
- Department of Neuroscience and Experimental Therapeutics, College of Medicine, Texas A&M Health Science Center, College Station, TX, USA
This review highlights the role of major endogenous neurosteroids in seizure disorders and the promise of neurosteroid replacement therapy in epilepsy. Neurosteroids are endogenous modulators of seizure susceptibility. Neurosteroids such as allopregnanolone (3α-hydroxy-5α-pregnane-20-one) and allotetrahydrodeoxycorticosterone (3α,21-dihydroxy-5α-pregnan-20-one) are positive modulators of GABA-A receptors. Aside from peripheral tissues, neurosteroids are synthesized within the brain, mostly in principal neurons. Neurosteroids potentiate synaptic GABA-A receptor function and also activate δ-subunit-containing extrasynaptic GABA-A receptors that mediate tonic currents and thus may play an important role in neuronal network excitability and seizure susceptibility. Our studies over the past decade have shown that neurosteroids are broad-spectrum anticonvulsants and confer seizure protection in various animal models. They protect against seizures induced by GABA-A receptor antagonists, 6-Hz model, pilocarpine-induced limbic seizures, and seizures in kindled animals. Unlike benzodiazepines, tolerance does not occur to their actions during chronic administration. Our recent studies provide compelling evidence that neurosteroids may have antiepileptogenic properties. There is emerging evidence that endogenous neurosteroids may play a key role in the pathophysiology of catamenial epilepsy, stress–sensitive seizure conditions, temporal lobe epilepsy, and alcohol-withdrawal seizures. It is suggested that neurosteroid replacement with natural or synthetic neurosteroids may be useful in the treatment of epilepsy. Synthetic analogs of neurosteroids that are devoid of hormonal side effects show promise in the treatment of diverse seizure disorders. Agents that stimulate endogenous production of neurosteroids may also be useful for treatment of epilepsy.
Introduction
Neurosteroids are steroids synthesized within the brain with unconventional rapid effects on neuronal excitability. It is well known that steroid hormones such as progesterone and deoxycorticosterone can exert anticonvulsant actions (Selye, 1941; Clarke et al., 1973). The anticonvulsant properties of progesterone and deoxycorticosterone are predominantly due to their conversion in the brain to neurosteroids allopregnanolone (3α-hydroxy-5α-pregnane-20-one) and allotetrahydrodeoxycorticosterone (3α,21-dihydroxy-5α-pregnan-20-one; THDOC), respectively (Reddy, 2003; Reddy et al., 2004; Figure 1). A variety of neurosteroids are known to be synthesized in the brain (Baulieu, 1981; Kulkarni and Reddy, 1995). The most widely studied are allopregnanolone, THDOC, and androstanediol. These neurosteroids are produced via sequential A-ring reduction of the steroid hormones by 5α-reductase and 3α-hydroxysteroid-oxidoreductase isoenzymes (Reddy, 2009a). The androgenic neurosteroid androstanediol (5α-androstan-3α,17β-diol; Figure 1) is synthesized from testosterone (Reddy, 2004a,b). Other neurosteroids such as 7α-hydroxypregnanolone have been reportedly synthesized in the brain (Tsutsui et al., 2010). In the periphery, the steroid precursors are mainly synthesized in the gonads, adrenal gland, and feto-placental unit, but synthesis of these neurosteroids likely occurs in the brain from cholesterol or from peripherally derived intermediates. Since neurosteroids are highly lipophilic and can readily cross the blood–brain barrier, neurosteroids synthesized in peripheral tissues accumulate in the brain (Reddy and Rogawski, 2010a).
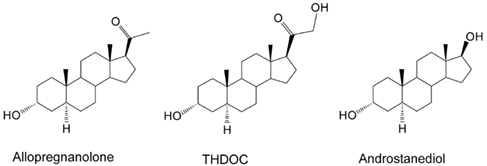
Figure 1. Chemical structures of major endogenous neurosteroids. THDOC differs from allopregnanolone by a 21β-hydroxyl group, while androstanediol differs from allopregnanolone by a 17β-hydroxyl group instead of 17β-methyl-carbonyl group. Synthetic analogs of neurosteroids are prepared by additional moieties at C3-position (e.g., ganaxolone), C2- and C11-positions (alphaxolone), and C11-position (minaxolone).
Recent evidence indicates that neurosteroids are present mainly in principal neurons in many brain regions that are relevant to focal epilepsies, including the hippocampus and neocortex (Agís-Balboa et al., 2006; Saalmann et al., 2007; Do Rego et al., 2009). The biosynthesis of neurosteroids is controlled by the translocator protein (18 kDa; TSPO), formerly called peripheral or mitochondrial benzodiazepine receptor (Rupprecht et al., 2009, 2010). Activation of TSPO by endogenous signals and ligands facilitates the intramitochondrial flux of cholesterol and thereby promotes neurosteroid synthesis. It is suggested that TSPO ligands might be an alternative approach for neurosteroid therapeutics (Nothdurfter et al., 2011). Currently, synthetic analogs of endogenous neurosteroids are under clinical trial for treatment of epilepsy (Reddy and Rogawski, 2010a).
This review describes the pathophysiological role of major endogenous neurosteroids in seizure disorders and the promise of neurosteroid replacement therapy in epilepsy. This review also summarizes the current status of synthetic neurosteroids and their therapeutic potentials. The main focus of the review is on GABAergic neurosteroids with anticonvulsant activity. Endogenous neurosteroids, such as pregnenolone, pregnenolone sulfate, and dehydroepiandrosterone sulfate, which promote neuronal excitability and seizures, are not discussed here because such description is beyond the scope of this article. The paradoxical effect of allopregnanolone are discussed elsewhere (Bäckström et al., 2011).
Overview of Neurosteroid Potentiation of GABA-A Receptors
Neurosteroids rapidly alter neuronal excitability through direct interaction with GABA-A receptors (Harrison and Simmonds, 1984; Majewska et al., 1986; Harrison et al., 1987; Gee et al., 1988; Hosie et al., 2007, 2009), which are the major receptors for the inhibitory neurotransmitter GABA. Activation of the GABA-A receptor by various ligands leads to an influx of chloride ions and to a hyperpolarization of the membrane that dampens the excitability. Allopregnanolone and other structurally related neurosteroids act as positive allosteric modulators and direct activators of GABA-A receptors (Figure 2). At low concentrations, neurosteroids potentiate GABA-A receptor currents, whereas at higher concentrations, they directly activate the receptor (Harrison et al., 1987; Reddy and Rogawski, 2002). Like barbiturates, neurosteroid enhancement of GABA-A receptors occurs through increases in both the channel open frequency and channel open duration (Twyman and Macdonald, 1992; Lambert et al., 2009; Ramakrishnan and Hess, 2010).
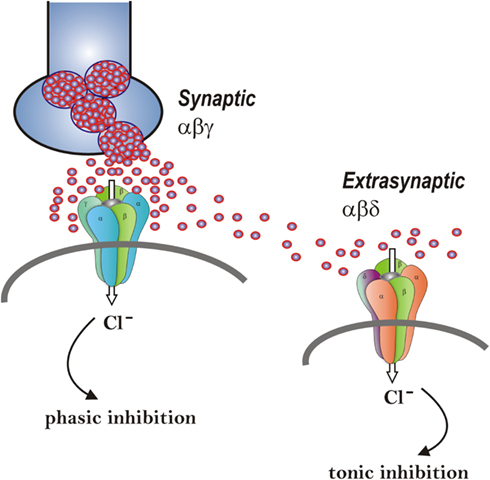
Figure 2. Neurosteroid modulation of synaptic and extrasynaptic GABA-A receptors. Postsynaptic GABA-A receptors, which are pentameric chloride channels composed of 2α2βγ subunits, mediate the phasic portion of GABAergic inhibition, while extrasynaptic GABA-A receptors, pentamers composed of 2α2βδ subunits, primarily contribute to tonic inhibition in the hippocampus. Neurosteroids activate both synaptic and extrasynaptic receptors and enhance the phasic and tonic inhibition. Therefore, they may promote effective inhibition of seizures. Two different strategies – concomitant augmentation of both phasic and tonic inhibition and selective augmentation of tonic inhibition − are being tested in epilepsy models.
The GABA-A receptor is a pentamer consisting of five subunits that form a chloride channel. Sixteen subunits (α1-6, β1-3, γ1-3, δ,ε,þ,θ, and π subunits) have been identified so far. The GABA site is located at the interface between α and β subunits. Benzodiazepines bind at the interface between α and γ subunits and they interact with subunit combinations α1,2,3,5β2γ2. The effect of neurosteroids on GABA-A receptors occurs by binding to discrete sites on the receptor–channel complex that are located within the transmembrane domains of the α- and β-subunits (Hosie et al., 2006, 2007), which they access by lateral membrane diffusion (Chisari et al., 2009, 2010). The binding sites for neurosteroids are distinct from the recognition sites for GABA, benzodiazepines, and barbiturates (Hosie et al., 2009). Androgenic neurosteroids such as androstanediol may interact with these sites, and a recent study indicates that this agent is a positive allosteric modulator of GABA-A receptors (Reddy and Jian, 2010). In whole-cell recordings from acutely dissociated hippocampus CA1 pyramidal cells in mice, androstanediol (but not its 3β-epimer) produced a concentration-dependent enhancement of GABA-activated currents (EC50, 5 μM). At 1 μM, androstanediol produced a 50% potentiation of GABA responses. In the absence of GABA, androstanediol has modest direct effects on GABA-A receptor-mediated currents even at high concentrations, indicating that it has lower direct efficacy than allopregnanolone and THDOC.
