 Archana Vijayakumar
Archana Vijayakumar
- Division of Endocrinology, Diabetes and Bone Diseases, Department of Medicine, Mount Sinai School of Medicine, New York, NY, USA
Growth hormone (GH), a master regulator of somatic growth, also regulates carbohydrate and lipid metabolism via complex interactions with insulin and insulin-like growth factor-1 (IGF-1). Data from human and rodent studies reveal the importance of GH in insulin synthesis and secretion, lipid metabolism and body fat remodeling. In this review, we will summarize the tissue-specific metabolic effects of GH, with emphasis on recent targets identified to mediate these effects. Furthermore, we will discuss what role GH plays in obesity and present possible mechanisms by which this may occur.
Introduction
Growth hormone (GH) is an anabolic hormone that has important functions in regulating somatic growth either directly or indirectly via effectors such as insulin-like growth factor-1 (IGF-1). However, another facet of the biological effects exerted by GH includes its ability to modulate metabolism and energy homeostasis. The metabolic actions of GH are diverse and tissue-specific, thus complicating our ability to understand them. Here we will describe the process of GH secretion and its regulation, signal transduction via the GH receptor (GHR), followed by a review of the available literature on the metabolic actions of GH in various tissues, including the liver, adipose tissue, skeletal muscle, and pancreas.
GH Secretion and Its Regulation
The human GH gene cluster is located on chromosome 17 and it includes five GH variants, amongst which is the pituitary GH-N (or GH-1) variant and four placental GH-V (or GH-2) variants. While the GH variants are expressed in a temporal manner during development, they all generate similar 22 kDa protein products. The pituitary GH-N transcript undergoes alternative splicing to yield two isoforms which are 22 and 20 kDa respectively, the former being the more predominant isoform in circulation, as has been reviewed elsewhere (Baumann, 2009).
GH is primarily expressed, synthesized, stored within secretory granules in the somatotrophic cells located in the anterior pituitary gland. Transcription of the GH gene is regulated by several transcription factors including Pit-1 (pituitary-specific transcription factor-1), Sp1 (specificity protein 1), activator protein 2, nuclear factor-1, and upstream stimulating factor. Glucocorticoids have been shown to increase GH transcription as well as mRNA stability; while thyroid hormone suppresses GH transcription (Strobl and Thomas, 1994).
GH is secreted in response to a rise in intracellular cyclic AMP (cAMP) or Ca2+ levels which result in membrane depolarization, translocation of GH granules to the membrane and the subsequent release of GH into circulation (Strobl and Thomas, 1994). GH secretion occurs in a pulsatile manner that is primarily regulated by the 180° out-of-phase secretion of two neuroendocrine hormones from the hypothalamus, namely, the GH releasing hormone (GHRH) and somatostatin that stimulate and repress GH secretion respectively. GHRH and somatostatin interact with their respective membrane-bound G-protein coupled receptors (GPCRs) on the somatotrophs and exert their biological effects via multiple mechanisms which are reviewed elsewhere (Anderson et al., 2004; Ben-Shlomo and Melmed, 2010), Ghrelin, identified in 1999 as a potent endogenous GH secretagogue, exerts its effects, independent of GHRH, and somatostatin, via the GH secretagogue receptor 1a (GHSR1a), which is also a GPCR (Anderson et al., 2004). Since its discovery, a wide range of effects on energy homeostasis and food intake have been identified for ghrelin, but these are out of the scope for this review (Pazos et al., 2008). GH secretion also exhibits sexual dimorphism which seems to be exerted by the differential effects of androgens and estrogens in the hypothalamus (Ohlsson et al., 2009).
Once in the circulation, GH binds to GH binding proteins (GHBPs), of which there are two classes. The high affinity GHBP is a soluble truncated form of the GHR and it preferentially binds the 22-kDa GH isoform; while the low affinity GHBP which is structurally unrelated to the GHR binds the 20-kDa GH isoform. The high affinity GHBP is generated by proteolytic cleavage of the extracellular domain (ECD) of the GHR in humans; while in rodents it is generated by alternative splicing of the GHR transcript resulting in a short-length GHR. The GHBPs are hypothesized to have two roles; one is to stabilize and prevent GH from degradation when in circulation, and the other is to control GH bioavailability by competing with the GHR for binding to GH (Baumann et al., 1994; Tuggle and Trenkle, 1996).
Growth hormone acts on the liver to stimulate the production and secretion of IGF-1 which is one of the most characterized targets of GH. The liver is the predominant source of circulating IGF-1; however, liver-specific igf-1 deficient (LID) mice display only a 75% reduction in circulating IGF-1 levels suggesting that extra-hepatic tissues contributed to the remaining 25% of the circulating IGF-1 pool (Liu et al., 1998). Circulating IGF-1 exists in a ternary complex with IGF binding proteins (IGFBPs), mainly IGFBP-3 and -5, and acid labile sub-unit (ALS). The ternary IGF-1-ALS-IGFBP-3 complex modulates the bioavailability of IGF-1, and also stabilizes IGF-1 in circulation (LeRoith, 1996). Interestingly, GH also regulates the expression of ALS and IGFBP-3 and thus, IGF-1 stability in serum (Elliott et al., 1992). Apart from the endocrine/circulating IGF-1, IGF-1 is also produced locally by tissues and exerts its effects in an autocrine/paracrine manner. IGF-1 plays a critical role in inhibiting GH secretion by feedback mechanisms by stimulating somatostatin and inhibiting GHRH release from the hypothalamus (Ohlsson et al., 2009). Furthermore, a recent study by Romero et al. (2010) using somatotroph-specific IGF-1 receptor (IGF-1R) knockout mice demonstrated that IGF-1 also has direct effects on GH secretion from the somatotrophs, independent of GHRH, and somatostatin.
GHR and Signal Transduction
GH mediates its intracellular effects via the GHR which is a one-pass transmembrane receptor belonging to the class 1 cytokine receptor family. It has an ECD which is connected to an intracellular/cytoplasmic domain (ICD) via a flexible linker. The GHR has no intrinsic kinase activity but the cytoplasmic kinase, Janus kinase 2 (Jak2) is constitutively associated with a Box1 region in the ICD of the GHR. In the inactive state, the Jak2 catalytic domain is masked by its pseudokinase domain. GH binding to pre-formed GHR dimers results in a conformational change in the receptors and associated Jak2 molecules. This event unmasks the catalytic domain of Jak2 and allows the adjacent Jak2 molecules to activate each other by transphosphorylation. Activated Jak2 then phosphorylates the cytoplasmic regions of the GHR which then recruits several downstream proteins (Lanning and Carter-Su, 2006).
The signal transducer and activator of transcription (STAT) family of transcription factors is a well-characterized downstream target recruited to the GHR. Once recruited to the GHR, STAT proteins are phosphorylated by Jak2 resulting in their dissociation from the receptor, homo- or hetero- dimerization, and translocation to the nucleus. Of the various STAT proteins, STAT5, and particularly STAT5b, mediates a majority of the biological effects of GH, including the transcription of IGF-1, and ALS. STAT5b has also been implicated in exerting the sexually dimorphic pattern of gene expression induced by GH (Holloway et al., 2007). Apart from STAT5, the GHR also recruits and activates STATs-1 and 3 in a Jak2-dependent manner (Ram et al., 1996; Smit et al., 1996).
Apart from Jak2, the GHR also interacts with Src kinase in a Jak2-independent manner, and can also activate the mitogen activated protein kinase (MAPK; or, extracellular signal regulated kinase, Erk) pathway downstream of both Jak2 and Src (Lanning and Carter-Su, 2006). Moreover, GHR localization to the lipid raft preferentially activates the MAPK pathway while cytosolic GHR localization activates the STAT5 pathway. Lipid raft targeted insulin receptor substrate-1 (IRS-1), an adaptor protein critical for insulin-and IGF-1- mediated signal transduction, seems to play a critical role in GHR-induced MAPK activation (Liang et al., 2000; Brooks et al., 2008; Ohlsson et al., 2009; Wang et al., 2009). Lastly, GHR signaling has also been associated with activation of the phosphatidylinositol-3 kinase (PI3K)/Akt pathway in a Jak2/IRS-1 dependent manner (Liang et al., 2000).
Down-regulation of GHR activation is mediated by the suppressors of cytokine signaling (SOCS) family of proteins, which include SOCS-1, -2, -3, -6, and cytokine-inducible SH2 containing protein (CIS), which are induced by the Jak/STAT pathway. GH itself has been shown to induce SOCS -2, and -3 (Greenhalgh et al., 2005; Nielsen et al., 2008). Moreover, the SOCS proteins can terminate GHR signaling via different mechanisms including inhibition of Jak2 kinase activity and competing with STAT5 for binding to GHR (Ram and Waxman, 1999; Dominici et al., 2005). Additionally, protein tyrosine phosphatases (PTPs) have also been implicated in terminating the GHR signal cascade (Pasquali et al., 2003; Choi et al., 2006; Pilecka et al., 2007). Furthermore, GHR can be desensitized by proteolysis. Jak2 seems to play a dual role in this process. On the one hand, GHR binding to Jak2 stabilizes the receptor and prevents it from degradation (He et al., 2005; Deng et al., 2007; Loesch et al., 2007). On the other hand, GH can induce desensitization of the GHR and this requires Jak2 kinase activity (Loesch et al., 2006; Deng et al., 2007).
Liver
It is well established that GH can stimulate hepatic glucose production which has two arms, gluconeogenesis (conversion of amino acids and intermediates of glucose metabolism to glucose) and glycogenolysis (breakdown of glycogen to glucose; Brooks et al., 2007; Lindberg-Larsen et al., 2007; Sakharova et al., 2008). However, it is still unclear whether GH preferentially stimulates gluconeogenesis or glycogenolysis and there is data to support either theory. High dose GH infusion (40 ng/kg/min for 4 h) into healthy individuals in a pituitary clamp study increased glycogenolysis but had no effect on gluconeogenesis (Ghanaat and Tayek, 2005). Also, pituitary microsurgery in acromegalic men resulted in a significant reduction in glycogenolysis with no impact on gluconeogenesis (Hoybye et al., 2008). In contrast to this, sub-cutaneous injections of recombinant human GH (rhGH; 0.05 mg/kg/day) in fasted nursing women for a week increased gluconeogenesis, but not glycogenolysis (Kaplan et al., 2008). 3 mg/day of rhGH administration for 6 months to HIV patients, who demonstrate significant visceral obesity, increased fasting gluconeogenesis but not glycogenolysis (Schwarz et al., 2002). Additionally, conditional deletion of the GHR in mouse livers did not result in differences in the mRNA expression of gluconeogenic genes compared to the control mice (Fan et al., 2009). Thus, the data seem to suggest that GH has a preferential effect on glycogenolysis compared to gluconeogenesis.
