not shown).
Antibodies used for (co-)IP and immunoblots were the following: anti-Stat5a/b (C-17, rabbit polyclonal, Santa Cruz Biotechnology, Santa Cruz, CA, USA) used at 0.4 μg/mL for IP and 0.02 μg/mL in blots; anti-HA (Roche, Basel, Switzerland, clone 12CA5), used at 0.8 μg/mL for IP and 0.08 μg/mL in blots, anti-phosphorylated Stat5a/b (AX1, mouse monoclonal, Advantex BioReagents, Conroe, TX, USA), used at 1:10,000 in blots; anti-human PRLR (Invitrogen-Zymed, 1A2B1, Carlsbad, CA, USA), used at 1:1,000 in blots.
Generation of hPRLRWT and hPRLRI146L Tagged Constructs
Fluorescence resonance energy transfer constructs were generated by introducing the coding sequence of the hPRLR long isoform (cDNA originally in pcDNA3.1 vector and containing the rat signal peptide, rSP; Lochnan et al., 1995) into pRK5-HA-ST plasmid, containing the two tags of interest (HA and ST). Briefly, an MluI site was inserted by site-directed mutagenesis (QuikChange Mutagenesis II kit, Stratagene, La Jolla, CA, USA) between the signal peptide and the ATG initiation codon of pRK5-rSP-hPRLRWT plasmid. Then, the PRLR coding sequence excised using SpeI and MluI enzymes was sub-cloned into linearized pRK5-mGluR5PS-HA-ST vector, produced by SpeI–MluI restriction of previously described pRK5-mGluR5PS-HA-ST-GB1R plasmid (Maurel et al., 2008). In the final pRK5-HA-ST-hPRLR vector, a linker of two amino acids connects the Snap tag to the N-terminus of the receptor. The HA-ST-hPRLRI146L encoding vector was generated by site-directed mutagenesis of pRK5-HA-ST-hPRLRWT using primers earlier reported (Bogorad et al., 2008). After transformation, Escherichia coli JM109 colonies were analyzed for their DNA content; recombinant plasmids were sequenced to assess sequence integrity and the presence of expected tags. FRET vectors encoding mGluR2 and CD4 receptors were described in a previous study (Maurel et al., 2008).
BRET constructs were generated by linking Rluc and YFP encoding sequence in 3′ of PRLR coding sequence contained in pcDNA3.1-hPRLRWT vector. Briefly, an EcoRV restriction site was substituted for the hPRLR stop codon by site-directed mutagenesis (generating pcDNA3.1-hPRLR(EcoRV)). Similarly, an EcoRV restriction site was introduced in 5′ of Rluc coding sequence contained in pcDNA3-CMV-hRluc plasmid (kindly provided by Dr. R. Jokers), generating pcDNA3-CMV-(EcoRV)hRluc. The Rluc fragment was extracted from pcDNA3-CMV-(EcoRV)hRluc by EcoRV-NotI digestion and sub-cloned into pcDNA3.1-hPRLR(EcoRV) plasmid linearized by EcoRV + NotI digestion. In the final construct (pcDNA3.1-hPRLRWT-Rluc), a sequence encoding 12 amino acids (DICRWDPPARAT) links PRLR and Rluc coding sequences. The YFP DNA fragment was obtained by EcoRV-NotI restriction of pcDNA-CMV-topaze (kindly provided by Dr. R. Jokers), then sub-cloned into EcoRV + NotI linearized pcDNA3.1-hPRLR(EcoRV) plasmid. In the final construct (pcDNA3.1-hPRLRWT-YFP), a sequence encoding 16 amino acids (DIKLAVPRARDPPVAT) links PRLR and YFP coding sequences. Both Rluc- and YFP-hPRLRWT encoding vectors were site-directed mutated to generate cognate hPRLRI146L vectors. All plasmids were verified by restriction enzyme mapping and sequencing in both directions to ensure that fusion protein cDNA were in frame and to discard any unexpected mutation.
Routine Cell Culture
HEK 293 and COS-7 cells were routinely cultured in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen), containing 10% FBS, 50 U/mL penicillin, 50 μg/mL streptomycin (Invitrogen, Grand Island, NY, USA), and 2 mM Glutamine, at 5% CO2 and 37°C. HL-5, a stable HEK 293 cell line that integrated in its genome the LHRE-luciferase reported gene and the hPRLRWT cDNA, was described in previous publications (Bernichtein et al., 2003a,b). They were cultured in the same media as described above, with the addition of 500 μg/mL Geneticin.
Ba/F3 cells are a murine pro-B cell line that is dependent on Interleukin-3 (IL-3) for growth. They were routinely maintained in RPMI 1640 medium supplemented with 10% heat-inactivated FBS, 2 mM glutamine, 50 U/mL penicillin, 50 μg/mL streptomycin, and 10% WEHI cell supernatant as source of IL-3 (Bernichtein et al., 2003a). Ba/F-LP is a selected population of Ba/F3 cells stably expressing the hPRLRWT (Bernichtein et al., 2003a); this particular cell line was cultured in the presence of 10 ng/mL hPRL instead of IL-3 (no addition of WEHI supernatant) and of 500 μg/mL Geneticin.
Transient and Stable Transfection of HEK 293 and COS-7 Cells
For transient transfections of untagged, FRET, or BRET PRLR constructs, we used lipofectamine 2,000 reagent (Invitrogen, Grand Island, NY, USA) and we strictly followed the manufacturer’s instructions. As pRK5 FRET vectors encoding PRLRWT or PRLRI146L contain no eukaryote antibiotic resistance, the generation of stable HEK 293 clones was performed by co-transfecting these plasmids together with empty pcDNA3.1 plasmid (containing geneticin resistance). We used Fugen reagent (Roche) and we followed manufacturer’s instructions with respect to DNA quantity ratio (10:1). After 48 h, Geneticin (500 μg/mL) was added for selection. After 2–3 weeks, clones were individually expanded and characterized for PRLR expression and PRL-responsiveness using immunoblot (see below). Two clones were selected for each receptor according to expression levels of the PRLR as determined by direct fluorescence at 620 nm (see below). These FRET clones were named PRLRhigh or PRLRlow (for both PRLRWT and PRLRI146L).
Western Blot and Co-Immunoprecipitation Analyses
HEK 293 cells (2 × 106) were plated in 10 cm Petri dishes and transfected using the plasmids of interest, alone or in combination. After 48 h, cells were washed twice in cold PBS then scratched in 1 mL PBS containing protease inhibitor cocktail (Complete Mini, Roche). After 5 min centrifugation at 800 g, cell pellets were resuspended in lysis buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1% Nonidet P40, 0.5% Deoxycholic acid, 0.1% SDS) in the presence of 2 mM Na-orthovanadate and protease inhibitor cocktail (30 min, 4°C). Lysates were centrifuged 13,000 g (4°C) and supernatants were collected before protein content was determined by Bradford assay.
Analysis of PRLR expression was performed by immunoblot. Cell lysates were resolved on 7.5% SDS-PAGE followed by transfer onto nitrocellulose membrane (mini-protean, Biorad, Hercules, CA, USA). After blocking (TBS 5% milk/0.1% Tween 20), membranes were incubated overnight with the appropriate primary antibody (diluted in TBS-T 3% BSA) at 4°C. Secondary anti-mouse (GE Healthcare) or anti-rabbit (Cell Signaling, Danvers, MA, USA) antibodies conjugated to horseradish peroxidase were incubated for 1 h at RT before detection of antigen-antibody complexes by enhanced chemiluminescence (GE Healthcare, Orsay, France).
Co-IP of untagged and HA-tagged PRLRs was performed by incubating cell lysates with anti-HA antibody (0.8 μg/mL) overnight at 4°C on rotary shaker. Protein A-Sepharose beads (GE Healthcare) were added for one additional hour (4°C under rotation). Immune complexes bound to the beads were washed twice in lysis buffer before resuspension in 20 μL loading buffer containing 2-mercaptoethanol. After heating (90°C, 5 min), samples were processed by SDS-PAGE and immunoblot as described above. Densitometric analyses were performed using ImageJ software. Data from two experiments were averaged. The [untagged PRLR/HA-PRLR] densitometric ratios in total lysates (bottom panels in Figure 2) were close to 1, indicating similar level of expression of both constructs. For each stimulation condition, this value was used to normalize band intensities calculated for co-IP, as follows: [untagged PRLR/HA-PRLR]co-IP/[untagged PRLR/HA-PRLR]total lysates.
Blue Native Page
Cells were stimulated at 37°C with hPRL (1 μg/mL) and harvested by scraping on ice. Then, they were collected by brief centrifugation (1,000 g, 5 min) and strongly homogenized using a tight fitting glass homogenizer in a hypotonic lysis buffer (10 mM Tris, pH 7.5, 0.2 M sucrose, 2 mM EDTA, 50 mM Na fluoride, 30 mM Na pyrophosphate, and protease inhibitor cocktail). The homogenates were then centrifuged at 5,000 g for 10 min to eliminate cell debris and nuclei. The supernatants were kept on ice while the pellet was resuspended, homogenized, and centrifuged again. Both step supernatants were combined then centrifuged at 120,000 g for 1 h at 4°C, and the final pellet containing crude membranes was kept frozen at −80°C until use.
To solubilize membrane proteins and complexes, the membrane pellet was vigorously resuspended in extraction buffer using a pipette (50 mM Bis–Tris–HCl pH 7.2, 50 mM NaCl, 10% w/v glycerol, and 0.001% Ponceau S, Native PAGE sample buffer, Invitrogen). Dodecyl-β-D-maltoside (DDM, Acros Organics) was then added to the membranes at a final concentration of 0.4% (or other concentration when indicated). After incubation on ice for 30 min (vortexing every 5 min), insoluble material was pelleted at 120,000 g for 30 min. Coomassie G250 was then added to the samples (0.5% final) for 5 min on ice before loading on blue native gels.
Blue Native PAGE (BN-PAGE) were performed according to a modified version of the protocol by Schagger and von Jagow (1991). A precast 4–16% polyacrylamide gradient gel was used to perform native (non-denaturing) electrophoresis (Native PAGE 4–16% Bis–Tris gel, Invitrogen). The migration was performed in a XCell SureLock Mini-Cell cuve (Invitrogen). The cathode buffer (50 mM Bis–Tris–HCl pH 6.8, 50 mM Tricine) containing 0.02% Coomassie G250 and the anode buffer (50 mM Bis–Tris–HCl, 50 mM Tricine pH 6.8) were chilled to 4°C before sample loading. Electrophoresis started at 4 mA (4°C). After 90 min, the cathode buffer was replaced by the same buffer containing 0.005% of the dye, and the electrophoresis was continued at 8 mA with voltage set at 120 V. After the dye front had run off the gel, the gel was washed with dissociating solution (0.1% SDS, 50 mM DTT) for 10 min at room temperature. Then, transfer onto PVDF membrane (Millipore) was performed as described above.
