 Pauline Gloaguen1,2,3,4
Pauline Gloaguen1,2,3,4 Pascale Crépieux1,2,3,4 Domitille Heitzler1,2,3,4
Pascale Crépieux1,2,3,4 Domitille Heitzler1,2,3,4 Anne Poupon1,2,3,4
Anne Poupon1,2,3,4 Eric Reiter1,2,3,4*
Eric Reiter1,2,3,4*- 1 BIOS Group, INRA, UMR85, Unité Physiologie de la Reproduction et des Comportements, Nouzilly, France
- 2 UMR6175, CNRS, Nouzilly, France
- 3 Université François Rabelais, Tours, France
- 4 L’Institut Français du Cheval et de l’Équitation, Nouzilly, France
Follicle-stimulating hormone (FSH) is a central regulator of male and female reproductive function. Over the last decade, there has been a growing perception of the complexity associated with FSH-induced cellular signaling. It is now clear that the canonical Gs/cAMP/PKA pathway is not the sole mechanism that must be considered in FSH biological actions. In parallel, consistent with the emerging concept of biased agonism, several examples of ligand-mediated selective signaling pathway activation by gonadotropin receptors have been reported. In this context, it is important to gain an integrative view of the signaling pathways induced by FSH and how they interconnect to form a network. In this review, we propose a first attempt at building topological maps of various pathways known to be involved in the FSH-induced signaling network. We discuss the multiple facets of FSH-induced signaling and how they converge to the hormone integrated biological response. Despite of their incompleteness, these maps of the FSH-induced signaling network represent a first step toward gaining a system-level comprehension of this hormone’s actions, which may ultimately facilitate the discovery of novel regulatory processes and therapeutic strategies for infertility and non-steroidal contraception.
Introduction
Follicle-stimulating hormone (FSH) plays a central role in the control of reproduction. FSH is a heterodimeric pituitary glycoprotein consisting of an α-subunit, which is common to other glycoprotein hormones, and a specific β-subunit (Papkoff and Ekblad, 1970). FSH binds to and activates the FSH receptor (FSHR), which belongs to the 7 transmembrane domains receptor (7TMR) family, also known as G protein-coupled receptors. The FSHR is expressed in Sertoli cells in testis and granulosa cells in ovaries (Simoni et al., 1997). FSH is required for normal growth and maturation of ovarian follicles in women and for normal spermatogenesis in men (Themmen and Huhtaniemi, 2000). Knock-out of the FSH β-subunit or the FSHR genes in mice result in significant reproductive defects in both sexes (Kumar et al., 1997; Dierich et al., 1998). Consistently, inactivating mutations in either FSH β subunit or FSHR led to similar reproductive defects (Matthews et al., 1993; Aittomaki et al., 1995; Layman et al., 1997; Huhtaniemi et al., 2006). Depending on the physiological situation, FSH has to control distinct, sometime opposite, integrated biological responses in its target cells, ranging from differentiation, cellular metabolism, steroidogenesis, proliferation, and apoptosis (Dias et al., 2010).
Due to its ability to control reproduction, either native or recombinant FSH preparations have been extensively used in reproductive medicine and animal breeding (Lunenfeld, 2004; Macklon et al., 2006). However, the use of FSH remains associated with significant drawbacks such as the risk of triggering ovarian hyperstimulation syndrome (OHSS; Vloeberghs et al., 2009) or heterogeneous responsiveness (Loutradis et al., 2003, 2004). Conversely, it has been proposed that FSHR blockers could potentially represent a novel non-steroidal approach for contraception (Naz et al., 2005). A causal role of FSH in the etiology of ovarian epithelial cancer has also been proposed many years ago (Choi et al., 2007). In addition, it has been recently reported that FSHR is ectopically expressed by endothelial cells associated with the angiogenesis of a wide panel of tumors, pointing out to the FSHR as a common marker for early diagnosis of most cancers (Radu et al., 2010). In addition, this finding also raises the intriguing possibility of a direct role played by FSH in promoting early tumoral angiogenesis. In this general context, selective pharmacological modulators of FSHR would be of great interest.
Noteworthy, it is increasingly accepted that certain ligands can lead to selective activation of signaling pathways by binding at their cognate 7TMRs in a process referred to as biased agonism (Kenakin, 2005; Rajagopal et al., 2010; Whalen et al., 2011). In line with this emerging concept, it has been recently reported that certain hormone glycosylation variants, potentiating antibodies and small molecule ligands can trigger biased responses at the FSHR (Arey et al., 2008; Wehbi et al., 2010a,b; Dias et al., 2011; Ulloa-Aguirre et al., 2011). It has also been shown that FSHR can be directly stimulated by antibodies, expending even more the potential to develop biased agonists at this receptor (Agrawal and Dighe, 2009). Paralleling the notion of biased agonism, it is now generally accepted that the canonical G protein-dependent coupling is not the unique mechanism leading to 7TMRs signaling (Lefkowitz and Shenoy, 2005; Reiter and Lefkowitz, 2006). The FSHR coupling to the classical Gαs/cAMP/protein kinase A (PKA) signaling pathway, which had been acknowledged as the sole effector mechanism of FSH for more than 20 years, is now viewed as one of several mechanisms contributing to the activation of a complex integrated signaling network. Deciphering the organization and dynamic functioning of this network is an important challenge that the field has now to address as it may help identify novel regulatory processes and therapeutic strategies for infertility and non-steroidal contraception.
In the present review paper, we attempted to integrate the data available on FSH-induced signaling by building a series of topological maps in the CellDesigner modeling environment (Kitano et al., 2005). CellDesigner is a structured diagram editor for drawing biochemical and gene-regulatory networks. Networks are drawn based on the graphical notation system proposed by Kitano et al. (2005). Models are stored using the Systems Biology Markup Language (SBML), a standard for representing models of biochemical and gene-regulatory networks. Because they are coded in SBML, CellDesigner models are easily shared or linked with simulation and other analysis packages through Systems Biology Workbench (SBW). For each reaction, the relevant references are indicated within the network representation, providing a unique and user-friendly integration of the available knowledge. For simplification purpose, we chose to only represent monomers in the maps throughout the paper although it is now well established that FSHR is expressed as dimers at the plasma membrane (Thomas et al., 2007; Guan et al., 2010). In the future, as the data become more abundant, it will be possible and more rigorous to draw separate models for each physiological situation (i.e., Sertoli vs. granulosa cells, young vs. mature, as a function of the species, endogenous vs. heterogeneously expressed receptors, etc.). However, in the present paper, in order to deal with the relative scarcity of available data for certain mechanisms, we decided to aggregate data even if they were generated in different cellular models.