The impact of GH on liver glucose uptake and utilization is not clear. Over-expression of the human GH (hGH) gene in rats increased basal hepatic glucose uptake and glycogen storage (Cho et al., 2006). Four week GHR antagonist (pegvisomant) treatment in acromegalic patients increased non-oxidative glucose disposal (Lindberg-Larsen et al., 2007). Thus, the main role of GH in the liver seems to be increasing hepatic glucose production while its role in glucose uptake is minor.
Growth hormone plays a major role in liver lipid metabolism. Sterol regulatory element-binding proteins (SREBPs) are a class of transcription factors that play a key role in lipid and cholesterol synthesis. In the human hepatocyte HepG2 cell line, GH was shown to phosphorylate SREBP-1a in a MAPK-dependent manner which resulted in increased transcription activity (Kotzka et al., 2010). Furthermore, GH also promotes triglyceride (TG) uptake into the liver by increasing lipoprotein lipase (LPL) and/or hepatic lipase expression and/or activity. GH deficiency is associated with decreased hepatic lipase mRNA expression and/or activity which are significantly improved with GH supplementation (Hoogerbrugge et al., 1993; Oscarsson et al., 1996; Neve et al., 1997). Additionally, mice over-expressing bovine GH (bGH) either in the whole-body (via the metallothionein promoter) or the liver (via the phosphoenolpyruvate carboxykinase, PEPCK promoter) have increased hepatic LPL expression (Frick et al., 2001; Wang et al., 2007).
In addition to TG uptake, data suggest that GH signaling also stimulates TG secretion. The GHRLD mice develop hepatic steatosis and have reduced TG secretion. Moreover adenoviral expression of IGF-1 in the GHRLD mice did not correct the hepatic steatosis, suggesting that GH modulates TG secretion in an IGF-1-independent manner (Fan et al., 2009). Further, liver-specific STAT5 knockout (STAT5LKO) mice also develop hepatic steatosis despite increased basal hepatic STAT1 and STAT3 phosphorylation suggesting that GH mediates TG secretion in a STAT5-dependent manner (Cui et al., 2007). Additionally, bGH transgenic mice have decreased TG secretion rate with lower serum TG levels (Frick et al., 2001; Wang et al., 2007). Loss of GH-induced STAT5 signaling in 391−/− mice increased liver TG levels under both lean and diet-induced obesity states. This was associated with an increase in the expression of the fatty acid transporter (CD36), and the peroxisome proliferator–activator receptor-Γ (PPAR-Γ). Moreover, over-expression of a dominant-negative STAT5 in hepatocytes also resulted in increased CD36, as well as fatty acid synthase expression. These suggest that loss of GH-induced STAT5 signaling increases TG accumulation in the liver by up-regulation of TG uptake as well as de novo lipogenesis (Barclay et al., 2011). Thus, while GH plays important roles in TG uptake and secretion in a STAT5-dependent manner, the underlying mechanisms and target genes involved in the process remain to be defined.
In addition to its effects on TG secretion, data suggest that GH may also stimulate hepatic fatty acid oxidation by regulating the expression of the adiponectin receptor 2 (AdipoR2) which is the predominant adiponectin receptor expressed in the liver. The biological actions of adiponectin include increased fatty acid oxidation, activation of the AMPK pathway, and glucose uptake (Kadowaki et al., 2006). Tail vein injection of a recombinant adenoviral vector containing GH into male mice increased AdipoR2 expression in the liver (Qin and Tian, 2010b). Similar adenoviral GH administration to obese rats, as well as mice with experimentally induced alcoholic hepatic steatosis prevented the development of hepatic steatosis which was associated with increased AdipoR2 expression, reduced inflammation, and increased PPARα phosphorylation (Qin and Tian, 2010a,c). Male lit/lit mice, which are a model of GH deficiency owing to a mutation in the GHRH receptor, have increased hepatic AdipoR1 expression (Arumugam et al., 2007). However, contrary to the data regarding the AdipoR2, studies have reported a repression of PPARα expression by GH. Hypophysectomy increased hepatic PPAR-α expression which was reversed with continuous GH infusion in both male and female rats (Jalouli et al., 2003). GHR knockout mice (GHRKO) also show an up-regulation of all three PPAR isoforms (PPAR-α, β/δ, and Γ) in the liver (Masternak et al., 2005). GH treatment of PPARα knockout mice resulted in a higher TG secretion rate than the GH-treated control mice, suggesting that suppression of PPARα function may facilitate the ability of GH to enhance TG secretion (Ljungberg et al., 2007).
In summary, GH stimulates hepatic glucose production, while it has modest effects on liver glucose uptake, utilization, or storage. GH plays a crucial role in TG secretion from the liver via as yet unknown mechanisms. Whether this is associated with changes in β-oxidation and liver TG uptake is still unclear. However, two factors must be considered while analyzing the metabolic effects of GH in the liver. The first is that there is convincing data that implicates GH signaling in proliferation of hepatocytes. Loss of GH signaling in multiple models decreases the rate of liver regeneration following partial hepatectomy (Pennisi et al., 2004; Cui et al., 2007; Zerrad-Saadi et al., 2011). Thus, altered cellular profile could also affect hepatic metabolism. Secondly, the metabolic effects of GH in the liver could also be secondary to GH-induced insulin resistance. Modulation of GH action in the liver results in decreased IGF-1 production and subsequently increased GH secretion which is further associated with insulin resistance. Thus, it is extremely difficult to tease apart the direct effects of GH on liver metabolism from its indirect effects via opposition of insulin action.
Adipose tissue
GH stimulates lipolysis in the adipose tissue, and particularly the visceral and sub-cutaneous depots (Chen et al., 2001; Nam et al., 2001; Berryman et al., 2004; Pasarica et al., 2007; Freda et al., 2008). Hormone-sensitive lipase (HSL or LIPE) is a crucial hormone implicated in this process. Activation of HSL in the adipose tissue occurs downstream of the Gs-coupled β-adrenergic receptors by activation of PKA/cAMP pathway. GH increases HSL activity in the adipose tissue both in humans (Samra et al., 1999) and rodents (Ng et al., 2000; Johansen et al., 2003), possibly by activating the β-adrenergic receptor (Yip and Goodman, 1999; Yang et al., 2004); however its effect on HSL gene expression is still unclear (Richelsen et al., 2000; Khalfallah et al., 2001). Moreover, studies have reported either a suppressive or no effect of GH on LPL expression and activity (Richelsen et al., 2000; Frick et al., 2001; Johansen et al., 2003) suggesting that GH has minimal effects on adipose tissue TG uptake.
GH may also modulate the expression of the lipid droplet associating protein, CIDE-A (cell-death-inducing DFF45-like effector). Loss of CIDE-A in mice results in increased metabolic rate and glucose disposal and protection from diet-induced obesity and insulin resistance (Lin and Li, 2004). CIDE proteins have also been associated with lipid droplets where they facilitate lipid accumulation and inhibit lipolysis. However, the data on GH regulation of CIDE-A are controversial. Treatment of GH-deficient (GHD) individuals with rhGH resulted in a down-regulation of CIDE-A expression in the sub-cutaneous adipose tissue (Zhao et al., 2011), while GHRKO mice had decreased expression of CIDE-A in the sub-cutaneous adipose tissue (Kelder et al., 2007). The reasons for the discrepancy in the data are not clear and bear further investigation.
Ten-month-old GH-resistant GHRKO and GH antagonist (GHa) mice have increased size of the brown adipose tissue (BAT) suggesting that GH also regulates BAT metabolism (Li et al., 2003). While the mechanisms by which GH affects the BAT are not well known, studies in the GHa mice suggest that GH increases uncoupling protein -1 (UCP-1) expression in the BAT (Li et al., 2003). Interestingly, CIDE-A is also expressed in high levels in the BAT where it localizes to the mitochondria, interacts with and modulates UCP-1 activity, and ultimately adaptive thermogenesis (Lin and Li, 2004). Thus, understanding the mechanism by which GH regulates CIDE-A not only in the white adipose tissue but also the BAT could be important for understanding how GH modulates lipid metabolism.
GH may also modulate glucocorticoid action in the adipose tissue. Recent studies have shown that GH down-regulates 11βHSD1 (11β-hydroxysteroid dehydrogenase type 1) expression in the adipose tissue (Morita et al., 2009; Zhao et al., 2011). 11βHSD1 amplifies glucocorticoid action by stimulating the conversion of inactive dehydrocorticosterone to active corticosterone. Activity of 11βHSD1 is increased in obesity. Rodents with adipose tissue-specific over-expression of 11βHSD1 are more insulin resistant; while knockout of 11βHSD1 protects mice from diet-induced obesity and insulin resistance (Kotelevtsev et al., 1997; Morton et al., 2001, 2004).