TR-FRET Analyses
Clones used for FRET studies (called PRLRhigh and PRLRlow) or transient transfected cells were plated in cultured medium at a density of 100,000 cells/well using 96 well white plate pre-coated with polyornithin (50 μL of 0.1 mg/mL polyornithin added per well for >1 h at 37°C). After 16 h (allowing cell adhesion), ST-PRLRs were labeled by SNAP-Lumi4 Tb (100 nM) in combination or not (control wells) with SNAP-acceptor [Red (500 nM) or Green (100 nM); Cisbio Bioassays, Codolet, France], in culture medium for 1 h at 37°C. Wells were washed twice with Tris-Krebs buffer (20 mM Tris–HCl pH 7.4, 188 mM NaCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 4.7 mM KCl, 1.8 mM CaCl2) and wells filled with 100 μL Tris-Krebs buffer. Plate reading was performed using a Mithras LB 940 reader (Berthold) upgraded with TR module: excitation at 340 nm, emission at 620 nm for Lumi4Tb detection, 665 nm for Red acceptor, and 520 nm for Green acceptor. After stimulation by hPRL or Del1-9-G129R-hPRL antagonist, measurements were repeated for the indicated time.
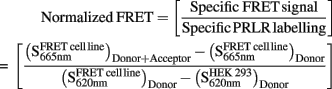
Normalized FRET was calculated as follows:
Signal was represented as ΔR signal = f(ligand concentration) or f(time) after data reduction with following calculation method:
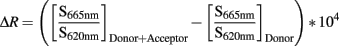
Results presented are representative of at least three independent experiments performed in triplicate. Representation and statistical analysis (t-test) were performed using GraphPad Prism software.
BRET Analyses
HEK 293 cells were plated at a density of 50,000 cell/well using white (luminescence and BRET measurements) or black (total YFP fluorescence) 96 well plates, pre-coated with polyornithin (see above). After 16 h, transfection was performed with DNA/lipofectamine 2,000 mix as recommended by the manufacturer, using DNA amount as indicated in the Legends. Medium was replaced 6–8 h post-transfection. After 36 additional hours (to ensure proper PRLR expression), cells were stimulated for 15 min by adding hPRL or Del1-9-G129R-hPRL antagonist, in the presence or in the absence of 0.015% of saponin, before BRET measurement. BRET signals were detected after addition of coelenterazine h (5μM final concentration) in each well (white plate) and followed in time. Signal detection was performed using Mithras LB 940 reader (Berthold, Thoiry, France). Bioluminescence emission of Renilla luciferase was measured at 485 nm, fluorescence emission of YFP at 530 nm. The relative expression level of PRLR–YFP fusion proteins was evaluated by measuring YFP fluorescence emitted at 530 nm after 485 nm lamp excitation using black plates.
Data reduction was done with following calculation method (Boute et al., 2001):
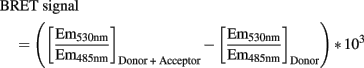
Results are representative of at least three independent experiments performed in triplicate and plotted using GraphPad Prism software to determine Bmax for each stimulation condition in each independent experiment. Data are represented as averaged percentages of Bmax or of basal BRET signal (i.e., non-stimulated condition). Statistical analysis was performed by using a two-tailed Student’s t-test when two groups were compared, which was confirmed by one-way ANOVA for comparison of the three stimulation conditions.
TR-FRET Binding Assays
FRET stable cell lines (PRLRhigh) were plated in cultured medium at a density 100,000 cells/well in 96 well white plates pre-coated with polyornithin. After 16 h, ST-PRLRs were labeled by SNAP-Lumi4 Tb (100 nM) in cultured medium for 1 h at 37°C. Wells were washed twice with Tris-Krebs buffer, then they were filled with 50 μL Tris-Krebs buffer supplemented with 0.1% BSA. After addition of various concentrations of hPRL-Red or Del1-9-G129R-hPRL-Red (added as 50 μL of [2×] concentrated protein diluted in Tris-Krebs buffer, 0.1% BSA), FRET signals were measured over 24 h. Plate reading was performed using Mithras LB 940 reader (Berthold) upgraded with TR module: excitation at 340 nm, emission at 620 nm for Lumi4Tb detection, or 665 nm for Red acceptor.
Representations correspond to 3 h of ligand stimulation at room temperature. Control wells were done with excess of non-labeled hPRL or Del1-9-G129R-hPRL antagonist.
Results are representative of at least three independent experiments performed in triplicate. The binding affinities of hPRL or antagonist were calculated by curve fitting with saturation binding equation (one site – specific binding) using GraphPad Prism software.
Results
Functional Characterization of TR-FRET and BRET PRLR Constructs
The various fusion constructs used for this study (Figure 1A) were generated from the coding sequence of long human (h) PRLR isoform, either PRLRWT or PRLRI146L (Bogorad et al., 2008). For constructs to be used in TR-FRET, we inserted two tandem N-terminal tags: HA (hemagglutinin) and SNAP tag (ST; Tag Lite technology, CisBio Bioassays). For BRET studies, Yellow Fluorescent Protein (YFP), or Renilla luciferase (Rluc) were added at the C-terminus of the PRLRs. Expression and activity of all PRLR variants were assessed using transiently transfected HEK 293 cells. As shown on Figure 1B, all expressed proteins exhibited the expected electrophoretic mobility as determined by αPRLR immunoblot. Whatever the tagged isoform considered, no difference in expression level was noticed between WT and I146L receptors. All constructs retained the ability to induce STAT5 phosphorylation, indicating that the various tags did not significantly alter the biological activity of the PRLR. For all but Rluc constructs, bioactivity was further confirmed using the classical PRL-responsive LHRE-Luciferase reporter plasmid assay previously described (Bernichtein et al., 2003a; data not shown). As earlier reported, the basal activity of hPRLRI146L constructs (including the untagged one) was hardly detectable using this readout in transient transfection (Goffin et al., 2010).
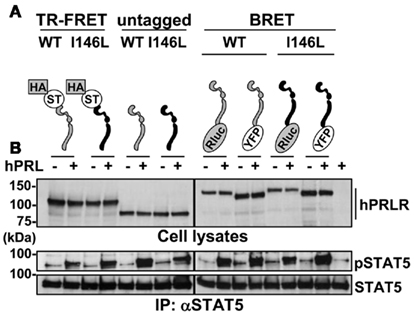
Figure 1. Functional validation of tagged PRLR constructs. (A) Schematic representation of different constructs used in this study. From left to right: tagged constructs used for TR-FRET experiments (insertion of HA and ST at PRLR N-terminus), untagged receptors and fusion receptors used for BRET experiments (insertion of Rluc or YFP at PRLR C-terminus). PRLRWT is represented in gray and PRLRI146L in black. Symbols: HA, hemagglutinin; ST, SNAP tag; Rluc, Renilla luciferase; YFP, yellow fluorescent protein. Note that the sizes of PRLR and tags as represented are not proportional to actual molecular masses. (B) PRLR expression in transient transfection (HEK 293 cells) was assessed by immunoblotting using a αPRLR mAb (non-transfected cells are shown on the right lane). Based on the electrophoretic mobility of untagged PRLR (∼90 kDa), the apparent size of tagged receptors was in agreement with the theoretical size of the various tags (21 kDa for HA–ST, 36 kDa for Rluc, and 27 kDa for YFP). The PRL-responsiveness of all receptors (hPRL 1 μg/mL added for 15 min at 37°C) was assessed by monitoring tyrosine phosphorylation of immunoprecipitated (IP) Stat5 using a specific phospho-Stat5 mAb.
Use of Biochemical Approaches to Characterize Basal and Ligand-Induced Homo- and Hetero-Dimerization of hPRLRWT and hPRLRI146L
Co-IP was used (i) to address the ability of unstimulated hPRLRWT and hPRLRI146L to form homo and/or heterodimers, and (ii) to test the effect of receptor triggering (using hPRL) or receptor blockade (using Del1-9-G129R-hPRL) on this property. For this purpose, we co-transfected plasmids encoding HA-ST-tagged or untagged PRLRs (WT and I146L, alone or together) in HEK 293 cells. Then receptor complexes were immunoprecipitated using anti-HA mAb and finally revealed using αPRLR immunoblotting. The specificity of HA mAb in IP experiments was demonstrated by its inability to pull-down untagged PRLR (data not shown). Immunoblotting of total cell lysates served as control of equivalent PRLR expression.
As shown on Figure 2A, IP of HA-ST-hPRLRWT pulled down untagged hPRLRWT in the absence of stimulation, confirming that this experimental procedure was appropriate to identify preformed receptor complexes earlier proposed to be homodimers (Gadd and Clevenger, 2006). As far as can be deduced using this semi-quantitative approach, stimulation by PRL, by a pure antagonist or both, reproducibly failed to consistently modify the amount of co-immunoprecipitated untagged PRLR. These results were confirmed by densitometric analysis performed as described in Section “Materials and Methods,” with co-IP/total lysate PRLRWT ratios equal to 0.74, 0.71, and 0.70 in basal, +PRL, and +antagonist conditions, respectively. The same experiments were repeated using PRLRI146L constructs (Figure 2B) and achieved very similar results (co-IP/total lysate ratios equal to 0.81, 0.77, and 0.78 in basal, +PRL, and +antagonist conditions, respectively). As PRLRI146L mutation was so far identified only in heterozygous patients (Bogorad et al., 2008), it was relevant to investigate its potential interaction with PRLRWT. This was performed by immunoprecipitating the potential heterocomplexes via HA-tagged PRLRWT (Figure 2C) or HA-tagged PRLRI146L (Figure 2D). In both configurations, co-IP of the untagged receptor was observed, which appeared qualitatively similar to what was observed in homodimerization experiments (Figures 2A,B). Although the antagonist seemed to result in a slight decrease of PRLRI146L co-IP by HA-PRLRWT (Figure 2C), this was not confirmed by the reverse experiment (Figure 2D).