Multiple Transduction Mechanisms are Activated by FSH
The Gαs/cAMP/PKA signaling pathway has been considered for more than 20 years as the key mechanism relaying FSH biological action inside target cells (Means et al., 1974; Dattatreyamurty et al., 1987). However, it is now amply documented that FSHR also engages other transduction mechanisms upon FSH binding. Figure 1 shows, in CellDesigner format, the known G protein-dependent and -independent transduction mechanisms engaged at the FSHR.
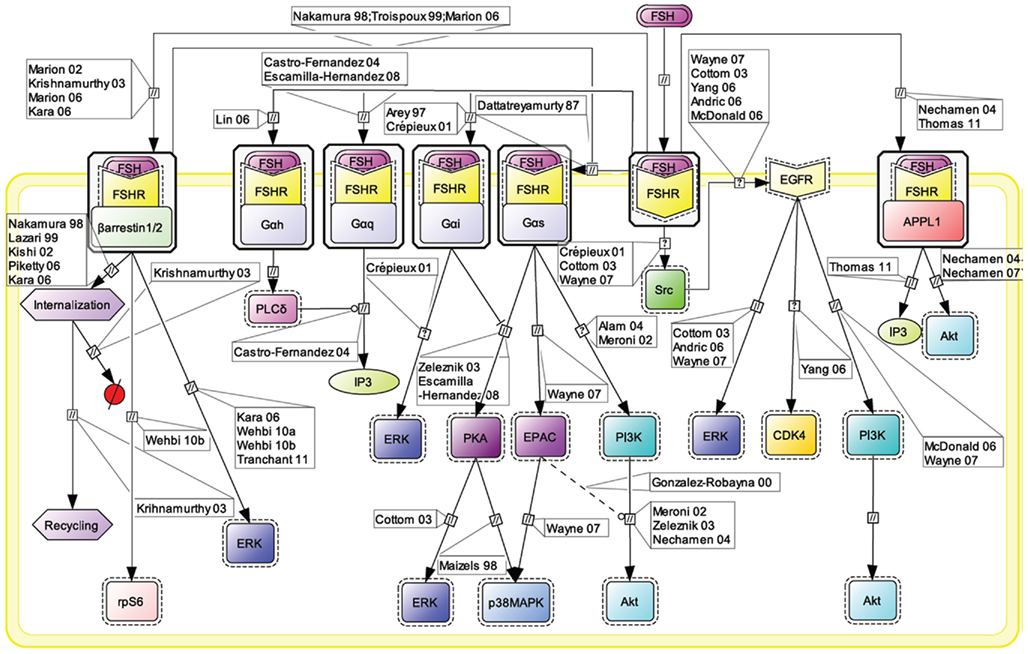
Figure 1. Multiple transduction mechanisms induced at the FSHR. The Gαs/cAMP/protein kinase A (PKA) signaling pathway has classically been considered as the key signaling mechanism triggered at the FSHR. Over the last decade, mounting evidence have shown that FSHR also engages other transduction mechanisms mainly coupling to other Gα subunits, β-arrestin-dependent signaling, EGFR transactivation, and APPL1-mediated signals. Bibliographic references are indicated on the map by the first author’s name with two digits for the year of publication. The CellDesigner program has been used to represent signaling pathways as shown in Figures 1–7. Complexes are surrounded by a box. Dashed lines indicate indirect reactions. The following semantic has been used: proteins ( ); active protein (
); active protein ( ); receptor (
); receptor ( ); transcript (
); transcript ( ); gene (
); gene ( ); catalysis (
); catalysis ( ); association (
); association ( ); dissociation (
); dissociation (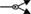 ); inhibition (
); inhibition ( ); phosphorylation (
); phosphorylation (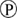 ); degradation (
); degradation (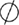 ); known transition omitted (
); known transition omitted (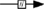 ); unknown transition (
); unknown transition (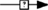 ).
).
Early assumption of Gαs/cAMP/PKA-independent signaling at the FSHR comes from the observation that adenovirus-mediated transduction of a constitutively active Gαs (Gαs Q227L) in undifferentiated rat granulosa cells was not capable of inducing the expression of differentiation markers such as aromatase or luteinizing hormone receptor (Zeleznik et al., 2003). Overexpression of a constitutively active form of protein kinase B (PKB/Akt) was required to fully restore the expression of these two markers (Zeleznik et al., 2003). Besides, PKB activation had been reported to depend in some instance on cAMP (Meroni et al., 2002; Alam et al., 2004), independently of PKA (Gonzalez-Robayna et al., 2000), suggesting that exchange protein directly activated by cAMP (EPAC) was also involved in the process. A role for EPAC in FSH biological action has been later demonstrated (Wayne et al., 2007). To conclude, PKB seems to be activated both by Gαs-dependent mechanisms, likely via EPAC, and by Gαs-independent mechanisms, in FSH-stimulated cells. Interestingly, quantitatively weaker gene expression was observed with a lentiviral vector overexpressing a constitutively active PKA (PKA-CQR), when compared to FSH (Escamilla-Hernandez et al., 2008a), suggesting again that both PKA-dependent and PKB-dependent signaling pathways are required for full FSH biological activity.
Consistent with the idea of Gαs-independent transduction mechanisms at the FSHR, it has been reported that this receptor couples to other G protein subtypes. Indeed, FSHR has been demonstrated to trigger pertussis toxin-sensitive pathways upon activation by certain hormonal variants (Arey et al., 1997) or in particular developmental stage of the target cells (Crépieux et al., 2001). In addition, the FSHR has also been shown to activate the inositol trisphosphate (IP3) signaling pathway, particularly at high doses of agonist (Quintana et al., 1994). Consistent with these data, FSHR directly interacts with Gαq subunit overexpressed in granulosa cells (Escamilla-Hernandez et al., 2008b). An alternative transduction mechanism has been reported in Sertoli cells and could also explain the observed FSH-induced IP3 response. According to this mechanism, FSHR functionally couples to Gαh, also known as tissue transglutaminase, which leads to PLCδ activation and IP3 accumulation (Lin et al., 2006).
Beside heterotrimeric G proteins, two protein families have been reported to specifically interact with the FSHR upon FSH stimulation: G protein-coupled receptor kinases (GRKs) and β-arrestins. Originally, GRKs and β-arrestins have been shown to control the desensitization, internalization, and recycling of FSHR (Nakamura et al., 1998; Lazari et al., 1999; Troispoux et al., 1999; Reiter et al., 2001; Kishi et al., 2002; Marion et al., 2002, 2006; Krishnamurthy et al., 2003a,b; Piketty et al., 2006). Over the last 10 years, the perception of β-arrestins’ functions has expended as they have been shown to act as G protein-independent signal transducers at many 7TMRs (Lefkowitz and Shenoy, 2005; Reiter and Lefkowitz, 2006) including the FSHR (Kara et al., 2006; Wehbi et al., 2010a,b; Tranchant et al., 2011). In the case of the FSHR, β-arrestin-dependent and G protein-independent activation of ERK and rpS6 have been reported so far. However, it is likely that β-arrestins are involved in the G protein-independent activation of a wide array of signaling pathways at the FSHR since they act as multifunctional scaffolds interacting with many protein partners (Xiao et al., 2007) and facilitating the phosphorylation of numerous intracellular targets (Xiao et al., 2010) at other 7TMRs (see Whalen et al., 2011 for a recent review).