In rodents models, GH deficiency has been associated with elevated adiponectin levels and GH excess with lower adiponectin levels (Berryman et al., 2004; Nilsson et al., 2005; del Rincon et al., 2007; Wang et al., 2007). However, the reports of adiponectin status in GHD or acromegalic patients have been inconsistent resulting from variabilities in the duration of GH deficiency, age of the subjects, dose of GH treatment, and low sample number (Ciresi et al., 2007; Joaquin et al., 2008; Andersson et al., 2009; Oliveira et al., 2010; Ueland et al., 2010). Circulating adiponectin levels in obese laron syndrome adults and young girls, who have a loss-of-function mutation in the GHR rendering them GH-resistant, is two- to five-fold higher than obese controls. Moreover, IGF-1 replacement in one sub-set of these patients did not affect adiponectin levels suggesting that regulation of adiponectin is an IGF-1-independent effect of GH (Kanety et al., 2009). Indeed, GH stimulated adiponectin secretion from differentiated 3T3-L1 adipocytes under normal and high glucose conditions (Wolfing et al., 2008). However, human adipocytes incubated with GH demonstrated decreased adiponectin secretion (Nilsson et al., 2005). Thus, the exact nature of GH regulation of adiponectin secretion and/or function remains to be determined.
bGH transgenic mice have increased adipose tissue expression of the p85α regulatory sub-unit of the PI3K while the opposite was found in lit/lit mice (del Rincon et al., 2007). The expression of the p85α sub-unit is believed to play an important role in the pathogenesis of obesity. Within the cell, the p85α sub-unit is in excess when compared to the p110 catalytic sub-unit of PI3K. Consequently there is a greater pool of p85α homodimers compared to the p85α–p110 heterodimers. The p85α homodimers can bind and sequester IRS-1, thereby preventing the activation of the PI3K (Mauvais-Jarvis et al., 2002; Ueki et al., 2002). GH-induced p85α in the 3T3-F442A adipocyte cell line (del Rincon et al., 2007). However, it is still unclear whether GH directly affects p85a expression in vivo. Moreover, it is also possible that the increased p85α expression in the bGH mice could be a manifestation of insulin resistance. Mice lacking all isoforms of p85 do not survive due to severe hypoglycemia (Fruman et al., 2000). Furthermore, heterozygous knockout of p85α in mice results in improved insulin sensitivity (Mauvais-Jarvis et al., 2002), and administration of antisense p85 oligonucleotide to diet-induced obese and ob/ob mice, that harbor a mutation in the leptin gene, improved their insulin sensitivity (Moriarty et al., 2009). Nevertheless, increased expression of p85α in the adipose tissue in GH excess could account for GH-induced insulin resistance.
In summary, the predominant effect of GH in the adipose tissue is the stimulation of lipolysis. This may occur by activation of HSL, a critical enzyme for lipolysis, or, as recent studies suggest by modulation of the expression of lipid droplet proteins such as CIDE-A. Other new targets for GH action include 11βHSD1 which is a key regulator of glucocorticoid action. Changes in GH action are also associated with alterations in adipokine profile which could arise due to direct effects of GH on the adipocyte or indirectly due to other biological effects of GH.
Skeletal muscle
Unlike in the adipose tissue, GH induces free fatty acid (FFA) uptake into skeletal muscle by up-regulation of LPL expression (Oscarsson et al., 1999; Khalfallah et al., 2001). There is also evidence suggesting that GH induces skeletal muscle HSL expression in GHD individuals (Trepp et al., 2008). GH treatment of lit/lit mice resulted in significant increase in PPAR-β/δ expression which is an important mediator of lipid metabolism in the skeletal muscle (Kim et al., 2008; Ehrenborg and Krook, 2009). The lit/lit mice also demonstrated an increase in the expression of the insulin-responsive transcription factor FOXO1 that has been previously shown to increase lipid uptake and oxidation in the C2C12 skeletal muscle cell line (Bastie et al., 2005; Kim et al., 2008). GH has also been shown to induce lipid accumulation in the muscle (Freda et al., 2008; Krag et al., 2008; Szendroedi et al., 2008; Trepp et al., 2008). The re-esterification of TG from FFAs results in generation of intermediates such as diacylglycerol and ceramides that activate PKC isoforms. PKC can down-regulate insulin signaling by several mechanisms (Samuel et al., 2010). Thus, in this manner, GH-induced increase in FFA uptake and TG synthesis could result in insulin resistance. These data also suggest that GH induces a shift in substrate utilization from glucose to lipids in the skeletal muscle.
A recent study using mice with skeletal muscle-specific knockout of the GHR reported worsening of glucose tolerance which was associated with decreased 2-deoxyglucose uptake in primary myoblast cultures and increased adipose tissue mass. Moreover, primary myoblasts isolated from these muscle-GHRKO mice demonstrated reduced IR protein content as well as increased serine phosphorylation of IRS-1 when compared to control myoblast cultures (Mavalli et al., 2010). These effects seem to be mediated by STAT5 as skeletal muscle-specific knockout of the STAT5 in mice results in slight increase in adiposity and worsening of insulin sensitivity (Klover and Hennighausen, 2007). These data, however, do not fit with the traditional view of GH antagonizing insulin action and in fact suggest that GH facilitates insulin signaling in the skeletal muscle. However, it should be mentioned that the promoters used to drive Cre recombinase expression in both these studies (the Mef2C and Myf5 promoters respectively) are active in the developing somite before differentiation of the myotome occurs (Ott et al., 1991; Naya et al., 1999). Thus, it is possible that early deletion of the GHR in regions other than the developing muscle could influence the phenotype of the mice. Additionally, there is also no convincing evidence to show that GH influences Glut4 translocation and subsequently glucose uptake (Khalfallah et al., 2001; Cho et al., 2006; Short et al., 2008) or glycogen synthase expression (Khalfallah et al., 2001) in the skeletal muscle.
Similar to the adipose tissue, states of GH excess have been associated with increased muscle expression of the regulatory p85α sub-unit of the PI3K (Barbour et al., 2004, 2005). LID mice have high circulating GH levels and are insulin resistant; they also have increased p85α expression in the skeletal muscle. Treatment of the LID mice with a GHRH antagonist normalized GH levels, ameliorated the insulin resistance, and also normalized p85α expression in the muscle (Barbour et al., 2005). Thus, increased muscle p85α expression could also account for GH-induced insulin resistance.
Thus, GH regulation of skeletal muscle metabolism is yet to be defined. Data suggest that GH induces lipid uptake and mobilization in the muscle; however whether and how it affects glucose uptake and metabolism remains unanswered.
Pancreas
While on the face of it, it may seem that the effects of GH on the pancreas may be secondary to its effects on peripheral insulin sensitivity, there is a lot of evidence to suggest that GH exerts direct effects in the pancreas and, especially in the β-cell which is the site of insulin synthesis and secretion.
The GHR and the closely similar prolactin receptor (PRLR) are expressed in the pancreatic β-cells where upon ligand stimulation they can stimulate insulin synthesis (Nielsen, 1982; Nielsen et al., 1989, 1990; Brelje et al., 1993, 2004). Moreover, the GHRKO mice have only 45% of the islet cell mass as normal mice (Robertson et al., 2006). Additionally, β-cell-specific GHRKO mice (βGHRKO) mice fail to demonstrate β-cell compensation when fed a HFD; this was also associated with reduced expression of cyclin D2 and Ki-67 (Wu et al., 2011). Accordingly, STAT5 stimulated cyclin D2 expression and thus, β-cell proliferation in the rat pancreatic β-cell line, the INS-1 cells (Friedrichsen et al., 2003). Similarly, GH down-regulated cytokine-induced β-cell apoptosis in a STAT5-dependent manner in the INS-1 cells (Jensen et al., 2005).
The GHRKO mice have low circulating insulin levels, reduced pancreatic insulin content and have a blunted glucose-stimulated insulin secretion (GSIS) response. Re-expression of IGF-1 in the pancreas alone did not improve insulin content but normalized islet area in the GHRKO mice (Guo et al., 2005). Similarly, investigators observed reduced pancreatic insulin content and lower circulating insulin levels in a mouse model of adult-onset of GH deficiency (Luque et al., 2011). We have recently shown that the βGHRKO mice have diminished GSIS response when fed a standard diet; this defect is exaggerated when the mice were challenged with a HFD making them insulin resistant. However, while GSIS was impaired in the βGHRKO mice, arginine stimulated insulin secretion was intact. Moreover, isolated islets from the obese βGHRKO mice responded to K+-channel blockers in a similar manner as islets isolated from the obese control mice. These data suggest that GH plays a role in insulin secretion, particularly in response to exogenous glucose. Indeed, we found that the βGHRKO mice had reduced expression of glucokinase which is the rate-limiting enzyme for glycolysis in the β-cell (Wu et al., 2011). Islet-specific knockout of STAT5 using the Cre recombinase driven by the rat insulin promoter (RIP) resulted in mice that were insulin resistant, and had diminished GSIS. However, mice lacking STAT5 in the β-cells via the Pdx-1 promoter did not display insulin resistance at 10 weeks of age, but aging and pregnancy were associated with worsening of glucose tolerance. Insulin secretion was not evaluated in the Pdx-1-STAT5 knockout mice. (Lee et al., 2007). The discrepancies between the two models was suggested to arise from the differential expression pattern of the RIP and Pdx -1 promoter; while the latter is localized only to the β-cell, the former is also expressed in certain regions of the brain. Another link between glucose metabolism and GH signaling comes from a study conducted with INS-1 cells, which showed that glucose stimulation results in cleavage and nuclear translocation of the cytoplasmic tail of the inactive phosphatase ICA512 which then binds to STAT5 and mediates the transcription of secretory granule genes (Mziaut et al., 2006). Thus, while these studies demonstrate an undeniable role of GH in GSIS, the underlying mechanisms, and role of STAT5 in this process remain to be clarified.
Another mechanism by which GH may affect insulin secretion is by modulating Ca2+ fluxes in the β-cell. In insulin secreting cells GH increased intracellular Ca2+ levels by up-regulating the ryanodine receptors (Zhang et al., 2004). Moreover, treatment of BRIN-BD11 β-cells with rhGH also increased intracellular Ca2+ levels in a Jak2 and Src-dependent manner. However, these effects were hypothesized to be acting through the PRLR rather than the GHR as ovine prolactin and not bGH mimicked the effects of the hGH (Zhang et al., 2006).
Thus, the data presented herein make an undeniable argument for the role of GH in insulin synthesis and secretion. Insulin secretion in response to exogenous glucose is dysregulated in insulin resistance and diabetes. Identifying the mechanisms by which GH affects β-cell function could help identify potentially new candidates to improve β-cell function in diabetes.
Reduced GH Production and Action in Obesity
GH secretion is consistently reduced in obesity (Makimura et al., 2008; Weltman et al., 2008). As a consequence, low GH secretion could further contribute to accumulation of abdominal fat. In fact, in a study with healthy volunteers 2 week over-eating resulted in significantly lower GH levels despite no changes in body weight suggesting that the reduction in GH secretion occurs prior to the manifestation of obesity (Cornford et al., 2011). However, despite the severe reduction in GH levels in obesity, there does not seem to be a proportional decrease in IGF-1 levels. In fact, most studies report either no change or only a modest change in total IGF-1 levels in obesity (Utz et al., 2008; Frystyk et al., 2009; Cornford et al., 2011). This has resulted in the hypothesis that the maintenance of IGF-1 levels can further reduce GH secretion by the negative feedback loop previously described. Additionally, it is also believed that free IGF-1 is elevated in obesity and this could further activate the negative feedback loop to suppress GH secretion. However, free IGF-1 levels measured in obese subjects are inconclusive (Rasmussen et al., 2006, 2007; Utz et al., 2008; Frystyk et al., 2009; Cornford et al., 2011). Moreover, reduced IGFBP-1 expression has also been reported in obese subjects, which could result in increased IGF-1 bioactivity (Frystyk et al., 2009; Cornford et al., 2011).