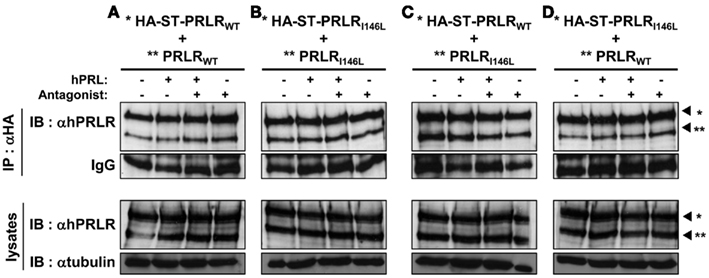
Figure 2. PRLRWT and PRLRI146L form homo and heterodimers. HEK 293 cells were co-transfected using various combinations of plasmids encoding HA-tagged (*) or untagged (**) PRLR (WT and I146L), as indicated in (A–D). Stimulation was performed using 500 ng/mL hPRL or 20 μg/mL Del1-9-G129R-hPRL, alone or in combination (15 min, 37°C). Co-IPs of receptor complexes from 1 mg cell lysates were performed using anti-HA antibody. The presence of tagged and untagged PRLR in immune complexes (black arrowheads) was determined by αhPRLR immublotting (IB) after 7.5% SDS-PAGE. These experiments show that PRLRWT and PRLRI146L homo and heterodimerize.
Taken together, these findings indicate that (i) I146L mutation does not induce consistently detectable modification of receptor stoichiometry, (ii) PRLRI146L can heterodimerize with PRLRWT, (iii) the antagonistic properties of Del1-9-G129R-hPRL are not mediated by a disruption of preformed PRLR homo/heterodimers, whatever the receptor involved.
In order to confirm the existence of receptor complexes in native conditions, we used blue native polyacrylamide gel electrophoresis (BN-PAGE) followed by αPRLR immunobloting using a monoclonal antibody targeting the ECD of the human receptor. In control conditions, the migration profile of PRLRWT expressed in so-called HL-5 clone (Bernichtein et al., 2003a) exhibited two major bands, which does not exclude that other (bigger) complexes not detected by this approach (e.g., due to masked epitopes) may also exist (Figure 3A). The profile of PRLRI146L was very similar to that of PRLRWT, therefore we focused on the latter to investigate the nature of these two bands. Solubilized membrane extracts were then treated using various denaturing conditions (2 M urea, 50 mM DTT, up to 0.8% DDM, 60°C heating), among which only heating modified the profile that now appeared as a single band (the faster migrating one), which was assigned to the monomeric form of the receptor (Figure 3A). This indicated that the upper band corresponded to a non-covalent complex, while the absence of effect of the other denaturing conditions suggested that this αPRLR-immunoreactive complex was very stable. Accordingly, urea concentration up to 4 M was necessary to disrupt this complex (data not shown). The exact nature of this larger complex could not be definitely demonstrated. Although it is likely that it corresponded to PRLR homodimers, one cannot exclude the possibility that this band also contained PRLR complexed to well identified receptor-associated-partners, e.g., JAK2, MAPK, or Stat proteins. However, re-blotting the membranes to identify any of these partners was unsuccessful.
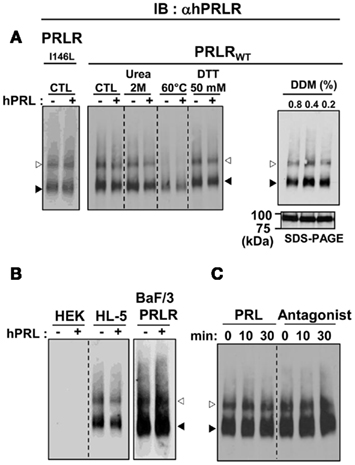
Figure 3. Blue native gel electrophoresis reveals that membrane-associated PRLRWT and PRLRI146L mainly exist in two oligomeric states. (A) Solubilized membrane extracts of HL-5 cells were resolved by BN-PAGE and PRLR complexes were identified by αPRLR immunoblotting. In control conditions (CTL), PRLRWT and PRLRI146L exhibited similar migration profiles composed of two main bands (labeled ▻ and ▶). Of the various denaturing conditions that were tested (urea, heating, DTT, and DDM), heating is the only one that significantly modified the migration profile as the PRLR migrated as a single band, assigning the latter to the monomeric form (▶). Samples treated with DDM were also analyzed by SDS-PAGE to ensure equal loading. (B) To determine whether the expression levels of the PRLR affected receptor stoichiometry, we compared stable clones expressing high (HL-5 clone) or low (Ba/F-LP population) levels of PRLR; non-transfected HEK 293 cells were used as negative control. Although the autoradiography shown for Ba/F 3 samples had to be over-exposed to detect the PRLR, the migration profile was qualitatively similar irrespective of PRLR expression levels. (C) The effect of ligand binding on the formation of receptor complexes was analyzed by adding PRL or Del1-9-G129R-hPRL antagonist (antago) to HL-5 cells. None of these conditions significantly altered the migration profile.
In order to assess that PRLR pre-association did not result from forced receptor clustering due to over-expression in transient transfections, we compared the HL-5 clone (60,000 hPRLRWT per cell; Kinet et al., 1999) to so-called Ba/F-LP, a previously described Ba/F3 cell population expressing less than 500 hPRLRWT per cell (Bernichtein et al., 2003a). As shown on Figure 3B, PRLR complexes extracted from both cell lines exhibited similar stoichiometries in native conditions, as the two major bands were again detected. Finally, in agreement with co-IP experiments, activation of PRLRWT by hPRL, or receptor blockade by Del1-9-G129R-hPRL antagonist, did not detectably modify the migration profile (Figure 3C).
Taken together, these experiments suggest that (i) membrane-anchored hPRLRWT exists in two main oligomeric states, presumably corresponding to monomeric and dimeric forms, (ii) the larger complex is highly stable and does not result from forced clustering, and finally (iii) neither triggering nor blockade of the receptor affect the apparent stoichiometry of PRLR complexes.
Use of BRET to Evaluate Distance/Orientation Modification in Preformed hPRLRWT and hPRLRI146L Homodimers Under hPRL or Antagonist Binding
As BN-PAGE analysis failed to detect any significant change in the native stoichiometry of membrane-associated PRLR under PRL or antagonist binding, we asked whether ligand binding could affect the conformation of preformed PRLR dimer, either WT or I146L, and whether the type of ligand could affect the results. As stated in the Introduction, recent BRET1 studies have suggested that PRL binding to PRLRWT does not induce detectable fluorescence changes (Qazi et al., 2006). We first aimed at confirming we were able to reproduce that data.
Saturation BRET experiments allow to determine BRET50 values, which discriminate receptor recruitment from a preformed dimer (Mercier et al., 2002). As shown in Figure 4A, no difference in BRET50 values could be observed for PRLRWT between basal (BRET50 = 2.0 ± 0.7) and activated states (BRET50 = 2.4 ± 0.8), in full agreement with previous reports (Qazi et al., 2006). We further showed that receptor blockade using the pure antagonist Del1-9-G129R-hPRL also failed to alter BRET50 value (2.7 ± 0.9). Further analyses of activation or blockade of homo- and hetero-paired PRLRWT or PRLRI146L were therefore performed in two transfection conditions only (4:1 and 8:1 PRLR–YFP/PRLR–Rluc ratios). In these conditions, neither PRL nor the antagonist significantly altered BRET signals compared to non-stimulated cells, whatever the receptor pairs involved (Figure 4B).
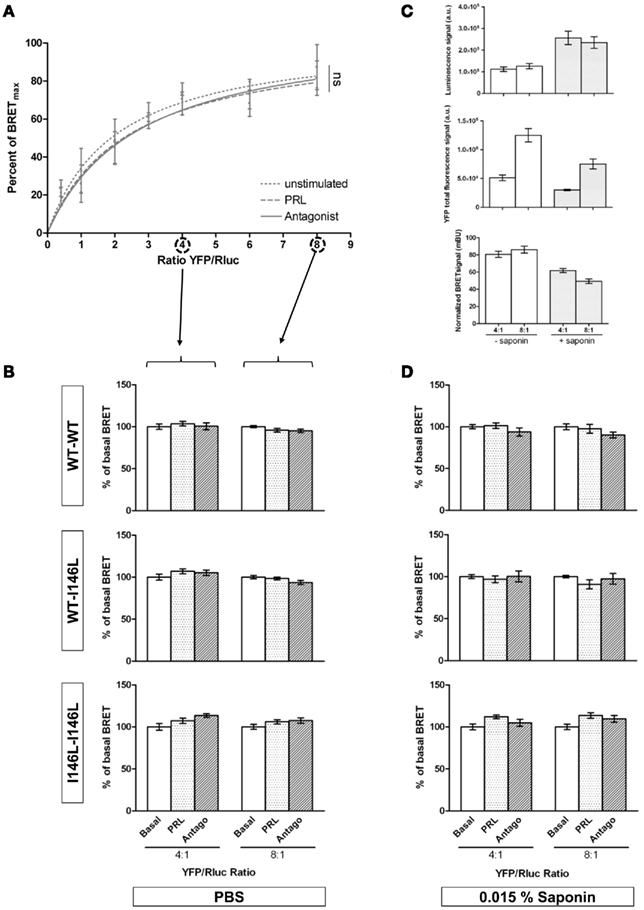
Figure 4. Analysis of PRLR activation and blockade using BRET1. (A) Saturation BRET assays of homo-paired PRLRWT (n = 3). HEK 293 cells were co-transfected using a constant amount of PRLR–Rluc plasmid (25 ng per well) and increasing amounts of PRLR–YFP plasmid (0–200 ng per well). Ratios are indicated on the X axis. BRET signals were detected in basal conditions and 15 min after stimulation using 1 μg/mL PRL or 10 μg/mL Del1-9-G129R-hPRL antagonist. Data are represented as the mean of percentage of maximal BRET signal (Bmax) calculated for each stimulation condition. (B) BRET assays were performed for homo and heterodimers of PRLRWT or PRLRI146L at 4:1 and 8:1 YFP/Rluc conditions. BRET signal was detected after 15 min of PRL or antagonist stimulation in PBS. Data are represented as the mean of percentage of basal condition (n = 4). (C) Luminescence of PRLR–Rluc (a.u. = arbitrary unit, top panel), total fluorescence of PRLR–YFP (middle panel) and normalized BRET (bottom panel) were measured in transfected HEK cells after a 15-min incubation in the presence or not of 0.015% saponin. As similar results were obtained for all receptor combinations, only data obtained using non-stimulated PRLRWT are shown. (D) Idem as in B, but for BRET assays performed in the presence of 0.015% saponin (n = 4).