The adaptor protein containing a PH domain, PTB domain, and leucine zipper motif 1 (APPL1) has also been reported to bind directly to the FSHR and trigger downstream signaling mechanisms. APPL1 has been suggested to interact with the first intracellular loop of the FSHR and to mediate FSH-dependent PI3K signaling (Nechamen et al., 2004). Recently, Thomas et al. (2011) have shown that the interaction of APPL1 with the FSHR is required for the activation of the inositol–phosphate calcium pathways upon FSH exposure. In addition, Src family members have also been reported to induce the PI3K pathway in granulosa cells (Wayne et al., 2007) and the ERK pathway in both Sertoli and granulosa cells (Crépieux et al., 2001; Cottom et al., 2003) upon FSH exposure.
Transactivation of the epithelial growth factor receptor (EGFR) also seems to play a role in the transmission of FSH signal within the target cells. Indeed, FSH has been shown to trigger EGFR autophosphorylation in granulosa cells through the activation of Src (Cottom et al., 2003; Wayne et al., 2007). Moreover, when EGFR is inhibited, a decrease in the ability of FSH to induce ERK or Akt phosphorylation and CDK4 activation has been observed in various models (Cottom et al., 2003; Andric and Ascoli, 2006; Shimada et al., 2006; Yang and Roy, 2006; Wayne et al., 2007).
In the remaining of this review, detailed topological maps for these different transduction mechanisms and their downstream signaling cascades will be presented and discussed.
The Canonical Gαs-Dependent Pathway
As already stated, the Gαs pathway has been the most studied and is associated with various intracellular events (Figure 2). Upon FSH activation, FSHR functionally couples to Gαs subunit which in turn induces adenylate cyclase activity (Northup et al., 1980). The adenylate cyclase-mediated cAMP production is counterbalanced by phosphodiesterase (PDE) activity (Fakunding et al., 1976). Accumulated cAMP binds to and activates two distinct downstream effectors: PKA and EPAC.
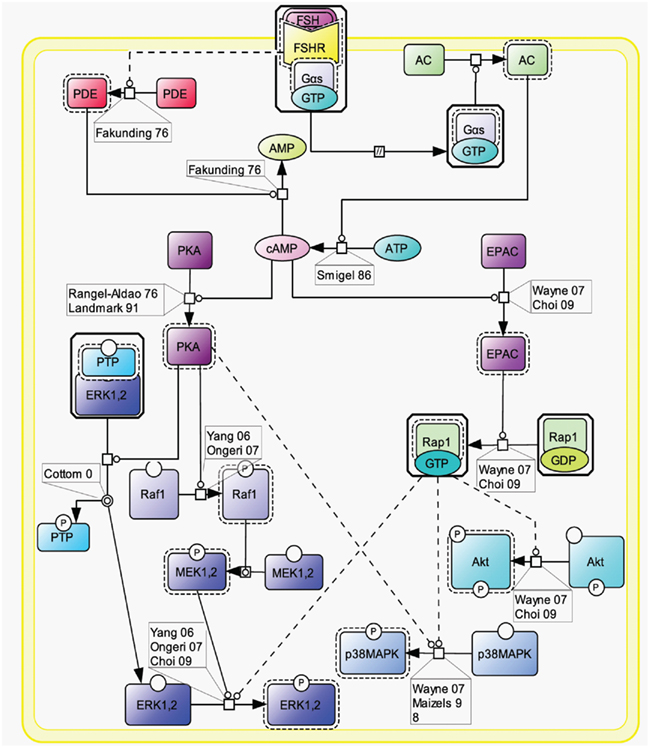
Figure 2. Canonical Gαs/cAMP/PKA pathway at the FSHR. The Gαs/cAMP/PKA pathway has been the most studied and is associated with various intracellular events. It is now acknowledged that PKA is not the sole target of cAMP accumulation. Indeed, EPAC is also activated upon FSH stimulation. Both PKA and EPAC contribute to the activation of MAPK ERK and p38, whereas EPAC also leads to Akt activation.
Upon cAMP binding, PKA’s catalytic subunits are released and activated (Rangel-Aldao and Rosen, 1976; Landmark et al., 1991). Active catalytic subunits of PKA subsequently phosphorylate a number of targets, either in the cytosol, or in the nucleus. Nuclear actions of PKA are treated in a separate section of this review. In the cytosol, FSH-induced ERK MAPK activation has been shown in several models to be sensitive to PKA inhibition (Crépieux et al., 2001; Kara et al., 2006). Two somehow contradictory mechanisms have been proposed to explain PKA-mediated activation of ERK upon FSH stimulation. First, PKA-mediated phosphorylation of a protein phosphatase (PTP) has been shown to disrupt the PTP/ERK complex, thereby leading to an increase in ERK phosphorylation with MEK being constitutively phosphorylated (Cottom et al., 2003). Second, it has been reported that PKA promotes Raf1 activation, which in turn activates MEK and finally ERK (Yang and Roy, 2006; Ongeri et al., 2007). Further studies will be necessary to clarify this situation. In addition, PKA inhibition has also been shown to impair FSH-induced MAPK p38 phosphorylation and the associated cell rounding and steroidogenesis in granulosa cells (Maizels et al., 1998; Yu et al., 2005).
Evidence have been provided that EPAC acts as an effector downstream of FSH-induced cAMP accumulation. Upon cAMP binding, EPAC promotes Rap1-GDP to GTP exchange and its subsequent activation in granulosa (Wayne et al., 2007) and in surface epithelium ovary (Choi et al., 2009) cells. Once activated, Rap1 relays FSH-dependent activation of Akt, p38, and ERK (Wayne et al., 2007; Choi et al., 2009). Again, further investigation will be necessary in order to delineate the exact contributions of PKA and EPAC in FSH signaling.
The β-Arrestin-Dependent Pathway
Initially, the β-arrestin pathway was viewed as controlling FSHR’s desensitization and recycling. Similar to many other 7TMRs, this perception has progressively evolved toward a more general role of β-arrestins as adapters and transducers (Reiter and Lefkowitz, 2006; Figure 3).