Although neurosteroids act on all GABA-A receptor isoforms, they have large effects on extrasynaptic δ-subunit-containing GABA-A receptors that mediate tonic currents (Belelli et al., 2002; Wohlfarth et al., 2002). The potentiation of δ-subunit-containing receptors by THDOC and other neurosteroids is selective for channels with low-efficacy gating characteristics marked by brief bursts and channel openings in conditions of both low and high GABA concentrations, and neurosteroids can thereby preferentially increase the efficacy of these receptors based on pharmacokinetics which are not yet fully understood (Bianchi and Macdonald, 2003). Neurosteroids therefore markedly enhance the current generated by δ-subunit-containing receptors even in the presence of saturating GABA concentrations. Consequently, GABA-A receptors that contain the δ-subunit are highly sensitive to neurosteroid potentiation and mice lacking δ-subunits show drastically reduced sensitivity to neurosteroids (Mihalek et al., 1999; Spigelman et al., 2002). Tonic current causes a steady inhibition of neurons and reduces their excitability. Neurosteroids therefore could play a role in setting the level of excitability by potentiation of tonic inhibition during seizures when ambient GABA rises (Stell et al., 2003). This phenomenon is being investigated to characterize the developmental and physiological implications of neurosteroid activation of tonic currents in the hippocampus and other areas.
The GABA-A receptor mediates two types of GABAergic inhibition, now stratified into synaptic (phasic) or extrasynaptic (tonic) inhibition (Figure 2). Although GABA activates synaptic (γ2-containing) GABA-A receptors with high efficacy, GABA activation of the extrasynaptic (δ-containing) GABA-A receptors are limited to low-efficacy activity characterized by minimal desensitization and brief openings. Physiological tonic currents of GABA receptors are dependent on the pentamer subunit composition and fairly independent of physiological levels of ambient, exogenous GABA (McCartney et al., 2007; Ransom et al., 2010). The high sensitivity of δ-containing receptor channels to neurosteroid modulation may be dependent on the δ-subunit or the low-efficacy channel function that it confers. There is evidence that neurosteroids preferentially enhance low-efficacy GABA-A receptor activity independent of subunit composition (Bianchi and Macdonald, 2003). Gaboxadol also modulates δ-subunit receptor isoforms at higher concentrations, acting as a superagonist, resulting in an induced higher efficacy gating pattern than either GABA or muscimol (Mortensen et al., 2010). Novel therapeutic approaches are being developed based on the emerging information on neurosteroid interaction with GABA-A receptors (Murashima and Yoshii, 2010).
Some endogenous neurosteroids are known to interact with GABA-A receptor and block its inhibitory function. Such neurosteroids include pregnenolone, pregnenolone sulfate (PS), dehydroepiandrosterone, and dehydroepiandrosterone sulfate (DHEAS). PS inhibits the GABA-A receptor function, and is also moderately potent allosteric agonist at NMDA receptors (Wu et al., 1991; Majewska, 1992). PS and DHEAS are proconvulsant steroids and can induce seizures when administered systemically or directly into the brain (Reddy and Kulkarni, 1998; Kokate et al., 1999; Williamson et al., 2004). The proconvulsant actions of PS are evident at high micromolar concentrations, which are 100- to 500-fold higher than its levels in the brain. Thus, it is highly unlikely that endogenous PS by itself can trigger seizures. However, PS can decrease GABAergic inhibitory transmission at physiological concentrations via a presynaptic action (Teschemacher et al., 1997; Mtchedlishvili and Kapur, 2003). Allopregnanolone blocks the seizure facilitating effects of PS and DHEAS, and consequently, these sulfated neurosteroids could contribute to seizure susceptibility when allopregnanolone and THDOC levels are low.
Anticonvulsant Activity of Neurosteroids
Allopregnanolone-like neurosteroids are powerful anticonvulsants. Exogenously administered neurosteroids, like other agents that act as positive GABA-A receptor modulators, exhibit broad-spectrum anticonvulsant effects in diverse rodent seizure models (Reddy, 2010). Neurosteroids protect against seizures induced by GABA-A receptor antagonists, including pentylenetetrazol and bicuculline, and are effective against pilocarpine-induced limbic seizures and seizures in kindled animals (Belelli et al., 1989; Kokate et al., 1994; Frye, 1995; Wieland et al., 1995; Reddy et al., 2004). Like other GABAergic agents, they may exacerbate generalized absence seizures (Snead, 1998; Citraro et al., 2006). As shown in Table 1, the potencies of neurosteroids in models where they confer seizure protection vary largely in accordance with their activities as positive allosteric modulators of GABA-A receptors. Thus, allopregnanolone is roughly equally potent as THDOC, but androstanediol and androsterone are somewhat less potent (Reddy, 2004a,b; Kaminski et al., 2005). Like other GABAergic agents, neurosteroids are inactive or only weakly active against seizures elicited by maximal electroshock. Neurosteroids are highly active in the 6-Hz model, a better paradigm in which limbic-like seizures are induced by electrical stimulation of lower frequency and longer duration than in the maximal electroshock test (Kaminski et al., 2004). Androstanediol, but not its 3β-epimer, produced a dose-dependent suppression of behavioral and electrographic seizures in the mouse hippocampus kindling (Reddy and Jian, 2010). The estimated concentrations of androstanediol producing 50% seizure protection in the kindling model (∼10 μM) are within the range of concentrations that potentiate GABA-A receptor function in CA1 pyramidal neurons.
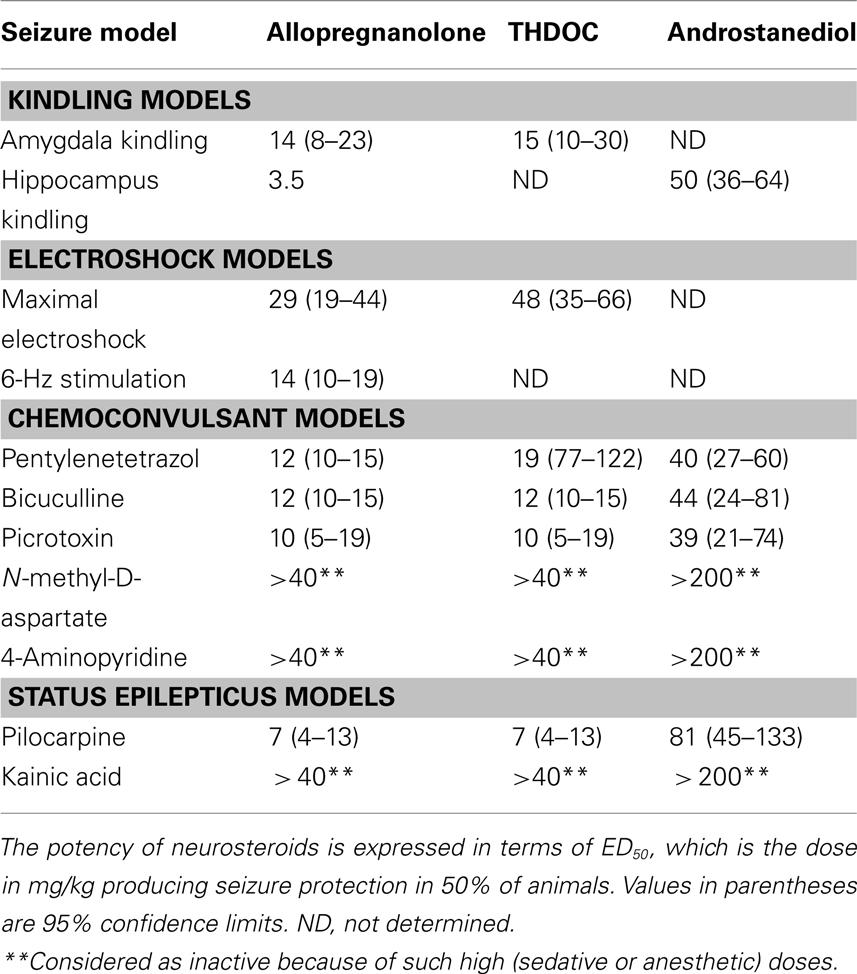
Table 1. Antiseizure profile (ED50 values) of endogenous neurosteroids in animal seizure models.
In addition, neurosteroids are also highly effective in suppressing seizures due to withdrawal of GABA-A receptor modulators including neurosteroids and benzodiazepines, as well as other types of agents such as ethanol and cocaine (Devaud et al., 1996; Tsuda et al., 1997; Reddy and Rogawski, 2001). In contrast to benzodiazepines, where utility in the chronic treatment of epilepsy is limited by tolerance, anticonvulsant tolerance is not evident with neurosteroids (Kokate et al., 1998; Reddy and Rogawski, 2000a). Thus, neurosteroids have the potential to be used in the chronic treatment of epilepsy. Unlike benzodiazepines, neurosteroids are able to modulate all isoforms of GABA-A receptors, including those that contain benzodiazepine-insensitive α4 and α6 subunits or lack the obligatory γ2 subunit required for benzodiazepine-sensitivity. Thus, it is clear that neurosteroids can act on GABA-A receptors where the proposed benzodiazepine tolerance mechanisms have been invoked by chronic GABAergic drug therapy or other endogenous conditions. Surprisingly, while chronic neurosteroid exposure does not lead to anticonvulsant tolerance, neurosteroid exposure does lead to tolerance for benzodiazepines (Reddy and Rogawski, 2000a). Thus, it appears that the same plastic changes that underlie benzodiazepine tolerance are brought into play by chronic neurosteroid exposure. However, neurosteroids acting at distinct sites on GABA-A receptors and exhibiting effects on the full range of GABA-A receptor isoforms, do not exhibit anticonvulsant tolerance. Overall, neurosteroids are more robust anticonvulsants than benzodiazepines.
Antiepileptogenic Activity of Neurosteroids
In addition to anticonvulsant activity, there is emerging evidence that endogenous neurosteroids play a role in regulating epileptogenesis (Edwards et al., 2001; Biagini et al., 2006, 2009, 2010; Reddy et al., 2010), The term “epileptogenesis” is used to describe the complex plastic changes in the brain that, following a precipitating event, convert a normal brain into a brain debilitated by recurrent seizures (Pitkänen et al., 2009). Limbic epilepsy is caused by diverse precipitating factors such as brain injury, stroke, infections, or prolonged seizures. Using the kindling model, we demonstrated that the development and persistence of limbic epileptogenesis are impaired in mice lacking progesterone receptors (Reddy and Mohan, 2011). To explore mechanisms underlying the observed seizure resistance, we investigated the role of neurosteroids using finasteride, a 5α-reductase inhibitor that blocks the synthesis of progesterone-derived neurosteroids. We determined the rate of rapid kindling in both control animals and those which had received injections of progesterone with or without concurrent finasteride treatment (Figure 3A). Progesterone produced a significant delay in the rate of kindling and pretreatment with finasteride blocked progesterone’s inhibition of kindling epileptogenesis. These findings are consistent with a contributory role of neurosteroids in limbic epileptogenesis. Thus, it is possible that inhibition of neurosteroids could incite mechanisms that may promote epileptogenesis.