It is well known that a large part of the PRLR pool resides inside the cell. We reasoned that BRET signals involving intracellular PRLR may partly mask BRET changes affecting cell surface receptors upon ligand binding. In order to increase the number of receptors accessible to the ligands, we repeated these experiments in the presence of 0.015% of saponin, a molecule known to permeabilize biological membranes (Couturier and Jockers, 2003). This effect of saponin was displayed by increased basal PRLR–Rluc signals presumably reflecting higher accessibility of intracellular receptors to coelenterazine (Figure 4C, top panel). As in the meantime we also observed that saponin intrinsically affected YFP fluorescence (Figure 4C, middle panel), this resulted in a slight decrease of absolute BRET levels (Figure 4C, lower panel). Despite membrane permeabilization, BRET assays of activation or blockade of either homo- or hetero-paired PRLR revealed no difference compared to the basal state (Figure 4D).
In conclusion, in agreement with previous studies as well as with all the experiments reported above, these data are not in favor of receptor recruitment as being the principal mechanism mediating PRLR activation. Rather, they suggest that ligand-mediated receptor activation or blockade involves rearrangements within preformed PRLR complexes upon ligand binding. These could involve conformational changes of cytoplasmic tails that could not be detected in this study due to the intrinsic limitations of BRET procedures.
Use of TR-FRET to Detect Preformed hPRLRWT and hPRLRI146L Homodimers
As both hPRLRI146L and hPRLRWT appeared to be equally and constantly dimerized in all tested conditions (unbound, activated, or blocked), we turned to a sensitive approach able to detect distance changes between interacting proteins, namely TR-FRET. The specific reactive fluorophore concentration used for ratiometric labeling of ST-PRLR was determined by fluorophore titration and FRET signal detection, as shown on Figure 5A. The ability of SNAP-fluorophores to specifically label cell surface ST-PRLR was also assessed by confocal imaging, e.g., using SNAP-Green (Figure 5B). As this was the first time this technology was applied to a member of the cytokine receptor superfamily, FRET signals obtained for the PRLRWT were compared to those obtained for a seven transmembrane domain GPCR (mGluR2) and a single transmembrane domain Immunoglobulin receptor (CD4) earlier characterized using this approach (Maurel et al., 2008). As shown in Figure 5C, COS-7 cells transiently expressing exogenous ST-PRLR exhibited FRET signals that were in the range of signals obtained for the two control receptors, suggesting the reliability of this method when applied to the PRLR. Finally, the specificity of PRLR FRET signals was assessed by co-expressing receptors lacking the SNAP tag. As shown on Figure 5D, expression of HA-PRLR markedly decreased FRET signals obtained using ST-PRLR due to the formation of HA-PRLR/ST-PRLR heterocomplexes. In contrast, high level expression of HA-CD4 had no effect on ST-PRLR FRET signals.
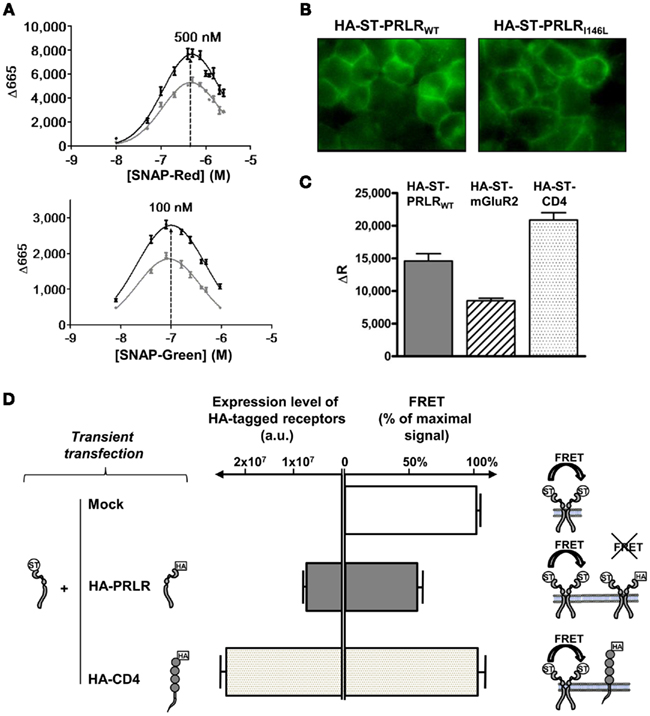
Figure 5. Application of TR-FRET technology to the PRLR. (A) Determination of fluorophore concentration for equimolar ratio of PRLR labeling using a given fluorophore pair. Variations in FRET signals using PRLRhigh stable cell lines (characterized on Figure 6) are shown as a function of acceptor fluorophore concentration. Cells were incubated 1 h at 37°C in the presence of 100 nM of SNAP-Lumi4 Tb and of increasing concentrations of SNAP-Red (left panel) or SNAP-Green (right panel). Maximal Δ665 signal [(S665)Donor + Acceptor − (S665)Donor] is representative of fluorophore concentration that gives same quantity of PRLR labeled with donor or acceptor. The optimal concentrations ratio was obtained for 100 nM of SNAP-Lumi4 Tb with 500 nM of SNAP-Red and 100 nM of SNAP-Lumi4 Tb with 100 nM of SNAP-Green. Data obtained for PRLRWT and PRLRI146L are represented in gray and black, respectively. (B) Confocal imaging of PRLRhigh stable clones after 1 h (4°C) labeling using SNAP-Green fluorophore. (C) COS-7 cells were transfected using plasmids encoding ST tagged receptors (PRLR, mGluR2, and CD4), then labeled 48 h later using 100 nM SNAP-Lumi4 Tb alone or in combination with 500 nM SNAP-Red. Signals at 620 or 665 nm were measured, and FRET signals (ΔR) were determined as detailed in Section “Materials and Methods.” The amplitude of FRET signals obtained for each receptor at similar expression level as determined by specific fluorescence values at 620 nm (S620) are represented. (D) COS-7 cells were co-transfected using plasmids encoding ST-PRLR (in all conditions) and, as indicated, empty vector (mock), HA-PRLR or HA-CD4 plasmid. Left bars represent the expression level of cell surface HA-receptors as determined using anti-HA ELISA (arbitrary units, a.u.). Right bars represent FRET signals (using 100 nM SNAP-Lumi4 Tb and 500 nM SNAP-Red) involving ST-PRLR homodimers in the three experimental conditions, as represented on the right. Values were normalized to the FRET signal obtained for ST-PRLR + empty vector condition.
Next, COS-7 cells were transiently transfected using increasing amounts of HA-ST-PRLRWT or HA-ST-PRLRI146L encoding vectors. Cell surface expression of PRLR proteins was quantified by labeling cell surface SNAP tags using SNAP-Lumi4 Tb alone and measuring fluorescence at 620 nm (Figure 6A, left panel). A good correlation could be observed between the amount of transfected plasmid and the fluorescence emitted by tagged PRLR (WT or I146L). FRET signals were then measured at 665 nm following double labeling of ST-PRLRs using SNAP-Lumi4 Tb and SNAP-Red fluorophore pair. Normalized FRET was calculated as the ratio between specific fluorescence at 665 nm (FRET signals) and the specific fluorescence at 620 nm (reflecting the number of cell surface PRLRs). As shown on Figure 6A (right panel), normalized FRET obtained in the absence of stimulation was not affected by the level of receptor expression. Since transient transfections could possibly mask minor variations of FRET signals due to heterogeneous expression levels of PRLR between the various wells, we aimed at confirming these findings using stable clones. Several geneticin-selected clones of HEK 293 cells expressing HA-ST-PRLRWT or HA-ST-PRLRI146L were characterized using (i) ST labeling and αPRLR western blotting to determine relative PRLR expression levels between the various clones, and (ii) phospho-Stat5 immunoblot to ensure PRL-responsiveness as shown in Figure 1B (data not shown). Based on these analyses, two stable clones to be used for further TR-FRET studies were selected for each receptor based on their high (PRLRhigh) versus low (PRLRlow) level of receptor expression (Figure 6B, left panel). As observed above using transient transfections, normalized FRET signals were very similar for all clones (Figure 6B, right panel), further confirming that the basal FRET detected for non-stimulated receptors (WT or I146L) was not affected by the expression level of the latter.
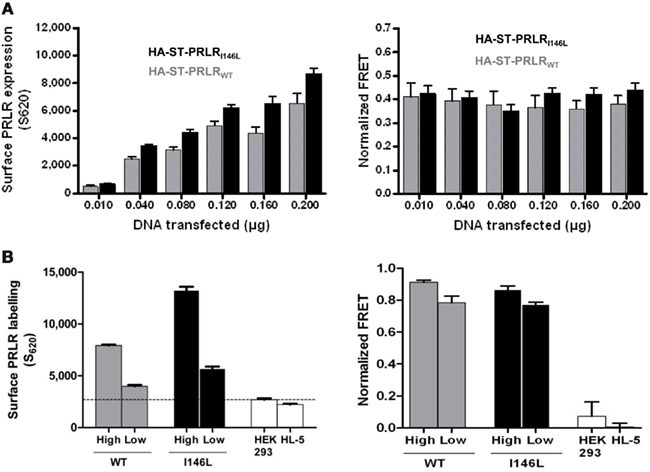
Figure 6. Constitutive dimerization of cell surface PRLRWT and PRLRI146L as determined by TR-FRET is independent of receptor expression level and cell type. (A) Increasing amounts of plasmids encoding PRLRWT or PRLRI146L TR-FRET constructs were transiently transfected into COS-7 cells. Increasing levels of PRLR protein expression was confirmed by measuring the level of fluorescence emitted by cells labeled with the fluorescence donor only (S620, left panel). Whatever the level of PRLR expressed at the cell surface, normalized FRET ratios measured after labeling with SNAP-Lumi4 Tb and SNAP-Red fluorophore combination were unchanged (right panel). (B) Similar experiments were performed using stable HEK 293 clones expressing either low (PRLRlow) or high (PRLRhigh) levels of HA-ST-PRLR (WT or I146L) as determined by 620 nm fluorescence [see (A)]; parental HEK 293 (undetectable levels of endogenous PRLR) and HL-5 clone (high level of untagged exogenous PRLR) were used as negative controls. Normalized FRET signals obtained after labeling these clones using SNAP-Lumi4 Tb and SNAP-Red fluorophore combination were similar, irrespective of PRLR expression levels (right panel). Results are representative of at least three independent experiments performed in triplicate.
Taken together, these results demonstrate that FRET signals observed for non-stimulated receptors reflect specific physical interactions and do not result from random collisions of labeled receptors due to over-expression. Therefore, for convenience of FRET detection, due to higher fluorescence levels, the experiments reported below were performed using the PRLRhigh clones.