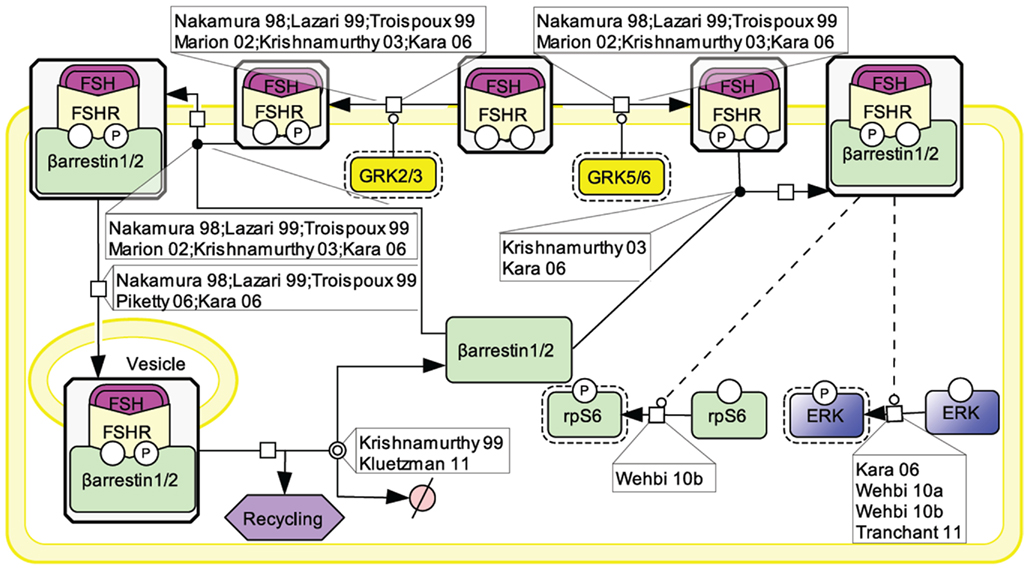
Figure 3. FSH-induced β-arrestin-dependent signaling. Initially, the perception of β-arrestins’ role was restricted to the control of FSHR’s desensitization and recycling. This view has progressively evolved toward a more general role of β-arrestins as adapters and transducers leading to the activation of MAPK ERK and rpS6 independently of G proteins upon FSH stimulation. GRK2/3 and GRK5/6 control the fate of the activated FSHR (i.e., desensitization vs. signaling) presumably through phosphorylation of distinct serines and threonines within the receptor’s C-tail.
The FSHR has been reported to be phosphorylated by GRKs 2, 3, 5, and 6 upon FSH binding to the receptor in various models (Nakamura et al., 1998; Lazari et al., 1999; Troispoux et al., 1999; Marion et al., 2002; Krishnamurthy et al., 2003a; Kara et al., 2006). A cluster of five serines and threonines located in the C terminus of the FSHR has been shown to account for the bulk of FSH-induced phosphorylation as a result of GRK2 action (Kara et al., 2006). It is also well documented that β-arrestins are recruited to the GRK-phosphorylated and agonist-occupied FSHR (Nakamura et al., 1998; Lazari et al., 1999; Troispoux et al., 1999; Marion et al., 2002; Krishnamurthy et al., 2003a; Kara et al., 2006). In addition to GRK2-dependent phosphorylation, which plays a major role in FSHR phosphorylation and β-arrestin recruitment, GRK5 and 6 have also been found to contribute to the same processes in HEK293 cells, though to a lesser extent (Kara et al., 2006). Interestingly, β-arrestins recruited to GRK2- or GRK5/6-phosphorylated FSHR have been suggested to exert distinct intracellular functions (Kara et al., 2006; Reiter and Lefkowitz, 2006). It is well established that β-arrestin 1 and 2 binding to GRK-phosphorylated FSHR leads to the internalization and recycling of the receptor (Nakamura et al., 1998; Lazari et al., 1999; Kishi et al., 2002; Kara et al., 2006; Piketty et al., 2006). In HEK293 cells, GRK2-phosphorylated FSHR has been reported to predominate in the β-arrestin-mediated internalization process (Kara et al., 2006). While most of the internalized FSHR is recycled back to the plasma membrane, a modest proportion of the receptor is routed to the lysosomal degradation pathway (Krishnamurthy et al., 2003b; Kluetzman et al., 2011).
A growing number of 7TMRs, including the FSHR, have been demonstrated to elicit signals independently of heterotrimeric G protein coupling, through direct interaction with β-arrestins (Kara et al., 2006; Reiter and Lefkowitz, 2006; Wehbi et al., 2010a,b; Tranchant et al., 2011). G protein-mediated ERK activation is rapid (reaching a maximum within ∼2–5 min) and transient. In contrast, ERK activation via β-arrestins is slower in onset (reaching a maximum within ∼5–10 min) but sustained (t1/2 > 1 h; Kara et al., 2006). In addition, β-arrestins have been shown to contribute to rpS6 phosphorylation in HEK293 cells upon FSHR activation (Wehbi et al., 2010b). As also reported for other 7TMRs, GRK5 and 6-induced phosphorylation of the activated FSHR is required for β-arrestin-dependent signaling pathway in HEK293 cells (Kara et al., 2006; Reiter and Lefkowitz, 2006). Interestingly, partially deglycosylated equine LH has recently been shown to preferentially activate β-arrestin-dependent signaling at the FSHR, presumably acting as a biased ligand at this receptor (Wehbi et al., 2010b; Ulloa-Aguirre et al., 2011).
The PI3K/mTOR Pathway
An array of evidence, gathered in various cell models, supports the notion that the PI3K/mTOR pathway plays an important role in FSH-induced actions including proliferation, regulation of gene transcription as well as of protein translation (Figure 4). FSH has been shown to lead to PI3K activation, PIP3 accumulation, and FSH-dependent proliferation in both granulosa cells and primary Sertoli cells from newborn rat (Park et al., 2005; Musnier et al., 2009; Dupont et al., 2010). Interestingly, in differentiating rat Sertoli cells, PIP3 accumulation is negatively regulated by PTEN whose expression is strongly and rapidly induced upon FSH stimulation, which results in a blockade of FSH-induced proliferation (Dupont et al., 2010).
