 Mélanie Bourque1,2
Mélanie Bourque1,2
- 1 Molecular Endocrinology and Genomic Research Center, Centre de recherche du CHUQ (CHUL), Quebec City, QC, Canada
- 2 Faculty of Pharmacy, Laval University, Quebec City, QC, Canada
- 3 Department of Anatomy and Neurobiology, Northeastern Ohio Universities College of Medicine and Pharmacy, Rootstown, OH, USA
The existence of a sex difference in Parkinson’s disease (PD) is observed as related to several variables, including susceptibility of the disease, age at onset, and symptoms. These differences between men and women represent a significant characteristic of PD, which suggest that estrogens may exert beneficial effects against the development and the progression of the disease. This paper reviews the neuroprotective and neuromodulator effects of 17β-estradiol and progesterone as compared to androgens in the nigrostriatal dopaminergic (NSDA) system of both female and male rodents. The 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mice model of PD and methamphetamine toxicity faithfully reproduce the sex differences of PD in that endogenous estrogen levels appear to influence the vulnerability to toxins targeting the NSDA system. Exogenous 17β-estradiol and/or progesterone treatments show neuroprotective properties against NSDA toxins while androgens fail to induce any beneficial effect. Sex steroid treatments show male and female differences in their neuroprotective action against methamphetamine toxicity. NSDA structure and function, as well as the distribution of estrogen receptors, show sex differences and may influence the susceptibility to the toxins and the response to sex steroids. Genomic and non-genomic actions of 17β-estradiol converge to promote survival factors and the presence of both estrogen receptors α and β are critical to 17β-estradiol neuroprotective action against MPTP toxicity.
Parkinson’s Disease
Sex Differences
Parkinson’s disease (PD) is a neurodegenerative disorder characterized by a progressive and selective loss of dopamine (DA) cell bodies in substantia nigra (Dauer and Przedborski, 2003). The death of nigral DA neurons results in striatal DA decrease, leading to a dysfunction in basal ganglia process, and the appearance of clinical symptoms such as resting tremor, rigidity, and bradykinesia (Obeso et al., 2008).
Several variables (including susceptibility to the disease, age at onset, symptoms) support the existence of a sex difference in PD (Miller and Cronin-Golomb, 2010). While most studies report a higher predisposition of PD in men (Van Den Eeden et al., 2003; Wooten et al., 2004; Shulman, 2007), sex is not considered a risk factor in a few studies (Granieri et al., 1991; de Rijk et al., 1995). Men have at least a 1.5-fold greater risk than women of developing PD (Van Den Eeden et al., 2003; Wooten et al., 2004). The age at onset in women occurs about 2 years later compared to men in the majority of studies (Twelves et al., 2003; Haaxma et al., 2007), while no difference has also been reported in one study (Baba et al., 2005). Moreover, differences in the profile of motor symptoms between men and women have also been reported. Total motor scores on the modified unified Parkinson’s disease rating scale (UPDRS) do not differ between men and women however, men have more advanced rigidity and women present with greater instability scores (Baba et al., 2005). When UPDRS motor scores were examined as a function of disease progression, sex differences were not observed in early PD (less than 5 years duration) however, women have better motor scores than men in advanced PD (disease duration more than 5 years; Lyons et al., 1998). Not only do men exhibit more severe parkinsonian motor features than women but a greater improvement of motor function following levodopa therapy is reported in women (Growdon et al., 1998; Lyons et al., 1998; Zappia et al., 2005). Haaxma et al. (2007) found that women have a higher frequency of presentation with the tremor dominant form of PD, which has been associated with a slower disease progression. [123I]FR-CIT single photon emission computed tomography (SPECT) measurements show that, at symptom onset, women had superior levels of striatal DA binding than men, suggesting that the development of symptomatic PD may be delayed by higher physiological striatal DA levels (Haaxma et al., 2007). In light of these findings, the authors suggested that the phenotype of PD in women is more benign (Haaxma et al., 2007). It should be noted however, that differences in disease presentation, for example, the initial symptom of tremor in women, was not consistently reported (Baba et al., 2005). Thus, sex differences represent a notable characteristic of PD, which suggests that estrogens may exert beneficial effects against the development and progression of the disease in women and/or that androgens may exert destructive effects upon the development and progression of the disease in men.
Influence of Endogenous and Exogenous Estrogens
Several studies have been conducted to investigate how endogenous estrogen status and estrogen therapy influence the risk of PD. A longer fertile lifespan (difference between age at menarche and age at menopause) was associated with a decreased risk of PD in women with natural menopause (>39 years of fertile lifespan) compared to the lowest fertile lifespan (<33 years) as reported among 83,482 women participating in the Observational Study of the Women’s Health Initiative (WHI-OS; Saunders-Pullman et al., 2009). A fertile lifespan shorter than 36 years was associated with an increased risk of PD and an earlier menopause was more common among women with PD (Ragonese et al., 2004). A case–control study reported that women with PD had undergone hysterectomy (with or without unilateral oophorectomy) more frequently than control subjects (Benedetti et al., 2001), however data from another report indicated that surgical menopause was associated with a decreased incidence of PD (Ragonese et al., 2004). Most findings suggest a beneficial effect of estrogen with regard to PD risk as the use of either postmenopausal estrogen (Currie et al., 2004; Popat et al., 2005) or oral contraceptives (Ascherio et al., 2003; Simon et al., 2009) were associated with a reduced risk of PD, and women with PD were less likely to use estrogen therapy (Benedetti et al., 2001); though there are also data relating that a similar risk exists between women who have or have not used estrogen therapy (Ascherio et al., 2003). The use of estrogen therapy on the risk of PD varies depending on the type of menopause, with increased risk in women with hysterectomy (Popat et al., 2005; Saunders-Pullman et al., 2009) and decreased risk in women with natural menopause (Popat et al., 2005). No association between exogenous or endogenous estrogens exposure on risk of PD has also been reported (Simon et al., 2009).
In women with PD experiencing regular menstrual cycles a worsening of parkinsonian symptoms was associated with premenstrual and menstrual periods, when estrogens and progesterone are at a low level (Quinn and Marsden, 1986; Kompoliti et al., 2000; Tolson et al., 2002). A reduction in levodopa effectiveness was also observed before and during the menstruation period in young PD women (Quinn and Marsden, 1986; Giladi and Honigman, 1995). Although it was thought that estrogen was the basis for these symptom fluctuations, a study conducted in a 5-week period in 10 PD women with menstrual cycles found that serum estrogens and progesterone levels were not related to PD severity (Kompoliti et al., 2000). Nevertheless, Horstink et al. (2003) report fluctuations of dyskinesias, a motor complication induced by levodopa treatment, as associated with estradiol levels in a young PD woman, with an increase in dyskinesia when estradiol levels were highest, followed by a diminution of dyskinesia and a worsening of parkinsonism in the premenstrual period. The majority of women with PD report a worsening of parkinsonian symptoms during pregnancy and into the postpartum period, while some women do not experience any deterioration of their symptoms (Golbe, 1987; Hagell et al., 1998; Shulman et al., 2000; Robottom et al., 2008). Case reports have documented an increase in total and motor UPDRS scores during and after pregnancy (Shulman et al., 2000), with a faster symptom progression than that observed in the comparison cohort (Robottom et al., 2008). The substantial variations in estrogen levels during and after pregnancy were proposed to be implicated in the worsening of parkinsonian symptoms during these periods (Rubin, 2007; Robottom et al., 2008).
An amelioration of PD symptoms and dyskinesia was reported to be present under conditions of estrogen therapy or high levels of endogenous estrogens (Villeneuve et al., 1978; Session et al., 1994; Giladi and Honigman, 1995). Postmenopausal women with early PD using estrogen therapy prior to initiation of levodopa have lower symptom severity scores (Saunders-Pullman et al., 1999) but estrogen therapy had no effect at later stages of the disease (Strijks et al., 1999). Results from double-blind studies reported a reduction of the dose of levodopa required to improve motor function in women receiving 17β-estradiol (Blanchet et al., 1999) and an improvement of motor disability in PD women with motor fluctuations when treated with estrogens (Tsang et al., 2000) whereas no effect of estrogen on motor function was also observed (Strijks et al., 1999).
While in some cases conflicting information is reported from the above human studies, a longer fertile lifespan seems to be consistently associated with a decreased risk of PD, suggesting that longer exposure to endogenous ovarian steroids exerts a beneficial effect against PD. PD symptoms seem to be modulated by endogenous variation of steroids as reported by the majority of women experiencing menstrual cycles and pregnancy. The most conflicting data comes from the use of estrogen therapy (most of the studies support a beneficial effect of estrogens) but many factors could influence the conclusion of these studies. First, a variety of hormonal therapy compounds are available and the studies do not always state the specific molecule used by women. Further, the dose, timing of initiation of treatment and duration of estrogen therapy can vary substantially. In addition, estrogen therapy could be used alone or in combination with progestin and the type of hormone therapy prescribed could differ between women with a hysterectomy and women with natural menopause. Indeed, women within the same cohort could have used different preparations and doses of estrogen therapy and could have experienced a different duration of treatment. Taken together, the results from these studies suggest that exposure to endogenous and exogenous estrogens seem to influence the risk and symptoms of PD while further research concerning the exact regimen of hormonal therapy will require careful analyses to enable a definitive conclusion.
Estrogens Receptor, Steroids, and Parkinson’s Disease
Estrogens receptor (ER) α and ERβ polymorphism were found to be unrelated with an increased risk of PD (Maraganore et al., 2002; Westberg et al., 2004; Li et al., 2009). Alternatively, ERβ polymorphism is reported more frequently in PD patients with an early age at onset (Westberg et al., 2004; Hakansson et al., 2005). A small number of studies have investigated alterations of neurosteroid synthesis in PD. Reduced levels of allopregnanolone and 5α-dehydroprogesterone were found in the cerebrospinal fluid of PD patients (di Michele et al., 2003). In the substantia nigra, reductions of 5α-reductase protein and mRNA were observed (Luchetti et al., 2010), which could lead to decreased synthesis of 5α-dehydroprogesterone and subsequently allopregnanolone (di Michele et al., 2003). Expression of sulfotransferase 2B1, an enzyme catalyzing the conversion of pregnenolone and dehydroepiandrosterone into their sulfated esters, in the substantia nigra was also downregulated and in the caudate nucleus, expression of 3α-hydroxysteroid dehydrogenase type 3, an enzyme catalyzing the synthesis of allopregnanolone, was upregulated (Luchetti et al., 2010), suggesting a compensatory mechanism in response to the loss of input from the substantia nigra. The authors suggest an involvement of neurosteroids in the neurodegenerative process of PD (Luchetti et al., 2010).
Influence of Sex Steroids in Experimental Models of Parkinson’s Disease
Sex Differences and Influence of Endogenous Hormones
Two animal models of induced degeneration affecting the nigrostriatal dopaminergic (NSDA) system are reviewed here as related to the influence of sex steroids – the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) model of PD and methamphetamine (MA)-induced DA toxicity. While both agents primarily target the NSDA system, they differ with regard to their mechanisms of action. For MPTP, conversion to its active metabolite, 1-methyl-4-phenylpyridinium (MPP+), is required followed by uptake into the DA neuron via the DA transporter (DAT). The interaction of MPP+ with mitochondrial complex 1 results in a disruption of the respiratory chain and eventual cell destruction. The exact mechanisms for toxicity resulting from MA remain elusive, but it is believed to produce a reversal in DAT function thus producing excessive amounts of DA which have the potential of generating free radicals and oxidative stress. Studies in these animal models have shown a greater neurotoxic effect in male than female mice, as observed by more extensive striatal DA reduction and a greater decrease in DAT specific binding in striatum (Wagner et al., 1993; Dluzen et al., 1996; Miller et al., 1998; Yu and Liao, 2000b; Bourque et al., 2009, 2011). Moreover, the levels of endogenous sex steroids have been reported to affect the susceptibility of female mice to the toxin. In female BALB/c mice treated with MA, a greater DA depletion was observed when the toxin was administered at diestrus (when estrogens levels are low) whereas lower DA loss occurs at proestrus (when estrogens levels are high; Yu and Liao, 2000b). This effect of estrous cycle variation seems to be strain dependent since this difference in MA sensibility was not observed in the C57BL/6J strain of mice (Yu and Liao, 2000b). These animal models faithfully reproduce the sex differences of PD in that endogenous estrogen levels appear to influence the vulnerability to toxins targeting the NSDA system and support a beneficial role of estrogens against NSDA neurodegeneration.