Use of TR-FRET to Monitor Distance Changes within hPRLRWT or hPRLRI146L Homodimers Following Receptor Triggering or Blockade
In order to assess TR-FRET methodology could be used to evaluate the effect of ligand binding on receptor conformation, we first monitored ligand–receptor interaction using TR-FRET binding assays. To that end, hPRL was randomly labeled on exposed lysines using Red fluorophores, and receptors (HA-ST-hPRLRWT or HA-ST-hPRLRI146L) were labeled on ST using SNAP-Lumi4 Tb as described above. Stimulations were performed at room temperature, and ligand-receptor FRET signals were recorded for 24 h. Although FRET signals reached the equilibrium (maximal values) after 17–20 h (data not shown), KD values could be calculated from much shorter times without being affected by longer incubations (dose–responses reported in Figure 7A were obtained after 3 h incubation). Curve fittings performed using the “one specific binding site model” revealed KD values of ∼4 nM for hPRL to both hPRLRWT and hPRLRI146L (Table 1), which is in good agreement with affinities calculated using classical 125I-hPRL receptor assays (Bernichtein et al., 2003b). This experiment demonstrated that (i) PRL does interact with tagged receptors with the same affinity as previously documented for untagged PRLRWT, and (ii) ligand/receptor interactions are detected shortly after ligand addition, and binding affinities can be reliably measured after 3 h incubation at RT, which is the condition that was used for the dose–response experiments described below.
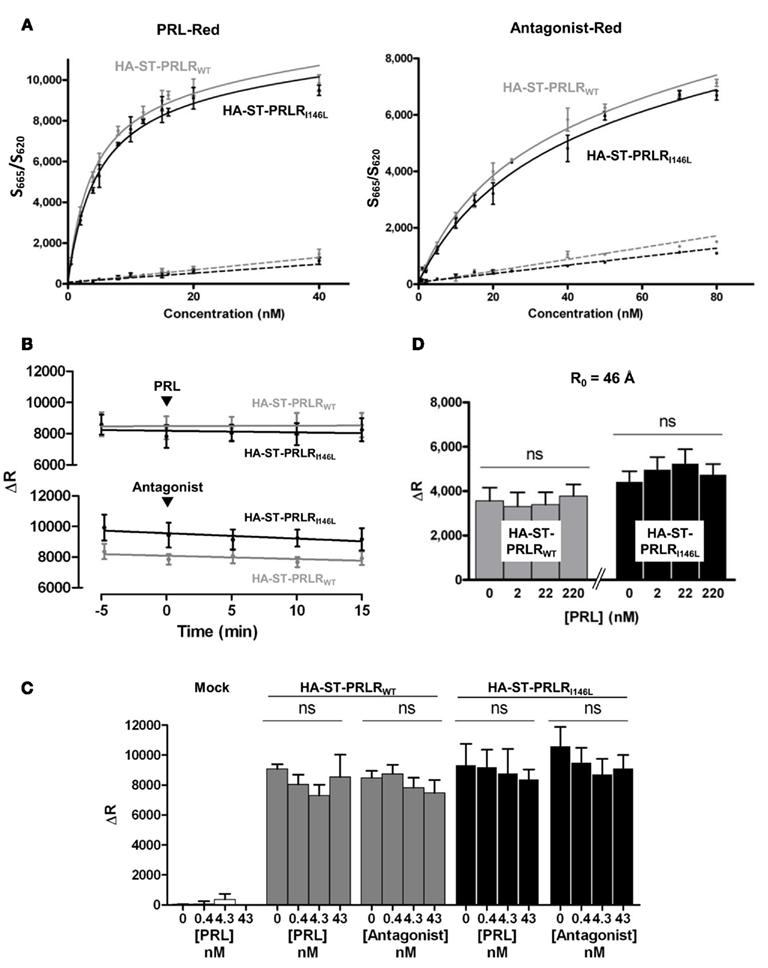
Figure 7. PRLRWT or PRLRI146L triggering does not affect receptor dimerization. (A) PRLRhigh stable cell lines were labeled using SNAP-Lumi4 Tb (100 nM), then stimulated with increasing doses of Red-labeled hPRL (left panel) or antagonist (right panel) alone (solid lines) or in the presence of excess non-labeled PRL (dotted lines), as indicated. FRET signal corresponding to ligand–receptor interaction was detected along time at RT (3 h stimulation is represented). Curve fitting was performed using a 1-site binding algorithm to determine KD values. (B,C) TR-FRET assay of homo-paired ST-PRLRWT and ST-PRLRI146L. COS-7 cells were transiently transfected using plasmids (0.2 μg/well) encoding either receptor as indicated (mock: empty plasmid). Forty-eight hours later, ST-PRLRs were labeled (1 h, 37°C) using SNAP-Lumi4 Tb (100 nM) and SNAP-Red (500 nM; R0 of 58Å). Time-course [PRL or antagonist 43 nM (B)] and PRL or antagonist dose-response [15 min (C)] stimulations were performed, which did not affect FRET signals for either PRLR. (D) TR-FRET signals of receptor triggering by addition of PRL (0–220 nM, 3 h, RT), obtained using 100 nM SNAP-Lumi4 Tb and 100 nM SNAP-Green fluorophores (R0 = 46 Å) failed to significantly affect FRET signals. Results are representative of at least three independent experiments performed in triplicate.
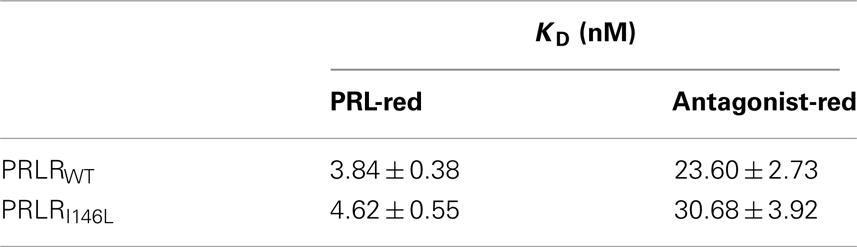
Table 1. Binding affinities of hPRL and Del1-9-G129R-hPRL to PRLRWT and PRLRI146L using TR-FRET binding assays.
We then used TR-FRET to investigate whether any distance change occurred between predimerized PRLR moieties upon PRL binding. Time-course and dose-dependent hPRL stimulations were performed for both receptors, first using transiently transfected COS-7 cells (Figures 7B,C). FRET signals (ΔR) obtained using SNAP-Lumi4 Tb + SNAP-Red fluorophore pair remained unchanged in comparison to baseline, irrespective of the time elapsed after stimulation (we tested 0–3 h at RT, 0–15 min timeframe with 43 nM of ligand is shown on Figure 7B) or the concentration of agonist or antagonist (0–43 nM), and this was true for both PRLRWT and PRLRI146L. The slightly lower values of ΔR signal obtained for hPRLRWT compared to hPRLRI146L clones presumably reflects the lower level of receptor expression in the former (Figure 6B). All these observations were strictly identical whether stimulations were performed at room temperature or 37°C (data not shown). We then aimed at repeating these experiments using stable cell lines in order to avoid any interference of possible heterogeneous PRLR expression in transient transfections. Using PRLRhigh stable clones described in Figure 6B, we fully confirmed that PRLR triggering failed to affect FRET signals for either receptors (Figure 7C). Finally, as subtle distance modifications resulting from conformational adjustments can sometimes be detected using one fluorophore pair and not with another, we aimed at using another fluorophore. Thanks to the spectral properties of terbium cryptate that exhibits a multi-peak emission spectrum, combination of various acceptors are possible with this lanthanide. The Förster distances (R0) of fluorophore pairs consisting of SNAP-Lumi4 Tb + SNAP-Red or SNAP-Green are 58 and 46 Å, respectively, which falls within the distance range of interacting proteins. The use of SNAP-Lumi4 Tb + SNAP-Green pair (Figure 7D) produced lower values of FRET ΔR compared to SNAP-Lumi4 Tb + SNAP-Red pair, which is strictly dependent on spectral properties of these fluorophores. No modification of ΔR was noticed under a wide range of hPRL concentration (Figure 7D), irrespective of the receptor involved.
We then conducted similar experiments using the pure PRLR antagonist Del1-9-G129R-hPRL as the ligand in order to determine whether the outcome of receptor blockade (i.e., using a ligand able to bind but not to activate the PRLR) differed from that described above for receptor triggering. TR-FRET binding assays confirmed the interaction between the antagonist and the ST-PRLRs (WT or I146L; Figure 7A, right panel), and again, affinity values calculated using this method were very similar to those obtained using classical radioreceptor assays, i.e., one log lower compared to hPRL (Table 1). Similar to hPRL binding, antagonist binding failed to affect TR-FRET signals, whether time-course (Figure 7B), dose–response (Figure 7C), or fluorophore pairs with different R0 (data not shown) were involved.
Taken together, the data reported in Figures 4–7 (i) confirm that no distance variation and/or reorientation of homodimerized PRLRWT intracellular domains (ICD) is detectable using BRET1 following receptor activation, and reveal that (ii) there is no major distance variation between the N-termini of unbound or liganded homodimerized PRLR chains. Importantly, these experiments discriminated neither PRL from the antagonist, nor PRLRI146L from the wild type receptor.
Discussion
Our group has been long involved in the development of PRLR antagonists (Goffin et al., 1996a, 2005; Bernichtein et al., 2003b). These studies led to the design of a pure PRLR antagonist (Del1-9-G129R-hPRL) that was shown to be a potent inhibitor of PRLR signaling mediated by endocrine as well as autocrine PRL, using both in vitro (for a review Bernichtein et al., 2010) and very recently, in vivo (Rouet et al., 2010) models. Furthermore, we reported in 2008 the first functional genetic PRLR variant associated with a human disease. This missense mutation encodes I146L substitution in the ECD, leading to a constitutive basal activity that is not always easily detected at the molecular level (Goffin et al., 2010), but anyway sufficient to immortalize stably transfected Ba/F3 cells (Bogorad et al., 2008; Courtillot et al., 2010). Interestingly, this ligand-independent basal activity is abrogated by Del1-9-G129R-hPRL, which then acts as a reverse agonist (Bogorad et al., 2008). The aim of this study was to progress in the understanding of the molecular mechanisms involved in agonist or I146L-mediated receptor activation, and antagonist-driven receptor blockade.