Increased circulating FFA levels in obesity may also have a suppressive effect on GH secretion. In GH3 rat pituitary tumor cells, incubation with cis-unsaturated fatty acids such as oleic acid reduced GH secretion. FFA interference with GH secretion was shown to occur by suppression of adenylate cyclase/cAMP/PKA pathway and impedance of Ca2+ influx which is critical for membrane depolarization (Perez et al., 1997, 1998). Indeed, acute lowering of FFA levels by administration of the lipolysis inhibitor acipimox in obese subjects increased GH secretion, both spontaneous and in response to treatment with GHRH (Cordido et al., 1996, 1998; Maccario et al., 1996; Kok et al., 2004; Koutkia et al., 2004; Scacchi et al., 2010).
The hyperinsulinemia associated with insulin resistance in obesity may also contribute to reduced GH secretion. Circulating insulin levels in lean, ob/ob, and diet-induced obese mice negatively correlated with pituitary mRNA expression of GH, GHRH receptor, and GHSR. Further the skeletal muscle and adipose tissue, but not the pituitaries, of obese mice demonstrated reduced response to an acute insulin stimulation (Luque and Kineman, 2006). Additionally, incubation of GH3 rat pituitary tumor cells with insulin suppressed GH secretion in a dose dependent manner (Melmed, 1984). Moreover, administration of an oral glucose challenge to healthy individuals or subjects with a non-functional pituitary tumor resulted in a significant fall in GH levels within the first hour (Kim et al., 2007; Verrua et al., 2011). Insulin treatment also inhibited GH release and reduced mRNA expression of GH, GHRH receptor and GHSR in primary pituitary cultures isolated from non-human primates (Luque et al., 2006). Obesity is also associated with hypoadiponectinemia and leptin resistance, both of which have been implicated in regulating GH secretion (Rasmussen et al., 2006; Jurimae et al., 2009; Myers et al., 2010). GH secretory cells in the pituitary gland express the adiponectin receptors (Rodriguez-Pacheco et al., 2007; Psilopanagioti et al., 2009; Steyn et al., 2009) and incubation of primary rat pituitary cells with adiponectin increased GH secretion (Rodriguez-Pacheco et al., 2007; Steyn et al., 2009). While selective deletion of leptin receptors from somatotrophic cells in mice did not affect total cell number in the somatotroph, there was a reduction in the number of cells expressing GH and consequently GH secretion suggesting that leptin affects GH expression and secretion, but not somatotroph development (Childs et al., 2011). Leptin treatment, but not pair-feeding, of ob/ob mice increased plasma GH levels, despite similar decreases in body weights. The leptin treatment was associated with an increase in ghrelin levels suggesting that leptin augmented ghrelin action (Luque et al., 2007). Thus, these studies suggest that obesity-induced changes in circulating levels of hormones and adipokines can also contribute to decreased GH secretion.
In addition to lower GH secretion in obesity, reduced GHR expression in the adipose tissue of obese subjects has also been observed (Erman et al., 2011a). Obesity is associated with an increase in systemic inflammation as seen with elevated circulating levels of TNF-α, IL-6, and other cytokines. An in vitro study reported that TNF-α decreased GHR expression in human adipocytes and HEK293 cells. In the same cell lines, glucocorticoids had a biphasic effect on GHR mRNA expression suggesting that as intracellular glucocorticoid levels increase, GHR mRNA expression decreases (Erman et al., 2011b). Alternative splicing of the GHR in the adipose tissue also results in a truncated GHR (ΔGHR) that lacks the ICD. The ΔGHR serves as a dominant-negative inhibitor of the functional full-length GHR, thereby terminating GHR signaling. An increased ratio of ΔGHR to full-length GHR in the omental and sub-cutaneous adipose tissue of obese women has been reported, providing another mechanism of GHR desensitization in obesity (Erman et al., 2011a). Thus, local GH resistance in the adipose tissue can also contribute to reduced GH action in obesity.
Thus, a vicious circle starting from the hyperinsulinemia, hypoadiponectinemia, leptin resistance, and increased free IGF-1 and FFA levels, associated with obesity, suppressing GH secretion which further results in fat accumulation could be an important factor in the pathogenesis of obesity. Furthermore, local GH resistance in the adipose tissue could further mediate fat accumulation and exacerbate the condition (Figure 1).
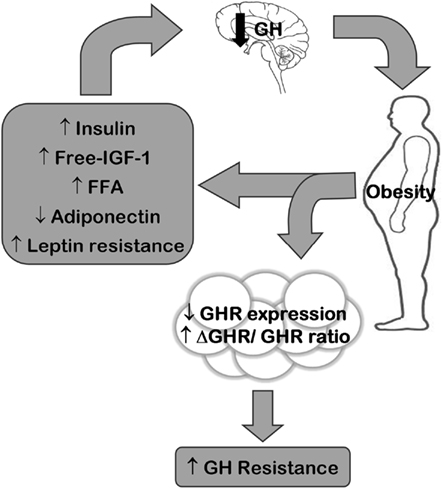
Figure 1. Possible mechanisms mediating reduced GH action in obesity. Obesity-induced hyperinsulinemia, hypoadiponectinemia, leptin resistance, and increased bioactive insulin-like growth factor-1 (IGF-1) and free fatty acid (FFA) levels could suppress GH secretion from the pituitary by various mechanisms (please refer to text). Reduced GH secretion further increases fat accumulation and, thus exacerbates the obesity condition. Moreover, reduced GH receptor (GHR) expression and increased expression of truncated GHR (ΔGHR) in the adipose tissue results in a GH-resistant state that also contributes to the complications associated with obesity.
Conclusion
GH exerts diverse effects on tissues. Many of the physiological effects of GH are still unknown. New targets of GHR signaling are steadily emerging, and the metabolic actions of GH may not be as clear as was initially believed. While GH opposes insulin action in peripheral tissues, it is also important for GSIS in the β-cells and for the maintenance of lipid homeostasis. Decreased GH action in obesity may in itself contribute to the associated metabolic abnormalities. Understanding the role of GH in physiological and pathological states could contribute to the development of new therapeutic strategies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Anderson, L. L., Jeftinija, S., and Scanes, C. G. (2004). Growth hormone secretion: molecular and cellular mechanisms and in vivo approaches. Exp. Biol. Med. (Maywood) 229, 291–302.
Andersson, B., Carlsson, L. M., Carlsson, B., Albertsson-Wikland, K., and Bjarnason, R. (2009). Decrease in adiponectin levels correlates to growth response in growth hormone-treated children. Horm. Res. 71, 213–218.
Arumugam, R., Fleenor, D., and Freemark, M. (2007). Effects of lactogen resistance and GH deficiency on mouse metabolism: pancreatic hormones, adipocytokines, and expression of adiponectin and insulin receptors. Endocrine 32, 182–191.
Barbour, L. A., Mizanoor Rahman, S., Gurevich, I., Leitner, J. W., Fischer, S. J., Roper, M. D., Knotts, T. A., Vo, Y., McCurdy, C. E., Yakar, S., Leroith, D., Kahn, C. R., Cantley, L. C., Friedman, J. E., and Draznin, B. (2005). Increased P85alpha is a potent negative regulator of skeletal muscle insulin signaling and induces in vivo insulin resistance associated with growth hormone excess. J. Biol. Chem. 280, 37489–37494.
Barbour, L. A., Shao, J., Qiao, L., Leitner, W., Anderson, M., Friedman, J. E., and Draznin, B. (2004). Human placental growth hormone increases expression of the p85 regulatory unit of phosphatidylinositol 3-kinase and triggers severe insulin resistance in skeletal muscle. Endocrinology 145, 1144–1150.
Barclay, J. L., Nelson, C. N., Ishikawa, M., Murray, L. A., Kerr, L. M., McPhee, T. R., Powell, E. E., and Waters, M. J. (2011). GH-dependent STAT5 signaling plays an important role in hepatic lipid metabolism. Endocrinology 152, 181–192.
Bastie, C. C., Nahle, Z., McLoughlin, T., Esser, K., Zhang, W., Unterman, T., and Abumrad, N. A. (2005). FoxO1 stimulates fatty acid uptake and oxidation in muscle cells through CD36-dependent and -independent mechanisms. J. Biol. Chem. 280, 14222–14229.
Baumann, G., Shaw, M., Amburn, K., Jan, T., Davila, N., Mercado, M., Stolar, M., and MacCart, J. (1994). Heterogeneity of circulating growth hormone. Nucl. Med. Biol. 21, 369–379.
Ben-Shlomo, A., and Melmed, S. (2010). Pituitary somatostatin receptor signaling. Trends Endocrinol. Metab. 21, 123–133.
Berryman, D. E., List, E. O., Coschigano, K. T., Behar, K., Kim, J. K., and Kopchick, J. J. (2004). Comparing adiposity profiles in three mouse models with altered GH signaling. Growth Horm. IGF Res. 14, 309–318.
Brelje, T. C., Scharp, D. W., Lacy, P. E., Ogren, L., Talamantes, F., Robertson, M., Friesen, H. G., and Sorenson, R. L. (1993). Effect of homologous placental lactogens, prolactins, and growth hormones on islet B-cell division and insulin secretion in rat, mouse, and human islets: implication for placental lactogen regulation of islet function during pregnancy. Endocrinology 132, 879–887.
Brelje, T. C., Stout, L. E., Bhagroo, N. V., and Sorenson, R. L. (2004). Distinctive roles for prolactin and growth hormone in the activation of signal transducer and activator of transcription 5 in pancreatic islets of langerhans. Endocrinology 145, 4162–4175.
Brooks, A. J., Wooh, J. W., Tunny, K. A., and Waters, M. J. (2008). Growth hormone receptor; mechanism of action. Int. J. Biochem. Cell Biol. 40, 1984–1989.