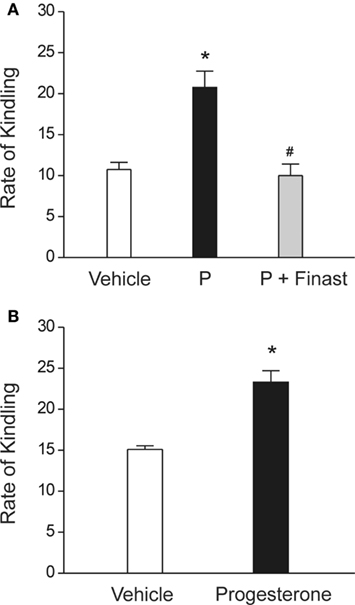
Figure 3. Role of neurosteroids in the limbic epileptogenesis. (A) Rapid kindling. Mice treated with the neurosteroid precursor progesterone (P) displayed marked retardation of rapid hippocampus epileptogenesis, as expressed by a slower rate of kindling development for stage 5 seizures. P’s inhibition of kindling epileptogenesis was blocked by finasteride. P (25 mg/kg, sc) was given 30-min prior to stimulation sessions and finasteride (50 mg/kg, ip) was injected 1 h before P treatment. *p < 0.01 vs. vehicle control, #p < 0.01 vs. P treatment alone. Adapted from Reddy and Mohan (2011). (B) Regular kindling. Rate of kindling (number of stimulations required to elicit behavioral stage 5 seizures) was significantly delayed in progesterone-treated animals. Progesterone (25 mg/kg, sc) was given 30-min prior to stimulation sessions. Adapted from Reddy et al. (2010).
Following pilocarpine-induced status epilepticus (SE) in the rat, the neurosteroidogenic enzyme P450scc is upregulated for several weeks, suggesting that it may be associated with promotion of neurosteroidogenesis (Biagini et al., 2009). Ordinarily, rats develop spontaneous recurrent seizures following a latent period of similar duration to the period during which P450scc is elevated. Inhibiting neurosteroid synthesis with finasteride accelerated the onset of spontaneous recurrent seizures (Biagini et al., 2006), suggesting that endogenous neurosteroids play a role in restraining epileptogenesis or at least that they inhibit the expression of seizures.
The development of epilepsy is linked to complex alterations in neuroplastic mechanisms. Dysregulation of neurosteroid synthesis may also play a role. This premise is being tested in various epileptogenic models (Reddy and Mohan, 2011). We investigated the role of the prototype endogenous neurosteroid allopregnanolone in controlling limbic epileptogenesis. Treatment with finasteride, a neurosteroid synthesis inhibitor, resulted in a significant increase in epileptogenesis in the hippocampus kindling model. Exogenous administration of allopregnanolone, at doses that produce levels similar to gonadotropins, markedly inhibited epileptogenesis. Based on these pilot studies, it is suggested that augmentation of neurosteroid synthesis may represent a unique strategy for preventing or retarding epileptogenesis.
Exogenous treatment with neurosteroids or with progesterone (P), which serves as a precursor for neurosteroid synthesis, has also been reported to delay the occurrence of epileptogenesis (Reddy et al., 2010). P targets multiple molecular and cellular mechanisms relevant to epileptogenesis including signaling cascades of inflammation, apoptosis, neurogenesis, and synaptic plasticity (Roof and Hall, 2000; Patel, 2004; Vezzani, 2005; Meffre et al., 2007; Stein and Sayeed, 2010). Therefore, P may be a natural disease-modifying agent. However, the potential disease-modifying effect of P in epileptogenic models is not widely investigated. Recently, we examined the effects of P on the development of hippocampus kindling in rodent models (Reddy et al., 2010). P significantly suppressed the rate of development of behavioral kindled seizure activity evoked by daily hippocampus stimulation at doses that do not significantly affect seizure expression and motor performance (Figure 3B), indicating a disease-modifying effect of P on limbic epileptogenesis. There was a significant increase in the rate of “rebound or withdrawal” kindling during drug-free stimulation sessions following abrupt discontinuation of P treatment. A washout period after termination of P treatment prevented such acceleration in kindling. These studies suggest that P exerts disease-modifying effects in the limbic epileptogenesis and it is likely that this effect of P may occur partly through neurosteroid mechanisms. Thus, it is likely that neurosteroids themselves or modulators of neurosteroid disposition could potentially have disease-modifying therapeutic activity.
Role of Endogenous Neurosteroids in Epilepsy
Neurosteroids may play a key role in the physiological regulation of seizure susceptibility in individuals with epilepsy. Endogenous neurosteroids may affect seizures situations in catamenial epilepsy, stress, temporal lobe epilepsy (TLE), and alcohol withdrawal (Reddy, 2009a; Kim et al., 2010). However, it is noteworthy that there is no evidence that alterations in neurosteroid levels in the absence of preexisting epilepsy can induce epileptogenesis. Indeed, 5α-reductase inhibitors such as finasteride, which are widely used clinically for the treatment of benign prostatic hypertrophy and male pattern hair loss, effectively inhibit neurosteroidogenesis. Increased incidence of seizures is not evident in patients taking finasteride. Nevertheless, it is possible that alterations in neurosteroidogenesis may play a role as inciting factors in the development and persistence of limbic epilepsy (Reddy and Mohan, 2011). Our recent work provides important new evidence that the availability of neurosteroids does indeed critically influence the propensity for seizures (Reddy and Zeng, 2007). We used epileptic female rats that had experienced SE. Spontaneous seizure activity was monitored for up to 5 months. The epileptic animals exhibited about two seizures per day, each lasting approximately a minute. Gonadotropin induced increase in neurosteroids was associated with reduced seizure intensity. However, when neurosteroids were withdrawn by using the neurosteroid synthesis inhibitor finasteride, a significant (two-fold) increase in seizure frequency was observed (Reddy, 2009a). These findings are confirmed in a recent study that utilized ovariectomized epileptic animals (Lawrence et al., 2010).
Catamenial Epilepsy
Catamenial epilepsy, the cyclical occurrence of seizure exacerbations during particular phases of the menstrual cycle in women with preexisting epilepsy, is a specific form of pharmacoresistant epilepsy. There are about 1.5 million women of child-bearing age with epilepsy in the United States; catamenial seizure exacerbations affect up to 70% of these women (Herzog et al., 2004; Bazan et al., 2005; Quigg et al., 2009; Reddy, 2009a; Kim et al., 2010; Verrotti et al., 2010). Although there are several forms of catamenial epilepsy, neurosteroids have been implicated only in the seizure exacerbations that occur in the most common situation, which is when women with normal menstrual cycles experience seizure exacerbations in the perimenstrual period. It is hypothesized that withdrawal of progesterone-derived neurosteroids leads to enhanced brain excitability predisposing to seizures. During the menstrual cycle, circulating progesterone levels are low in the follicular phase but rise in the mid-luteal phase for about 10–11 days, before declining in the late luteal phase. Circulating allopregnanolone levels parallel those of its parent progesterone (Tuveri et al., 2008). In addition to neurosteroid fluctuations, plasticity in GABA-A receptor subunits could play a role in the enhanced seizure susceptibility in perimenstrual catamenial epilepsy. Animal studies have shown that prolonged exposure to allopregnanolone followed by withdrawal such as that occurs during menstruation causes a marked increase in expression of the α4-subunit, a key subunit linked to enhanced neuronal excitability, seizure susceptibility, and benzodiazepine resistance (Gulinello et al., 2001; Maguire et al., 2005; Shen et al., 2005; Smith and Gong, 2005; Smith et al., 2007; Gangisetty and Reddy, 2010). Although α4 can coassemble with γ2 to form synaptic GABA-A receptors, it preferentially coassembles with δ to form extrasynaptic GABA-A receptors. Overall, these neuroendocrine changes can result in reduced inhibition resulting in enhanced excitability, which, among other effects, predisposes to catamenial seizures.
We have developed a rodent model of perimenstrual catamenial epilepsy (Reddy et al., 2001; Reddy and Zeng, 2007). Rodents have a 4- to 5-day estrous cycle and studies of fluctuations in seizure susceptibility in cycling female rodents have not led to results that are relevant to the human menstrual cycle. In order to provide a model that more closely mimics the human situation, a condition of elevated progesterone was created in rats by gonadotropin treatment. This resulted in prolonged high circulating levels of estrogen and progesterone similar to those that occur in the luteal phase of the menstrual cycle. Then, to simulate the withdrawal of allopregnanolone that occurs at the time of menstruation, the animals were treated with finasteride 11 days after the initiation of gonadotropin treatment. Withdrawal of neurosteroids had led to decreased seizure threshold and increased seizure activity (Reddy et al., 2001). We have conducted additional studies using this paradigm in female rats with spontaneous recurrent seizures (Reddy and Zeng, 2007). In epileptic animals, neurosteroid withdrawal was associated with a marked increase in seizure frequency. In women with epilepsy, finasteride therapy had led to an increase in seizure frequency and severity (Herzog and Frye, 2003), suggesting that endogenous neurosteroids do modulate seizure susceptibility.