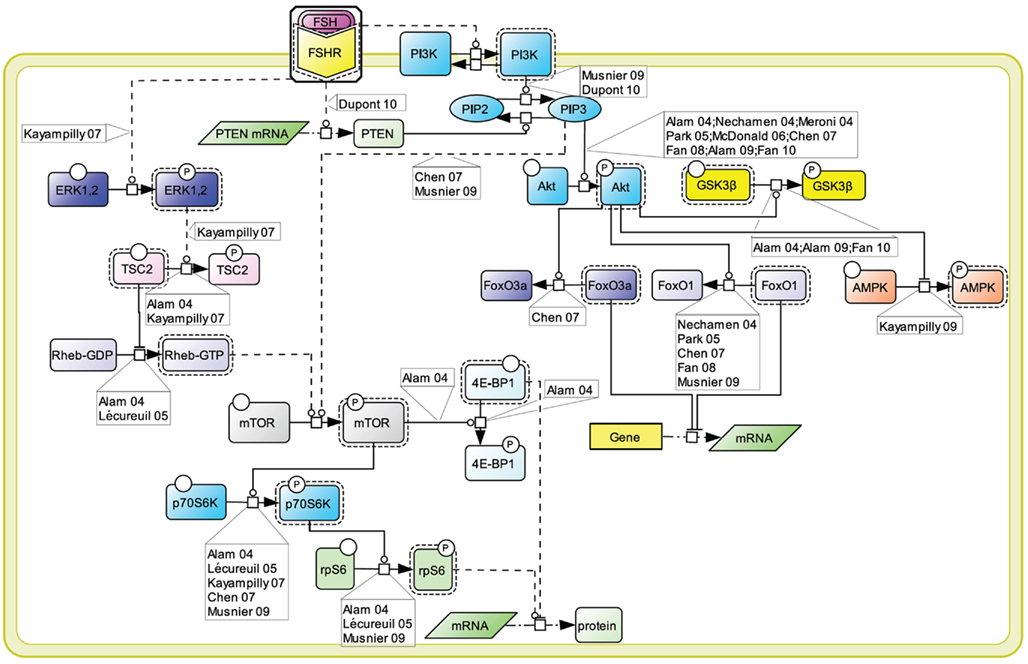
Figure 4. PI3K/mTOR signaling at the FSHR. The PI3K/mTOR pathway plays an important role in FSH-induced actions, including proliferation, regulation of gene transcription as well as of protein translation.
This FSH-induced PI3K/PIP3 pathway has been reported by numerous authors to trigger Akt phosphorylation and activation (Gonzalez-Robayna et al., 2000; Alam et al., 2004, 2009; Meroni et al., 2004; Nechamen et al., 2004; Park et al., 2005; McDonald et al., 2006; Chen et al., 2007; Fan et al., 2008, 2010). In turn Akt leads to GSK3β phosphorylation and deactivation (Alam et al., 2009; Fan et al., 2010). It also triggers AMPK phosphorylation and its deactivation (Kayampilly and Menon, 2009). In addition, Akt has been reported to inactivate FoxO3a (Chen et al., 2007) and FoxO1 (Cunningham et al., 2003; Nechamen et al., 2004; Park et al., 2005; Chen et al., 2007; Fan et al., 2008; Musnier et al., 2009), two transcription factors which differentially impact on gene regulation.
The PI3K/PIP3 pathway has also been shown to induce mTOR phosphorylation and activation (Chen et al., 2007; Musnier et al., 2009). Another mechanism contributing to the full activation of mTOR upon FSH stimulation involves ERK activation which leads to TSC2 phosphorylation, thereby releasing its negative regulation on Rheb. Rheb-GTP in turn induces mTOR activity (Alam et al., 2004; Lécureuil et al., 2005; Kayampilly and Menon, 2007).
Once activated, mTOR controls p70S6K activity (Alam et al., 2004; Lécureuil et al., 2005; Chen et al., 2007; Kayampilly and Menon, 2007; Musnier et al., 2009). Active mTOR also leads to the phosphorylation and inactivation of 4E-BP1, a factor negatively controlling protein translation (Alam et al., 2004). In parallel, FSH-induced p70S6K activity triggers a robust phosphorylation of rpS6 (Alam et al., 2004; Lécureuil et al., 2005; Musnier et al., 2009). The combination of rpS6 activation and 4E-BP1 inhibition support the notion that FSH controls and activates protein translation in addition to its well-known effects on gene expression.
FSHR Interacting Proteins
A number of proteins has been reported to interact with the FSHR and to impact on FSH-induced signaling pathways (Dias et al., 2005). One of the alternative transduction mechanism used by the FSHR involves the APPL1 (Figure 5). APPL1 has been shown to interact with the first and second intracellular loop of the FSHR, and has been proposed to mediate FSH-dependent PI3K and Akt signaling (Nechamen et al., 2004). Furthermore, recent studies in HEK293 cells and KGN granulosa cells have shown that residues K376, L377, and F382 in the first intracellular loop of the FSHR are implicated in the interaction of the receptor with APPL1 and that K376 particularly, links the activated FSHR to the inositol–phosphate pathway and FSH-stimulated intracellular calcium mobilization (Thomas et al., 2011). Other studies have reported calcium accumulation upon FSH stimulation in different cell models (Flores et al., 1990; Jayes et al., 2000; Lin et al., 2006; Lai et al., 2008). Further studies will be necessary to determine to what extent APPL1/FSHR interaction is a general requirement for FSH-induced calcium signaling.
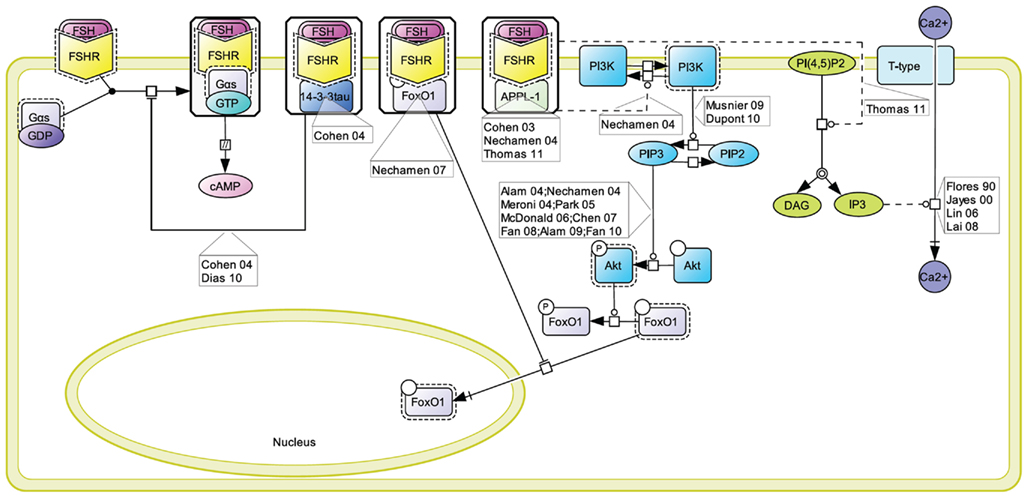
Figure 5. Proteins interacting at the FSHR and their role in signaling. A number of proteins have been reported to interact with the FSHR and to impact on FSH-induced signaling pathways. These include 14-3-3τ, FoxO1, and APPL1, the latter two being involved in the control of the PI3K/Akt pathway downstream of the FSHR.