It is currently accepted that the active form of the PRLR is a homodimer. This model relies on structural (Elkins et al., 2000; Broutin et al., 2010), biophysical (Sivaprasad et al., 2004; Jomain et al., 2007), biochemical (Gadd and Clevenger, 2006), and mutagenesis/functional (Goffin et al., 1994, 2005; Gadd and Clevenger, 2006; Langenheim et al., 2006) evidence that agonist ligands interact with two PRLR moieties. Accordingly, experiments involving ECDs (SPR, crystallogenesis; Jomain et al., 2007; Svensson et al., 2008) showed that functional abolition of binding site 2 prevented antagonists from interacting with the second receptor moiety (they only form 1:1 complexes via binding site 1). Recent studies have addressed whether dimerization of membrane-anchored PRLR was constitutive or ligand-induced. In other words, they investigated whether receptor activation involved (i) clustering of two monomeric receptors to form a homodimer, following the sequential dimerization model initially suggested (Fuh et al., 1992; Goffin et al., 1994; Figure 8A), or (ii) conformational rearrangements within preformed dimers (Figures 8B–D). Clearly, the current consensus is in favor of the latter model (Gadd and Clevenger, 2006; Qazi et al., 2006). However, the actual mechanism of receptor triggering is still uncertain as the putative rearrangements induced by agonists are yet to be identified. BRET2 studies involving C-terminal tagged PRLR have indicated that agonist binding (natural or engineered ligands) was required to generate detectable BRET signals, while mild antagonists failed to do so (Tan et al., 2005; Langenheim et al., 2006). According to the predimerized model, such data can be interpreted as evidence that agonists induce conformational changes propagating from the ECD to the intracellular part of the receptor, leading both C-terminal tails to orient in sufficient proximity to permit energy transfer (see below). However, such data are not incompatible with a model of sequential dimerization in which agonists, but not antagonists, would be able to bring together two receptor moieties. Otherwise, BRET1 studies involving any isoform of the PRLR (Qazi et al., 2006; Xie et al., 2009) have shown that basal BRET signals were detectable without hormone stimulation, suggesting that C-terminal tails of unbound receptors are actually at distance compatible with energy transfer, which is in favor of the predimerized model. In addition, as classical FRET/BRET signals are directed by two distinct parameters – distance and respective dipole orientation of fluorophores (Pfleger and Eidne, 2006) – simultaneous modifications of both parameters can theoretically compensate each other and mask conformational changes. These few examples highlight the difficulty of interpreting data obtained using classical resonance energy transfer technologies.
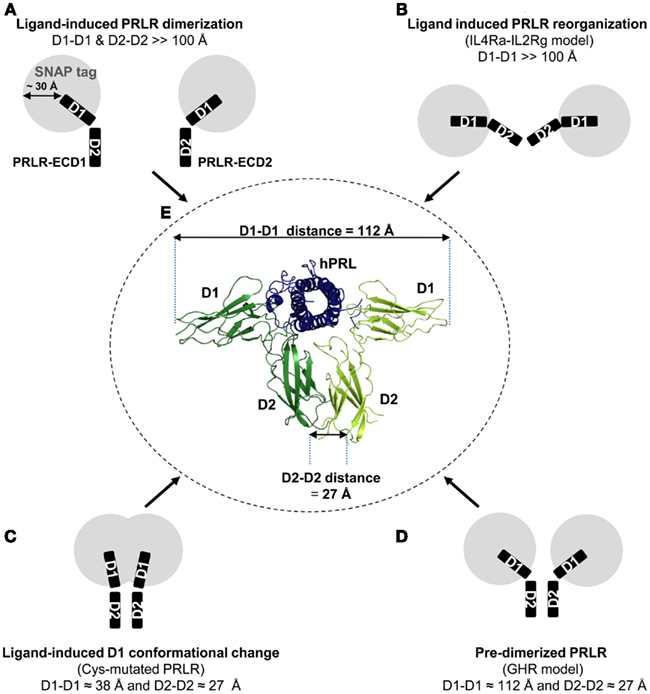
Figure 8. Hypothetic models of preformed PRLR homodimer triggering by PRL. The various models of cytokine receptor activation discussed in the text are illustrated. (A–D) Represent the unbound states, while panel E corresponds to the liganded receptor. In (A–D), two PRLR-ECDs are represented each as two black rectangles, corresponding to subdomains D1 and D2. The potential space occupancy of the SNAP tag is represented by a gray circle whose size was predicted based on the 3D structure of O6-benzyl-guanine transferase (PDB: 2JEG), the enzyme from which SNAP tag is derived. The 30-Å value corresponds to the distance between the active site of the enzyme (which traps the fluorophore in the case of the SNAP tag) and the C-terminus, which is fused to the N-terminus of the PRLR-ECD. (A) Sequential dimerization of PRLR induced by the ligand. In this model, the distance separating the two PRLR moieties before ligand binding is expected to be much higher than 100 Å, i.e., beyond the detection limit of RET technologies. (B) This model of predimerized receptor was proposed for the IL-4R. Although D2 domains of each monomer are in close proximity, D1 domains are distant from each other. After ligand binding, D1 domains are supposed to undergo conformational changes leading to reduced D1–D1 distance within the receptor dimer. (C) This model was proposed for an engineered cysteine-mutated SF1b PRLR variant. In this model, the PRLR exists as a preformed dimer in which D1 and D2 domains are aligned. In contrast to model B, ligand binding is assumed to move both D1 domains away from each other. (D) This final model of predimerized receptor has been proposed for the GHR. In this model, the distance between D1 domains is relatively constant irrespective of ligand binding. Receptor activation involves conformational and subtle intrinsic structural changes that are not detectable by RET approaches. (E) The crystal structure of the trimeric hPRL/rPRLR-ECD2 complex (PDB: 3NPZ) is represented. Distances were determined using the PyMOL Molecular Graphics System (DeLano, 2002; LLC, San Carlos, CA, USA) and the program Coot (Emsley and Cowtan, 2004).
The results of our co-IP experiments (Figure 2) are in good agreement with those earlier reported by others (Gadd and Clevenger, 2006), who suggested that receptor dimers exist prior to ligand binding. Our observations further strengthened the hypothesis that the activation state of the PRLR is not related to its dimerization state, at least from what can be deduced by denaturing electrophoresis (SDS-PAGE). Indeed, our data definitely discarded the possibility that the pure antagonist Del1-9-G129R-hPRL acted by disrupting receptor homodimerization (while its antagonistic effect was assessed in control phospho-Stat5 experiments, data not shown). According to a previous study showing that the partial antagonist G129R-hPRL failed to induce BRET signals (while PRL did; Tan et al., 2005), our findings support the hypothesis that antagonists induce either no, or different conformational changes compared to those accompanying activation of the receptor by agonists (see below). We also demonstrated that the ability of PRLRI146L to homodimerize was undistinguishable from that of PRLRWT (in the presence or in the absence of ligand), suggesting that I146L mutation does not detectably alter the frequency and/or stability of receptor–receptor interactions. Finally, we provided evidence that PRLRI146L and PRLRWT are able to heterodimerize. The functional properties of such heterodimers are at present unknown. We can notice that constitutive signaling was observed in a reconstituted “heterozygous” experimental model involving MCF-7 cells expressing endogenous PRLRWT and exogenous PRLRI146L (Bogorad et al., 2008). This model provides evidence that basal activity of PRLRI146L is not abolished in the presence of PRLRWT, but is certainly not a proof that it is mediated by heterodimers. Further investigations are warranted since all patients identified to date exhibited heterozygous profile regarding the mutation (Bogorad et al., 2008; Courtillot et al., 2010).
Antagonist-mediated receptor blockade and I146L-mediated receptor triggering are two activity-related events that originate from the ECD of the PRLR. As none of these situations seemed to affect receptor dimerization per se, it was relevant to investigate how the conformation of the ECD could potentially be affected. To address this question, we used TR-FRET technology, which presents two major advantages over classical FRET/BRET approaches. First, as TR-FRET signals are only directed by distances between fluorescent tags, any FRET variation can be translated as a distance change between fluorophores, without any risk of misinterpretation. Second, as fluorophores specifically label cell surface receptors, TR-FRET is particularly appropriate to follow up distance modifications between homodimerized ECDs that would be induced by ligand binding. The existence of FRET signals in the absence of stimulation first indicated that SNAP tags linked to receptor ECDs are at distances compatible with energy transfer (<100 Å). Relative FRET signals were independent of PRLR expression levels, arguing for the specificity of the signal observed. PRLRI146L and PRLRWT produced similar levels of FRET, suggesting they do not distinguish from each other by the distance separating ECDs of homodimerized receptor. All these observations agree with, and complement biochemical studies confirming that unbound receptors (WT and I146L) are in close proximity at the cell surface, and supporting their existence as preformed homodimers. Second, and more important, the addition of hPRL or Del1-9-G129R-hPRL failed to modify the intensity of FRET signals in all the conditions tested, including time-course and dose-dependent experiments, and this was true for both receptors. Furthermore, we used two different fluorophore pairs (with different R0) to rule out the possibility that the absence of FRET signal modification could be related to the intrinsic characteristics of fluorophores (Figures 7C,D). In these studies, the actual occurrence of ligand–receptor interactions was confirmed in control binding experiments involving energy transfer between labeled ligands and receptors, ruling out any technical issue (Figure 7A).
Taken together, these data indicate that receptor activation and blockade do not correlate with detectable distance changes between the homodimerized ECDs compared to the unbound state. As TR-FRET involves cell surface labeling, i.e., the population of receptor that specifically interacts with the ligands, these data definitely discard the sequential model of PRLR dimerization (Figure 8A). The two crystal structures of PRL/PRLR-ECD2 complexes that we recently reported (Broutin et al., 2010; Van Agthoven et al., 2010) indicate that the N-termini of the two ECDs are 112 Å away from each other in the crystals (Figure 8E). It could be tempting to speculate that this distance applies to the ECDs of the membrane-anchored receptor, either bound or unbound. However, the actual distance between membrane PRLR-ECDs could not be experimentally determined in this study, since FRET signals are mediated by fluorophores that are covalently linked to SNAP tags, the exact position of which relative to PRLR-ECDs is unknown. However, it is clear that these tags fold outside ligand binding sites, since binding affinities (Figure 7A) and signaling capacities (Figure 1B) of ST-PRLRs were not affected by the presence of the tag. Regarding binding, the affinities determined by ligand–receptor TR-FRET (Figure 7A) perfectly matched previously reported values (Kinet et al., 1999; Bernichtein et al., 2003b), which brings a proof-of-concept that this non-radioactive technology involving labeled PRL could be used as an alternative methodology to classical binding assays involving 125I-PRL.
Various models of structural ECD flexibility have been proposed for cytokine receptors (Figures 8B–D), that can be used to speculate on the mechanism of PRLR activation. Three of them are shortly discussed below. The type I IL-4 receptor is an heterocomplex involving the cytokine-specific IL-4 Rα chain and the common Γ chain. Weidemann et al. (2007) have proposed a model (Figure 8B) in which the tryptophans of the highly conserved WS motif of each receptor moiety interact with the lipid membrane in the unbound state, providing an “open” conformation (“off-state”) of the ECD heterocomplex. This conformation is assumed to be released upon ligand binding to IL-4 Rα before bringing the Γ chain ECD into a “closed” conformation locked by the cytokine (“on-state”). As such a model involves large modifications of the ECD–ECD distance which should result in big variations of TR-FRET signals, we believe it does not apply to the PRLR.