Effects of Estrogens and Progesterone in Female and Male Rodents
Experimental studies have been conducted in order to investigate the potential beneficial effect of exogenous sex steroid treatments against NSDA toxins. A summary of these studies showing steroids, dose, toxin, and DA markers investigated is listed in Tables 1– 4. These studies are separated by sex to highlight the observed sex differences and we refer the reader to the tables for details and a complete reference to relevant studies. Beneficial effects of 17β-estradiol and estradiol benzoate on DA concentrations against MPTP toxicity in female and male mice (intact or gonadectomized) are shown when physiological concentrations of this gonadal steroid are administered prior to the toxin (Tables 1 and 3; Dluzen et al., 2001b; D’Astous et al., 2004b; Liu et al., 2008). A protective effect of 17β-estradiol or estradiol benzoate against MA-induced DA loss was also obtained with a pre-treatment in female mice (Dluzen et al., 2002; D’Astous et al., 2005a). In contrast, 17β-estradiol failed to show any protective effect on DA content in gonadectomized male mice treated with MA (Anderson et al., 2005) and produced a severe acute toxicity to MA in intact male mice (Dluzen et al., 2002). Timing of treatment with 17β-estradiol or estradiol benzoate seems to be critical to reach a maximal beneficial effect, with at least 24 h required for the pre-treatment whereas shorter time intervals fail to achieve optimal protection (Gajjar et al., 2003). In support of this finding are data showing that treatment with a high dose of 17β-estradiol shortly prior to and after MPTP injection failed to protect striatal DA depletion whereas some effects were observed on DAT and tyrosine hydroxylase proteins levels (Ookubo et al., 2008). Moreover, in a study investigating several markers of dopaminergic function, the time required to obtain maximal protection against MA toxicity was observed with a 24-h pre-treatment of estradiol benzoate while pre-treatments of 30 min or 12 h showed incomplete and varying degrees of neuroprotection (Gajjar et al., 2003), suggesting different mechanisms of estradiol action (D’Astous et al., 2004a). Since estradiol is a neuromodulator of DA metabolism (Sanchez et al., 2010), investigations of the integrity of DA neurons using other DA markers to demonstrate the beneficial effect of estradiol are critical. The DAT and the vesicular monoamine transporter 2 (VMAT2) are both important modulators of DA neurotransmission (Sotnikova et al., 2006; Guillot and Miller, 2009) and both play a pivotal role in MPTP and MA toxicities (Gainetdinov et al., 1997, 1998; Fumagalli et al., 1998, 1999). A decrease in DAT and VMAT2 binding in DA neurons of the PD brain is associated with the loss of DA (Wilson et al., 1996). The protective effect of 17β-estradiol on DA content observed in MPTP and MA mice has also been shown on DAT and VMAT2 specific binding (Callier et al., 2001; D’Astous et al., 2004a, 2005a; Jourdain et al., 2005). Moreover, positive correlations exist between DA concentrations and DAT and VMAT2 levels, suggesting that binding to the transporters reflects the degree of degeneration or protection (Jourdain et al., 2005).
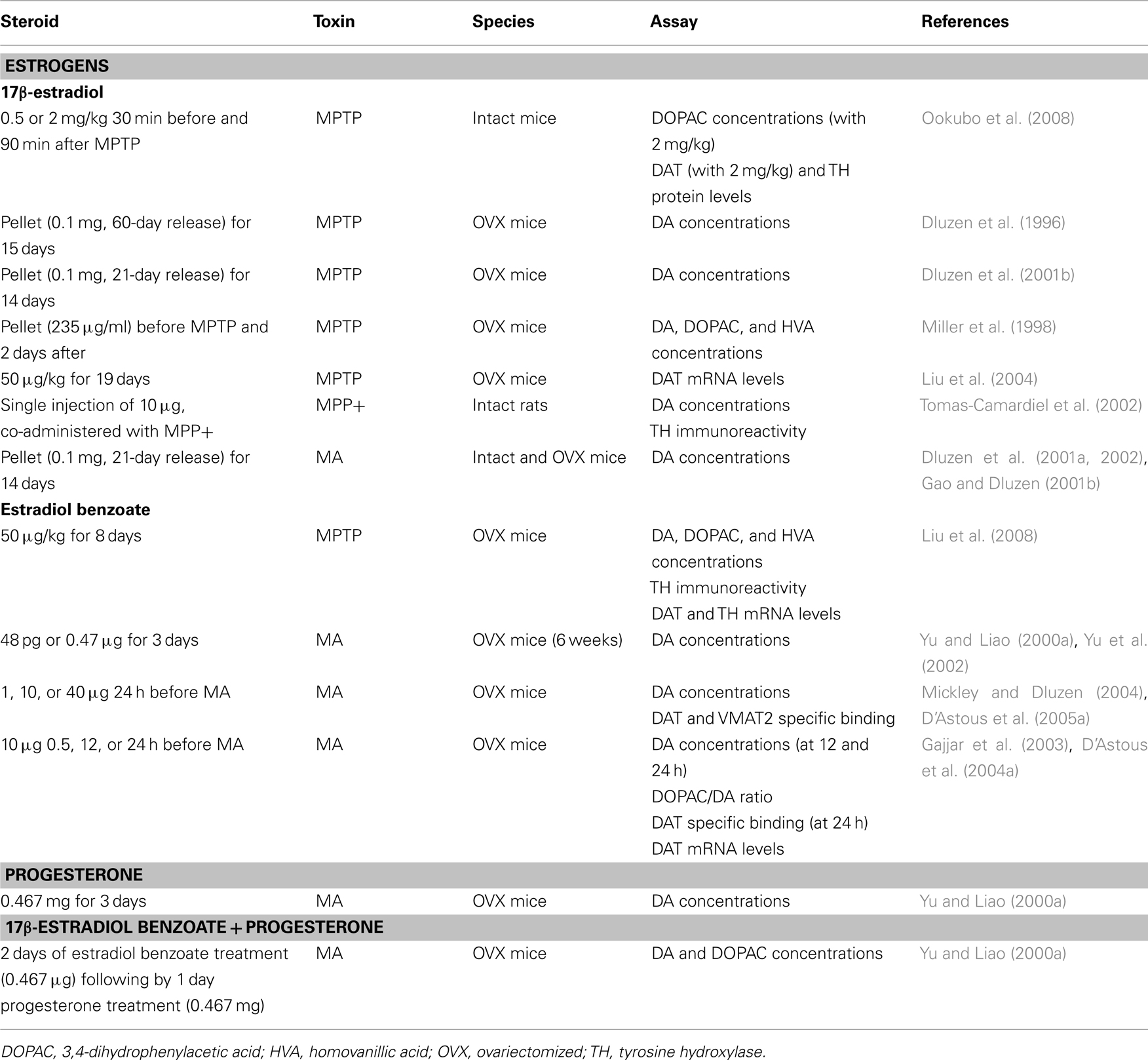
Table 1. In vivo studies and neuroprotection in female rodents: studies showing protection of DA markers.
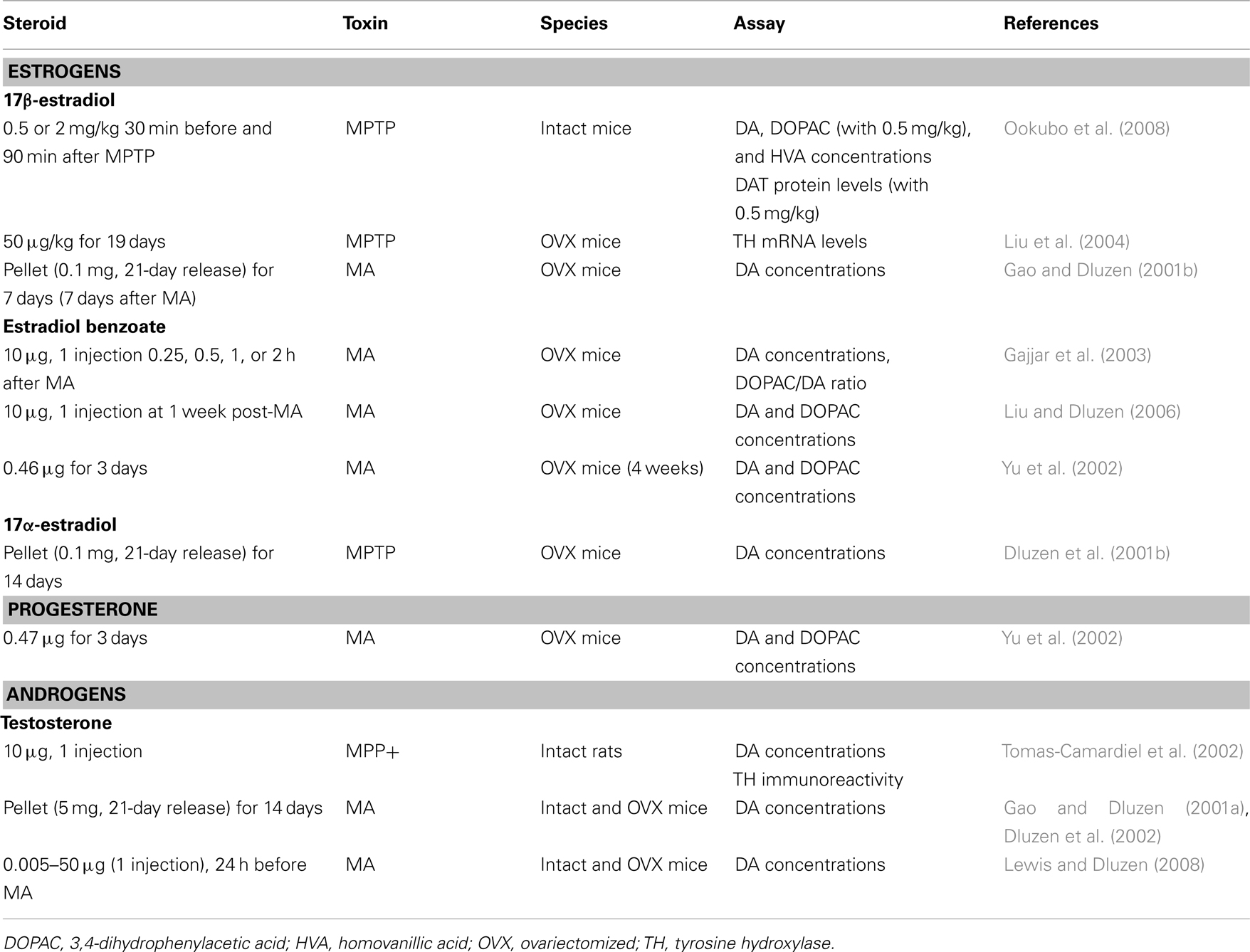
Table 2. In vivo studies and neuroprotection in female rodents: studies showing no protection of DA markers.
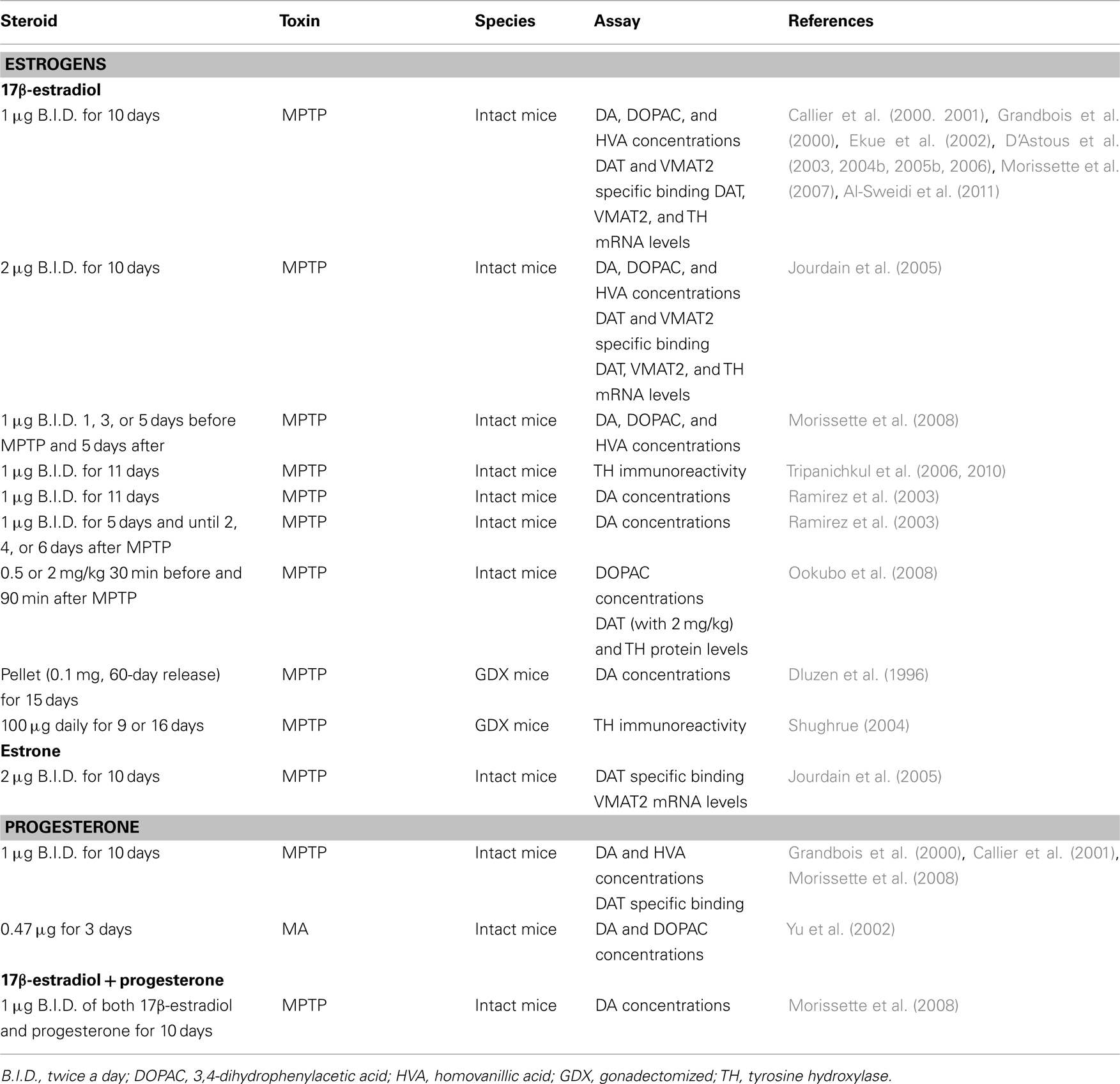
Table 3. In vivo studies and neuroprotection in male rodents: studies showing protection of DA markers.