A direct interaction between the transcription factor FoxO1a and the FSHR has been evidenced (Nechamen et al., 2007). It has been suggested that, while interacting with the FSHR, FoxO1a cannot translocate to the nucleus and affect gene transcription (Dias et al., 2010). Interestingly, APPL1 also potentially contributes to the nuclear exclusion of FoxO1a though a distinct mechanism. Indeed, APPL1-mediated PI3K activation leads to Akt-dependent phosphorylation of FoxO1a, impairing its nuclear localization. It has been proposed that FoxO1a phosphorylation could occur within FSHR-containing signalosome since APPL1 is well-known to interact with Akt (Dias et al., 2010).
The FSHR interacts with the scaffolding protein 14-3-3τ (Cohen et al., 2004). This interaction has been mapped in the second intracellular loop, in a region encompassing the ERW motif, where it is predicted to compete with Gαs coupling. Consistently, when overexpressed, 14-3-3τ has been reported to dampen FSH-induced cAMP accumulation (Cohen et al., 2004; Dias et al., 2010).
EGFR Transactivation
Similar to other GPCRs, many lines of evidence point to EGFR transactivation as an important transduction mechanism used by FSHR (Cottom et al., 2003; McDonald et al., 2006; Figure 6). Two mechanisms have been shown to contribute to EGFR activation upon FSH stimulation. First, the metalloprotease ADAM17 has been shown to be phosphorylated in response to FSH stimulation (Yamashita et al., 2007, 2009, 2010). Once activated, ADAM17 splits a pro-EGF-like protein, releasing an EGF-like ligand which subsequently binds to and activates EGFR (Yamashita et al., 2007). The mechanism by which FSHR induces ADAM17 phosphorylation is not yet identified. However, data from other receptors suggest that either p38 (Xu and Derynck, 2010) or Src (Zhang et al., 2006) could be involved in this process. A second mechanism of EGFR transactivation by the FSHR involves Src which has been proposed to be activated upon FSH exposure and to directly phosphorylate EGFR thereby eliciting EGF-independent activation of this receptor (Wayne et al., 2007).
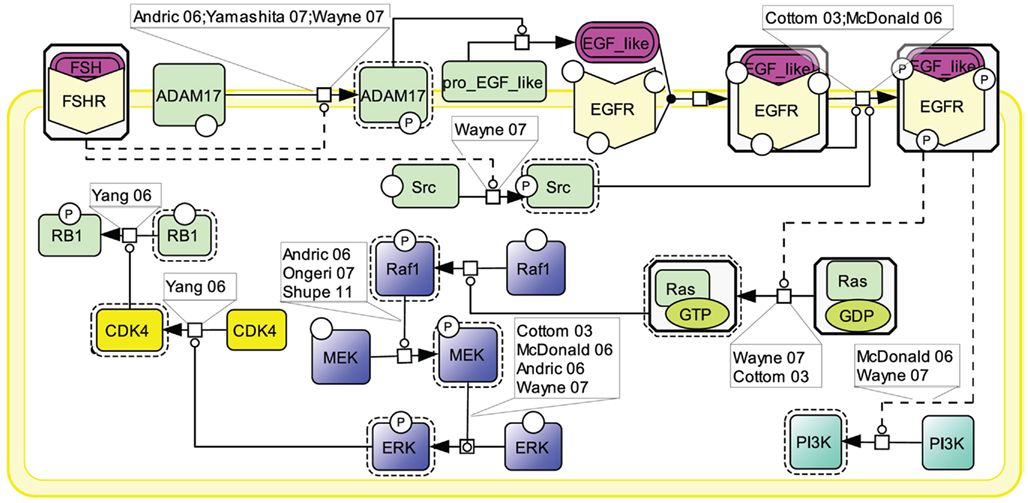
Figure 6. Transactivation of the EGFR by FSH. Many lines of evidence point to EGFR transactivation as an important transduction mechanism used by FSHR. Two mechanisms have been reported: first, FSH stimulates the ADAM17 metalloprotease that leads to the release of an EGF-like ligand, which subsequently binds to and activates EGFR; second, FSH activates Src which directly phosphorylates EGFR, thereby eliciting EGF-independent activation of this receptor. MAPK ERK and PI3K pathways have been shown to be activated as a result of EGFR transactivation by FSHR.
When EGFR activation is pharmacologically inhibited, a significant decrease in phosphorylated ERK is observed (Cottom et al., 2003; Andric and Ascoli, 2006; McDonald et al., 2006; Wayne et al., 2007; Shupe et al., 2011). EGFR transactivation activates Ras which in turn induces the Raf1/MEK/ERK MAP kinase module, ultimately resulting in ERK phosphorylation (Cottom et al., 2003; Andric and Ascoli, 2006; McDonald et al., 2006; Ongeri et al., 2007; Wayne et al., 2007; Shupe et al., 2011). In granulosa cells, it has been shown that, upon sustained FSH exposure, a self-activation loop involving the MAP kinase module as well as phospholipase A2 and PKC, leads to CDK4 stimulation, retinoblastoma (RB1) inactivation, and ultimately DNA synthesis (Yang and Roy, 2006). EGFR transactivation has also been reported to trigger PI3K (Wayne et al., 2007).
Nuclear Events Controlled by FSH
For simplification purpose, we decided to aggregate all the knowledge accumulated in the literature about FSH-induced nuclear events in a separate map (Figure 7). Gene transcription has long been known to be affected by FSH, as the hormone is known to control for instance genes implicated in steroidogenesis (Escamilla-Hernandez et al., 2008a) or in cell cycle via Smad proteins (Wang et al., 2011).