Brooks, N. L., Trent, C. M., Raetzsch, C. F., Flurkey, K., Boysen, G., Perfetti, M. T., Jeong, Y. C., Klebanov, S., Patel, K. B., Khodush, V. R., Kupper, L. L., Carling, D., Swenberg, J. A., Harrison, D. E., and Combs, T. P. (2007). Low utilization of circulating glucose after food withdrawal in Snell dwarf mice. J. Biol. Chem. 282, 35069–35077.
Chen, X. L., Lee, K., Hartzell, D. L., Dean, R. G., Hausman, G. J., McGraw, R. A., Della-Fera, M. A., and Baile, C. A. (2001). Adipocyte insensitivity to insulin in growth hormone-transgenic mice. Biochem. Biophys. Res. Commun. 283, 933–937.
Childs, G. V., Akhter, N., Haney, A., Syed, M., Odle, A., Cozart, M., Brodrick, Z., Gaddy, D., Suva, L. J., Akel, N., Crane, C., Benes, H., Charlesworth, A., Luque, R., Chua, S., and Kineman, R. D. (2011). The somatotrope as a metabolic sensor: deletion of leptin receptors causes obesity. Endocrinology 152, 69–81.
Cho, Y., Ariga, M., Uchijima, Y., Kimura, K., Rho, J. Y., Furuhata, Y., Hakuno, F., Yamanouchi, K., Nishihara, M., and Takahashi, S. (2006). The novel roles of liver for compensation of insulin resistance in human growth hormone transgenic rats. Endocrinology 147, 5374–5384.
Choi, J. H., Kim, H. S., Kim, S. H., Yang, Y. R., Bae, Y. S., Chang, J. S., Kwon, H. M., Ryu, S. H., and Suh, P. G. (2006). Phospholipase Cgamma1 negatively regulates growth hormone signalling by forming a ternary complex with Jak2 and protein tyrosine phosphatase-1B. Nat. Cell Biol. 8, 1389–1397.
Ciresi, A., Amato, M. C., Criscimanna, A., Mattina, A., Vetro, C., Galluzzo, A., D’Acquisto, G., and Giordano, C. (2007). Metabolic parameters and adipokine profile during GH replacement therapy in children with GH deficiency. Eur. J. Endocrinol. 156, 353–360.
Cordido, F., Fernandez, T., Martinez, T., Peñalva, A., Peinó, R., Casanueva, F. F., and Dieguez, C. (1998). Effect of acute pharmacological reduction of plasma free fatty acids on growth hormone (GH) releasing hormone-induced GH secretion in obese adults with and without hypopituitarism. J. Clin. Endocrinol. Metab. 83, 4350–4354.
Cordido, F., Peino, R., Peñalva, A., Alvarez, C. V., Casanueva, F. F., and Dieguez, C. (1996). Impaired growth hormone secretion in obese subjects is partially reversed by acipimox-mediated plasma free fatty acid depression. J. Clin. Endocrinol. Metab. 81, 914–918.
Cornford, A. S., Barkan, A. L., and Horowitz, J. F. (2011). Rapid suppression of growth hormone concentration by overeating: potential mediation by hyperinsulinemia. J. Clin. Endocrinol. Metab. 96, 824–830.
Cui, Y., Hosui, A., Sun, R., Shen, K., Gavrilova, O., Chen, W., Cam, M. C., Gao, B., Robinson, G. W., and Hennighausen, L. (2007). Loss of signal transducer and activator of transcription 5 leads to hepatosteatosis and impaired liver regeneration. Hepatology 46, 504–513.
del Rincon, J. P., Iida, K., Gaylinn, B. D., McCurdy, C. E., Leitner, J. W., Barbour, L. A., Kopchick, J. J., Friedman, J. E., Draznin, B., and Thorner, M. O. (2007). Growth hormone regulation of p85alpha expression and phosphoinositide 3-kinase activity in adipose tissue: mechanism for growth hormone-mediated insulin resistance. Diabetes 56, 1638–1646.
Deng, L., He, K., Wang, X., Yang, N., Thangavel, C., Jiang, J., Fuchs, S. Y., and Frank, S. J. (2007). Determinants of growth hormone receptor down-regulation. Mol. Endocrinol. 21, 1537–1551.
Dominici, F. P., Argentino, D. P., Muñoz, M. C., Miquet, J. G., Sotelo, A. I., and Turyn, D. (2005). Influence of the crosstalk between growth hormone and insulin signalling on the modulation of insulin sensitivity. Growth Horm. IGF Res. 15, 324–336.
Ehrenborg, E., and Krook, A. (2009). Regulation of skeletal muscle physiology and metabolism by peroxisome proliferator-activated receptor delta. Pharmacol. Rev. 61, 373–393.
Elliott, M. J., Faulkner-Jones, B. E., Stanton, H., Hamilton, J. A., and Metcalf, D. (1992). Plasminogen activator in granulocyte-macrophage-CSF transgenic mice. J. Immunol. 149, 3678–3681.
Erman, A., Veilleux, A., Tchernof, A., and Goodyer, C. G. (2011a). Human growth hormone receptor (GHR) expression in obesity: I. GHR mRNA expression in omental and subcutaneous adipose tissues of obese women. Int. J. Obes. (Lond.) [Epub ahead of print].
Erman, A., Wabitsch, M., and Goodyer, C. G. (2011b). Human growth hormone receptor (GHR) expression in obesity: II. Regulation of the human GHR gene by obesity-related factors. Int. J. Obes. (Lond.) [Epub ahead of print]
Fan, Y., Menon, R. K., Cohen, P., Hwang, D., Clemens, T., DiGirolamo, D. J., Kopchick, J. J., Le Roith, D., Trucco, M., and Sperling, M. A. (2009). Liver-specific deletion of the growth hormone receptor reveals essential role of GH signaling in hepatic lipid metabolism. J. Biol. Chem. 284, 19937–19944.
Freda, P. U., Shen, W., Heymsfield, S. B., Reyes-Vidal, C. M., Geer, E. B., Bruce, J. N., and Gallagher, D. (2008). Lower visceral and subcutaneous but higher intermuscular adipose tissue depots in patients with growth hormone and insulin-like growth factor I excess due to acromegaly. J. Clin. Endocrinol. Metab. 93, 2334–2343.
Frick, F., Bohlooly, Y. M., Lindén, D., Olsson, B., Törnell, J., Edén, S., and Oscarsson, J. (2001). Long-term growth hormone excess induces marked alterations in lipoprotein metabolism in mice. Am. J. Physiol. Endocrinol. Metab. 281, E1230–E1239.
Friedrichsen, B. N., Richter, H. E., Hansen, J. A., Rhodes, C. J., Nielsen, J. H., Billestrup, N., and Møldrup, A. (2003). Signal transducer and activator of transcription 5 activation is sufficient to drive transcriptional induction of cyclin D2 gene and proliferation of rat pancreatic beta-cells. Mol. Endocrinol. 17, 945–958.
Fruman, D. A., Mauvais-Jarvis, F., Pollard, D. A., Yballe, C. M., Brazil, D., Bronson, R. T., Kahn, C. R., and Cantley, L. C. (2000). Hypoglycaemia, liver necrosis and perinatal death in mice lacking all isoforms of phosphoinositide 3-kinase p85 alpha. Nat. Genet. 26, 379–382.
Frystyk, J., Brick, D. J., Gerweck, A. V., Utz, A. L., and Miller, K. K. (2009). Bioactive insulin-like growth factor-I in obesity. J. Clin. Endocrinol. Metab. 94, 3093–3097.
Ghanaat, F., and Tayek, J. A. (2005). Growth hormone administration increases glucose production by preventing the expected decrease in glycogenolysis seen with fasting in healthy volunteers. Metab. Clin. Exp. 54, 604–609.
Greenhalgh, C. J., Rico-Bautista, E., Lorentzon, M., Thaus, A. L., Morgan, P. O., Willson, T. A., Zervoudakis, P., Metcalf, D., Street, I., Nicola, N. A., Nash, A. D., Fabri, L. J., Norstedt, G., Ohlsson, C., Flores-Morales, A., Alexander, W. S., and Hilton, D. J. (2005). SOCS2 negatively regulates growth hormone action in vitro and in vivo. J. Clin. Invest. 115, 397–406.
Guo, Y., Lu, Y., Houle, D., Robertson, K., Tang, Z., Kopchick, J. J., Liu, Y. L., and Liu, J. L. (2005). Pancreatic islet-specific expression of an insulin-like growth factor-I transgene compensates islet cell growth in growth hormone receptor gene-deficient mice. Endocrinology 146, 2602–2609.
He, K., Loesch, K., Cowan, J. W., Li, X., Deng, L., Wang, X., Jiang, J., and Frank, S. J. (2005). Janus kinase 2 enhances the stability of the mature growth hormone receptor. Endocrinology 146, 4755–4765.
Holloway, M. G., Cui, Y., Laz, E. V., Hosui, A., Hennighausen, L., and Waxman, D. J. (2007). Loss of sexually dimorphic liver gene expression upon hepatocyte-specific deletion of Stat5a-Stat5b locus. Endocrinology 148, 1977–1986.
Hoogerbrugge, N., Jansen, H., Staels, B., Seip, M. J., and Birkenhäger, J. C. (1993). Growth hormone normalizes hepatic lipase in hypothyroid rat liver. Metab. Clin. Exp. 42, 669–671.
Hoybye, C., Chandramouli, V., Efendic, S., Hulting, A. L., Landau, B. R., Schumann, W. C., and Wajngot, A. (2008). Contribution of gluconeogenesis and glycogenolysis to hepatic glucose production in acromegaly before and after pituitary microsurgery. Horm. Metab. Res. 40, 498–501.
Jalouli, M., Carlsson, L., Améen, C., Lindén, D., Ljungberg, A., Michalik, L., Edén, S., Wahli, W., and Oscarsson, J. (2003). Sex difference in hepatic peroxisome proliferator-activated receptor alpha expression: influence of pituitary and gonadal hormones. Endocrinology 144, 101–109.
Jensen, J., Galsgaard, E. D., Karlsen, A. E., Lee, Y. C., and Nielsen, J. H. (2005). STAT5 activation by human GH protects insulin-producing cells against interleukin-1beta, interferon-gamma and tumour necrosis factor-alpha-induced apoptosis independent of nitric oxide production. J. Endocrinol. 187, 25–36.