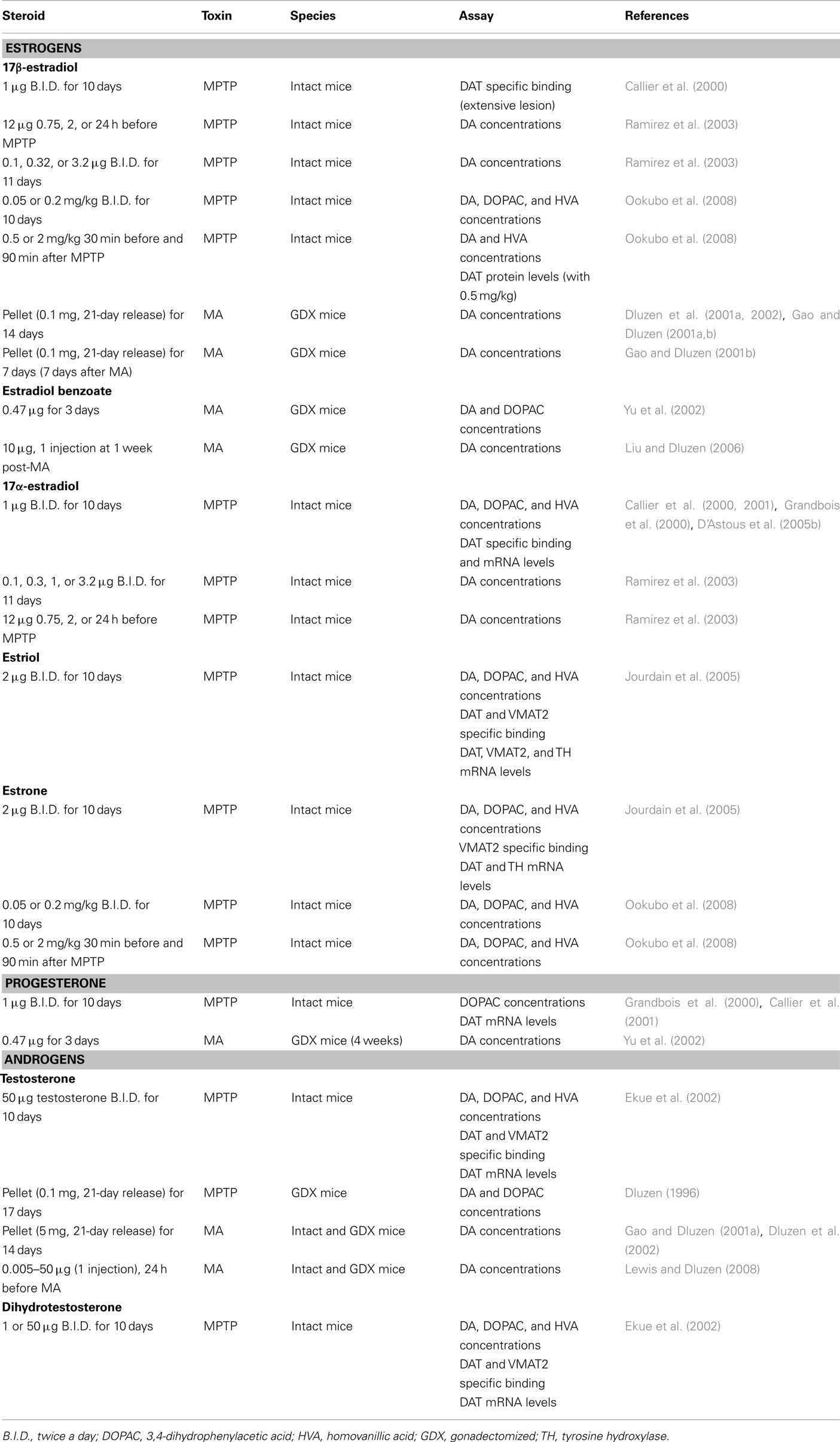
Table 4. In vivo studies and neuroprotection in male rodents: studies showing no protection of DA markers.
While it has received less attention than 17β-estradiol, the neuroprotective effect of the other major ovarian steroid progesterone has also been investigated in animal models of PD. Like 17β-estradiol, progesterone shows beneficial effects against MPTP and MA toxicities (Grandbois et al., 2000; Yu and Liao, 2000a; Callier et al., 2001; Yu et al., 2002; Morissette et al., 2008). In contrast to 17β-estradiol, progesterone is neuroprotective in male mice treated with MA (Yu et al., 2002). Sex differences in the dose of progesterone used to be effective as a neuroprotectant against MA are also reported, with male mice requiring lower doses than females (Yu et al., 2002). Interestingly, co-administration of 17β-estradiol and progesterone (Morissette et al., 2008), or administration of progesterone following 17β-estradiol treatment (Yu and Liao, 2000a) does not oppose the beneficial effect of 17β-estradiol, as has been observed with medroxyprogesterone acetate (Nilsen and Brinton, 2002).
17β-estradiol clearly shows a neuroprotective capacity when administered as a pre-treatment, that is, under conditions of a non-injured brain. Whether 17β-estradiol could retain this capacity within an impaired dopaminergic system has also been investigated. Estradiol treatment after a MA-induced lesion has been introduced lacks a protective capacity against MA toxicity in female mice (Gao and Dluzen, 2001b; Gajjar et al., 2003) and could even worsen the extent of observed damage (Liu and Dluzen, 2006). This is, in part, consistent with the healthy cell bias of estrogen’s effect, which proposes that if neurons are healthy at the time of estrogen treatment, their response to estrogen is beneficial for both survival and neurological functions whereas in the presence of impaired function, estrogen exposure over time exacerbates brain injury (Chen et al., 2006; Brinton et al., 2008). Accordingly, 17β-estradiol can prevent but does not seem to have the capacity of regeneration nor is it protective under conditions of an impaired system.
Effects of Androgens in Female and Male Rodents
Androgenic compounds have also received attention in order to investigate their effect in neuroprotective studies. Steroids could be synthesized in the brain, as supported by the presence and distribution of neurosteroidogenic enzymes (Do Rego et al., 2009). Thus, estradiol and testosterone could be formed in the brain. Aromatase, the enzyme converting testosterone to estradiol and androstenedione to estrone, and 5α-reductase, the enzyme converting testosterone into dihydrotestosterone, are present in the brain (Do Rego et al., 2009). While testosterone is considered an important precursor of estradiol biosynthesis and could also be 5α-reduced into dihydrotestosterone (important in testis and ovary), a direct effect of testosterone by itself is also possible as has been described in intracrine tissues (Luu-The and Labrie, 2010). As a result of such testosterone biotransformation it is oftentimes not certain whether any modulatory effects upon toxicity from this steroid result from testosterone, its metabolites and/or its possible conversion into estradiol. To investigate specifically the androgenic potential in neuroprotective activity, dihydrotestosterone, the most potent androgen, is a more appropriate compound since it is not aromatized.
In MPTP-treated male mice, testosterone treatment fails to show any protective effect (Dluzen, 1996; Ekue et al., 2002), suggesting that testosterone is not biotransformed into estradiol in the brain, more specifically in the basal ganglia, in adequate concentrations to reach protective levels. The lack of effect of testosterone is not sex-dependent since this steroid does not protect against MA toxicity in either female or male mice (Gao and Dluzen, 2001a; Lewis and Dluzen, 2008). Not only does testosterone lack a neuroprotective function but further exacerbates DA depletion in male mice receiving a chronic (Dluzen et al., 2002) or acute (Lewis and Dluzen, 2008) administration of testosterone. These findings of an exacerbation of MA-induced DA toxicity with testosterone are supported by data from the 6-hydroxydopamine rat model which shows that greater amounts of striatal DA depletion are obtained in intact versus gonadectomized male rats subjected to 6-hydroxydopamine (Murray et al., 2003; Gillies et al., 2004). Dihydrotestosterone also lacks any beneficial effect against loss of striatal DA concentrations in MPTP-lesioned male mice (Ekue et al., 2002), suggesting that stimulation of androgen receptors was not effective in inducing a protective effect. Indeed, these studies show that androgens do not play a protective role but may actually intensify toxicity in the NSDA pathway. Such effects may be relatively specific to the NSDA system as testosterone is reported to exert neuroprotective effects in others models (Bialek et al., 2004).
Comparison between Clinical and Animal Reports
There is a relatively good consistence between clinical and animal data concerning the neuroprotective activity of ovarian steroids. Longer exposure to ovarian steroids is associated with a reduce risk of PD in women as presented in Section “Influence of Endogenous and Exogenous Estrogens.” In animal models, pre-treatment with both 17β-estradiol and progesterone shows neuroprotective activity against MPTP and MA, while 17β-estradiol lacks neuroprotective effect when administered under condition of impaired NSDA system (see Effects of Estrogens and Progesterone in Female and Male Rodents).
While animal studies show that 17β-estradiol does not exert any neuroprotective effect when administered after the lesion, clinical reports suggest that estrogens can act as a neuromodulator of DA system on PD symptoms. Women with PD experience worsening of their symptoms during periods of low endogenous steroids exposure and estrogen therapy has been reported to improve PD symptom (see Influence of Endogenous and Exogenous Estrogens). To our knowledge no study performed in MPTP-treated rodents has investigated the effect of estrogen treatment on symptoms. In female ovariectomized hemiparkinsonian MPTP monkeys where clear PD symptoms are present we have shown that acutely 17β-estradiol and dehydroepiandrosterone (DHEA, a neurosteroid and precursor of 17β-estradiol) potentiate the motor response obtained with a low dose of levodopa the most common treatment for PD (Belanger et al., 2003, 2006). Moreover, after a long washout of the acute treatments, a chronic 17β-estradiol treatment in these hemiparkinsonian MPTP monkeys increased striatal DA and metabolites concentrations in the intact and lesioned side while the DAT was only increased in the intact side of these very extensively denervated monkeys (Morissette and Di Paolo, 2009). The prodopaminergic effects of estradiol within the NSDA system were observed on DA release and metabolism, DA receptor, DAT, tyrosine hydroxylase, and monoamine oxidase (Sanchez et al., 2010).
Mechanism of Action of Estradiol in Female and Male Rodents
Dopaminergic system
Sex differences in brain structure and function may affect the susceptibility to the toxins. Striatal DA concentration shows no difference between females and males whereas the number of tyrosine hydroxylase immunoreactive cells in the substantia nigra pars compacta were observed to be higher in male rodents, with a sex difference in the topographical distribution (McArthur et al., 2007a; Gillies and McArthur, 2010). In female rats, nigrostriatal DA neurotransmission seems to be more regulated by autoreceptor and transporter mechanisms (Walker et al., 2006). Moreover, although DAT affinity is observed to be the same in males and females, in female rodents superior DA uptake and vesicular storage is present (Morissette and Di Paolo, 1993b; Walker et al., 2000; Bhatt and Dluzen, 2005; Ji et al., 2007; Dluzen et al., 2008), suggesting a greater functional activity of DAT and VMAT2 in females. Further evidence demonstrating a sex difference in DAT function has been indicated from data showing that DA responses to MA infusion in the presence of the DAT inhibitor nomifensine were not abolished in female striatal tissue whereas the response was eliminated in males (Kunnathur et al., 2006). The DAT is considered a critical component of MPTP and MA toxicities since this transporter is the predominant site by which MA and MPP+ enter DA cells (Sotnikova et al., 2006). The importance of the DAT in MA and MPTP toxicities has been revealed in DAT knockout mice that show no striatal DA neurotoxicity when treated with MA or MPTP (Gainetdinov et al., 1997; Fumagalli et al., 1998), thus substantiating that decreased DAT activity is beneficial against toxins. This finding may seem inconsistent with the decreased susceptibility of female mice to the toxins and the greater efficiency of their DAT activity. In contrast to the effects resulting from a decreased number DAT, that is, a reduced potential for uptake of neurotoxins, a decreased number of VMAT2, has the potential for increasing toxicity responses to MA and MPTP (Gainetdinov et al., 1998; Fumagalli et al., 1999; Guillot et al., 2008). With fewer VMAT2, there would be increased amounts of MPP+ available to interact with mitochondrial complex 1, as less sequestering of MPP+ would occur (Guillot and Miller, 2009). Similarly, the high amount of extra- and intra-cellular DA release by MA favors the production of reactive species (Fleckenstein et al., 2007) which could be sequestered by the VMAT2. Thus, a more efficient DA uptake as well as sequestering of excessive DA and MPP+ into vesicular storage via the VMAT2, as reported in females, could decrease the degree of oxidative stress and favor a more efficient protection of DA terminals when exposed to toxins that utilize these transporters (Guillot and Miller, 2009).