Dufau’s group recently proposed a somehow opposite model for a short PRLR (SF1b) mutant lacking the two internal disulfide bonds within the D1 subdomain of the ECD (Figure 8C). Given the general importance of intramolecular disulfides in protein folding, such a mutant is expected to exhibit marked conformational alteration (its actual 3D structure is unknown). Accordingly, it is no longer able to bind PRL (Rozakis-Adcock and Kelly, 1991; Xie et al., 2009). In addition, this mutant was found to have lost its dominant-negative activity toward the long isoform, which correlated with its inability to heteromerize with the latter (Xie et al., 2009). Computer modeling based on the oPL-PRLR2 crystal structure (Elkins et al., 2000) suggested that structural changes affecting D1, although not drastic, could lead to a reorientation of D1 with respect to D2 domains (basically, they are predicted to align), which could result in stabilization of the mutant homodimer due to interactions between D1 domains. At first sight, such a model could have applied to PRLRI146L, since receptor homodimer stabilization was a reasonable starting hypothesis to explain the higher basal activity of this natural variant (Bogorad et al., 2008). It is very unlikely, however, as PRLRI146L exhibits two major differences with this SF1b cysteine mutant: it binds PRL with high affinity (Figure 7A) and it heterodimerizes with PRLRWT. Even if one hypothesized that PRLRI146L could adopt such an aligned D1–D2 conformation in the unbound state while remaining able to interact with PRL, hormone binding would imply the opening of the ECD homodimer to reach the known structure of the ternary complex (Figure 8C). This would likely produce significant FRET signal modifications that were not experimentally observed.
Finally, the third model is the one proposed for GHR (Brown et al., 2005) and EPOR (Remy et al., 1999), two mono-type cytokine receptors. Like the PRLR, both have been proposed to be dimerized prior to ligand binding. Concerning the GHR, Waters and colleagues proposed that receptor–receptor clustering in the unbound state is mainly mediated by transmembrane domain interactions, resulting in a close proximity of the D2 domains of the two monomers (Figure 8D). In this model, the ligand is assumed to induce relative rotational/translational movement of subunits within the receptor dimer, without detectable changes in ECD conformation as determined using classical FRET. These rotational movements involving no major distance alteration between both receptor chains would propagate toward the cytoplasmic domain, leading receptor-associated JAK2 kinases into close proximity (Brown et al., 2005). The EPOR model slightly differs from the latter as D2 domains have been proposed not to interact in the unbound state. Ligand binding is assumed to bring both D2 and downstream transmembrane/cytoplasmic domains at shorter distances, in a scissor movement, allowing Jak kinases to interact, and to transphosphorylate (Remy et al., 1999).
Our results for PRLR suggest some similarities with the activation mechanisms proposed for these two closely related receptors. In particular, it has been proposed that transmembrane domains play an essential role in stabilizing unliganded homodimers (Gadd and Clevenger, 2006). Nevertheless, as genetic manipulations of the PRLR similar to those performed in the GHR to support the rotational/translational model failed to achieve similar results, this suggests that these two receptors exhibit some differences in their mechanism of activation (Liu and Brooks, 2011). Comparison of free and bound 3D structures of PRLR agonists/antagonist released within the past 3 years have provided clues to the subtle changes occurring in the hormone and the ECD upon receptor activation (Jomain et al., 2007; Svensson et al., 2008; Broutin et al., 2010; Van Agthoven et al., 2010). The 3D structure of PRLRI146L is hoped to reveal whether some of these ligand-induced structural changes are intrinsically present in the receptor mutant. Also, determining how these minor structural rearrangements propagate to the cytoplasmic domain needs to be addressed for the PRLR. In this respect, a general trait of BRET1 studies involving C-terminal tagged cytokine receptors (OBR, GHR, PRLR) is that BRET signals are higher for short/truncated compared to long receptor isoforms (Couturier and Jockers, 2003; Brown et al., 2005; Qazi et al., 2006). In addition, for OBR, an effect of ligand binding is observed using short isoforms only (Couturier and Jockers, 2003). As the constitutive activity of the I146L genetic variant has only been demonstrated for the long receptor (Bogorad et al., 2008), our study exclusively focused on that isoform. Therefore, it may not be surprising that we detected no significant changes in BRET signals upon activation. The larger size of the long receptor cytoplasmic domains may indeed offer a higher degree of flexibility to C-terminal tags, which could interfere with the detection of conformational changes driven by the ECD. As no 3D structure of any cytokine receptor cytoplasmic domain is available, it is very difficult to speculate on distance parameters, and variation thereof, between two receptor chains. For all these reasons, it would certainly be misleading to interpret the absence of strong BRET signal variation as the evidence for the absence of structural rearrangement in the cytoplasmic domain of PRLRWT and PRLRI146L.
Although structural and functional data provide evidence that the PRLR is a homodimer, the occurrence of higher order oligomers was still to be investigated. We used non-denaturing blue native electrophoresis to address this question. This approach revealed the existence of two major forms, whose molecular mass could not be definitely assessed using reference membrane proteins of known molecular mass. However, the fact that only the faster migrating band persisted after heat denaturation supported that the latter corresponded to the monomeric receptor. The slow-migrating complex was tentatively assigned to the homodimer evidenced by FRET and co-IP studies. One could have expected complexes involving the receptor associated with signaling proteins to be detected using this experimental approach. However, membrane blotting using Stat5 or JAK2 antibodies failed to reveal these proteins, suggesting either that such interactions are too transient or not sufficiently stable to persist along sample preparation, or that such complexes represent a minority of membrane-bound PRLR that are below detection threshold, or perhaps that epitopes of these native receptor-associated proteins were not accessible. Taken together, our result do not provide evidence for the existence of receptor oligomers (tetramers, …), although this negative result should not be interpreted as a proof of their non-occurrence. Further studies involving alternative TR-FRET approaches, e.g., the use of receptor constructs involving two different tags (Doumazane et al., 2010), are necessary to specifically address this question.
Conclusion
In conclusion, this study (i) shows that preformed PRLR homodimers identified by co-IP can be detected using TR-FRET, (ii) demonstrates that receptor blockade does not involve disruption of preformed PRLR dimers, (iii) suggests that receptor activation or blockade induce neither detectable change of receptor stoichiometry nor significant movement leading to a change in the distance between the ECDs, and (iv) indicates that none of the parameters investigated in this study allowed to discriminate PRLRWT from PRLRI146L. This latter observation suggests that the basal activity of PRLRI146L may involve subtle intramolecular peculiarities that await the determination of its 3D structure to be identified.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Peter van Endert, Fabrice Maurin, Jurriaan Zwier, and Michel Laget for their help in performing and analyzing TR-FRET studies. They are also grateful to Florence Borot, Aleksander Edelman, Isabelle Broutin, and Patrick England for helpful advices and discussions. Estelle Tallet, Isabelle Fernandez, and Chi Zhang are recipients of support from the Ministère de l’Education Nationale, de la Recherche et de la Technologie. This work was supported in part by INSERM, University Paris Descartes, ANR (Grant 07-PCVI-0029), and CisBio Bioassays.
References
Bernichtein, S., Jeay, S., Vaudry, R., Kelly, P. A., and Goffin, V. (2003a). New homologous bioassays for human lactogens show that agonism or antagonism of various analogs is a function of assay sensitivity. Endocrine 20, 177–190.
Bernichtein, S., Kayser, C., Dillner, K., Moulin, S., Kopchick, J. J., Martial, J. A., Norstedt, G., Isaksson, O., Kelly, P. A., and Goffin, V. (2003b). Development of pure prolactin receptor antagonists. J. Biol. Chem. 278, 35988–35999.
Bernichtein, S., Touraine, P., and Goffin, V. (2010). New concepts in prolactin biology. J. Endocrinol. 206, 1–11.
Bogorad, R. L., Courtillot, C., Mestayer, C., Bernichtein, S., Harutyunyan, L., Jomain, J. B., Bachelot, A., Kuttenn, F., Kelly, P. A., Goffin, V., and Touraine, P. (2008). Identification of a gain-of-function mutation of the prolactin receptor in women with benign breast tumors. Proc. Natl. Acad. Sci. U.S.A. 105, 14533–14538.
Boulay, J. L., O’Shea, J. J., and Paul, W. E. (2003). Molecular phylogeny within type I cytokines and their cognate receptors. Immunity 19, 159–163.
Boute, N., Pernet, K., and Issad, T. (2001). Monitoring the activation state of the insulin receptor using bioluminescence resonance energy transfer. Mol. Pharmacol. 60, 640–645.
Broutin, I., Jomain, J. B., Tallet, E., Van Agthoven, J., Raynal, B., Hoos, S., Kragelund, B. B., Kelly, P. A., Ducruix, A., England, P., and Goffin, V. (2010). Crystal structure of an affinity-matured prolactin complexed to its dimerized receptor reveals the topology of hormone binding site 2. J. Biol. Chem. 285, 8422–8433.
Brown, R. J., Adams, J. J., Pelekanos, R. A., Wan, Y., McKinstry, W. J., Palethorpe, K., Seeber, R. M., Monks, T. A., Eidne, K. A., Parker, M. W., and Waters, M. J. (2005). Model for growth hormone receptor activation based on subunit rotation within a receptor dimer. Nat. Struct. Mol. Biol. 12, 814–821.
Clevenger, C. V., Furth, P. A., Hankinson, S. E., and Schuler, L. A. (2003). The role of prolactin in mammary carcinoma. Endocr. Rev. 24, 1–27.
Courtillot, C., Chakhtoura, Z., Bogorad, R., Genestie, C., Bernichtein, S., Badachi, Y., Janaud, G., Akakpo, J. P., Bachelot, A., Kuttenn, F., Goffin, V., and Touraine, P. (2010). Characterization of two constitutively active prolactin receptor variants in a cohort of 95 women with multiple breast fibroadenomas. J. Clin. Endocrinol. Metab. 95, 271–279.
Couturier, C., and Jockers, R. (2003). Activation of the leptin receptor by a ligand-induced conformational change of constitutive receptor dimers. J. Biol. Chem. 278, 26604–26611.
Dagvadorj, A., collins, S., Jomain, J. B., Abdulghani, J., Karras, J., Zellweger, T., Li, H., Nurmi, M., Alanen, K., Mirtti, T., Visakorpi, T., Bubendorf, L., Goffin, V., and Nevalainen, M. T. (2007). Autocrine prolactin promotes prostate cancer cell growth via Janus kinase-2-signal transducer and activator of transcription-5a/b signaling pathway. Endocrinology 148, 3089–3101.