Joaquin, C., Aguilera, E., Granada, M. L., Pastor, M. C., Salinas, I., Alonso, N., and Sanmarti, A. (2008). Effects of GH treatment in GH-deficient adults on adiponectin, leptin and pregnancy-associated plasma protein-A. Eur. J. Endocrinol. 158, 483–490.
Johansen, T., Richelsen, B., Hansen, H. S., Din, N., and Malmlöf, K. (2003). Growth hormone-mediated breakdown of body fat: effects of GH on lipases in adipose tissue and skeletal muscle of old rats fed different diets. Horm. Metab. Res. 35, 243–250.
Jurimae, J., Jurimae, T., Ring-Dimitriou, S., LeMura, L. M., Arciero, P. J., and von Duvillard, S. P. (2009). Plasma adiponectin and insulin sensitivity in overweight and normal-weight middle-aged premenopausal women. Metab. Clin. Exp. 58, 638–643.
Kadowaki, T., Yamauchi, T., Kubota, N., Hara, K., Ueki, K., and Tobe, K. (2006). Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J. Clin. Invest. 116, 1784–1792.
Kanety, H., Hemi, R., Ginsberg, S., Pariente, C., Yissachar, E., Barhod, E., Funahashi, T., and Laron, Z. (2009). Total and high molecular weight adiponectin are elevated in patients with Laron syndrome despite marked obesity. Eur. J. Endocrinol. 161, 837–844.
Kaplan, W., Sunehag, A. L., Dao, H., and Haymond, M. W. (2008). Short-term effects of recombinant human growth hormone and feeding on gluconeogenesis in humans. Metab. Clin. Exp. 57, 725–732.
Kelder, B., Berryman, D. E., Clark, R., Li, A., List, E. O., and Kopchick, J. J. (2007). CIDE-A gene expression is decreased in white adipose tissue of growth hormone receptor/binding protein gene disrupted mice and with high-fat feeding of normal mice. Growth Horm. IGF Res. 17, 346–351.
Khalfallah, Y., Sassolas, G., Borson-Chazot, F., Vega, N., and Vidal, H. (2001). Expression of insulin target genes in skeletal muscle and adipose tissue in adult patients with growth hormone deficiency: effect of one year recombinant human growth hormone therapy. J. Endocrinol. 171, 285–292.
Kim, D. S., Itoh, E., Iida, K., and Thorner, M. O. (2008). Growth hormone increases mRNA Levels of PPARdelta and Foxo1 in skeletal muscle of growth hormone deficient lit/lit Mice. Endocr. J. 56, 141–147.
Kim, S. W., Kim, K. W., Shin, C. S., Park do, J., Park, K. S., Cho, B. Y., Lee, H. K., and Kim, S. Y. (2007). Acylated ghrelin secretion is acutely suppressed by oral glucose load or insulin-induced hypoglycemia independently of basal growth hormone secretion in humans. Horm. Res. 67, 211–219.
Klover, P., and Hennighausen, L. (2007). Postnatal body growth is dependent on the transcription factors signal transducers and activators of transcription 5a/b in muscle: a role for autocrine/paracrine insulin-like growth factor I. Endocrinology 148, 1489–1497.
Kok, P., Buijs, M. M., Kok, S. W., Van Ierssel, I. H., Frölich, M., Roelfsema, F., Voshol, P. J., Meinders, A. E., and Pijl, H. (2004). Acipimox enhances spontaneous growth hormone secretion in obese women. Am. J. Physiol. Regul. Integr. Comp. Physiol. 286, R693–R698.
Kotelevtsev, Y., Holmes, M. C., Burchell, A., Houston, P. M., Schmoll, D., Jamieson, P., Best, R., Brown, R., Edwards, C. R., Seckl, J. R., and Mullins, J. J. (1997). 11beta-hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc. Natl. Acad. Sci. U.S.A. 94, 14924–14929.
Kotzka, J., Knebel, B., Avci, H., Jacob, S., Nitzgen, U., Jockenhovel, F., Heeren, J., Haas, J., and Muller-Wieland, D. (2010). Phosphorylation of sterol regulatory element-binding protein (SREBP)-1a links growth hormone action to lipid metabolism in hepatocytes. Atherosclerosis 213, 156–165.
Koutkia, P., Meininger, G., Canavan, B., Breu, J., and Grinspoon, S. (2004). Metabolic regulation of growth hormone by free fatty acids, somatostatin, and ghrelin in HIV-lipodystrophy. Am. J. Physiol. Endocrinol. Metab. 286, E296–E303.
Krag, M. B., Nielsen, S., Guo, Z., Pedersen, S. B., Schmitz, O., Christiansen, J. S., and Jørgensen, J. O. (2008). Peroxisome proliferator-activated receptor gamma agonism modifies the effects of growth hormone on lipolysis and insulin sensitivity. Clin. Endocrinol. (Oxf.) 69, 452–461.
Lanning, N. J., and Carter-Su, C. (2006). Recent advances in growth hormone signaling. Rev. Endocr. Metab. Disord. 7, 225–235.
Lee, J. Y., Gavrilova, O., Davani, B., Na, R., Robinson, G. W., and Hennighausen, L. (2007). The transcription factors Stat5a/b are not required for islet development but modulate pancreatic beta-cell physiology upon aging. Biochim. Biophys. Acta 1773, 1455–1461.
LeRoith, D. (1996). Insulin-like growth factor receptors and binding proteins. Baillieres Clin. Endocrinol. Metab. 10, 49–73.
Li, Y., Knapp, J. R., and Kopchick, J. J. (2003). Enlargement of interscapular brown adipose tissue in growth hormone antagonist transgenic and in growth hormone receptor gene-disrupted dwarf mice. Exp. Biol. Med. (Maywood) 228, 207–215.
Liang, L., Jiang, J., and Frank, S. J. (2000). Insulin receptor substrate-1-mediated enhancement of growth hormone-induced mitogen-activated protein kinase activation. Endocrinology 141, 3328–3336.
Lin, S. C., and Li, P. (2004). CIDE-A, a novel link between brown adipose tissue and obesity. Trends Mol. Med. 10, 434–439.
Lindberg-Larsen, R., Moller, N., Schmitz, O., Nielsen, S., Andersen, M., Orskov, H., and Jørgensen, J. O. (2007). The impact of pegvisomant treatment on substrate metabolism and insulin sensitivity in patients with acromegaly. J. Clin. Endocrinol. Metab. 92, 1724–1728.
Liu, J. L., Grinberg, A., Westphal, H., Sauer, B., Accili, D., Karas, M., and LeRoith, D. (1998). Insulin-like growth factor-I affects perinatal lethality and postnatal development in a gene dosage-dependent manner: manipulation using the Cre/loxP system in transgenic mice. Mol. Endocrinol. 12, 1452–1462.
Ljungberg, A., Linden, D., Améen, C., Bergström, G., and Oscarsson, J. (2007). Importance of PPAR alpha for the effects of growth hormone on hepatic lipid and lipoprotein metabolism. Growth Horm. IGF Res. 17, 154–164.
Loesch, K., Deng, L., Cowan, J. W., Wang, X., He, K., Jiang, J., Black, R. A., and Frank, S. J. (2006). Janus kinase 2 influences growth hormone receptor metalloproteolysis. Endocrinology 147, 2839–2849.
Loesch, K., Deng, L., Wang, X., He, K., Jiang, J., and Frank, S. J. (2007). Endoplasmic reticulum-associated degradation of growth hormone receptor in Janus kinase 2-deficient cells. Endocrinology 148, 5955–5965.
Luque, R. M., Gahete, M. D., Valentine, R. J., and Kineman, R. D. (2006). Examination of the direct effects of metabolic factors on somatotrope function in a non-human primate model, Papio anubis. J. Mol. Endocrinol. 37, 25–38.
Luque, R. M., Huang, Z. H., Shah, B., Mazzone, T., and Kineman, R. D. (2007). Effects of leptin replacement on hypothalamic-pituitary growth hormone axis function and circulating ghrelin levels in ob/ob mice. Am. J. Physiol. Endocrinol. Metab. 292, E891–E899.
Luque, R. M., and Kineman, R. D. (2006). Impact of obesity on the growth hormone axis: evidence for a direct inhibitory effect of hyperinsulinemia on pituitary function. Endocrinology 147, 2754–2763.
Luque, R. M., Lin, Q., Córdoba-Chacón, J., Subbaiah, P. V., Buch, T., Waisman, A., Vankelecom, H., and Kineman, R. D. (2011). Metabolic impact of adult-onset, isolated, growth hormone deficiency (AOiGHD) due to destruction of pituitary somatotropes. PLoS ONE 6, e15767.
Maccario, M., Procopio, M., Grottoli, S., Oleandri, S. E., Boffano, G. M., Taliano, M., Camanni, F., and Ghigo, E. (1996). Effects of acipimox, an antilipolytic drug, on the growth hormone (GH) response to GH-releasing hormone alone or combined with arginine in obesity. Metab. Clin. Exp. 45, 342–346.
Makimura, H., Stanley, T., Mun, D., You, S. M., and Grinspoon, S. (2008). The effects of central adiposity on growth hormone (GH) response to GH-releasing hormone-arginine stimulation testing in men. J. Clin. Endocrinol. Metab. 93, 4254–4260.
Masternak, M. M., Al-Regaiey, K. A., Del Rosario Lim, M. M., Bonkowski, M. S., Panici, J. A., Przybylski, G. K., and Bartke, A. (2005). Caloric restriction results in decreased expression of peroxisome proliferator-activated receptor superfamily in muscle of normal and long-lived growth hormone receptor/binding protein knockout mice. J. Gerontol. A Biol. Sci. Med. Sci. 60, 1238–1245.
Mauvais-Jarvis, F., Ueki, K., Ueki, K., Fruman, D. A., Hirshman, M. F., Sakamoto, K., Goodyear, L. J., Iannacone, M., Accili, D., Cantley, L. C., and Kahn, C. R. (2002). Reduced expression of the murine p85alpha subunit of phosphoinositide 3-kinase improves insulin signaling and ameliorates diabetes. J. Clin. Invest. 109, 141–149.