Aromatase activity and mRNA expression show no sex difference in the mouse striatum (Kuppers and Beyer, 1998). However, a sex difference in the expression and activity of aromatase is reported in astrocytes, suggesting that astrocytes of females possess the potential to produce more estradiol than astrocytes of males (Liu et al., 2007). Moreover, aromatase expression in astrocytes is induced following lesion and this aromatase activity has been shown to be neuroprotective (Garcia-Segura, 2008). Male rats treated with an aromatase inhibitor show increased susceptibility to a dopaminergic toxin (McArthur et al., 2007b). In addition, increased vulnerability to MPTP has been reported in aromatase knockout female mice as compared to ovariectomized wild-type females, indicating the contribution of extra-gonadal 17β-estradiol synthesis in the neuroprotection of the NSDA pathway (Morale et al., 2008). The induction of aromatase expression, and its neuroprotective effect, following lesion seem contradictory with the increase toxicity in female mice when 17β-estradiol is given after MA and the lack of protective effect when 17β-estradiol is given shortly after toxins. It should be noted that increased aromatase expression seems to be localized close to the lesion site (Carswell et al., 2005), promoting local synthesis of estradiol, as compared to systemic administration of 17β-estradiol given after toxins. Furthermore, the “amount/degree” of the initial lesion seems to be critical since the capacity for estrogen to enhance the neurotoxicity response in the previously lesioned MA-treated mice was more prominent under conditions where less initial damage was present (Liu and Dluzen, 2006). In addition, the timing/duration of the initial lesion could also influence the effect of estrogen on neurotoxicity.
The finding that estradiol protects DAT specific binding in MPTP- and MA-treated mice could also be considered contradictory since increased susceptibility to toxins is associated with higher DAT levels. Inconsistencies in the literature have been reported concerning the effect of estradiol treatment on DAT. Our group reported that 17β-estradiol treatment in ovariectomized female rats left the affinity of [3H]GBR 12935 binding unchanged whereas DAT density increased (Morissette and Di Paolo, 1993a). A study using [3H]WIN35,428 has also reported unchanged affinity for DAT binding by 17β-estradiol treatment in the striatum of gonadectomized male rats (Meyers and Kritzer, 2009). In contrast, decreased DA uptake in ovariectomized female rats treated with 17β-estradiol has been reported using [3H]DA and a decrease of DAT density measured by [3H]BTCP binding (Attali et al., 1997). [3H]DA uptake from striatal synaptosomes of ovariectomized rats was also shown to be dose-dependently inhibited by 17β-estradiol (Disshon et al., 1998). While the issue regarding these effects of estrogen upon the DAT remains controversial, a more consistent result that emerges from studies of several laboratories is that 17β-estradiol treatment of ovariectomized rats restores DA uptake and DAT density to levels observed in intact female rats. Ovariectomized female rats treated with 17β-estradiol have equivalent DA uptake and DAT density as that of intact females not treated with 17β-estradiol (Attali et al., 1997; Le Saux and Di Paolo, 2006; McArthur et al., 2007b). Considering that females have a superior function of DAT and VMAT2 (Morissette and Di Paolo, 1993b; Walker et al., 2000; Bhatt and Dluzen, 2005; Ji et al., 2007; Dluzen et al., 2008) and are less susceptible to NSDA toxins than males (Miller et al., 1998; Bourque et al., 2011), the preservation of a integral and optimal DA system seems to be important in the neuroprotection process. In addition, DAT affinity and density were not modulated by 17β-estradiol treatment in male rodents (Jourdain et al., 2005; Meyers and Kritzer, 2009) but protection against MPTP is observed (Bourque et al., 2009). While it has been suggested that inhibition of the DAT by estradiol could be an important mechanism for neuroprotection in females (Disshon and Dluzen, 1999), the findings that 17β-estradiol can be effective as a neuroprotectant against MPTP in both males and females, in the apparent absence of any effects upon the DAT in males, suggests alternative or supplementary mechanisms for neuroprotection by this gonadal steroid hormone. As one possibility, there may exist an important interaction among 17β-estradiol, its binding with ERs and the DAT that contributes to this ability to display neuroprotection. Specifically, data resulting from an in vitro study have demonstrated that a physiological concentration of 17β-estradiol does not change the membrane and total DAT levels, whereas estrone and estriol cause removal of membrane DAT, with a reduction of total cellular DAT content also being observed with estriol (Alyea and Watson, 2009). Furthermore, 17β-estradiol and estrone, but not estriol, differently changed the subcellular localization of the ERs (Alyea and Watson, 2009). Despite the reduction in membrane DAT caused by estrone and estriol, estrone shows some weak protective activity against MPTP whereas estriol lacks a neuroprotective effect (Jourdain et al., 2005) and these two estrogens are weak agonist of ERs (Kuiper et al., 1997), supporting a close link between affinity for ER binding and a neuroprotective effect. Whereas DAT knockout mice have shown an important role for the DAT in MPTP and MA toxicities (Gainetdinov et al., 1997; Fumagalli et al., 1998), the effect of 17β-estradiol in preserving the affinity and the density of the DAT seems to be an important aspect of the neuroprotective effect. Furthermore, higher DAT density has been reported in females than males (Morissette and Di Paolo, 1993b) and females are less affected by toxins than males (Bourque et al., 2009). Clinical imaging studies in healthy participants have shown that striatal DAT binding is higher in women than men (Lavalaye et al., 2000; Staley et al., 2001) and a lower incidence and prevalence of PD is observed in women. Studies in healthy postmenopausal women reported that a 6-week period of estrogen therapy increases [99mTc]TRODAT-1 binding to DAT in the anterior putamen (Gardiner et al., 2004), that long-term use of estrogen therapy increases dopaminergic function (Craig et al., 2004), and estrogen therapy has been associated with a decreased risk of PD (Currie et al., 2004; Popat et al., 2005).
An intriguing sex specific effect of estradiol is observed in MA-treated male mice. While male rodents demonstrate a neuroprotective action of 17β-estradiol in response to MPTP (Bourque et al., 2009) a lack of any apparent beneficial effect from estrogen is seen in MA-treated mice (Dluzen et al., 2002). MPTP and MA mechanisms of action differ markedly, with MPTP affecting the mitochondrial complex 1 (Smeyne and Jackson-Lewis, 2005) and MA producing excessive amounts of DA release likely leading to reactive species production (Fleckenstein et al., 2007). The failure of 17β-estradiol to function as a neuroprotectant against MA in the male mouse is not readily obvious. A critical component of MA toxicity is body temperature, which influences the oxidation process (LaVoie and Hastings, 1999). MA itself produces hyperthermia and variations of body temperature have been reported to influence the extent of degeneration (LaVoie and Hastings, 1999). While female mice experience reductions of body temperature in response to estradiol treatment (Gao and Dluzen, 2001b), this effect was not present in male mice (Dluzen et al., 2002), where no change in body temperature was observed. The differential between female and male mice with regard estradiol’s effect on body temperature could be involved in the sex difference response to the toxin. Interestingly, MPTP or MPP+ also produce a brief initial period of hyperthermia in mice, but this is followed by a more prolonged period of hypothermia (Satoh et al., 1987). Moreover, mice maintained at 4°C show a greater accumulation of striatal MPP+ along with greater depletions of striatal DA as compared with mice maintained at 22°C (Moy et al., 1998). Essentially opposite results are obtained with MA, where greater striatal DA concentration depletions are obtained in mice maintained at 22°C (Moy et al., 1998) and hypothermia diminished MA-induced striatal DA toxicity in the rat (Bowyer et al., 1992). Accordingly, the inability for estradiol to decrease body temperature may represent a particularly critical variable with regard to moderating MA-, but not MPTP-, induced striatal DA toxicity in the male mouse.
A critical point to consider is whether developmental effects of steroids (organizational effects) influence the neuroprotective response against toxin observed in adults. Anderson et al. (2005) attempted to address this issue of organizational effects in the MA model. Briefly, female mice gonadectomized at 3–5 days of age and immediately treated with testosterone propionate (1.25 mg), that is, masculinized females, continued to show an estrogen neuroprotection response to MA when tested as adults. Male mice gonadectomized at 3–5 days of age and immediately treated with sesame oil, that is, feminized males, failed to show an estrogen neuroprotection response when tested as adults. Therefore, attempts to masculinize female mice or feminize male mice did not alter the sexually dimorphic effect of estrogen (i.e., neuroprotection in females and no effect in males) upon MA-induced neurotoxicity responses. These results suggest that the long-term and even organizational effects of steroids may not necessarily (or adversely) affect the responses obtained in adults.
Estrogens receptors
The presence of 17β-estradiol within the brain at the time of injury is a critical component of the neuroprotective effect of this steroid since post-treatment fails to protect DA neurons (Gajjar et al., 2003; Liu and Dluzen, 2006). The timing of 17β-estradiol treatment also seems to influence the extent of this response since different durations of treatment produce varying degrees of neuroprotection (Gajjar et al., 2003), with increased effectiveness being associated with a longer treatment intervals (24 > 12 > 0.5 h), suggesting that different mechanisms of 17β-estradiol are operating. Estradiol produces its actions by genomic and non-genomic effects. Genomic mechanisms involve gene transcription mediated by activation of nuclear receptors, ERα and ERβ, and require periods of hours to days to exert their effects (Vasudevan and Pfaff, 2008). Non-genomic actions are defined by rapid effects (within minutes even seconds) of 17β-estradiol initiated by interaction with membrane ER and/or G protein-coupled estrogen receptor 1 (GPER1), leading to activation of signaling pathways (Vasudevan and Pfaff, 2008). Genomic and non-genomic actions of 17β-estradiol are known to act together to potentiate transcriptional activity (Vasudevan and Pfaff, 2008). 17β-estradiol can activate Akt and extracellular signal-regulated kinase (ERK1/2) signaling; both of which have been implicated in 17β-estradiol neuroprotective effects (Bryant et al., 2006; Raz et al., 2008). 17β-estradiol action can promote the up-regulation of neurotrophic factors such as brain-derived neurotrophic factor, the anti-apoptotic molecule Bcl-2 and/or inhibition of pro-apoptotic proteins such as BAD and Bax (Kipp et al., 2006; Brann et al., 2007). Interactions with growth factors, such as insulin-like growth factor 1, can also contribute to the protective effects of estradiol (Garcia-Segura et al., 2010) since these actions promote survival and have been implicated in the neuroprotective effect of 17β-estradiol.
Non-genomic actions of 17β-estradiol show sex differences in the activation of intracellular mechanisms in the mouse brain (Abraham and Herbison, 2005). Moreover, a sex-related difference in ERK1/2 activation by 17β-estradiol is reported in male and female rat astrocytes (Zhang et al., 2002b). Differential distributions of ERs are reported between female and male rodents. ERα has been detected in the striatum of both female and male rodents (Merchenthaler et al., 2004; Shughrue, 2004; Rodriguez-Navarro et al., 2008; Schultz et al., 2009) whereas the levels of ERα, as quantified by Western blot, are higher in the striatum of female mice (Rodriguez-Navarro et al., 2008). Furthermore, ERα seems to be primarily associated with the membrane fraction rather than the nuclear part when extracted from homogenized striatal tissue of female rats (Schultz et al., 2009). ERβ is not found in the striatum of male mice but both presence and absence have been reported in females (Mitra et al., 2003; Merchenthaler et al., 2004; Shughrue, 2004). Both ERα and ERβ are present in female mice substantia nigra pars compacta and striatum (Mitra et al., 2003) but are absent in male mice (Shughrue, 2004) whereas their presence has been detected in male rats (Zhang et al., 2002a). In female mice, the immunoreactivity of ERβ in substantia nigra pars compacta seems to be more prominent than ERα whereas the opposite seems true of the striatum (Mitra et al., 2003). GPER1 has been detected in the striatum and substantia nigra pars compacta with a similar pattern of distribution between female and male rodents (Brailoiu et al., 2007; Hazell et al., 2009; Bourque et al., 2011). The literature seems to be more consistent concerning ERα showing only species difference between male rats and mice in substantia nigra. More discrepancies appear for the distribution of ERβ. Differences in species, the antibody used and cellular localization (nuclear; Mitra et al., 2003; Merchenthaler et al., 2004 versus extranuclear; Mitra et al., 2003) could provide some explanation for the divergent distribution of the ERβ described. Thus, when referring only to studies with mice, a different distribution seems to be present between females and males, as ERs are present in female mice substantia nigra pars compacta but absent in males. ERα has been detected in the striatum of both female and male mice, whereas the levels of this receptor seem higher in females. ERβ is not found in the striatum of male mice but both presence and absence have been reported in females. Colocalization of ERβ and tyrosine hydroxylase in substantia nigra pars compacta has been observed in both female and male rats (Creutz and Kritzer, 2004; Quesada et al., 2007). Studies combining double-label immunocytochemistry for ERβ and tyrosine hydroxylase positive substantia nigra pars compacta neurons with retrograde tract tracing revealed a defined topographical organization strongly favoring projection to the ventral striatum, whereas few ERβ and tyrosine hydroxylase positive substantia nigra pars compacta neurons were found to project to the dorsal striatum (Creutz and Kritzer, 2004). No androgen receptor and tyrosine hydroxylase positive substantia nigra pars compacta neurons were found to project to the striatum (Creutz and Kritzer, 2004). It seems reasonable to postulate that the sex differences observed in the distribution and levels of ERs could influence the dopaminergic system and mechanisms of 17β-estradiol action. It remains to be investigated whether estradiol activates the same signaling molecules in females and males and/or if a sexual dimorphism in downstream signaling proteins is linked with the ERs that are present in the dopaminergic system. Neuroprotective effects of 17β-estradiol against toxins can involve different signaling pathways between females and males.