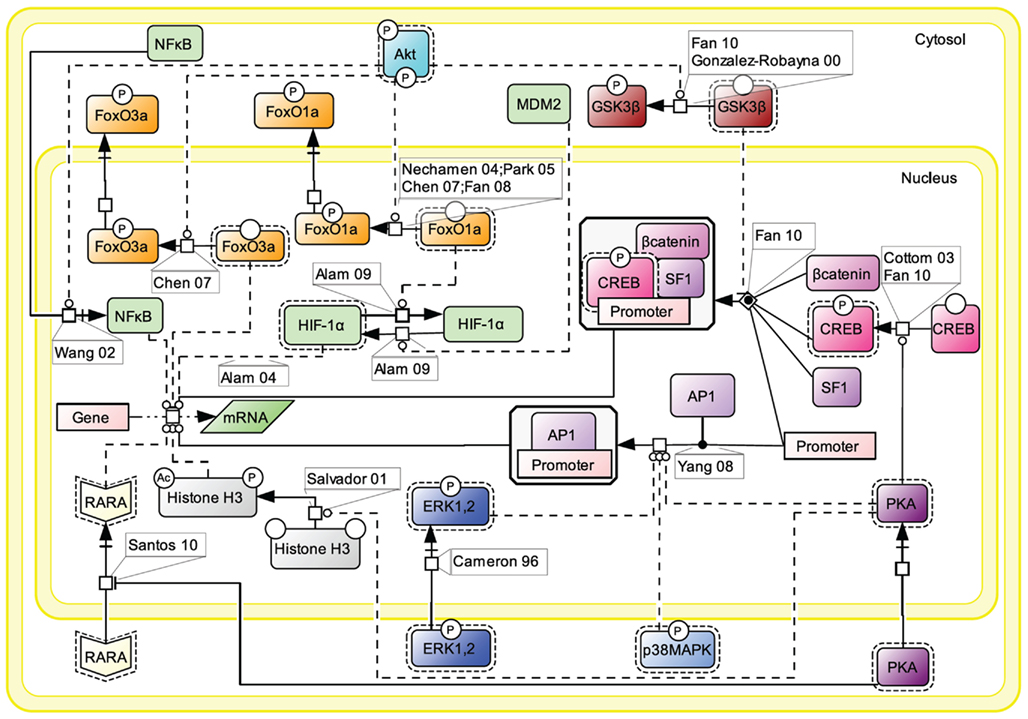
Figure 7. Nuclear events controlled by FSH. Gene transcription has long been known to be affected by FSH. Multiple signaling pathways that are activated upon stimulation (i.e., PKA, p38, ERK, and Akt) subsequently trigger the activation or suppression of various transcription factors’ activities at the nuclear level.
Upon FSH activation, PKA catalytic subunit translocates to the nucleus, where it activates CREB through phosphorylation on S133, thereby controlling cAMP response element (CRE)-containing genes (Cottom et al., 2003; Fan et al., 2010). It also promotes the recruitment of the activator protein 1 (AP1) transcription factor to their cognate promoter regions (Yang et al., 2008). Nuclear PKA also leads to histone H3 phosphorylation and acetylation (Salvador et al., 2001). Histone H3 phosphorylation is known to favor cell division (Hans and Dimitrov, 2001) and ChIP experiments have revealed FSH-induced interaction between phosphorylated, acetylated histone H3 and the promoter region of c-Fos, a well-known signal transducer of cell proliferation and differentiation (Salvador et al., 2001). Interestingly, it has recently been reported that cytosolic PKA is able to bind the retinoic acid receptor alpha (RARA) and by doing so, to inhibit RARA translocation to the nucleus, hence the effects this nuclear receptor exerts on gene transcription (Santos and Kim, 2010).
Akt controls the activity of several transcription factors through PKA-independent mechanisms. Akt has been reported to suppress the inhibition of some transcription factors and to inhibit the activity of some others. For example, upon FSH exposure, Akt phosphorylates FoxO1a which is then excluded from the nucleus and is not able to inhibit MDM2-dependent HIF-1α activation anymore (Nechamen et al., 2004; Park et al., 2005; Chen et al., 2007; Fan et al., 2008; Alam et al., 2009). Akt also phosphorylates FoxO3a provoking its exclusion from the nucleus, blocking the pro-apoptotic effects of this transcription factor (Chen et al., 2007). In addition, Akt has been reported to promote GSK3β phosphorylation and deactivation which indirectly favors LEF-dependent transcription (Gonzalez-Robayna et al., 2000; Fan et al., 2010). The nuclear translocation of NFκB is also promoted by Akt (Wang et al., 2002).
Once phosphorylated, MAP kinases ERK and p38 affect various transcriptional regulations in the nucleus (Cameron et al., 1996), participating to the regulation of AP1 and CREB activities.
Future Directions
Reconstructing the very complex intracellular networks triggered by FSH from the literature represents a huge challenge, partly due to the tremendous heterogeneity of experimental approaches and models, and partly because of the subjectivity and “bias” which cannot be avoided when manually assembling such network topologies. In the future, automated methods will probably allow building complex networks from the available data in a completely unbiased manner. In parallel, mass-spectrometry-based high-throughput methods now allow to grasp the phosphoproteome with an unprecedented power and in an unbiased fashion. Data generated using such experimental approaches will soon become available for FSH-responsive cells and will probably deliver an exhaustive coverage of the FSH-mediated signaling mechanisms induced in different cellular context. It will also be of importance to continue to bridge the gap between intracellular signaling pathways and integrated cellular responses such as gene regulation, metabolism, differentiation, proliferation, or apoptosis. Once the architecture of the FSH-induced signaling network will be deciphered, dynamical modeling will allow numerical simulations to be carried out and functional prediction to be made. In that context, the generation of high quality dynamical data of the key FSH-induced signaling events will likely become a prerequisite in order to calibrate and validate the dynamical models.
Together, these technological breakthroughs will likely help reconstructing the global FSH-induced signaling mechanisms, predicting the way it processes information and delivers adapted physiological outcomes. It is expected that all these computational resources will help rationalize the development of pathway-selective pharmacological approaches at the FSHR. In addition, several genetic studies have reported a number of mutations and polymorphisms in the FSHR. Until now, the functional consequences of these genetic alterations were evaluated solely by measuring their effects on the classical Gs/cAMP/PKA pathway. However, as we have recently shown for the A189V mutation of the FSHR, multiplexed assessment of the mutants’ and/or variants’ functionality can allow uncovering subtle perturbations/imbalances within the signaling networks (Tranchant et al., 2011). Therefore, with the availability of computational and experimental tools, it will become possible to achieve better assessment of the real functional impact of genetic alterations encountered, not only in the FSH or FSHR but also anywhere within the activated networks.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Action d’Envergure (AE) INRIA/INRA Regate, and the ASAM project (INRA/INRIA). Pauline Gloaguen is recipient of a thesis fellowship from Région Centre.
References
Agrawal, G., and Dighe, R. R. (2009). Critical involvement of the hinge region of the follicle-stimulating hormone receptor in the activation of the receptor. J. Biol. Chem. 284, 2636–2647.
Aittomaki, K., Lucena, J. L., Pakarinen, P., Sistonen, P., Tapanainen, J., Gromoll, J., Kaskikari, R., Sankila, E. M., Lehvaslaiho, H., Engel, A. R., Nieschlag, E., Huhtaniemi, I., and De La Chapelle, A. (1995). Mutation in the follicle-stimulating hormone receptor gene causes hereditary hypergonadotropic ovarian failure. Cell 82, 959–968.
Alam, H., Maizels, E. T., Park, Y., Ghaey, S., Feiger, Z. J., Chandel, N. S., and Hunzicker-Dunn, M. (2004). Follicle-stimulating hormone activation of hypoxia-inducible factor-1 by the phosphatidylinositol 3-kinase/AKT/Ras homolog enriched in brain (Rheb)/mammalian target of rapamycin (mTOR) pathway is necessary for induction of select protein markers of follicular differentiation. J. Biol. Chem. 279, 19431–19440.