The neurosteroid allopregnanolone is involved in the pathophysiology of catamenial epilepsy. It is suggested that seizure susceptibility decreases when neurosteroid levels are high (mid-luteal phase), and increases during their withdrawal (perimenstrual periods) in close association with specific changes in GABA-A receptor subunits expression in the hippocampus. This premise has been investigated using a mouse hippocampus kindling model of catamenial epilepsy. Our results show that fully kindled mice undergoing neurosteroid withdrawal have increased generalized seizure frequency and intensity; enhanced seizure susceptibility; and, similar to the clinical catamenial seizure phenotype, reduced benzodiazepine-sensitivity, and enhanced neurosteroid potency (Gangisetty and Reddy, 2010; Reddy and Gould, 2011). The increased susceptibility to seizures and alterations in antiseizure drug responses are associated with increased abundance of the δ and α4-subunits of GABA-A receptors in the hippocampus. However, the molecular mechanisms underlying the upregulation of α4-subunit expression remain unclear. The role of PRs and the transcription factor early growth response factor-3 (Egr3) in regulation of the GABA-A receptor α4-subunit expression in the hippocampus was investigated in a mouse neurosteroid withdrawal paradigm (Gangisetty and Reddy, 2010). Neurosteroid withdrawal-induced a threefold increase in α4-subunit expression in WT mice, but this upregulation was undiminished in PR knockout mice. The expression of the transcription factor early growth response factor-3 (Egr3), which controls α4-subunit transcription, was increased significantly following neurosteroid withdrawal in WT and PR knockout mice. Neurosteroid withdrawal-induced α4-subunit upregulation was completely suppressed by antisense Egr3 inhibition. These results support that neurosteroid withdrawal-induced upregulation of GABA-A receptor α4-subunit expression is mediated by the Egr3 via a PR-independent signaling pathway.
The neurosteroid withdrawal model of catamenial epilepsy was used to investigate therapies for perimenstrual catamenial epilepsy (Reddy and Rogawski, 2000b, 2001). A key result is that conventional antiepileptic drugs, including benzodiazepines and valproate, are less potent in protecting against seizures during the period of enhanced seizure susceptibility following neurosteroid withdrawal. This pharmacoresistance appears to mimic the situation in women with catamenial epilepsy where breakthrough seizures occur despite treatment with antiepileptic drugs. In contrast to the results with conventional antiepileptic drugs, neurosteroids, including allopregnanolone, THDOC, and their 5β-isomers, were found to have enhanced activity in the catamenial epilepsy model (Reddy and Rogawski, 2001). This suggested a “neurosteroid replacement” approach to treat catamenial seizure exacerbations (Reddy and Rogawski, 2009). A neurosteroid could be administered in a “pulse” prior to menstruation and then withdrawn, or continuously administered throughout the month. The neurosteroid would be administered at low doses to avoid sedative side effects. Such low doses are expected to contribute little anticonvulsant activity during most of the menstrual cycle. Patients would still require treatment with conventional antiepileptic medications. However, during the period of enhanced seizure susceptibility at the time of menstruation, the increased potency of the neurosteroid would confer protection against perimenstrual seizure exacerbations. It is noteworthy that while the anticonvulsant activity of neurosteroids increases in conjunction with neurosteroid withdrawal, there is no corresponding increase in side effects (Reddy and Rogawski, 2009). Therefore, side effects would not be expected to be enhanced negating the potential of the therapeutic approach.
Although neurosteroids seem to be the most direct approach to the treatment of catamenial epilepsy, there is only limited anecdotal data is available to support their use (McAuley et al., 2001). No neurosteroid is currently approved by the United States Food and Drug Administration (FDA). In contrast, two open-label trials have shown that adjunctive progesterone therapy produces significant reductions in seizure occurrence (Herzog, 2009). It is recommended that the hormone be administered during the entire second half of the menstrual cycle and tapered gradually as it is believed that abrupt discontinuation can result in rebound seizure exacerbation. Synthetic analogs of neurosteroids may overcome certain obstacles and side effects associated with natural progesterone therapy (see below, Neurosteroid Therapy section). Other therapeutic approaches may also be helpful for managing catamenial seizures (Vilos et al., 2011).
Stress–Sensitive Seizure Conditions
Neurosteroids are released during physiological stress. Stress results in the hypothalamic release of corticotropin-releasing hormone, which liberates ACTH from the anterior pituitary. Along with cortisol, ACTH also enhances the synthesis of adrenal deoxycorticosterone (Tan and Mulrow, 1975; Kater et al., 1989), which is released into the circulation and can serve as a precursor for synthesis of the neurosteroid THDOC (Figure 1). In contrast to allopregnanolone, which is present in the brain even after adrenalectomy and gonadectomy, THDOC appears to be derived nearly exclusively from adrenal sources (Purdy et al., 1990; Reddy, 2003). Plasma and brain levels of THDOC and allopregnanolone rise rapidly following acute stress (Purdy et al., 1991; Concas et al., 1998; Reddy and Rogawski, 2002). Acute stressors such as swimming, foot shock or carbon dioxide exposure elicit an increase in allopregnanolone and THDOC concentrations in plasma and in brain (Barbaccia et al., 1996, 1997; Vallee et al., 2000).
Stress-induced neurosteroids have been demonstrated to elevate seizure threshold (Reddy and Rogawski, 2002). Stress-induced seizure protection could be due to circulating neurosteroids synthesized in peripheral tissues or to those produced locally in the brain. However, the effect of swim stress-induced increases in seizure threshold and THDOC levels in rats were abolished in adrenalectomized animals, implicating adrenal-derived THDOC. Despite stress-induced seizure protection in animals, patients, and clinicians are not likely to recognize a reduction in seizure frequency associated with stress. Indeed, stress has been reported to trigger seizure activity in persons with epilepsy (Temkin and Davis, 1984; Frucht et al., 2000). During stressful episodes adrenal hormone levels are expected to fluctuate and it may simply be the withdrawal of THDOC during such fluctuations that is associated with seizure provocation. Alternatively, other unidentified hormonal factors with proconvulsant activity may be responsible for stress-induced increases in seizures. However, chronic stress of the type experienced by patients with epilepsy likely has different endocrinological consequences than acute stress. The effects on seizures of fluctuations in neurosteroids in chronic stress remain to be studied.
Temporal Lobe Epilepsy
Neurosteroids may play a role in limbic epilepsy. Sexual and reproductive dysfunction are common among persons with epilepsy (Edwards et al., 2000). In particular, men with TLE often have diminished libido and sexual potency that is associated with low testosterone levels (Herzog et al., 1986; Brunet et al., 1995; Herzog, 2002). This hypogonadal state has been attributed to the effects of certain hepatic enzyme-inducing antiepileptic drugs, or alternatively – given the extensive connections between temporal lobe structures such as the amygdala and hypothalamic nuclei that govern the production and secretion of gonadotropin releasing hormone – to suppression of the hypothalamic–pituitary–gonadal axis by limbic seizures. There is evidence that serum androgens normalize after temporal lobe surgery which results in successful seizure control but not in those that continue to have seizures. This supports the view that seizures are responsible for the hypoandrogenic state (Bauer et al., 2000). Testosterone, as noted previously, is a precursor for at least three neurosteroids with anticonvulsant properties: 5α-androstanediol, androsterone, and etiocholanolone (Reddy, 2004a,b; Kaminski et al., 2005; Reddy and Jian, 2010). There is evidence that serum levels of at least two of these steroids (androsterone and etiocholanolone) are reduced in men with epilepsy compared with control subjects (Brunet et al., 1995). It is conceivable that reduced levels of such anticonvulsant neurosteroids leads to enhanced propensity for seizures and thus neurosteroid replacement might be a useful therapeutic approach.
Certain biological factors in TLE may influence the sensitivity to endogenous neurosteroids and could have an impact on the efficacy of exogenous neurosteroids used in epilepsy therapy. Studies in a SE model of TLE have shown a striking reduction in δ-subunit-containing GABA-A receptors in the dentate gyrus (Peng et al., 2004; Zhang et al., 2007), suggesting that neurosteroid effects on non-synaptic GABA-A receptors may be reduced. In addition, in dentate gyrus granule cells neurosteroid modulation of synaptic currents is diminished and α4-subunit-containing receptors are present at synapses (Sun et al., 2007). All of these changes may exacerbate seizures in epileptic animals but may reduce the efficacy of endogenous neurosteroids. The expression of neurosteroidogenic enzymes such as P450scc and 3α-HSOR appears to be elevated in the hippocampus in animals and human subjects affected by TLE (Stoffel-Wagner et al., 2000, 2003; Biagini et al., 2009). If local neurosteroidogenesis is enhanced, this may in part counteract the epileptogenesis-induced changes.
Alcohol-Withdrawal Seizures
Alcohol withdrawal is known to be associated with seizures. Systemic administration of moderate doses (1–2.5 g/kg) of ethanol causes increases in plasma and brain neurosteroids that may contribute to many of the behavioral effects of ethanol in rodents (Morrow et al., 2006). This effect of ethanol is believed to be due to activation of the hypothalamic–pituitary–adrenal axis. As is the case in the catamenial epilepsy model, chronic ethanol-induced elevations in neurosteroids lead to an enhancement in the anticonvulsant actions of the neurosteroids allopregnanolone and THDOC (Devaud et al., 1996). These effects are associated with increases in the sensitivity of GABA-A receptors to neurosteroids (Morrow et al., 2006). Endogenous neurosteroids may protect against ethanol withdrawal seizures. However, ethanol induction of allopregnanolone is diminished in tolerant and dependent animals. Reduced availability of allopregnanolone under such circumstances may be a factor that predisposes to alcohol-withdrawal seizures. As is the case with catamenial epilepsy, neurosteroid replacement could conceivably be useful in the treatment of alcohol-withdrawal seizures, given that current pharmacological approaches are not entirely satisfactory.