In vitro studies have shown that ERs subtypes (including GPER1) have different effects in 17β-estradiol-mediated DA efflux (Alyea et al., 2008) and that ERα and ERβ, but not GPER1, are associated with the plasma membrane DAT (Alyea and Watson, 2009). Moreover, modulation of tyrosine hydroxylase transcription by 17β-estradiol is regulated in opposite directions depending on the ER subtype (Maharjan et al., 2005). Using ERα and ERβ knockout male mice, our group and others have investigated the role of each ER on NSDA markers in the neuroprotective effect of 17β-estradiol against MPTP toxicity (Morissette et al., 2007; Al-Sweidi et al., 2011). While ERα and ERβ knockout mice show normal striatal DA concentrations, DAT specific binding was increased in ERα knockout mice and normal VMAT2 specific binding was measured (Morissette et al., 2007; Al-Sweidi et al., 2011). By contrast, ERβ knockout mice display lower DA turnover as well as a reduction in striatal DAT and VMAT2 specific binding (Morissette et al., 2007; Al-Sweidi et al., 2011). Female ERα knockout mice exhibit higher D1 DA receptor expression levels and reduced expression of tyrosine hydroxylase and brain-derived neurotrophic factor in the midbrain of both female and male ERα knockout mice (Kuppers et al., 2008). Whereas both ERα and ERβ male knockout mice display normal serum 17β-estradiol levels, higher levels of testosterone, dihydrotestosterone, and 3β-diol were measured in ERα male knockout mice (Al-Sweidi et al., 2011). The highest susceptibility to MPTP was observed in ERα male knockout mice and the levels of testosterone and 3β-diol were inversely correlated with the loss of DA concentration (Al-Sweidi et al., 2011). The significance of these findings remains to be elucidated, however it is interesting to note that not only does testosterone fail to induce a neuroprotective effect in MPTP and MA-treated mice (Dluzen, 1996; Ekue et al., 2002; Lewis and Dluzen, 2008) but may exacerbate NSDA neurotoxicity responses as described below in Section “Mechanism of Action of Androgens in Female and Male Rodents.” Exogenous 17β-estradiol does not protect ERα or ERβ male knockout mice from MPTP toxicity, showing that both ERs are necessary for neuroprotection (Morissette et al., 2007; Al-Sweidi et al., 2011). Moreover, the levels of ERs were not modulated by MPTP lesion (Shughrue, 2004). In contrast, we have recently reported increased GPER1 levels in male mice with a moderate MA-induced lesion, while the levels of this receptor remains unchanged in MA-treated female mice (Bourque et al., 2011). Thus, the sex difference in ERs distribution as well as the different roles for each of the ERs in the DA system could be associated with the sex differences present in the susceptibility to toxin and also influence their responses to 17β-estradiol.
Mechanism of Action of Androgens in Female and Male Rodents
In contrast to that of estradiol, there exists relatively little information on the mechanisms of testosterone action within the NSDA system as related to striatal DA toxicity. To a large extent this disinterest stems from the apparent absence of any neuroprotectant effects of this gonadal steroid. However, with the advent of data suggesting that this male gonadal steroid may contribute to an aggravation of toxins that target the NSDA system (Gao and Dluzen, 2001a; Dluzen et al., 2002; Murray et al., 2003; Gillies et al., 2004; Lewis and Dluzen, 2008), it might be worthwhile to re-consider these testosterone effects and mechanisms.
With regard to the NSDA system, treatment of male rats with the anabolic-androgen steroid, nandrolone decanoate, leads to decreases in D1-like receptor labeling and an increase in D2-like binding sites within the caudate putamen (Kindlundh et al., 2001), which creates a type of hypodopaminergic condition. Moreover, a reduction in locomotor behavior and rearing are also observed in these rats treated with nandrolone decanoate (Johansson et al., 2000). Further support for this attenuation in striatal dopaminergic function by testosterone is provided from data showing that spontaneous (Dluzen and Ramirez, 1989) and amphetamine-stimulated locomotor and stereotyped behaviors (Menniti and Baum, 1981; Savageau and Beatty, 1981; Beatty et al., 1982; Dluzen et al., 1986) are decreased in the presence of testosterone. Moreover, basal (Dluzen and Ramirez, 1989) and stimulated (Hernandez et al., 1994; Shemisa et al., 2006) striatal DA release are decreased in testosterone treated rodents. Collating these parameters leads to the conclusion that the presence of testosterone is associated with a generalized reduction in NSDA activity, like that seen in PD.
The exact mechanisms involved in producing this testosterone-dependent reduction in NSDA function are not know, but some tangential data are available that can provide some perspective. Testosterone treatment of neuroblastoma cells induces apoptosis through activation of a Ca2+ signaling pathway, an effect that cannot be attributable to conversion into estrogens (Estrada et al., 2006). With the use of N27 cells, which might represent a more relevant model for testing toxicity on dopaminergic neurons, it was also demonstrated that testosterone contributes to an apoptotic cascade, impairing mitochondrial function and increasing oxidative stress to produce a caspase-3-dependent cleavage which would activate protein kinase Cδ (Cunningham et al., 2009). Additional work with this model revealed that this testosterone effect involved an intracellular androgen receptor and was not attributable to conversion into estradiol. The fact that, at least in the rat, androgen receptors are mainly expressed in the substantia nigra pars compacta versus the substantia nigra pars reticulata (Kritzer, 1997), combined with data from the cortex of the mouse that androgen receptors show increased phosphorylation levels to testosterone in aged males (Thakur et al., 2000), provides particularly relevant implications regarding the capacity for testosterone to exert age-related, adverse consequences within the NSDA system as related to PD. This testosterone-dependent increase in oxidative stress and free radical production may, in part, result from modulation of VMAT2 function by this gonadal steroid. Significantly greater amounts of reserpine-evoked DA release are obtained from the striatum of orchidectomized mice treated with testosterone versus those not receiving testosterone (Shemisa et al., 2006). Such results suggest that testosterone may be working like and/or synergistically with reserpine to inhibit VMAT2 function, thereby producing excessive DA levels available for metabolism to free radicals. Testosterone produced a non-significant decrease in vesicular DA uptake in cocaine-treated orchidectomized rats (Chen et al., 2003), and the VMAT2 of males is more sensitive to the toxic effects of MA, as substantia nigra VMAT2 mRNA is significantly decreased in male, but not female, mice receiving either low (20 mg/kg) or high (40 mg/kg) doses of this NSDA toxin (Bourque et al., 2011).
Taken together, the capacity for testosterone to induce a generalized hypodopaminergic state combined with an age-related enhanced potential for oxidative stress in critical NSDA sites, suggests a role and mechanism for this gonadal steroid in PD. In this way, the sex difference in the incidence of PD may involve a combination of neuroprotective effects of estrogens within women and neurodestructive effects of testosterone in men. While much work remains to be performed on this topic, these bidirectional sex and hormonal responses to NSDA toxins may comprise a rewarding direction of investigation to understand the pathology of conditions like PD.
Conclusion
Clinical and epidemiological reports on PD as well as animal models show a sex difference in neurodegeneration of the NSDA system. Most of the experimental studies focused on a 17β-estradiol neuroprotective effect, showing that low doses, but not high doses, are effective to protect the NSDA pathway against toxins. Furthermore, increased effectiveness of 17β-estradiol is associated with a longer treatment interval. While having received less attention, progesterone shows interesting neuroprotective actions in both male and female mice and does not oppose the effect of 17β-estradiol on the NSDA system when these two steroids were co-administered. Androgens lack neuroprotective properties in NSDA pathway and may even worsen the extent of the lesion. Genomic and non-genomic actions of 17β-estradiol are implicated in neuroprotection of NSDA system and the presence of both ERα and ERβ are critical for a beneficial effect. The contribution of GPER1 in the neuroprotective effect of 17β-estradiol in the NSDA pathway remains to be investigated but recent in vitro (Gingerich et al., 2010) and in vivo (Lebesgue et al., 2010) studies have reported a neuroprotective action of GPER1 activation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by a grant from the Canadian Institutes of Health Research (CIHR) to Thérèse Di Paolo and a NEOUCOM Research Incentive Grant to Dean E. Dluzen. Mélanie Bourque received a studentship from the Fonds de la Recherche en Santé du Québec (FRSQ).
References
Abraham, I. M., and Herbison, A. E. (2005). Major sex differences in non-genomic estrogen actions on intracellular signaling in mouse brain in vivo. Neuroscience 131, 945–951.
Al-Sweidi, S., Morissette, M., Bourque, M., and Di Paolo, T. (2011). Estrogen receptors and gonadal steroids in vulnerability and protection of dopamine neurons in a mouse model of Parkinson’s disease. Neuropharmacology 61, 583–591.
Alyea, R. A., Laurence, S. E., Kim, S. H., Katzenellenbogen, B. S., Katzenellenbogen, J. A., and Watson, C. S. (2008). The roles of membrane estrogen receptor subtypes in modulating dopamine transporters in PC-12 cells. J. Neurochem. 106, 1525–1533.
Alyea, R. A., and Watson, C. S. (2009). Nongenomic mechanisms of physiological estrogen-mediated dopamine efflux. BMC Neurosci. 10, 59. doi: 10.1186/1471-2202-10-59
Anderson, L. I., Leipheimer, R. E., and Dluzen, D. E. (2005). Effects of neonatal and prepubertal hormonal manipulations upon estrogen neuroprotection of the nigrostriatal dopaminergic system within female and male mice. Neuroscience 130, 369–382.
Ascherio, A., Chen, H., Schwarzschild, M. A., Zhang, S. M., Colditz, G. A., and Speizer, F. E. (2003). Caffeine, postmenopausal estrogen, and risk of Parkinson’s disease. Neurology 60, 790–795.
Attali, G., Weizman, A., Gil-Ad, I., and Rehavi, M. (1997). Opposite modulatory effects of ovarian hormones on rat brain dopamine and serotonin transporters. Brain Res. 756, 153–159.
Baba, Y., Putzke, J. D., Whaley, N. R., Wszolek, Z. K., and Uitti, R. J. (2005). Gender and the Parkinson’s disease phenotype. J. Neurol. 252, 1201–1205.
Beatty, W. W., Dodge, A. M., and Traylor, K. L. (1982). Stereotyped behavior elicited by amphetamine in the rat: influences of the testes. Pharmacol. Biochem. Behav. 16, 565–568.
Belanger, N., Gregoire, L., Bedard, P., and Di Paolo, T. (2003). Estradiol and dehydroepiandrosterone potentiate levodopa-induced locomotor activity in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine monkeys. Endocrine 21, 97–101.
Belanger, N., Gregoire, L., Bedard, P. J., and Di Paolo, T. (2006). DHEA improves symptomatic treatment of moderately and severely impaired MPTP monkeys. Neurobiol. Aging 27, 1684–1693.
Benedetti, M. D., Maraganore, D. M., Bower, J. H., Mcdonnell, S. K., Peterson, B. J., Ahlskog, J. E., Schaid, D. J., and Rocca, W. A. (2001). Hysterectomy, menopause, and estrogen use preceding Parkinson’s disease: an exploratory case-control study. Mov. Disord. 16, 830–837.
Bhatt, S. D., and Dluzen, D. E. (2005). Dopamine transporter function differences between male and female CD-1 mice. Brain Res. 1035, 188–195.
Bialek, M., Zaremba, P., Borowicz, K. K., and Czuczwar, S. J. (2004). Neuroprotective role of testosterone in the nervous system. Pol. J. Pharmacol. 56, 509–518.
Blanchet, P. J., Fang, J., Hyland, K., Arnold, L. A., Mouradian, M. M., and Chase, T. N. (1999). Short-term effects of high-dose 17beta-estradiol in postmenopausal PD patients: a crossover study. Neurology 53, 91–95.
Bourque, M., Dluzen, D. E., and Di Paolo, T. (2009). Neuroprotective actions of sex steroids in Parkinson’s disease. Front. Neuroendocrinol. 30, 142–157.
Bourque, M., Liu, B., Dluzen, D. E., and Di Paolo, T. (2011). Sex differences in methamphetamine toxicity in mice: effect on brain dopamine signaling pathways. Psychoneuroendocrinology 36, 955–969.