Degorce, F., Card, A., Soh, S., Trinquet, E., Knapik, G. P., and Xie, B. (2009). HTRF: a technology tailored for drug discovery – a review of theoretical aspects and recent applications. Curr. Chem. Genomics 3, 22–32.
Doumazane, E., Scholler, P., Zwier, J. M., Trinquet, E., Rondard, P., and Pin, J. P. (2010). A new approach to analyze cell surface protein complexes reveals specific heterodimeric metabotropic glutamate receptors. FASEB J. 25, 66–77.
Elkins, P. A., Christinger, H. W., Sandowski, Y., Sakal, E., Gertler, A., De Vos, A. M., and Kossiakoff, A. A. (2000). Ternary complex between placental lactogen and the extracellular domain of the prolactin receptor. Nat. Struct. Biol. 7, 808–815.
Emsley, P., and Cowtan, K. (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132.
Fuh, G., Colosi, P., Wood, W. I., and Wells, J. A. (1993). Mechanism-based design of prolactin receptor antagonists. J. Biol. Chem. 268, 5376–5381.
Fuh, G., Cunningham, B. C., Fukunaga, R., Nagata, S., Goeddel, D. V., and Wells, J. A. (1992). Rational design of potent antagonists to the human growth hormone receptor. Science 256, 1677–1680.
Gadd, S. L., and Clevenger, C. V. (2006). Ligand-independent dimerization of the human prolactin receptor isoforms: functional implications. Mol. Endocrinol. 20, 2734–2746.
Goffin, V., Bernichtein, S., Touraine, P., and Kelly, P. A. (2005). Development and potential clinical uses of human prolactin receptor antagonists. Endocr. Rev. 26, 400–422.
Goffin, V., Bogorad, R. L., and Touraine, P. (2010). Identification of gain-of-function variants of the human prolactin receptor. Meth. Enzymol. 484, 329–355.
Goffin, V., Kinet, S., Ferrag, F., Binart, N., Martial, J. A., and Kelly, P. A. (1996a). Antagonistic properties of human prolactin analogs that show paradoxical agonistic activity in the Nb2 bioassay. J. Biol. Chem. 271, 16573–16579.
Goffin, V., Shiverick, K. T., Kelly, P. A., and Martial, J. A. (1996b). Sequence-function relationships within the expanding family of prolactin, growth hormone, placental lactogen and related proteins in mammals. Endocr. Rev. 17, 385–410.
Goffin, V., Norman, M., and Martial, J. A. (1992). Alanine-scanning mutagenesis of human prolactin: importance of the 58-74 region for bioactivity. Mol. Endocrinol. 6, 1381–1392.
Goffin, V., Struman, I., Mainfroid, V., Kinet, S., and Martial, J. A. (1994). Evidence for a second receptor binding site on human prolactin. J. Biol. Chem. 269, 32598–32606.
Goffin, V., Touraine, P., Culler, M. D., and Kelly, P. A. (2006). Drug Insight: prolactin-receptor antagonists, a novel approach to treatment of unresolved systemic and local hyperprolactinemia? Nat. Clin. Pract. Endocrinol. Metab. 2, 571–581.
Jomain, J. B., Tallet, E., Broutin, I., Hoos, S., Van Agthoven, J., Ducruix, A., Kelly, P. A., Kragelund, B. B., England, P., and Goffin, V. (2007). Structural and thermodynamical bases for the design of pure prolactin receptor antagonists. X-ray structure of Del1-9-G129R-hPRL. J. Biol. Chem. 282, 33118–33131.
Kelly, P. A., Djiane, J., Postel-Vinay, M. C., and Edery, M. (1991). The prolactin/growth hormone receptor family. Endocr. Rev. 12, 235–251.
Kinet, S., Bernichtein, S., Kelly, P. A., Martial, J. A., and Goffin, V. (1999). Biological properties of human prolactin analogs depend not only on global hormone affinity, but also on the relative affinities of both receptor binding sites. J. Biol. Chem. 274, 26033–26043.
Kinet, S., Goffin, V., Mainfroid, V., and Martial, J. A. (1996). Characterization of lactogen receptor binding site 1 of human prolactin. J. Biol. Chem. 271, 14353–14360.
Langenheim, J. F., Tan, D., Walker, A. M., and Chen, W. Y. (2006). Two wrongs can make a right: dimers of prolactin and growth hormone receptor antagonists behave as agonists. Mol. Endocrinol. 20, 661–674.
Li, H., Ahonen, T. J., Alanen, K., Xie, J., LeBaron, M. J., Pretlow, T. G., Ealley, E. L., Zhang, Y., Nurmi, M., Singh, B., Martikainen, P. M., and Nevalainen, M. T. (2004). Activation of signal transducer and activator of transcription 5 in human prostate cancer is associated with high histological grade. Cancer Res. 64, 4774–4782.
Liu, W., and Brooks, C. L. (2011). Functional impact of manipulation on the relative orientation of human prolactin receptor domains. Biochemistry 50, 5333–5344.
Lochnan, H. A., Buteau, H., Richards, S., Edery, M., and Kelly, P. A. (1995). Functional activity of the human prolactin receptor and its ligands. Mol. Cell. Endocrinol. 114, 91–99.
Maurel, D., Comps-Agrar, L., Brock, C., Rives, M. L., Bourrier, E., Ayoub, M. A., Bazin, H., Tinel, N., Durroux, T., Prezeau, L., Trinquet, E., and Pin, J. P. (2008). Cell-surface protein-protein interaction analysis with time-resolved FRET and snap-tag technologies: application to GPCR oligomerization. Nat. Methods 5, 561–567.
McHale, K., Tomaszewski, J. E., Puthiyaveettil, R., Livolsi, V. A., and Clevenger, C. V. (2008). Altered expression of prolactin receptor-associated signaling proteins in human breast carcinoma. Mod. Pathol. 21, 565–571.
Mercier, J. F., Salahpour, A., Angers, S., Breit, A., and Bouvier, M. (2002). Quantitative assessment of beta 1- and beta 2-adrenergic receptor homo- and heterodimerization by bioluminescence resonance energy transfer. J. Biol. Chem. 277, 44925–44931.
Pfleger, K. D., and Eidne, K. A. (2006). Illuminating insights into protein-protein interactions using bioluminescence resonance energy transfer (BRET). Nat. Methods 3, 165–174.
Qazi, A. M., Tsai-Morris, C. H., and Dufau, M. L. (2006). Ligand-independent homo- and hetero-dimerization of human prolactin receptor variants: inhibitory action of the short forms by heterodimerization. Mol. Endocrinol. 20, 1912–1923.
Remy, I., Wilson, I. A., and Michnick, S. W. (1999). Erythropoietin receptor activation by a ligand-induced conformation change. Science 283, 990–993.
Rouet, V., Bogorad, R. L., Kayser, C., Kessal, K., Genestie, C., Bardier, A., Grattan, D. R., Kelder, B., Kopchick, J. J., Kelly, P. A., and Goffin, V. (2010). Local prolactin is a target to prevent expansion of basal/stem cells in prostate tumors. Proc. Natl. Acad. Sci. U.S.A. 107, 15199–15204.
Rozakis-Adcock, M., and Kelly, P. A. (1991). Mutational analysis of the ligand binding domain of the prolactin receptor. J. Biol. Chem. 266, 16472–16477.
Schagger, H., and von Jagow, G. (1991). Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal. Biochem. 199, 223–231.
Sivaprasad, U., Canfield, J. M., and Brooks, C. L. (2004). Mechanism for ordered receptor binding by human prolactin. Biochemistry 43, 13755–13765.
Svensson, L. A., Bondensgaard, K., Norskov-Lauritsen, L., Christensen, L., Becker, P., Andersen, M. D., Maltesen, M. J., Rand, K. D., and Breinholt, J. (2008). Crystal structure of a prolactin receptor antagonist bound to the extracellular domain of the prolactin receptor. J. Biol. Chem. 283, 19085–19094.
Tallet, E., Rouet, V., Jomain, J. B., Kelly, P. A., Bernichtein, S., and Goffin, V. (2008). Rational design of competitive prolactin/growth hormone receptor antagonists. J. Mammary Gland Biol. Neoplasia 13, 105–117.
Tan, D., Johnson, D. A., Wu, W., Zeng, L., Chen, Y. H., Chen, W. Y., Vonderhaar, B. K., and Walker, A. M. (2005). Unmodified prolactin (PRL) and S179D PRL-initiated bioluminescence resonance energy transfer between homo- and hetero-pairs of long and short human PRL receptors in living human cells. Mol. Endocrinol. 19, 1291–1303.
Teilum, K., Hoch, J. C., Goffin, V., Kinet, S., Martial, J. A., and Kragelund, B. B. (2005). Solution structure of human prolactin. J. Mol. Biol. 351, 810–823.
Van Agthoven, J., Zhang, C., Tallet, E., Raynal, B., Hoos, S., Baron, B., England, P., Goffin, V., and Broutin, I. (2010). Structural characterization of the stem-stem dimerization interface between prolactin receptor chains complexed with the natural hormone. J. Mol. Biol. 404, 112–126.
Weidemann, T., Hofinger, S., Muller, K., and Auer, M. (2007). Beyond dimerization: a membrane-dependent activation model for interleukin-4 receptor-mediated signalling. J. Mol. Biol. 366, 1365–1373.
Keywords: dimerization, TR-FRET, PRLRI146L, antagonist
Citation: Tallet E, Fernandez I, Zhang C, Salsac M, Gregor N, Ayoub MA, Pin JP, Trinquet E and Goffin V (2011) Investigation of prolactin receptor activation and blockade using time-resolved fluorescence resonance energy transfer. Front. Endocrin. 2:29. doi: 10.3389/fendo.2011.00029
Received: 24 February 2011;
Accepted: 26 August 2011;
Published online: 16 September 2011.
Edited by:
Ralf Jockers, University of Paris, FranceReviewed by:
Eric Reiter, Institut National de la Recherche Agronomique, FranceCyril Couturier, U761 INSERM, Université Lille2, France
Copyright: © 2011 Tallet, Fernandez, Zhang, Salsac, Gregor, Ayoub, Pin, Trinquet and Goffin. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Vincent Goffin, INSERM U845, Faculté de Médecine Necker, Centre de Recherche “Croissance et Signalisation”, Equipe “Physiopathologie des hormones PRL/GH”, 156 Rue de Vaugirard, 75730 Paris CEDEX 15, France. e-mail:dmluY2VudC5nb2ZmaW5AaW5zZXJtLmZy