Status Epilepticus
Novel therapies are desperately needed for refractory SE, an emergency neurological condition characterized by persistent seizures lasting more than 30 min, progressive internalization of synaptic GABA-A receptors, and benzodiazepine resistance. The extrasynaptic δ-subunit-containing GABA-A receptors that generate “tonic” inhibition do not internalize during SE, so that neurosteroids, which are positive modulators of extrasynaptic and synaptic GABA-A receptors with robust anticonvulsant activity, could be more effective treatments for SE. Neurosteroids have been tested in animal models of SE (Kokate et al., 1996; Reddy, 2009b). Our recent studies indicate that THDOC therapy can effectively terminate electrographic and behavioral SE induced chemically by lithium-pilocarpine in rats (Kuruba and Reddy, 2011). We found that with early and late administration after SE onset, THDOC successfully aborted seizures with sustained suppression of SE, a profile superior to the benzodiazepine diazepam. In addition, THDOC therapy may confer significant neuroprotection by diminishing the neuronal cell death associated with SE. Further studies are needed to clarify whether neurosteroids might be valuable in the treatment of SE.
Neurosteroid Therapy of Epilepsy
Despite intense research on neurosteroids, there is no neurosteroid-based drug available for patients. Ganaxolone, the synthetic 3β-methyl derivative of allopregnanolone, is the only neurosteroid that has been evaluated for the treatment of epilepsy in humans (Monaghan et al., 1999; Carter et al., 1997). Unlike allopregnanolone and related natural neurosteroids that can undergo back conversion by 3α-HSOR isoenzymes to hormonally active intermediates, the 3β-methyl substituent of ganaxolone eliminates such metabolism and thereby avoids hormonal side effects. Ganaxolone has similar pharmacological properties to the natural neurosteroids such as allopregnanolone (Reddy and Woodward, 2004). It has protective activity in diverse rodent seizure models, including clonic seizures induced by the chemoconvulsants pentylenetetrazol, bicuculline, flurothyl, aminophylline; limbic seizures in the 6-Hz model; amygdala and cocaine-kindled seizures; and corneal kindled seizures (Gasior et al., 2000; Liptáková et al., 2000; Reddy and Rogawski, 2000a, 2010b; Kaminski et al., 2003, 2004). During prolonged daily treatment, tolerance does not develop with the anticonvulsant activity of ganaxolone (Kokate et al., 1998; Reddy and Rogawski, 2000a). In our recent study in female amygdala kindled mice, ganaxolone elicited strong suppression of behavioral and electrographic seizures with ED50 of 6.6 mg/kg (Reddy and Rogawski, 2010b). Ganaxolone treatment was associated with significant reduction in the afterdischarge duration. As expected, there was a substantial suppression of behavioral and electrographic seizures in mice treated with clonazepam. While clonazepam was more potent than ganaxolone, the overall maximal efficacy of both drugs was similar. These studies provide strong evidence that the synthetic neurosteroid analog ganaxolone is highly effective antiseizure agent in the amygdala kindling model, which is a clinically relevant model of complex partial epilepsy.
Clinical Studies
Ganaxolone has been tested in various clinical trials to assess efficacy in the treatment of epilepsy (Reddy and Woodward, 2004; Rogawski et al., 2010). More than 900 subjects have received the drug at doses up to 1875 mg/day in adults and up to 54 mg/kg/day in children in phase 1 normal volunteer studies, epilepsy trials, and also clinical trials for migraine. Overall, the drug is safe and well tolerated. The most common side effect is reversible dose-dependent sedation. One epilepsy trial used the inpatient presurgical study design in adults with partial seizures (Laxer et al., 2000). A second study was an open-label, add-on trial in pediatric patients with a history of infantile spasms (Kerrigan et al., 2000). A third study was an open-label non-randomized, dose-escalation add-on trial in highly refractory pediatric and adolescent patients; three patients in this latter study were followed in an extension phase over 3.5 years (Pieribone et al., 2007). As discussed previously, there is limited anecdotal information supporting the efficacy of ganaxolone in the treatment of catamenial seizure exacerbations (McAuley et al., 2001). Recently, a double-blind, randomized, placebo controlled study was completed in adults with partial seizures (Rogawski et al., 2010). A separate trial was completed in children with infantile spasms. In this study, there was no clear statistically significant treatment effect although some subjects did appear to demonstrate a treatment-related reduction in spasm clusters. The adult trial included 147 subjects with partial onset seizures with or without secondary generalization who were refractory to conventional antiepileptic drugs. Ganaxolone treatment produced an 18% decrease in mean weekly seizure frequency, compared with a 2% increase for placebo over the 10-week treatment period. Results from the open-label extension phase of the study indicated that ganaxolone maintains its efficacy over time.
Advantages of Neurosteroid Therapy
An ideal new drug for epilepsy should have a rapid or intermediate onset of action that is effective against a broad range of convulsive and con-convulsive seizures. It should also be effective in SE even when given late after seizure onset, should exhibit no tolerance, exhibit no pharmacokinetic or teratogenic effects, be relatively safe, and readily available for clinical development. Neurosteroids partly meet or exceed these expectations, and possess several advantages: (i) Neurosteroids can be effective for broad seizure types, even in diazepam-refractory seizures because they can activate most GABA-A receptor isoforms; (ii) Unlike benzodiazepines, neurosteroids lack tolerance upon repeated or chronic treatment which has been proven in clinical trials; (iii) They show a rapid onset and intermediate duration of action; (iv) Well established mechanism of action at GABA-A receptors; (v) Maximal efficacy is expected even in resistant seizures, due to their positive and direct (non-allosteric) actions in promoting GABAergic inhibition at high dosage (vi); They promote tonic inhibition that does not rely on interneurons that may be damaged in some patients with TLE; and (vii) They are under clinical trials for epilepsy indications.
Conclusion
Neurosteroids that enhance the GABAergic neurotransmission are potent anticonvulsants and may regulate various neuronal excitability networks. Neurosteroids are believed to play a role in the regulation of seizure susceptibility in the setting of preexisting epilepsy. Menstrual and stress related fluctuations in seizures may be related to alterations in brain neurosteroids. Additionally, men with TLE who have a suppressed hypothalamic–pituitary–gonadal axis may have a reduction in testosterone-derived neurosteroids that could worsen seizures. New information on the role of neurosteroids in limbic epileptogenesis is emerging. Treatment with synthetic neurosteroids may be beneficial to patients with partial seizures. Further studies are required to determine whether “neurosteroid replacement” is a useful approach for epileptic seizures related to endogenous neurosteroid fluctuations, such as in catamenial epilepsy and stress. Novel agents that increase the brain synthesis of neurosteroids, such as TSPO ligands, may find utility in the treatment of epilepsy.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The original research described in this article was supported in part by the NIH grants NS051398, NS052158, and NS071597 (to Doodipala Samba Reddy). The author thanks Chase Carver for reading the manuscript.
References
Agís-Balboa, R. C., Pinna, G., Zhubi, A., Maloku, E., Veldic, M., Costa, E., and Guidotti, A. (2006). Characterization of brain neurons that express enzymes mediating neurosteroid biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 103, 14602–14607.
Bäckström, T., Haage, D., Löfgren, M., Johansson, I. M., Strömberg, J., Nyberg, S., Andréen, L., Ossewaarde, L., Wingen, G. A., Turkmen, S., and Bengtsson, S. K. (2011). Paradoxical effects of GABA-A modulators may explain sex steroid induced negative mood symptoms in some persons. Neuroscience 191, 46–54.
Barbaccia, M. L., Roscetti, G., Trabucchi, M., Mostallino, M. C., Concas, A., Purdy, R. H., and Biggio, G. (1996). Time-dependent changes in rat brain neuroactive steroid concentrations and GABAA receptor function after acute stress. Neuroendocrinology 63, 166–172.
Barbaccia, M. L., Roscetti, G., Trabucchi, M., Purdy, R. H., Mostallino, M. C., Concas, A., and Biggio, G. (1997). The effects of inhibitors of GABAergic transmission and stress on brain and plasma allopregnanolone concentrations. Br. J. Pharmacol. 120, 1582–1588.
Bauer, J., Stoffel-Wagner, B., Flügel, D., Kluge, M., Schramm, J., Bidlingmaier, F., and Elger, C. E. (2000). Serum androgens return to normal after temporal lobe epilepsy surgery in men. Neurology 55, 820–824.
Baulieu, E.-E. (1981). “Steroid hormones in the brain: several mechanisms?” in Steroid Hormone Regulation of the Brain, eds F. Fuxe, J. A. Gustafsson, and L. Wetterberg (Oxford: Pergamon Press), 3–14.
Bazan, A. C., Montenegro, M. A., Cendes, F., Min, L. L., and Guerreiro, C. A. (2005). Menstrual cycle worsening of epileptic seizures in women with symptomatic focal epilepsy. Arq. Neuropsiquiatr. 63, 751–756.
Belelli, D., Bolger, M. B., and Gee, K. W. (1989). Anticonvulsant profile of the progesterone metabolite 5α-pregnan-3α-ol-20-one. Eur. J. Pharmacol. 166, 325–329.
Belelli, D., Casula, A., Ling, A., and Lambert, J. J. (2002). The influence of subunit composition on the interaction of neurosteroids with GABAA receptors. Neuropharmacology 43, 651–661.
Biagini, G., Baldelli, E., Longo, D., Pradelli, L., Zini, I., Rogawski, M. A., and Avoli, M. (2006). Endogenous neurosteroids modulate epileptogenesis in a model of temporal lobe epilepsy. Exp. Neurol. 201, 519–524.
Biagini, G., Longo, D., Baldelli, E., Zoli, M., Rogawski, M. A., Bertazzoni, G., and Avoli, M. (2009). Neurosteroids and epileptogenesis in the pilocarpine model: evidence for a relationship between P450scc induction and length of the latent period. Epilepsia 50(Suppl. 1), 53–58.
Biagini, G., Panuccio, G., and Avoli, M. (2010). Neurosteroids and epilepsy. Curr. Opin. Neurol. 23, 170–176.
Bianchi, M. T., and Macdonald, R. L. (2003). Neurosteroids shift partial agonist activation of GABA-A receptor channels from low- to high-efficacy gating patterns. J. Neurosci. 23, 10934–10943.
Brunet, M., Rodamilans, M., Martinez-Osaba, M. J., Santamaria, J., To-Figueras, J., Torra, M., Corbella, J., and Rivera, F. (1995). Effects of long-term antiepileptic therapy on the catabolism of testosterone. Pharmacol. Toxicol. 76, 371–375.