Mavalli, M. D., DiGirolamo, D. J., Fan, Y., Riddle, R. C., Campbell, K. S., van Groen, T., Frank, S. J., Sperling, M. A., Esser, K. A., Bamman, M. M., and Clemens, T. L. (2010). Distinct growth hormone receptor signaling modes regulate skeletal muscle development and insulin sensitivity in mice. J. Clin. Invest. 120, 4007–4020.
Melmed, S. (1984). Insulin suppresses growth hormone secretion by rat pituitary cells. J. Clin. Invest. 73, 1425–1433.
Moriarty, M. W., McCurdy, C. E., Janssen, R. C., Shaw, T., Leitner, J. W., Friedman, J. E., and Draznin, B. (2009). In vivo knockdown of p85alpha with an antisense oligonucleotide improves insulin sensitivity in Lep(ob/ob) and diet-induced obese mice. Horm. Metab. Res. 41, 757–761.
Morita, J., Hakuno, F., Hizuka, N., Takahashi, S., and Takano, K. (2009). Growth hormone (GH) or insulin-like growth factor (IGF)-I represses 11beta-hydroxysteroid dehydrogenase type 1 (HSD1) mRNA expression in 3T3-L1 cells and its activity in their homogenates. Endocr. J. 56, 561–570.
Morton, N. M., Holmes, M. C., Fiévet, C., Staels, B., Tailleux, A., Mullins, J. J., and Seckl, J. R. (2001). Improved lipid and lipoprotein profile, hepatic insulin sensitivity, and glucose tolerance in 11beta-hydroxysteroid dehydrogenase type 1 null mice. J. Biol. Chem. 276, 41293–41300.
Morton, N. M., Ramage, L., and Seckl, J. R. (2004). Down-regulation of adipose 11beta-hydroxysteroid dehydrogenase type 1 by high-fat feeding in mice: a potential adaptive mechanism counteracting metabolic disease. Endocrinology 145, 2707–2712.
Myers, M. G. Jr., Leibel, R. L., Seeley, R. J., and Schwartz, M. W. (2010). Obesity and leptin resistance: distinguishing cause from effect. Trends Endocrinol. Metab. 21, 643–651.
Mziaut, H., Trajkovski, M., Kersting, S., Ehninger, A., Altkrüger, A., Lemaitre, R. P., Schmidt, D., Saeger, H. D., Lee, M. S., Drechsel, D. N., Müller, S., and Solimena, M. (2006). Synergy of glucose and growth hormone signalling in islet cells through ICA512 and STAT5. Nat. Cell Biol. 8, 435–445.
Nam, S. Y., Kim, K. R., Cha, B. S., Song, Y. D., Lim, S. K., Lee, H. C., and Huh, K. B. (2001). Low-dose growth hormone treatment combined with diet restriction decreases insulin resistance by reducing visceral fat and increasing muscle mass in obese type 2 diabetic patients. Int. J. Obes. Relat. Metab. Disord. 25, 1101–1107.
Naya, F. J., Wu, C., Richardson, J. A., Overbeek, P., and Olson, E. N. (1999). Transcriptional activity of MEF2 during mouse embryogenesis monitored with a MEF2-dependent transgene. Development 126, 2045–2052.
Neve, B. P., Hoogerbrugge, N., Verhoeven, A. J., Birkenhäger, J. C., and Jansen, H. (1997). Growth hormone restores hepatic lipase mRNA levels but the translation is impaired in hepatocytes of hypothyroid rats. Biochim. Biophys. Acta 1345, 172–179.
Ng, F. M., Jiang, W. J., Gianello, R., Pitt, S., and Roupas, P. (2000). Molecular and cellular actions of a structural domain of human growth hormone (AOD9401) on lipid metabolism in Zucker fatty rats. J. Mol. Endocrinol. 25, 287–298.
Nielsen, C., Gormsen, L. C., Jessen, N., Pedersen, S. B., Møller, N., Lund, S., and Jørgensen, J. O. (2008). Growth hormone signaling in vivo in human muscle and adipose tissue: impact of insulin, substrate background, and growth hormone receptor blockade. J. Clin. Endocrinol. Metab. 93, 2842–2850.
Nielsen, J. H. (1982). Effects of growth hormone, prolactin, and placental lactogen on insulin content and release, and deoxyribonucleic acid synthesis in cultured pancreatic islets. Endocrinology 110, 600–606.
Nielsen, J. H., Linde, S., Welinder, B. S., Billestrup, N., and Madsen, O. D. (1989). Growth hormone is a growth factor for the differentiated pancreatic beta-cell. Mol. Endocrinol. 3, 165–173.
Nielsen, J. H., Moldrup, A., and Billestrup, N. (1990). Expression of the growth hormone receptor gene in insulin producing cells. Biomed. Biochim. Acta 49, 1151–1155.
Nilsson, L., Binart, N., Bohlooly-Y, M., Bramnert, M., Egecioglu, E., Kindblom, J., Kelly, P. A., Kopchick, J. J., Ormandy, C. J., Ling, C., and Billig, H. (2005). Prolactin and growth hormone regulate adiponectin secretion and receptor expression in adipose tissue. Biochem. Biophys. Res. Commun. 331, 1120–1126.
Ohlsson, C., Mohan, S., Sjögren, K., Tivesten, A., Isgaard, J., Isaksson, O., Jansson, J. O., and Svensson, J. (2009). The role of liver-derived insulin-like growth factor-I. Endocr. Rev. 30, 494–535.
Oliveira, C. R., Salvatori, R., Meneguz-Moreno, R. A., Aguiar-Oliveira, M. H., Pereira, R. M., Valença, E. H., Araujo, V. P., Farias, N. T., Silveira, D. C., Vieira, J. G., and Barreto-Filho, J. A. (2010). Adipokine profile and urinary albumin excretion in isolated growth hormone deficiency. J. Clin. Endocrinol. Metab. 95, 693–698.
Oscarsson, J., Ottosson, M., Johansson, J. O., Wiklund, O., Mårin, P., Björntorp, P., and Bengtsson, B. A. (1996). Two weeks of daily injections and continuous infusion of recombinant human growth hormone (GH) in GH-deficient adults. II. Effects on serum lipoproteins and lipoprotein and hepatic lipase activity. Metabolism 45, 370–377.
Oscarsson, J., Ottosson, M., Vikman-Adolfsson, K., Frick, F., Enerbäck, S., Lithell, H., and Edén, S. (1999). GH but not IGF-I or insulin increases lipoprotein lipase activity in muscle tissues of hypophysectomised rats. J. Endocrinol. 160, 247–255.
Ott, M. O., Bober, E., Lyons, G., Arnold, H., and Buckingham, M. (1991). Early expression of the myogenic regulatory gene, myf-5, in precursor cells of skeletal muscle in the mouse embryo. Development 111, 1097–1107.
Pasarica, M., Zachwieja, J. J., Dejonge, L., Redman, S., and Smith, S. R. (2007). Effect of growth hormone on body composition and visceral adiposity in middle-aged men with visceral obesity. J. Clin. Endocrinol. Metab. 92, 4265–4270.
Pasquali, C., Curchod, M. L., Wälchli, S., Espanel, X., Guerrier, M., Arigoni, F., Strous, G., and Hooft van Huijsduijnen, R. (2003). Identification of protein tyrosine phosphatases with specificity for the ligand-activated growth hormone receptor. Mol. Endocrinol. 17, 2228–2239.
Pazos, Y., Casanueva, F. F., and Camiña, J. P. (2008). Basic aspects of ghrelin action. Vitam. Horm. 77, 89–119.
Pennisi, P. A., Kopchick, J. J., Thorgeirsson, S., LeRoith, D., and Yakar, S. (2004). Role of growth hormone (GH) in liver regeneration. Endocrinology 145, 4748–4755.
Perez, F. R., Camina, J. P., Menéndez, C., Beiras, A., Casabiell, X., and Casanueva, F. F. (1998). Cis-unsaturated free fatty acids block VIP-mediated GH and PRL secretion by perturbing the cAMP/protein kinase A pathway. Pituitary 1, 25–32.
Perez, F. R., Camina, J. P., Zugaza, J. L., Lage, M., Casabiell, X., and Casanueva, F. F. (1997). cis-FFA do not alter membrane depolarization but block Ca2+ influx and GH secretion in KCl-stimulated somatotroph cells. Suggestion for a direct cis-FFA perturbation of the Ca2+ channel opening. Biochim. Biophys. Acta 1329, 269–277.
Pilecka, I., Patrignani, C., Pescini, R., Curchod, M. L., Perrin, D., Xue, Y., Yasenchak, J., Clark, A., Magnone, M. C., Zaratin, P., Valenzuela, D., Rommel, C., and Hooft van Huijsduijnen, R. (2007). Protein-tyrosine phosphatase H1 controls growth hormone receptor signaling and systemic growth. J. Biol. Chem. 282, 35405–35415.
Psilopanagioti, A., Papadaki, H., Kranioti, E. F., Alexandrides, T. K., and Varakis, J. N. (2009). Expression of adiponectin and adiponectin receptors in human pituitary gland and brain. Neuroendocrinology 89, 38–47.
Qin, Y., and Tian, Y. P. (2010a). Exploring the molecular mechanisms underlying the potentiation of exogenous growth hormone on alcohol-induced fatty liver diseases in mice. J. Transl. Med. 8, 120.
Qin, Y., and Tian, Y. P. (2010b). Hepatic adiponectin receptor R2 expression is up-regulated in normal adult male mice by chronic exogenous growth hormone levels. Mol. Med. Report 3, 525–530.
Qin, Y., and Tian, Y. P. (2010c). Preventive effects of chronic exogenous growth hormone levels on diet-induced hepatic steatosis in rats. Lipids Health Dis. 9, 78.
Ram, P. A., Park, S. H., Choi, H. K., and Waxman, D. J. (1996). Growth hormone activation of Stat 1, Stat 3, and Stat 5 in rat liver. Differential kinetics of hormone desensitization and growth hormone stimulation of both tyrosine phosphorylation and serine/threonine phosphorylation. J. Biol. Chem. 271, 5929–5940.
Ram, P. A., and Waxman, D. J. (1999). SOCS/CIS protein inhibition of growth hormone-stimulated STAT5 signaling by multiple mechanisms. J. Biol. Chem. 274, 35553–35561.