Bowyer, J. F., Tank, A. W., Newport, G. D., Slikker, W. Jr., Ali, S. F., and Holson, R. R. (1992). The influence of environmental temperature on the transient effects of methamphetamine on dopamine levels and dopamine release in rat striatum. J. Pharmacol. Exp. Ther. 260, 817–824.
Brailoiu, E., Dun, S. L., Brailoiu, G. C., Mizuo, K., Sklar, L. A., Oprea, T. I., Prossnitz, E. R., and Dun, N. J. (2007). Distribution and characterization of estrogen receptor G protein-coupled receptor 30 in the rat central nervous system. J. Endocrinol. 193, 311–321.
Brann, D. W., Dhandapani, K., Wakade, C., Mahesh, V. B., and Khan, M. M. (2007). Neurotrophic and neuroprotective actions of estrogen: basic mechanisms and clinical implications. Steroids 72, 381–405.
Brinton, R. D., Thompson, R. F., Foy, M. R., Baudry, M., Wang, J., Finch, C. E., Morgan, T. E., Pike, C. J., Mack, W. J., Stanczyk, F. Z., and Nilsen, J. (2008). Progesterone receptors: form and function in brain. Front. Neuroendocrinol. 29, 313–339.
Bryant, D. N., Sheldahl, L. C., Marriott, L. K., Shapiro, R. A., and Dorsa, D. M. (2006). Multiple pathways transmit neuroprotective effects of gonadal steroids. Endocrine 29, 199–207.
Callier, S., Morissette, M., Grandbois, M., and Di Paolo, T. (2000). Stereospecific prevention by 17beta-estradiol of MPTP-induced dopamine depletion in mice. Synapse 37, 245–251.
Callier, S., Morissette, M., Grandbois, M., Pelaprat, D., and Di Paolo, T. (2001). Neuroprotective properties of 17beta-estradiol, progesterone, and raloxifene in MPTP C57Bl/6 mice. Synapse 41, 131–138.
Carswell, H. V., Dominiczak, A. F., Garcia-Segura, L. M., Harada, N., Hutchison, J. B., and Macrae, I. M. (2005). Brain aromatase expression after experimental stroke: topography and time course. J. Steroid Biochem. Mol. Biol. 96, 89–91.
Chen, R., Osterhaus, G., Mckerchar, T., and Fowler, S. C. (2003). The role of exogenous testosterone in cocaine-induced behavioral sensitization and plasmalemmal or vesicular dopamine uptake in castrated rats. Neurosci. Lett. 351, 161–164.
Chen, S., Nilsen, J., and Brinton, R. D. (2006). Dose and temporal pattern of estrogen exposure determines neuroprotective outcome in hippocampal neurons: therapeutic implications. Endocrinology 147, 5303–5313.
Craig, M. C., Cutter, W. J., Wickham, H., Van Amelsvoort, T. A., Rymer, J., Whitehead, M., and Murphy, D. G. (2004). Effect of long-term estrogen therapy on dopaminergic responsivity in post-menopausal women–a preliminary study. Psychoneuroendocrinology 29, 1309–1316.
Creutz, L. M., and Kritzer, M. F. (2004). Mesostriatal and mesolimbic projections of midbrain neurons immunoreactive for estrogen receptor beta or androgen receptors in rats. J. Comp. Neurol. 476, 348–362.
Cunningham, R. L., Giuffrida, A., and Roberts, J. L. (2009). Androgens induce dopaminergic neurotoxicity via caspase-3-dependent activation of protein kinase Cdelta. Endocrinology 150, 5539–5548.
Currie, L. J., Harrison, M. B., Trugman, J. M., Bennett, J. P., and Wooten, G. F. (2004). Postmenopausal estrogen use affects risk for Parkinson disease. Arch. Neurol. 61, 886–888.
D’Astous, M., Gajjar, T. M., Dluzen, D. E., and Di Paolo, T. (2004a). Dopamine transporter as a marker of neuroprotection in methamphetamine-lesioned mice treated acutely with estradiol. Neuroendocrinology 79, 296–304.
D’Astous, M., Morissette, M., and Di Paolo, T. (2004b). Effect of estrogen receptor agonists treatment in MPTP mice: evidence of neuroprotection by an ER alpha agonist. Neuropharmacology 47, 1180–1188.
D’Astous, M., Mendez, P., Morissette, M., Garcia-Segura, L. M., and Di Paolo, T. (2006). Implication of the phosphatidylinositol-3 kinase/protein kinase B signaling pathway in the neuroprotective effect of estradiol in the striatum of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mice. Mol. Pharmacol. 69, 1492–1498.
D’Astous, M., Mickley, K. R., Dluzen, D. E., and Di Paolo, T. (2005a). Differential protective properties of estradiol and tamoxifen against methamphetamine-induced nigrostriatal dopaminergic toxicity in mice. Neuroendocrinology 82, 111–120.
D’Astous, M., Morissette, M., Callier, S., and Di Paolo, T. (2005b). Regulation of striatal preproenkephalin mRNA levels in MPTP-lesioned mice treated with estradiol. J. Neurosci. Res. 80, 138–144.
D’Astous, M., Morissette, M., Tanguay, B., Callier, S., and Di Paolo, T. (2003). Dehydroepiandrosterone (DHEA) such as 17beta-estradiol prevents MPTP-induced dopamine depletion in mice. Synapse 47, 10–14.
Dauer, W., and Przedborski, S. (2003). Parkinson’s disease: mechanisms and models. Neuron 39, 889–909.
de Rijk, M. C., Breteler, M. M., Graveland, G. A., Ott, A., Grobbee, D. E., Van Der Meche, F. G., and Hofman, A. (1995). Prevalence of Parkinson’s disease in the elderly: the Rotterdam Study. Neurology 45, 2143–2146.
di Michele, F., Longone, P., Romeo, E., Lucchetti, S., Brusa, L., Pierantozzi, M., Bassi, A., Bernardi, G., and Stanzione, P. (2003). Decreased plasma and cerebrospinal fluid content of neuroactive steroids in Parkinson’s disease. Neurol. Sci. 24, 172–173.
Disshon, K. A., Boja, J. W., and Dluzen, D. E. (1998). Inhibition of striatal dopamine transporter activity by 17beta-estradiol. Eur. J. Pharmacol. 345, 207–211.
Disshon, K. A., and Dluzen, D. E. (1999). Use of in vitro superfusion to assess the dynamics of striatal dopamine clearance: influence of estrogen. Brain Res. 842, 399–407.
Dluzen, D. E. (1996). Effects of testosterone upon MPTP-induced neurotoxicity of the nigrostriatal dopaminergic system of C57/B1 mice. Brain Res. 715, 113–118.
Dluzen, D. E., Anderson, L. I., and Pilati, C. F. (2002). Methamphetamine-gonadal steroid hormonal interactions: effects upon acute toxicity and striatal dopamine concentrations. Neurotoxicol. Teratol. 24, 267–273.
Dluzen, D. E., Bhatt, S., and McDermott, J. L. (2008). Differences in reserpine-induced striatal dopamine output and content between female and male mice: implications for sex differences in vesicular monoamine transporter 2 function. Neuroscience 154, 1488–1496.
Dluzen, D. E., Green, M. A., and Ramirez, V. D. (1986). The effect of hormonal condition on dose-dependent amphetamine-stimulated behaviors in the male rat. Horm. Behav. 20, 1–6.
Dluzen, D. E., McDermott, J. L., and Anderson, L. I. (2001a). Tamoxifen diminishes methamphetamine-induced striatal dopamine depletion in intact female and male mice. J. Neuroendocrinol. 13, 618–624.
Dluzen, D. E., McDermott, J. L., and Anderson, L. I. (2001b). Tamoxifen eliminates estrogen’s neuroprotective effect upon MPTP-induced neurotoxicity of the nigrostriatal dopaminergic system. Neurotox. Res. 3, 291–300.
Dluzen, D. E., McDermott, J. L., and Liu, B. (1996). Estrogen as a neuroprotectant against MPTP-induced neurotoxicity in C57/B1 mice. Neurotoxicol. Teratol. 18, 603–606.
Dluzen, D. E., and Ramirez, V. D. (1989). Effects of orchidectomy on nigro-striatal dopaminergic function: behavioral and physiological evidence. J. Neuroendocrinol. 1, 285–290.
Do Rego, J. L., Seong, J. Y., Burel, D., Leprince, J., Luu-the, V., Tsutsui, K., Tonon, M. C., Pelletier, G., and Vaudry, H. (2009). Neurosteroid biosynthesis: enzymatic pathways and neuroendocrine regulation by neurotransmitters and neuropeptides. Front. Neuroendocrinol. 30, 259–301.
Ekue, A., Boulanger, J. F., Morissette, M., and Di Paolo, T. (2002). Lack of effect of testosterone and dihydrotestosterone compared to 17beta-oestradiol in 1-methyl-4-phenyl-1,2,3,6, tetrahydropyridine-mice. J. Neuro-endocrinol. 14, 731–736.
Estrada, M., Varshney, A., and Ehrlich, B. E. (2006). Elevated testosterone induces apoptosis in neuronal cells. J. Biol. Chem. 281, 25492–25501.
Fleckenstein, A. E., Volz, T. J., Riddle, E. L., Gibb, J. W., and Hanson, G. R. (2007). New insights into the mechanism of action of amphetamines. Annu. Rev. Pharmacol. Toxicol. 47, 681–698.
Fumagalli, F., Gainetdinov, R. R., Valenzano, K. J., and Caron, M. G. (1998). Role of dopamine transporter in methamphetamine-induced neurotoxicity: evidence from mice lacking the transporter. J. Neurosci. 18, 4861–4869.
Fumagalli, F., Gainetdinov, R. R., Wang, Y. M., Valenzano, K. J., Miller, G. W., and Caron, M. G. (1999). Increased methamphetamine neurotoxicity in heterozygous vesicular monoamine transporter 2 knock-out mice. J. Neurosci. 19, 2424–2431.
Gainetdinov, R. R., Fumagalli, F., Jones, S. R., and Caron, M. G. (1997). Dopamine transporter is required for in vivo MPTP neurotoxicity: evidence from mice lacking the transporter. J. Neurochem. 69, 1322–1325.
Gainetdinov, R. R., Fumagalli, F., Wang, Y. M., Jones, S. R., Levey, A. I., Miller, G. W., and Caron, M. G. (1998). Increased MPTP neurotoxicity in vesicular monoamine transporter 2 heterozygote knockout mice. J. Neurochem. 70, 1973–1978.
Gajjar, T. M., Anderson, L. I., and Dluzen, D. E. (2003). Acute effects of estrogen upon methamphetamine induced neurotoxicity of the nigrostriatal dopaminergic system. J. Neural Transm. 110, 1215–1224.
Gao, X., and Dluzen, D. E. (2001a). The effect of testosterone upon methamphetamine neurotoxicity of the nigrostriatal dopaminergic system. Brain Res. 892, 63–69.
Gao, X., and Dluzen, D. E. (2001b). Tamoxifen abolishes estrogen’s neuroprotective effect upon methamphetamine neurotoxicity of the nigrostriatal dopaminergic system. Neuroscience 103, 385–394.
Garcia-Segura, L. M. (2008). Aromatase in the brain: not just for reproduction anymore. J. Neuroendocrinol. 20, 705–712.
Garcia-Segura, L. M., Arevalo, M. A., and Azcoitia, I. (2010). Interactions of estradiol and insulin-like growth factor-I signalling in the nervous system: new advances. Prog. Brain Res. 181, 251–272.
Gardiner, S. A., Morrison, M. F., Mozley, P. D., Mozley, L. H., Brensinger, C., Bilker, W., Newberg, A., and Battistini, M. (2004). Pilot study on the effect of estrogen replacement therapy on brain dopamine transporter availability in healthy, postmenopausal women. Am. J. Geriatr. Psychiatry 12, 621–630.
Gillies, G. E., and McArthur, S. (2010). Estrogen actions in the brain and the basis for differential action in men and women: a case for sex-specific medicines. Pharmacol. Rev. 62, 155–198.
Gillies, G. E., Murray, H. E., Dexter, D., and McArthur, S. (2004). Sex dimorphisms in the neuroprotective effects of estrogen in an animal model of Parkinson’s disease. Pharmacol. Biochem. Behav. 78, 513–522.
Gingerich, S., Kim, G. L., Chalmers, J. A., Koletar, M. M., Wang, X., Wang, Y., and Belsham, D. D. (2010). Estrogen receptor alpha and G-protein coupled receptor 30 mediate the neuroprotective effects of 17beta-estradiol in novel murine hippocampal cell models. Neuroscience 170, 54–66.
Grandbois, M., Morissette, M., Callier, S., and Di Paolo, T. (2000). Ovarian steroids and raloxifene prevent MPTP-induced dopamine depletion in mice. Neuroreport 11, 343–346.
Granieri, E., Carreras, M., Casetta, I., Govoni, V., Tola, M. R., Paolino, E., Monetti, V. C., and De Bastiani, P. (1991). Parkinson’s disease in Ferrara, Italy, 1967 through 1987. Arch. Neurol. 48, 854–857.