Carter, R. B., Wood, P. L., Wieland, S., Hawkinson, J. E., Belelli, D., Lambert, J. J., White, H. S., Wolf, H. H., Mirsadeghi, S., Tahir, S. H., Bolger, M. B., Lan, N. C., and Gee, K. W. (1997). Characterization of the anticonvulsant properties of ganaxolone (CCD 1042; 3α-hydroxy-3β-methyl-5α-pregnan-20-one), a selective, high-affinity, steroid modulator of the γ-aminobutyric acidA receptor. J. Pharmacol. Exp. Ther. 280, 1284–1295.
Chisari, M., Eisenman, L. N., Covey, D. F., Mennerick, S., and Zorumski, C. F. (2010). The sticky issue of neurosteroids and GABAA receptors. Trends Neurosci. 33, 299–306.
Chisari, M., Eisenman, L. N., Krishnan, K., Bandyopadhyaya, A. K., Wang, C., Taylor, A., Benz, A., Covey, D. F., Zorumski, C. F., and Mennerick, S. (2009). The influence of neuroactive steroid lipophilicity on GABAA receptor modulation: evidence for a low-affinity interaction. J. Neurophysiol. 102, 1254–1264.
Citraro, R., Russo, E., Di Paola, E. D., Ibbadu, G. F., Gratteri, S., Marra, R., and De Sarro, G. (2006). Effects of some neurosteroids injected into some brain areas of WAG/Rij rats, an animal model of generalized absence epilepsy. Neuropharmacology 50, 1059–1071.
Clarke, R. S., Dundee, J. W., and Carson, I. W. (1973). Proceedings: a new steroid anaesthetic-althesin. Proc. R. Soc. Med. 66, 1027–1030.
Concas, A., Mostallino, M. C., Porcu, P., Folesa, P., Barbaccia, M. L., Trabucchi, M., Purdy, R. H., Grisenti, P., and Biggio, G. (1998). Role of brain allopregnanolone in the plasticity of γ-aminobutyric acid type A receptor in rat brain during pregnancy and after delivery. Proc. Natl. Acad. Sci. U.S.A. 95, 13284–13289.
Devaud, L. L., Purdy, R. H., Finn, D. A., and Morrow, A. L. (1996). Sensitization of γ-aminobutyric acidA receptors to neuroactive steroids in rats during ethanol withdrawal. J. Pharmacol. Exp. Ther. 278, 510–517.
Do Rego, J. L., Seong, J. Y., Burel, D., Leprince, J., Luu-The, V., Tsutsui, K., Tonon, M. C., Pelletier, G., and Vaudry, H. (2009). Neurosteroid biosynthesis: enzymatic pathways and neuroendocrine regulation by neurotransmitters and neuropeptides. Front. Neuroendocrinol. 30, 259–301.
Edwards, H. E., MacLusky, N. J., and Burnham, W. M. (2000). Epileptic seizures: do they cause reproductive dysfunction? Univ. Toronto Med. J. 104–111.
Edwards, H. E., Mo, V., Burnham, W. M., and MacLusky, N. J. (2001). Gonadectomy unmasks an inhibitory effect of progesterone on amygdala kindling in male rats. Brain Res. 889, 260–263.
Frucht, M. M., Quigg, M., Schwaner, C., and Fountain, N. B. (2000). Distribution of seizure precipitants among epilepsy syndromes. Epilepsia 41, 1534–1539.
Frye, C. A. (1995). The neuroactive steroid 3α,5α-THP has anti-seizure and possible neuroprotective effects in an animal model of epilepsy. Brain Res. 696, 113–120.
Gangisetty, O., and Reddy, D. S. (2010). Neurosteroid withdrawal regulates GABA-A receptor α4-subunit expression and seizure susceptibility by activation of progesterone receptor-independent early growth response factor-3 pathway. Neuroscience 170, 865–880.
Gasior, M., Ungard, J. T., Beekman, M., Carter, R. B., and Witkin, J. M. (2000). Acute and chronic effects of the synthetic neuroactive steroid, ganaxolone, against the convulsive and lethal effects of pentylenetetrazol in seizure-kindled mice: comparison with diazepam and valproate. Neuropharmacology 39, 1184–1196.
Gee, K. W., Bolger, M. B., Brinton, R. E., Coirini, H., and McEwen, B. S. (1988). Steroid modulation of the chloride ionophore in rat brain: structure-activity requirements, regional dependence and mechanism of action. J. Pharmacol. Exp. Ther. 246, 803–812.
Gulinello, M., Gong, Q. H., Li, X., and Smith, S. S. (2001). Short-term exposure to a neuroactive steroid increases α4 GABAA receptor subunit levels in association with increased anxiety in the female rat. Brain Res. 910, 55–66.
Harrison, N. L., and Simmonds, M. A. (1984). Modulation of the GABA receptor complex by a steroid anaesthetic. Brain Res. 323, 287–292.
Harrison, N. L., Majewska, M. D., Harrington, J. W., and Barker, J. L. (1987). Structure-activity relationships for steroid interactions with the γ-aminobutyric acidA receptor complex. J. Pharmacol. Exp. Ther. 241, 346–353.
Herzog, A. G. (2002). Altered reproductive endocrine regulation in men with epilepsy: implications for reproductive function and seizures. Ann. Neurol. 51, 539–542.
Herzog, A. G., and Frye, C. A. (2003). Seizure exacerbation associated with inhibition of progesterone metabolism. Ann. Neurol. 53, 390–391.
Herzog, A. G., Seibel, M. M., Schomer, D. L., Vaitukaitis, J. L., and Geschwind, N. (1986). Reproductive endocrine disorders in men with partial seizures of temporal lobe origin. Arch. Neurol. 43, 347–350.
Herzog, A. G., Harden, C. L., Liporace, J., Pennell, P., Schomer, D. L., Sperling, M., Fowler, K., Nikolov, B., Shuman, S., and Newman, M. (2004). Frequency of catamenial seizure exacerbation in women with localization-related epilepsy. Ann. Neurol. 56, 431–434.
Hosie, A. D., Wilkins, M. E., da Silva, H. M. A., and Smart, T. G. (2006). Endogenous neurosteroids regulate GABAA receptors through two discrete transmembrane sites. Nature 444, 486–489.
Hosie, A. M., Clarke, L., da Silva, H., and Smart, T. G. (2009). Conserved site for neurosteroid modulation of GABAA receptors. Neuropharmacology 56, 149–154.
Hosie, A. M., Wilkins, M. E., and Smart, T. G. (2007). Neurosteroid binding sites on GABAA receptors. Pharmacol. Ther. 116, 7–19.
Kaminski, R. M., Gasior, M., Carter, R. B., and Witkin, J. M. (2003). Protective efficacy of neuroactive steroids against cocaine kindled-seizures in mice. Eur. J. Pharmacol. 474, 217–222.
Kaminski, R. M., Livingood, M. R., and Rogawski, M. A. (2004). Allopregnanolone analogs that positively modulate GABA receptors protect against partial seizures induced by 6-Hz electrical stimulation in mice. Epilepsia 45, 864–877.
Kaminski, R. M., Marini, H., Kim, W. J., and Rogawski, M. A. (2005). Anticonvulsant activity of androsterone and etiocholanolone. Epilepsia 46, 819–827.
Kater, C. E., Biglieri, E. G., Brust, N., Chang, B., Hirai, J., and Irony, I. (1989). Stimulation and suppression of the mineralocorticoid hormones in normal subjects and adrenocortical disorder. Endocr. Rev. 10, 149–164.
Kerrigan, J. F., Shields, W. D., Nelson, T. Y., Bluestone, D. L., Dodson, W. E., Bourgeois, B. F., Pellock, J. M., Morton, L. D., and Monaghan, E. P. (2000). Ganaxolone for treating intractable infantile spasms: a multicenter, open-label, add-on trial. Epilepsy Res. 42, 133–139.
Kim, G. H., Lee, H. W., Park, H., Lee, S. K., Lee, S. A., Kim, Y. I., Song, H. K., Shin, D. J., and Hong, S. B. (2010). Seizure exacerbation and hormonal cycles in women with epilepsy. Epilepsy Res. 90, 214–220.
Kokate, T. G., Cohen, A. L., Karp, E., and Rogawski, M. A. (1996). Neuroactive steroids protect against pilocarpine- and kainic acid-induced limbic seizures and status epilepticus in mice. Neuropharmacology 35, 1049–1056.
Kokate, T. G., Juhng, K. N., Kirkby, R. D., Llamas, J., Yamaguchi, S., and Rogawski, M. A. (1999). Convulsant actions of the neurosteroid pregnenolone sulfate in mice. Brain Res. 831, 119–124.
Kokate, T. G., Svensson, B. E., and Rogawski, M. A. (1994). Anticonvulsant activity of neuroactive steroids: correlation with γ-aminobutyric acid-evoked chloride current potentiation. J. Pharmacol. Exp. Ther. 270, 1223–1229.
Kokate, T. G., Yamaguchi, S., Pannell, L. K., Rajamani, U., Carroll, D. M., Grossman, A. B., and Rogawski, M. A. (1998). Lack of anticonvulsant tolerance to the neuroactive steroid pregnanolone in mice. J. Pharmacol. Exp. Ther. 287, 553–558.
Kulkarni, S. K., and Reddy, D. S. (1995). Neurosteroids: a new class of neuromodulators. Drugs Today 31, 433–455.
Kuruba, R., and Reddy, D. S. (2011). Neuroprotective effects of GABAergic agents in the rat model of refractory status epilepticus. Soc. Neurosci. Abstr. PN 338.08.
Lambert, J. J., Cooper, M. A., Simmons, R. D., Weir, C. J., and Belelli, D. (2009). Neurosteroids: endogenous allosteric modulators of GABA-A receptors. Psychoneuroendocrinology 34(Suppl. 1), S48–S58.
Lawrence, C., Martin, B. S., Sun, C., Williamson, J., and Kapur, J. (2010). Endogenous neurosteroid synthesis modulates seizure frequency. Ann. Neurol. 67, 689–693.