Rasmussen, M. H., Juul, A., and Hilsted, J. (2007). Effect of weight loss on free insulin-like growth factor-I in obese women with hyposomatotropism. Obesity (Silver Spring) 15, 879–886.
Rasmussen, M. H., Juul, A., Kjems, L. L., and Hilsted, J. (2006). Effects of short-term caloric restriction on circulating free IGF-I, acid-labile subunit, IGF-binding proteins (IGFBPs)-1-4, and IGFBPs-1-3 protease activity in obese subjects. Eur. J. Endocrinol. 155, 575–581.
Richelsen, B., Pedersen, S. B., Kristensen, K., Børglum, J. D., Nørrelund, H., Christiansen, J. S., and Jørgensen, J. O. (2000). Regulation of lipoprotein lipase and hormone-sensitive lipase activity and gene expression in adipose and muscle tissue by growth hormone treatment during weight loss in obese patients. Metab. Clin. Exp. 49, 906–911.
Robertson, K., Kopchick, J. J., and Liu, J. L. (2006). Growth hormone receptor gene deficiency causes delayed insulin responsiveness in skeletal muscles without affecting compensatory islet cell overgrowth in obese mice. Am. J. Physiol. Endocrinol. Metab. 291, E491–E498.
Rodriguez-Pacheco, F., Martinez-Fuentes, A. J., Tovar, S., Pinilla, L., Tena-Sempere, M., Dieguez, C., Castaño, J. P., and Malagon, M. M. (2007). Regulation of pituitary cell function by adiponectin. Endocrinology 148, 401–410.
Romero, C. J., Ng, Y., Luque, R. M., Kineman, R. D., Koch, L., Bruning, J. C., and Radovick, S. (2010). Targeted deletion of somatotroph insulin-like growth factor-I signaling in a cell-specific knockout mouse model. Mol. Endocrinol. 24, 1077–1089.
Sakharova, A. A., Horowitz, J. F., Surya, S., Goldenberg, N., Harber, M. P., Symons, K., and Barkan, A. (2008). Role of growth hormone in regulating lipolysis, proteolysis, and hepatic glucose production during fasting. J. Clin. Endocrinol. Metab. 93, 2755–2759.
Samra, J. S., Clark, M. L., Humphreys, S. M., MacDonald, I. A., Bannister, P. A., Matthews, D. R., and Frayn, K. N. (1999). Suppression of the nocturnal rise in growth hormone reduces subsequent lipolysis in subcutaneous adipose tissue. Eur. J. Clin. Invest. 29, 1045–1052.
Samuel, V. T., Petersen, K. F., and Shulman, G. I. (2010). Lipid-induced insulin resistance: unravelling the mechanism. Lancet 375, 2267–2277.
Scacchi, M., Orsini, F., Cattaneo, A., Grasso, A., Filippini, B., Pecori Giraldi, F., Fatti, L. M., Moro, M., and Cavagnini, F. (2010). The diagnosis of GH deficiency in obese patients: a reappraisal with GHRH plus arginine testing after pharmacological blockade of lipolysis. Eur. J. Endocrinol. 163, 201–206.
Schwarz, J. M., Mulligan, K., Lee, J., Lo, J. C., Wen, M., Noor, M. A., Grunfeld, C., and Schambelan, M. (2002). Effects of recombinant human growth hormone on hepatic lipid and carbohydrate metabolism in HIV-infected patients with fat accumulation. J. Clin. Endocrinol. Metab. 87, 942.
Short, K. R., Moller, N., Bigelow, M. L., Coenen-Schimke, J., and Nair, K. S. (2008). Enhancement of muscle mitochondrial function by growth hormone. J. Clin. Endocrinol. Metab. 93, 597–604.
Smit, L. S., Meyer, D. J., Billestrup, N., Norstedt, G., Schwartz, J., and Carter-Su, C. (1996). The role of the growth hormone (GH) receptor and JAK1 and JAK2 kinases in the activation of Stats 1, 3, and 5 by GH. Mol. Endocrinol. 10, 519–533.
Steyn, F. J., Boehme, F., Vargas, E., Wang, K., Parkington, H. C., Rao, J. R., and Chen, C. (2009). Adiponectin regulate growth hormone secretion via adiponectin receptor mediated Ca(2+) signalling in rat somatotrophs in vitro. J. Neuroendocrinol. 21, 698–704.
Szendroedi, J., Zwettler, E., Schmid, A. I., Chmelik, M., Pacini, G., Kacerovsky, G., Smekal, G., Nowotny, P., Wagner, O., Schnack, C., Schernthaner, G., Klaushofer, K., and Roden, M. (2008). Reduced Basal ATP synthetic flux of skeletal muscle in patients with previous acromegaly. PLoS ONE 3, e3958.
Trepp, R., Fluck, M., Stettler, C., Boesch, C., Ith, M., Kreis, R., Hoppeler, H., Howald, H., Schmid, J. P., Diem, P., and Christ, E. R. (2008). Effect of GH on human skeletal muscle lipid metabolism in GH deficiency. Am. J. Physiol. Endocrinol. Metab. 294, E1127–E1134.
Tuggle, C. K., and Trenkle, A. (1996). Control of growth hormone synthesis. Domest. Anim. Endocrinol. 13, 1–33.
Ueki, K., Fruman, D. A., Brachmann, S. M., Tseng, Y. H., Cantley, L. C., and Kahn, C. R. (2002). Molecular balance between the regulatory and catalytic subunits of phosphoinositide 3-kinase regulates cell signaling and survival. Mol. Cell Biol. 22, 965–977.
Ueland, T., Fougner, S. L., Godang, K., Lekva, T., Schurgers, L. J., Scholz, H., Halvorsen, B., Schreiner, T., Aukrust, P., and Bollerslev, J. (2010). Associations between body composition, circulating interleukin-1 receptor antagonist, osteocalcin, and insulin metabolism in active acromegaly. J. Clin. Endocrinol. Metab. 95, 361–368.
Utz, A. L., Yamamoto, A., Sluss, P., Breu, J., and Miller, K. K. (2008). Androgens may mediate a relative preservation of IGF-I levels in overweight and obese women despite reduced growth hormone secretion. J. Clin. Endocrinol. Metab. 93, 4033–4040.
Verrua, E., Filopanti, M., Ronchi, C. L., Olgiati, L., Ferrante, E., Giavoli, C., Sala, E., Mantovani, G., Arosio, M., Beck-Peccoz, P., Lania, A. G., and Spada, A. (2011). GH response to oral glucose tolerance test: a comparison between patients with acromegaly and other pituitary disorders. J. Clin. Endocrinol. Metab. 96, E83–E88.
Wang, X., Yang, N., Deng, L., Li, X., Jiang, J., Gan, Y., and Frank, S. J. (2009). Interruption of growth hormone signaling via SHC and ERK in 3T3-F442A preadipocytes upon knockdown of insulin receptor substrate-1. Mol. Endocrinol. 23, 486–496.
Wang, Z., Masternak, M. M., Al-Regaiey, K. A., and Bartke, A. (2007). Adipocytokines and the regulation of lipid metabolism in growth hormone transgenic and calorie-restricted mice. Endocrinology 148, 2845–2853.
Weltman, A., Weltman, J. Y., Watson Winfield, D. D., Frick, K., Patrie, J., Kok, P., Keenan, D. M., Gaesser, G. A., and Veldhuis, J. D. (2008). Effects of continuous versus intermittent exercise, obesity, and gender on growth hormone secretion. J. Clin. Endocrinol. Metab. 93, 4711–4720.
Wolfing, B., Neumeier, M., Buechler, C., Aslanidis, C., Schölmerich, J., and Schäffler, A. (2008). Interfering effects of insulin, growth hormone and glucose on adipokine secretion. Exp. Clin. Endocrinol. Diabetes 116, 47–52.
Wu, Y., Liu, C., Sun, H., Vijayakumar, A., Giglou, P. R., Qiao, R., Oppenheimer, J., Yakar, S., and LeRoith, D. (2011). Growth hormone receptor regulates beta cell hyperplasia and glucose-stimulated insulin secretion in obese mice. J. Clin. Invest. 121, 2422–2426.
Yang, S., Mulder, H., Holm, C., and Edén, S. (2004). Effects of growth hormone on the function of beta-adrenoceptor subtypes in rat adipocytes. Obes. Res. 12, 330–339.
Yip, R. G., and Goodman, H. M. (1999). Growth hormone and dexamethasone stimulate lipolysis and activate adenylyl cyclase in rat adipocytes by selectively shifting Gi alpha2 to lower density membrane fractions. Endocrinology 140, 1219–1227.
Zerrad-Saadi, A., Lambert-Blot, M., Mitchell, C., Bretes, H., Collin de l’Hortet, A., Baud, V., Chereau, F., Sotiropoulos, A., Kopchick, J. J., Liao, L., Xu, J., Gilgenkrantz, H., and Guidotti, J. E. (2011). GH receptor plays a major role in liver regeneration through the control of EGFR and ERK1/2 activation. Endocrinology 152, 2731–2741.
Zhang, F., Zhang, Q., Tengholm, A., and Sjöholm, A. (2006). Involvement of JAK2 and Src kinase tyrosine phosphorylation in human growth hormone-stimulated increases in cytosolic free Ca2+ and insulin secretion. Am. J. Physiol. Cell Physiol. 291, C466–C475.
Keywords: growth hormone, metabolism, lipids, carbohydrates, tissue-specific, insulin resistance
Citation: Vijayakumar A, Yakar S and LeRoith D (2011) The intricate role of growth hormone in metabolism. Front. Endocrin. 2:32. doi: 10.3389/fendo.2011.00032
Received: 26 July 2011;
Paper pending published: 08 August 2011;
Accepted: 30 August 2011;
Published online: 27 September 2011.
Edited by:
Sally Radovick, Johns Hopkins University, USAReviewed by:
Rhonda D. Kineman, University of Illinois at Chicago, USARoberto Salvatori, Johns Hospital University, USA
Copyright: © 2011 Vijayakumar, Yakar and LeRoith. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Derek LeRoith, Division of Endocrinology, Diabetes and Bone Disease, Mount Sinai School of Medicine, One Gustav Levy Place, Box 1055, New York, NY 10029-6574, USA. e-mail:ZGVyZWsubGVyb2l0aEBtc3NtLmVkdQ==