Growdon, J. H., Kieburtz, K., McDermott, M. P., Panisset, M., and Friedman, J. H. (1998). Levodopa improves motor function without impairing cognition in mild non-demented Parkinson’s disease patients. Parkinson Study Group. Neurology 50, 1327–1331.
Guillot, T. S., and Miller, G. W. (2009). Protective actions of the vesicular monoamine transporter 2 (VMAT2) in monoaminergic neurons. Mol. Neurobiol. 39, 149–170.
Guillot, T. S., Shepherd, K. R., Richardson, J. R., Wang, M. Z., Li, Y., Emson, P. C., and Miller, G. W. (2008). Reduced vesicular storage of dopamine exacerbates methamphetamine-induced neurodegeneration and astrogliosis. J. Neurochem. 106, 2205–2217.
Haaxma, C. A., Bloem, B. R., Borm, G. F., Oyen, W. J., Leenders, K. L., Eshuis, S., Booij, J., Dluzen, D. E., and Horstink, M. W. (2007). Gender differences in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatr. 78, 819–824.
Hagell, P., Odin, P., and Vinge, E. (1998). Pregnancy in Parkinson’s disease: a review of the literature and a case report. Mov. Disord. 13, 34–38.
Hakansson, A., Westberg, L., Nilsson, S., Buervenich, S., Carmine, A., Holmberg, B., Sydow, O., Olson, L., Johnels, B., Eriksson, E., and Nissbrandt, H. (2005). Interaction of polymorphisms in the genes encoding interleukin-6 and estrogen receptor beta on the susceptibility to Parkinson’s disease. Am. J. Med. Genet. B Neuropsychiatr. Genet. 133B, 88–92.
Hazell, G. G., Yao, S. T., Roper, J. A., Prossnitz, E. R., O’Carroll, A. M., and Lolait, S. J. (2009). Localisation of GPR30, a novel G protein-coupled oestrogen receptor, suggests multiple functions in rodent brain and peripheral tissues. J. Endocrinol. 202, 223–236.
Hernandez, L., Gonzalez, L., Murzi, E., Paez, X., Gottberg, E., and Baptista, T. (1994). Testosterone modulates mesolimbic dopaminergic activity in male rats. Neurosci. Lett. 171, 172–174.
Horstink, M. W., Strijks, E., and Dluzen, D. E. (2003). Estrogen and Parkinson’s disease. Adv. Neurol. 91, 107–114.
Ji, J., McDermott, J. L., and Dluzen, D. E. (2007). Sex differences in K+-evoked striatal dopamine output from superfused striatal tissue fragments of reserpine-treated CD-1 mice. J. Neuroendocrinol. 19, 725–731.
Johansson, P., Lindqvist, A., Nyberg, F., and Fahlke, C. (2000). Anabolic androgenic steroids affects alcohol intake, defensive behaviors and brain opioid peptides in the rat. Pharmacol. Biochem. Behav. 67, 271–279.
Jourdain, S., Morissette, M., Morin, N., and Di Paolo, T. (2005). Oestrogens prevent loss of dopamine transporter (DAT) and vesicular monoamine transporter (VMAT2) in substantia nigra of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mice. J. Neuroendocrinol. 17, 509–517.
Kindlundh, A. M., Lindblom, J., Bergstrom, L., Wikberg, J. E., and Nyberg, F. (2001). The anabolic-androgenic steroid nandrolone decanoate affects the density of dopamine receptors in the male rat brain. Eur. J. Neurosci. 13, 291–296.
Kipp, M., Karakaya, S., Pawlak, J., Araujo-Wright, G., Arnold, S., and Beyer, C. (2006). Estrogen and the development and protection of nigrostriatal dopaminergic neurons: concerted action of a multitude of signals, protective molecules, and growth factors. Front. Neuroendocrinol. 27, 376–390.
Kompoliti, K., Comella, C. L., Jaglin, J. A., Leurgans, S., Raman, R., and Goetz, C. G. (2000). Menstrual-related changes in motoric function in women with Parkinson’s disease. Neurology 55, 1572–1575.
Kritzer, M. F. (1997). Selective colocalization of immunoreactivity for intracellular gonadal hormone receptors and tyrosine hydroxylase in the ventral tegmental area, substantia nigra, and retrorubral fields in the rat. J. Comp. Neurol. 379, 247–260.
Kuiper, G. G., Carlsson, B., Grandien, K., Enmark, E., Haggblad, J., Nilsson, S., and Gustafsson, J. A. (1997). Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology 138, 863–870.
Kunnathur, V., Shemisa, K., Liu, B., Salvaterra, T. J., and Dluzen, D. E. (2006). Sex differences in methamphetamine-evoked striatal dopamine of mice are reversed by nomifensine. Neurotoxicol. Teratol. 28, 557–562.
Kuppers, E., and Beyer, C. (1998). Expression of aromatase in the embryonic and postnatal mouse striatum. Brain Res. Mol. Brain Res. 63, 184–188.
Kuppers, E., Krust, A., Chambon, P., and Beyer, C. (2008). Functional alterations of the nigrostriatal dopamine system in estrogen receptor-alpha knockout (ERKO) mice. Psychoneuroendocrinology 33, 832–838.
Lavalaye, J., Booij, J., Reneman, L., Habraken, J. B., and Van Royen, E. A. (2000). Effect of age and gender on dopamine transporter imaging with [123I]FP-CIT SPET in healthy volunteers. Eur. J. Nucl. Med. 27, 867–869.
LaVoie, M. J., and Hastings, T. G. (1999). Dopamine quinone formation and protein modification associated with the striatal neurotoxicity of methamphetamine: evidence against a role for extracellular dopamine. J. Neurosci. 19, 1484–1491.
Le Saux, M., and Di Paolo, T. (2006). Influence of oestrogenic compounds on monoamine transporters in rat striatum. J. Neuroendocrinol. 18, 25–32.
Lebesgue, D., Traub, M., De Butte-Smith, M., Chen, C., Zukin, R. S., Kelly, M. J., and Etgen, A. M. (2010). Acute administration of non-classical estrogen receptor agonists attenuates ischemia-induced hippocampal neuron loss in middle-aged female rats. PLoS ONE 5, e8642. doi: 10.1371/journal.pone.0008642
Lewis, C., and Dluzen, D. E. (2008). Testosterone enhances dopamine depletion by methamphetamine in male, but not female, mice. Neurosci. Lett. 448, 130–133.
Li, X. L., Cheng, W. D., Li, J., Zheng, Y. F., Guo, C. J., Sun, S. G., and Wang, L. X. (2009). Does estrogen receptor gene polymorphism play a role in Parkinson’s disease? Biomed. Pharmacother. 63, 599–602.
Liu, B., and Dluzen, D. E. (2006). Effect of estrogen upon methamphetamine-induced neurotoxicity within the impaired nigrostriatal dopaminergic system. Synapse 60, 354–361.
Liu, B., Xie, J. X., Rowlands, D. K., Gou, Y. L., Leung, C. C., Chung, Y. W., and Chan, H. C. (2004). Neuroprotective effects of Bak Foong Pill in 1-methyl-4-phenyl-1,2,3,6-tetrahyrdropyridine (MPTP)-induced Parkinson’s disease model mice. Biol. Pharm. Bull. 27, 1245–1250.
Liu, L. X., Chen, W. F., Xie, J. X., and Wong, M. S. (2008). Neuroprotective effects of genistein on dopaminergic neurons in the mice model of Parkinson’s disease. Neurosci. Res. 60, 156–161.
Liu, M., Hurn, P. D., Roselli, C. E., and Alkayed, N. J. (2007). Role of P450 aromatase in sex-specific astrocytic cell death. J. Cereb. Blood Flow Metab. 27, 135–141.
Luchetti, S., Bossers, K., Frajese, G. V., and Swaab, D. F. (2010). Neurosteroid biosynthetic pathway changes in substantia nigra and caudate nucleus in Parkinson’s disease. Brain Pathol. 20, 945–951.
Luu-The, V., and Labrie, F. (2010). The intracrine sex steroid biosynthesis pathways. Prog. Brain Res. 181, 177–192.
Lyons, K. E., Hubble, J. P., Troster, A. I., Pahwa, R., and Koller, W. C. (1998). Gender differences in Parkinson’s disease. Clin. Neuropharmacol. 21, 118–121.
Maharjan, S., Serova, L., and Sabban, E. L. (2005). Transcriptional regulation of tyrosine hydroxylase by estrogen: opposite effects with estrogen receptors alpha and beta and interactions with cyclic AMP. J. Neurochem. 93, 1502–1514.
Maraganore, D. M., Farrer, M. J., McDonnell, S. K., Elbaz, A., Schaid, D. J., Hardy, J. A., and Rocca, W. A. (2002). Case-control study of estrogen receptor gene polymorphisms in Parkinson’s disease. Mov. Disord. 17, 509–512.
McArthur, S., Mchale, E., and Gillies, G. E. (2007a). The size and distribution of midbrain dopaminergic populations are permanently altered by perinatal glucocorticoid exposure in a sex- region- and time-specific manner. Neuropsychopharmacology 32, 1462–1476.
McArthur, S., Murray, H. E., Dhankot, A., Dexter, D. T., and Gillies, G. E. (2007b). Striatal susceptibility to a dopaminergic neurotoxin is independent of sex hormone effects on cell survival and DAT expression but is exacerbated by central aromatase inhibition. J. Neurochem. 100, 678–692.
Menniti, F. S., and Baum, M. J. (1981). Differential effects of estrogen and androgen on locomotor activity induced in castrated male rats by amphetamine, a novel environment, or apomorphine. Brain Res. 216, 89–107.
Merchenthaler, I., Lane, M. V., Numan, S., and Dellovade, T. L. (2004). Distribution of estrogen receptor alpha and beta in the mouse central nervous system: in vivo autoradiographic and immunocytochemical analyses. J. Comp. Neurol. 473, 270–291.
Meyers, B., and Kritzer, M. F. (2009). In vitro binding assays using (3)H nisoxetine and (3)H WIN 35,428 reveal selective effects of gonadectomy and hormone replacement in adult male rats on norepinephrine but not dopamine transporter sites in the cerebral cortex. Neuroscience 159, 271–282.
Mickley, K. R., and Dluzen, D. E. (2004). Dose-response effects of estrogen and tamoxifen upon methamphetamine-induced behavioral responses and neurotoxicity of the nigrostriatal dopaminergic system in female mice. Neuroendocrinology 79, 305–316.
Miller, D. B., Ali, S. F., O’Callaghan, J. P., and Laws, S. C. (1998). The impact of gender and estrogen on striatal dopaminergic neurotoxicity. Ann. N. Y. Acad. Sci. 844, 153–165.
Miller, I. N., and Cronin-Golomb, A. (2010). Gender differences in Parkinson’s disease: clinical characteristics and cognition. Mov. Disord. 25, 2695–2703.
Mitra, S. W., Hoskin, E., Yudkovitz, J., Pear, L., Wilkinson, H. A., Hayashi, S., Pfaff, D. W., Ogawa, S., Rohrer, S. P., Schaeffer, J. M., Mcewen, B. S., and Alves, S. E. (2003). Immunolocalization of estrogen receptor beta in the mouse brain: comparison with estrogen receptor alpha. Endocrinology 144, 2055–2067.
Morale, M. C., L’Episcopo, F., Tirolo, C., Giaquinta, G., Caniglia, S., Testa, N., Arcieri, P., Serra, P. A., Lupo, G., Alberghina, M., Harada, N., Honda, S., Panzica, G. C., and Marchetti, B. (2008). Loss of aromatase cytochrome P450 function as a risk factor for Parkinson’s disease? Brain Res. Rev. 57, 431–443.
Morissette, M., and Di Paolo, T. (1993a). Effect of chronic estradiol and progesterone treatments of ovariectomized rats on brain dopamine uptake sites. J. Neurochem. 60, 1876–1883.
Morissette, M., and Di Paolo, T. (1993b). Sex and estrous cycle variations of rat striatal dopamine uptake sites. Neuroendocrinology 58, 16–22.
Morissette, M., and Di Paolo, T. (2009). Effect of estradiol on striatal dopamine activity of female hemiparkinsonian monkeys. J. Neurosci. Res. 87, 1634–1644.
Morissette, M., Jourdain, S., Al Sweidi, S., Menniti, F. S., Ramirez, A. D., and Di Paolo, T. (2007). Role of estrogen receptors in neuroprotection by estradiol against MPTP toxicity. Neuropharmacology 52, 1509–1520.
Morissette, M., Sweidi, S. A., Callier, S., and Di Paolo, T. (2008). Estrogen and SERM neuroprotection in animal models of Parkinson’s disease. Mol. Cell. Endocrinol. 290, 60–69.
Moy, L. Y., Albers, D. S., and Sonsalla, P. K. (1998). Lowering ambient or core body temperature elevates striatal MPP+ levels and enhances toxicity to dopamine neurons in MPTP-treated mice. Brain Res. 790, 264–269.