Laxer, K., Blum, D., Abou-Khalil, B. W., Morrell, M. J., Lee, D. A., Data, J. L., and Monaghan, E. P. (2000). Assessment of ganaxolone’s anticonvulsant activity using a randomized, double-blind, presurgical trial design. Ganaxolone presurgical study group. Epilepsia 41, 1187–1194.
Liptáková, S., Velísek, L., Velísková, J., and Moshé, S. L. (2000). Effect of ganaxolone on flurothyl seizures in developing rats. Epilepsia 41, 788–793.
Maguire, J. L., Stell, B. M., Rafizadeh, M., and Mody, I. (2005). Ovarian cycle-linked changes in GABAA receptors mediating tonic inhibition alter seizure susceptibility and anxiety. Nat. Neurosci. 8, 797–804.
Majewska, M. D. (1992). Neuroactive steroids: endogenous bimodal modulators of the GABAA receptor. Mechanism of action and physiological significance. Prog. Neurobiol. 38, 379–395.
Majewska, M. D., Harrison, N. L., Schwartz, R. D., Barker, J. L., and Paul, S. M. (1986). Steroid hormone metabolites are barbiturate-like modulators of the GABA receptor. Science 232, 1004–1007.
McAuley, J. W., Reeves, A. L., Flyak, J., Monaghan, E. P., and Data, J. (2001). A pilot study of the neurosteroid ganaxolone in catamenial epilepsy: clinical experience in two patients. Epilepsia 42(Suppl. 7), :85.
McCartney, M. R., Deeb, T. Z., Henderson, T. N., and Hales, T. G. (2007). Tonically active GABA-A receptors in hippocampal pyramidal neurons exhibit constitutive GABA-independent gating. Mol. Pharmacol. 71, 539–548.
Meffre, D., Pianos, A., Liere, P., Eychenne, B., Cambourg, A., Schumacher, M., Stein, D. G., and Guennoun, R. (2007). Steroid profiling in brain and plasma of male and pseudopregnant female rats after traumatic brain injury: analysis by gas chromatography/mass spectrometry. Endocrinology 148, 2505–2517.
Mihalek, R. M., Banerjee, P. K., Korpi, E. R., Quinlan, J. J., Firestone, L. L., Mi, Z. P., Lagenaur, C., Tretter, V., Sieghart, W., Anagnostaras, S. G., Sage, J. R., Fanselow, M. S., Guidotti, A., Spigelman, I., Li, Z., DeLorey, T. M., Olsen, R. W., and Homanics, G. E. (1999). Attenuated sensitivity to neuroactive steroids in γ-aminobutyrate type A receptor δ subunit knockout mice. Proc. Natl. Acad. Sci. U.S.A. 96, 12905–12910.
Monaghan, E. P., McAuley, J. W., and Data, J. L. (1999). Ganaxolone: a novel positive allosteric modulator of the GABAA receptor complex for the treatment of epilepsy. Expert Opin. Investig. Drugs 8, 1663–1671.
Morrow, A. L., Porcu, P., Boyd, K. N., and Grant, K. A. (2006). Hypothalamic-pituitary-adrenal axis modulation of GABAergic neuroactive steroids influences ethanol sensitivity and drinking behavior. Dialogues Clin. Neurosci. 8, 463–477.
Mortensen, M., Ebert, B., Wafford, K., and Smart, T. G. (2010). Distinct activities of GABA agonists at synaptic- and extrasynaptic-type GABA-A receptors. J. Physiol. (Lond.) 588.8, 1251–1268.
Mtchedlishvili, Z., and Kapur, J. (2003). A presynaptic action of the neurosteroid pregnenolone sulfate on GABAergic synaptic transmission. Mol. Pharmacol. 64, 857–864.
Murashima, Y. L., and Yoshii, M. (2010). New therapeutic approaches for epilepsies, focusing on reorganization o f the GABA-A receptor subunits by neurosteroids. Epilepsia 51(Suppl. 3), 131–134.
Nothdurfter, C., Rammes, G., Baghai, T. C., Schüle, C., Schumacher, M., Papadopoulos, V., and Rupprecht, R. (2011). TSPO (18 kDa) as a target for novel anxiolytics with a favourable side-effect profile. J. Neuroendocrinol. doi: 10.1111/j.1365-2826.2011.02166.x [Epub ahead of print].
Patel, M. (2004). Mitochondrial dysfunction and oxidative stress: cause and consequences of epileptic seizures. Free Radic. Biol. Med. 37, 1951–1962.
Peng, Z., Huang, C. S., Stell, B. M., Mody, I., and Houser, C. R. (2004). Altered expression of the δ subunit of the GABAA receptor in a mouse model of temporal lobe epilepsy. J. Neurosci. 24, 8629–8639.
Pieribone, V. A., Tsai, J., Soufflet, C., Rey, E., Shaw, K., Giller, E., and Dulac, O. (2007). Clinical evaluation of ganaxolone in pediatric and adolescent patients with refractory epilepsy. Epilepsia 48, 1870–1874.
Pitkänen, A., Immonen, R. J., Gröhn, O. H., and Kharatishvili, I. (2009). From traumatic brain injury to posttraumatic epilepsy: what animal models tell us about the process and treatment options. Epilepsia 50(Suppl. 2), 21–29.
Purdy, R. H., Morrow, A. L., Blinn, J. R., and Paul, S. M. (1990). Synthesis, metabolism, and pharmacological activity of 3α-hydroxy steroids which potentiate GABA-receptor-mediated chloride ion uptake in rat cerebral cortical synaptoneurosomes. J. Med. Chem. 33, 1572–1581.
Purdy, R. H., Morrow, A. L., Moore, P. H. Jr., and Paul, S. M. (1991). Stress-induced elevations of γ-aminobutyric acid type A receptor-active steroids in the rat brain. Proc. Natl. Acad. Sci. U.S.A. 88, 4553–4557.
Quigg, M., Smithson, S. D., Fowler, K. M., Sursal, T., and Herzog, A. G. (2009). NIH Progesterone trial study group: laterality and location influence catamenial seizure expression in women with partial epilepsy. Neurology 73, 223–227.
Ramakrishnan, L., and Hess, G. P. (2010). Mechanism of potentiation of a dysfunctional epilepsy-linked mutated GABA-A receptor by a neurosteroid (3alpha, 21-dihydroxy-5alpha-pregnan-20-one): transient kinetic investigations. Biochemistry 49, 7892–7901.
Ransom, C. B., Wu, Y., and Richerson, G. B. (2010). Postdepolarization potentiation of GABA-A receptors: a novel mechanism regulating tonic conductance in hippocampal neurons. J. Neurosci. 30, 7672–7684.
Reddy, D. S. (2003). Is there a physiological role for the neurosteroid THDOC in stress-sensitive conditions? Trends Pharmacol. Sci. 24, 103–106.
Reddy, D. S. (2004a). Anticonvulsant activity of the testosterone-derived neurosteroid 3α-androstanediol. Neuroreport 15, 515–518.
Reddy, D. S. (2004b). Testosterone modulation of seizure susceptibility is mediated by neurosteroids 3α-androstanediol and 17β-estradiol. Neuroscience 129, 195–207.
Reddy, D. S. (2009a). The role of neurosteroids in the pathophysiology and treatment of catamenial epilepsy. Epilepsy Res. 85, 1–30.
Reddy, D. S. (2009b). Gender differences in antiseizure sensitivity of neurosteroids in the pilocarpine model of status epilepticus. Epilepsia 50(Suppl.11), :126.
Reddy, D. S. (2010). Neurosteroids: endogenous role in the human brain and therapeutic potentials. Prog. Brain Res. 186, 113–137.
Reddy, D. S., Castaneda, D. C., O’Malley, B. W., and Rogawski, M. A. (2004). Anticonvulsant activity of progesterone and neurosteroids in progesterone receptor knockout mice. J. Pharmacol. Exp. Ther. 310, 230–239.
Reddy, D. S., Gangisetty, O., and Briyal, S. (2010). Disease-modifying activity of progesterone in the hippocampus kindling model of epileptogenesis. Neuropharmacology 59, 573–581.
Reddy, D. S., and Gould, J. (2011). A mouse hippocampus kindling model of catamenial epilepsy. Epilepsia (in press).
Reddy, D. S., and Jian, K. (2010). The testosterone-derived neurosteroid androstanediol is a positive allosteric modulator of GABAA receptors. J. Pharmacol. Exp. Ther. 334, 1031–1041.
Reddy, D. S., Kim, H. Y., and Rogawski, M. A. (2001). Neurosteroid withdrawal model of perimenstrual catamenial epilepsy. Epilepsia 42, 328–336.
Reddy, D. S., and Kulkarni, S. K. (1998). Proconvulsant effects of neurosteroids pregnenolone sulfate and dehydroepiandrosterone sulfate in mice. Eur. J. Pharmacol. 345, 55–59.
Reddy, D. S., and Mohan, A. (2011). Development and persistence of limbic epileptogenesis are impaired in mice lacking progesterone receptors. J. Neurosci. 31, 650–658.
Reddy, D. S., and Rogawski, M. A. (2000a). Enhanced anticonvulsant activity of ganaxolone after neurosteroid withdrawal in a rat model of catamenial epilepsy. J. Pharmacol. Exp. Ther. 294, 909–915.
Reddy, D. S., and Rogawski, M. A. (2000b). Chronic treatment with the neuroactive steroid ganaxolone in the rat induces anticonvulsant tolerance to diazepam but not to itself. J. Pharmacol. Exp. Ther. 295, 1241–1248.
Reddy, D. S., and Rogawski, M. A. (2001). Enhanced anticonvulsant activity of neuroactive steroids in a rat model of catamenial epilepsy. Epilepsia 42, 303–310.
Reddy, D. S., and Rogawski, M. A. (2002). Stress-induced deoxycorticosterone-derived neuro-active steroids modulates GABAA re-ceptor function and seizure suscep-tibility. J. Neurosci. 42, 3795–3805.
Reddy, D. S., and Rogawski, M. A. (2009). Neurosteroid replacement therapy for catamenial epilepsy. Neurotherapeutics 6, 392–401.
Reddy, D. S., and Rogawski, M. A. (2010a). Neurosteroids as endogenous regulators of seizure susceptibility and use in the treatment of epilepsy. Epilepsia 51(Suppl. 5), :84.
Reddy, D. S., and Rogawski, M. A. (2010b). Ganaxolone suppression of behavioral and electrographic seizures in the mouse amygdala kindling model. Epilepsy Res. 89, 254–260.
Reddy, D. S., and Woodward, R. (2004). Ganaxolone: a prospective overview. Drugs Future 29, 227–242.
Reddy, D. S., and Zeng, Y. C. (2007). Effect of neurosteroid withdrawal on spontaneous recurrent seizures in a rat model of catamenial epilepsy. FASEB J. 21, A1179.
Rogawski, M. A., Nohria, V., Tsai, J., Shaw, K., Farfel, G., and Pieribone, V. A. (2010). Ganaxolone, In: Bialer M, Johannessen SI, Levy RH, Perucca E, Tomson T, White HS. Progress report on new antiepileptic drugs: a summary of the Tenth Eilat Conference (EILAT X). Epilepsy Res. 92, 89–124.
Roof, R. L., and Hall, E. D. (2000). Gender differences in acute CNS trauma and stroke: neuroprotective effects of estrogen and progesterone. J. Neurotrauma 17, 367–388.
Rupprecht, R., Papadopoulos, V., Rammes, G., Baghai, T. C., Fan, J., Akula, N., Groyer, G., Adams, D., and Schumacher, M. (2010). Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat. Rev. Drug Discov. 9, 971–988.
Rupprecht, R., Rammes, G., Eser, D., Baghai, T. C., Schüle, C., Nothdurfter, C., Troxler, T., Gentsch, C., Kalkman, H. O., Chaperon, F., Uzunov, V., McAllister, K. H., Bertaina-Anglade, V., La Rochelle, C. D., Tuerck, D., Floesser, A., Kiese, B., Schumacher, M., Landgraf, R., Holsboer, F., and Kucher, K. (2009). Translocator protein (18 kD) as target for anxiolytics without benzodiazepine-like side effects. Science 325, 490–493.
Saalmann, Y. B., Kirkcaldie, M. T., Waldron, S., and Calford, M. B. (2007). Cellular distribution of the GABA-A receptor-modulating 3a-hydroxy, 5a-reduced pregnane steroids in the adult rat brain. J. Neuroendocrinol. 19, 272–284.
Shen, H., Gong, Q. H., Yuan, M., and Smith, S. S. (2005). Short-term steroid treatment increases δ GABAA receptor subunit expression in rat CA1 hippocampus: pharmacological and behavioral effects. Neuropharmacology 49, 573–586.
Smith, S. S., and Gong, Q. H. (2005). Neurosteroid administration and withdrawal alter GABAA receptor kinetics in CA1 hippocampus of female rats. J. Physiol. (Lond.) 564, 421–436.
Smith, S. S., Shen, H., Gong, Q. H., and Zhou, X. (2007). Neurosteroid regulation of GABA-A receptors: focus on the alpha4 and delta subunits. Pharmacol. Ther. 116, 58–76.
Snead, O. C. III. (1998). Ganaxolone, a selective, high-affinity steroid modulator of the γ-aminobutyric acid-A receptor, exacerbates seizures in animal models of absence. Ann. Neurol. 44, 688–691.
Spigelman, I., Li, Z., Banerjee, P. K., Mihalek, R. M., Homanics, G. E., and Olsen, R. W. (2002). Behavior and physiology of mice lacking the GABAA-receptor δ subunit. Epilepsia 43(Suppl. 5), 3–8.
Stein, D. G., and Sayeed, I. (2010). Is progesterone worth consideration as a treatment for brain injury. Am. J. Roentgenol. 194, 20–22.
Stell, B. M., Brickley, S. G., Tang, C. Y., Farrant, M., and Mody, I. (2003). Neuroactive steroids reduce neuronal excitability by selectively enhancing tonic inhibition mediated by δ subunit-containing GABAA receptors. Proc. Natl. Acad. Sci. U.S.A. 100, 14439–14444.
Stoffel-Wagner, B., Beyenburg, S., Watzka, M. S., Blumcke, I., Bauer, J., Schramm, J., Bidlingmaier, F., and Elger, C. E. (2000). Expression of 5α-reductase and 3α-hydroxysteroid oxidoreductase in the hippocampus of patients with chronic temporal lobe epilepsy. Epilepsia 41, 140–147.
Stoffel-Wagner, B., Watzka, M., Steckelbroeck, S., Ludwig, M., Clusmann, H., Bidlingmaier, F., Casarosa, E., Luisi, S., Elger, C. E., and Beyenburg, S. (2003). Allopregnanolone serum levels and expression of 5α-reductase and 3α-hydroxysteroid dehydrogenase isoforms in hippocampal and temporal cortex of patients with epilepsy. Epilepsy Res. 54, 11–19.
Sun, C., Mtchedlishvili, Z., Erisir, A., and Kapur, J. (2007). Diminished neurosteroid sensitivity of synaptic inhibition and altered location of the α4 subunit of GABAA receptors in an animal model of epilepsy. J. Neurosci. 27, 12641–12650.
Tan, S. Y., and Mulrow, P. J. (1975). The contribution of the zona fasciculata and glomerulosa to plasma 11-deoxycorticosterone levels in man. J. Clin. Endocrinol. Metab. 41, 126–130.
Temkin, N. R., and Davis, G. R. (1984). Stress as a risk factor for seizures among adults with epilepsy. Epilepsia 25, 450–456.
Teschemacher, A., Kasparov, S., Kravitz, E. A., and Rahamimoff, R. (1997). Presynaptic action of the neurosteroid pregnenolone sulfate on inhibitory transmitter release in cultured hippocampal neurons. Brain Res. 772, 226–232.
Tsuda, M., Suzuki, T., and Misawa, M. (1997). Modulation of the decrease in the seizure threshold of pentylenetetrazole in diazepam-withdrawn mice by the neuroactive steroid 5α-pregnan-3α,21-diol-20-one (alloTHDOC). Addict. Biol. 2, 455–460.
Tsutsui, K., Haraguchi, S., Matsunaga, M., Koyama, T., Do Rego, J. L., and Vaudry, H. (2010). Identification of 7alpha-hydroxypregnenolone, a novel bioactive amphibian neurosteroid stimulating locomotor activity, and its physiological roles in the regulation of locomotion. Gen. Comp. Endocrinol. 168, 275–279.
Tuveri, A., Paoletti, A. M., Orrù, M., Melis, G. B., Marotto, M. F., Zedda, P., Marrosu, F., Sogliano, C., Marra, C., Biggio, G., and Concas, A. (2008). Reduced serum level of THDOC, an anticonvulsant steroid, in women with perimenstrual catamenial epilepsy. Epilepsia 49, 1221–1229.
Twyman, R. E., and Macdonald, R. L. (1992). Neuroactive steroid regulation of GABAA receptor single-channel kinetic properties of mouse spinal cord neurons in culture. J. Physiol. (Lond.) 456, 215–245.
Vallee, M., Rivera, J. D., Koob, G. F., Purdy, R. H., and Fitzgerald, R. L. (2000). Quantification of neurosteroids in rat plasma and brain following swim stress and allopregnanolone administration using negative chemical ionization gas chromatography/mass spectrometry. Anal. Biochem. 287, 153–166.
Verrotti, A., Laus, M., Coppola, G., Parisi, P., Mohn, A., and Chiarelli, F. (2010). Catamenial epilepsy: hormonal aspects. Gynecol. Endocrinol. 26, 783–790.
Vilos, G. A., Hollett-Caines, J., Abu-Rafea, B., Ahmad, R., and Mazurek, M. F. (2011). Resolution of catamenial epilepsy after goserelin therapy and oophorectomy: case report of presumed cerebral endometriosis. J. Minim. Invasive Gynecol. 18, 128–130.
Wieland, S., Belluzzi, J. D., Stein, L., and Lan, N. C. (1995). Comparative behavioral characterization of the neuroactive steroids 3α-OH,5α-pregnan-20-one and 3α-OH,5β-pregnan-20-one in rodents. Psychopharmacology (Berl.) 118, 65–71.
Williamson, J., Mtchedlishvili, Z., and Kapur, J. (2004). Characterization of the convulsant action of pregnenolone sulfate. Neuropharmacology 46, 56–864.
Wohlfarth, K. M., Bianchi, M. T., and Macdonald, R. L. (2002). Enhanced neurosteroid potentiation of ternary GABAA receptors containing the (d subunit. J. Neurosci. 22, 1541–1549.
Wu, F. S., Gibbs, T. T., and Farb, D. H. (1991). Pregnenolone sulfate: a positive allosteric modulator at the N-methyl-D-aspartate receptor. Mol. Pharmacol. 40, 333–336.
Keywords: allopregnanolone, THDOC, neurosteroids, seizure, epilepsy, catamenial epilepsy, epileptogenesis, GABA-A receptor
Citation: Reddy DS (2011) Role of anticonvulsant and antiepileptogenic neurosteroids in the pathophysiology and treatment of epilepsy. Front. Endocrin. 2:38. doi: 10.3389/fendo.2011.00038
Received: 08 July 2011; Paper pending published: 09 August 2011;
Accepted: 06 September 2011; Published online: 05 October 2011.
Edited by:
Kazuyoshi Tsutsui, Waseda University, JapanReviewed by:
Paolo Magni, Università degli Studi di Milano, ItalyGianluca Tosini, Morehouse School of Medicine, USA
Mingde Wang, Umeå University, Sweden
Copyright: © 2011 Reddy. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Doodipala Samba Reddy, Department of Neuroscience and Experimental Therapeutics, College of Medicine, Texas A&M Health Science Center, 228 Reynolds Medical Building, College Station, TX 77843, USA. e-mail:cmVkZHlAbWVkaWNpbmUudGFtaHNjLmVkdQ==