Murray, H. E., Pillai, A. V., McArthur, S. R., Razvi, N., Datla, K. P., Dexter, D. T., and Gillies, G. E. (2003). Dose- and sex-dependent effects of the neurotoxin 6-hydroxydopamine on the nigrostriatal dopaminergic pathway of adult rats: differential actions of estrogen in males and females. Neuroscience 116, 213–222.
Nilsen, J., and Brinton, R. D. (2002). Impact of progestins on estrogen-induced neuroprotection: synergy by progesterone and 19-norprogesterone and antagonism by medroxyprogesterone acetate. Endocrinology 143, 205–212.
Obeso, J. A., Marin, C., Rodriguez-Oroz, C., Blesa, J., Benitez-Temino, B., Mena-Segovia, J., Rodriguez, M., and Olanow, C. W. (2008). The basal ganglia in Parkinson’s disease: current concepts and unexplained observations. Ann. Neurol. 64(Suppl. 2), S30–S46.
Ookubo, M., Yokoyama, H., Takagi, S., Kato, H., and Araki, T. (2008). Effects of estrogens on striatal damage after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) neurotoxicity in male and female mice. Mol. Cell. Endocrinol. 296, 87–93.
Popat, R. A., Van Den Eeden, S. K., Tanner, C. M., Mcguire, V., Bernstein, A. L., Bloch, D. A., Leimpeter, A., and Nelson, L. M. (2005). Effect of reproductive factors and postmenopausal hormone use on the risk of Parkinson disease. Neurology 65, 383–390.
Quesada, A., Romeo, H. E., and Micevych, P. (2007). Distribution and localization patterns of estrogen receptor-beta and insulin-like growth factor-1 receptors in neurons and glial cells of the female rat substantia nigra: localization of ERbeta and IGF-1R in substantia nigra. J. Comp. Neurol. 503, 198–208.
Quinn, N. P., and Marsden, C. D. (1986). Menstrual-related fluctuations in Parkinson’s disease. Mov. Disord. 1, 85–87.
Ragonese, P., D’Amelio, M., Salemi, G., Aridon, P., Gammino, M., Epifanio, A., Morgante, L., and Savettieri, G. (2004). Risk of Parkinson disease in women: effect of reproductive characteristics. Neurology 62, 2010–2014.
Ramirez, A. D., Liu, X., and Menniti, F. S. (2003). Repeated estradiol treatment prevents MPTP-induced dopamine depletion in male mice. Neuroendocrinology 77, 223–231.
Raz, L., Khan, M. M., Mahesh, V. B., Vadlamudi, R. K., and Brann, D. W. (2008). Rapid estrogen signaling in the brain. Neurosignals 16, 140–153.
Robottom, B. J., Mullins, R. J., and Shulman, L. M. (2008). Pregnancy in Parkinson’s disease: case report and discussion. Expert Rev. Neurother. 8, 1799–1805.
Rodriguez-Navarro, J. A., Solano, R. M., Casarejos, M. J., Gomez, A., Perucho, J., De Yebenes, J. G., and Mena, M. A. (2008). Gender differences and estrogen effects in parkin null mice. J. Neurochem. 106, 2143–2157.
Sanchez, M. G., Bourque, M., Morissette, M., and Di Paolo, T. (2010). Steroids-dopamine interactions in the pathophysiology and treatment of CNS disorders. CNS Neurosci. Ther. 16, e43–e71.
Satoh, N., Yonezawa, A., Tadano, T., Kisara, K., Arai, Y., and Kinemuchi, H. (1987). Acute effects of a parkinsonism-inducing neurotoxin, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) on mouse body temperature. Life Sci. 41, 1415–1424.
Saunders-Pullman, R., Derby, C., Santoro, N., Bressman, S., Chiu, B., Lipton, R. B., and Wassertheil-Smoller, S. (2009). “Role of endogenous and exogenous hormone exposure on the risk of Parkinson disease,” in Annual meeting of American Academy of Neurology, Seattle, WA, Abstract S23.002.
Saunders-Pullman, R., Gordon-Elliott, J., Parides, M., Fahn, S., Saunders, H. R., and Bressman, S. (1999). The effect of estrogen replacement on early Parkinson’s disease. Neurology 52, 1417–1421.
Savageau, M. M., and Beatty, W. W. (1981). Gonadectomy and sex differences in the behavioral responses to amphetamine and apomorphine of rats. Pharmacol. Biochem. Behav. 14, 17–21.
Schultz, K. N., Von Esenwein, S. A., Hu, M., Bennett, A. L., Kennedy, R. T., Musatov, S., Toran-Allerand, C. D., Kaplitt, M. G., Young, L. J., and Becker, J. B. (2009). Viral vector-mediated overexpression of estrogen receptor-alpha in striatum enhances the estradiol-induced motor activity in female rats and estradiol-modulated GABA release. J. Neurosci. 29, 1897–1903.
Session, D. R., Pearlstone, M. M., Jewelewicz, R., and Kelly, A. C. (1994). Estrogens and Parkinson’s disease. Med. Hypotheses 42, 280–282.
Shemisa, K., Kunnathur, V., Liu, B., Salvaterra, T. J., and Dluzen, D. E. (2006). Testosterone modulation of striatal dopamine output in orchidectomized mice. Synapse 60, 347–353.
Shughrue, P. J. (2004). Estrogen attenuates the MPTP-induced loss of dopamine neurons from the mouse SNc despite a lack of estrogen receptors (ERalpha and ERbeta). Exp. Neurol. 190, 468–477.
Shulman, L. M., Minagar, A., and Weiner, W. J. (2000). The effect of pregnancy in Parkinson’s disease. Mov. Disord. 15, 132–135.
Simon, K. C., Chen, H., Gao, X., Schwarzschild, M. A., and Ascherio, A. (2009). Reproductive factors, exogenous estrogen use, and risk of Parkinson’s disease. Mov. Disord. 24, 1359–1365.
Smeyne, R. J., and Jackson-Lewis, V. (2005). The MPTP model of Parkinson’s disease. Brain Res. Mol. Brain Res. 134, 57–66.
Sotnikova, T. D., Beaulieu, J. M., Gainetdinov, R. R., and Caron, M. G. (2006). Molecular biology, pharmacology and functional role of the plasma membrane dopamine transporter. CNS Neurol. Disord. Drug Targets 5, 45–56.
Staley, J. K., Krishnan-Sarin, S., Zoghbi, S., Tamagnan, G., Fujita, M., Seibyl, J. P., Maciejewski, P. K., O’malley, S., and Innis, R. B. (2001). Sex differences in [123I]beta-CIT SPECT measures of dopamine and serotonin transporter availability in healthy smokers and nonsmokers. Synapse 41, 275–284.
Strijks, E., Kremer, J. A., and Horstink, M. W. (1999). Effects of female sex steroids on Parkinson’s disease in postmenopausal women. Clin. Neuropharmacol. 22, 93–97.
Thakur, M. K., Asaithambi, A., and Mukherjee, S. (2000). Synthesis and phosphorylation of androgen receptor of the mouse brain cortex and their regulation by sex steroids during aging. Mol. Cell. Biochem. 203, 95–101.
Tolson, D., Fleming, V., and Schartau, E. (2002). Coping with menstruation: understanding the needs of women with Parkinson’s disease. J. Adv. Nurs. 40, 513–521.
Tomas-Camardiel, M., Sanchez-Hidalgo, M. C., Sanchez Del Pino, M. J., Navarro, A., Machado, A., and Cano, J. (2002). Comparative study of the neuroprotective effect of dehydroepiandrosterone and 17beta-estradiol against 1-methyl-4-phenylpyridium toxicity on rat striatum. Neuroscience 109, 569–584.
Tripanichkul, W., Gerdprasert, O., and Jaroensuppaperch, E. O. (2010). Estrogen reduces BDNF level, but maintains dopaminergic cell density in the striatum of MPTP mouse model. Int. J. Neurosci. 120, 489–495.
Tripanichkul, W., Sripanichkulchai, K., and Finkelstein, D. I. (2006). Estrogen down-regulates glial activation in male mice following 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine intoxication. Brain Res. 1084, 28–37.
Tsang, K. L., Ho, S. L., and Lo, S. K. (2000). Estrogen improves motor disability in parkinsonian postmenopausal women with motor fluctuations. Neurology 54, 2292–2298.
Twelves, D., Perkins, K. S., and Counsell, C. (2003). Systematic review of incidence studies of Parkinson’s disease. Mov. Disord. 18, 19–31.
Van Den Eeden, S. K., Tanner, C. M., Bernstein, A. L., Fross, R. D., Leimpeter, A., Bloch, D. A., and Nelson, L. M. (2003). Incidence of Parkinson’s disease: variation by age, gender, and race/ethnicity. Am. J. Epidemiol. 157, 1015–1022.
Vasudevan, N., and Pfaff, D. W. (2008). Non-genomic actions of estrogens and their interaction with genomic actions in the brain. Front. Neuroendocrinol. 29, 238–257.
Villeneuve, A., Langlier, P., and Bedard, P. (1978). Estrogens, dopamine and dyskinesias. Can. Psychiatr. Assoc. J. 23, 68–70.
Wagner, G. C., Tekirian, T. L., and Cheo, C. T. (1993). Sexual differences in sensitivity to methamphetamine toxicity. J. Neural Transm. Gen. Sect. 93, 67–70.
Walker, Q. D., Ray, R., and Kuhn, C. M. (2006). Sex differences in neurochemical effects of dopaminergic drugs in rat striatum. Neuropsychopharmacology 31, 1193–1202.
Walker, Q. D., Rooney, M. B., Wightman, R. M., and Kuhn, C. M. (2000). Dopamine release and uptake are greater in female than male rat striatum as measured by fast cyclic voltammetry. Neuroscience 95, 1061–1070.
Westberg, L., Hakansson, A., Melke, J., Shahabi, H. N., Nilsson, S., Buervenich, S., Carmine, A., Ahlberg, J., Grundell, M. B., Schulhof, B., Klingborg, K., Holmberg, B., Sydow, O., Olson, L., Johnels, E. B., Eriksson, E., and Nissbrandt, H. (2004). Association between the estrogen receptor beta gene and age of onset of Parkinson’s disease. Psychoneuroendocrinology 29, 993–998.
Wilson, J. M., Levey, A. I., Rajput, A., Ang, L., Guttman, M., Shannak, K., Niznik, H. B., Hornykiewicz, O., Pifl, C., and Kish, S. J. (1996). Differential changes in neurochemical markers of striatal dopamine nerve terminals in idiopathic Parkinson’s disease. Neurology 47, 718–726.
Wooten, G. F., Currie, L. J., Bovbjerg, V. E., Lee, J. K., and Patrie, J. (2004). Are men at greater risk for Parkinson’s disease than women? J. Neurol. Neurosurg. Psychiatr. 75, 637–639.
Yu, L., Kuo, Y., Cherng, C. G., Chen, H. H., and Hsu, C. H. (2002). Ovarian hormones do not attenuate methamphetamine-induced dopaminergic neurotoxicity in mice gonadectomized at 4 weeks postpartum. Neuroendocrinology 75, 282–287.
Yu, L., and Liao, P. C. (2000a). Estrogen and progesterone distinctively modulate methamphetamine-induced dopamine and serotonin depletions in C57BL/6J mice. J. Neural Transm. 107, 1139–1147.
Yu, L., and Liao, P. C. (2000b). Sexual differences and estrous cycle in methamphetamine-induced dopamine and serotonin depletions in the striatum of mice. J. Neural Transm. 107, 419–427.
Zappia, M., Annesi, G., Nicoletti, G., Arabia, G., Annesi, F., Messina, D., Pugliese, P., Spadafora, P., Tarantino, P., Carrideo, S., Civitelli, D., De Marco, E. V., Ciro-Candiano, I. C., Gambardella, A., and Quattrone, A. (2005). Sex differences in clinical and genetic determinants of levodopa peak-dose dyskinesias in Parkinson disease: an exploratory study. Arch. Neurol. 62, 601–605.
Zhang, J. Q., Cai, W. Q., Zhou, D. S., and Su, B. Y. (2002a). Distribution and differences of estrogen receptor beta immunoreactivity in the brain of adult male and female rats. Brain Res. 935, 73–80.
Keywords: 17β-estradiol, androgens, dopamine, neuroprotection, neuromodulation, sex difference, MPTP, methamphetamine
Citation: Bourque M, Dluzen DE and Di Paolo T (2011) Male/female differences in neuroprotection and neuromodulation of brain dopamine. Front. Endocrin. 2:35. doi: 10.3389/fendo.2011.00035
Received: 29 June 2011;
Accepted: 02 September 2011;
Published online: 30 September 2011.
Edited by:
Hubert Vaudry, University of Rouen, FranceReviewed by:
Charlotte A. Cornil, University of Liege, BelgiumLuis Miguel Garcia-Segura, Consejo Superior de Investigaciones Científicas, Spain
Copyright: © 2011 Bourque, Dluzen and Di Paolo. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Thérèse Di Paolo, Molecular Endocrinology and Genomic Research Center, Centre de recherche du CHUQ (CHUL), 2705 Laurier Boulevard, Quebec City, QC, Canada G1V 4G2. e-mail:dGhlcmVzZS5kaXBhb2xvQGNyY2h1bC51bGF2YWwuY2E=