Alam, H., Weck, J., Maizels, E., Park, Y., Lee, E. J., Ashcroft, M., and Hunzicker-Dunn, M. (2009). Role of the phosphatidylinositol-3-kinase and extracellular regulated kinase pathways in the induction of hypoxia-inducible factor (HIF)-1 activity and the HIF-1 target vascular endothelial growth factor in ovarian granulosa cells in response to follicle-stimulating hormone. Endocrinology 150, 915–928.
Andric, N., and Ascoli, M. (2006). A delayed gonadotropin-dependent and growth factor-mediated activation of the extracellular signal-regulated kinase 1/2 cascade negatively regulates aromatase expression in granulosa cells. Mol. Endocrinol. 20, 3308–3320.
Arey, B. J., Stevis, P. E., Deecher, D. C., Shen, E. S., Frail, D. E., Negro-Vilar, A., and Lopez, F. J. (1997). Induction of promiscuous G protein coupling of the follicle-stimulating hormone (FSH) receptor: a novel mechanism for transducing pleiotropic actions of FSH isoforms. Mol. Endocrinol. 11, 517–526.
Arey, B. J., Yanofsky, S. D., Claudia Perez, M., Holmes, C. P., Wrobel, J., Gopalsamy, A., Stevis, P. E., Lopez, F. J., and Winneker, R. C. (2008). Differing pharmacological activities of thiazolidinone analogs at the FSH receptor. Biochem. Biophys. Res. Commun. 368, 723–728.
Cameron, M. R., Foster, J. S., Bukovsky, A., and Wimalasena, J. (1996). Activation of mitogen-activated protein kinases by gonadotropins and cyclic adenosine 5′-monophosphates in porcine granulosa cells. Biol. Reprod. 55, 111–119.
Chen, Y.-J., Hsiao, P.-W., Lee, M.-T., Mason, J. I., Ke, F.-C., and Hwang, J.-J. (2007). Interplay of PI3K and cAMP/PKA signaling, and rapamycin-hypersensitivity in TGFbeta1 enhancement of FSH-stimulated steroidogenesis in rat ovarian granulosa cells. J. Endocrinol. 192, 405–419.
Choi, J.-H., Chen, C.-L., Poon, S. L., Wang, H.-S., and Leung, P. C. K. (2009). Gonadotropin-stimulated epidermal growth factor receptor expression in human ovarian surface epithelial cells: involvement of cyclic AMP-dependent exchange protein activated by cAMP pathway. Endocr. Relat. Cancer 16, 179–188.
Choi, J. H., Wong, A. S., Huang, H. F., and Leung, P. C. (2007). Gonadotropins and ovarian cancer. Endocr. Rev. 28, 440–461.
Cohen, B. D., Nechamen, C. A., and Dias, J. A. (2004). Human follitropin receptor (FSHR) interacts with the adapter protein 14-3-3tau. Mol. Cell. Endocrinol. 220, 1–7.
Cottom, J., Salvador, L. M., Maizels, E. T., Reierstad, S., Park, Y., Carr, D. W., Davare, M. A., Hell, J. W., Palmer, S. S., Dent, P., Kawakatsu, H., Ogata, M., and Hunzicker-Dunn, M. (2003). Follicle-stimulating hormone activates extracellular signal-regulated kinase but not extracellular signal-regulated kinase kinase through a 100-kDa phosphotyrosine phosphatase. J. Biol. Chem. 278, 7167–7179.
Crépieux, P., Marion, S., Martinat, N., Fafeur, V., Vern, Y. L., Kerboeuf, D., Guillou, F., and Reiter, E. (2001). The ERK-dependent signalling is stage-specifically modulated by FSH, during primary Sertoli cell maturation. Oncogene 20, 4696–4709.
Cunningham, M. A., Zhu, Q., Unterman, T. G., and Hammond, J. M. (2003). Follicle-stimulating hormone promotes nuclear exclusion of the forkhead transcription factor FoxO1a via phosphatidylinositol 3-kinase in porcine granulosa cells. Endocrinology 144, 5585–5594.
Dattatreyamurty, B., Figgs, L. W., and Reichert, L. E. (1987). Physical and functional association of follitropin receptors with cholera toxin-sensitive guanine nucleotide-binding protein. J. Biol. Chem. 262, 11737–11745.
Dias, J. A., Bonnet, B., Weaver, B. A., Watts, J., Kluetzman, K., Thomas, R. M., Poli, S., Mutel, V., and Campo, B. (2011). A negative allosteric modulator demonstrates biased antagonism of the follicle stimulating hormone receptor. Mol. Cell. Endocrinol. 333, 143–150.
Dias, J. A., Mahale, S. D., Nechamen, C. A., Davydenko, O., Thomas, R. M., and Ulloa-Aguirre, A. (2010). Emerging roles for the FSH receptor adapter protein APPL1 and overlap of a putative 14-3-3tau interaction domain with a canonical G-protein interaction site. Mol. Cell. Endocrinol. 329, 17–25.
Dias, J. A., Nechamen, C. A., and Atari, R. (2005). Identifying protein interactors in gonadotropin action. Endocrine 26, 241–247.
Dierich, A., Sairam, M. R., Monaco, L., Fimia, G. M., Gansmuller, A., Lemeur, M., and Sassone-Corsi, P. (1998). Impairing follicle-stimulating hormone (FSH) signaling in vivo: targeted disruption of the FSH receptor leads to aberrant gametogenesis and hormonal imbalance. Proc. Natl. Acad. Sci. U.S.A. 95, 13612–13617.
Dupont, J., Musnier, A., Decourtye, J., Boulo, T., Lécureuil, C., Guillou, H., Valet, S., Fouchécourt, S., Pitetti, J.-L., Nef, S., Reiter, E., and Crépieux, P. (2010). FSH-stimulated PTEN activity accounts for the lack of FSH mitogenic effect in prepubertal rat Sertoli cells. Mol. Cell. Endocrinol. 315, 271–276.
Escamilla-Hernandez, R., Little-Ihrig, L., Orwig, K. E., Yue, J., Chandran, U., and Zeleznik, A. J. (2008a). Constitutively active protein kinase A qualitatively mimics the effects of follicle-stimulating hormone on granulosa cell differentiation. Mol. Endocrinol. 22, 1842–1852.
Escamilla-Hernandez, R., Little-Ihrig, L., and Zeleznik, A. J. (2008b). Inhibition of rat granulosa cell differentiation by overexpression of Galphaq. Endocrine 33, 21–31.
Fakunding, J. L., Tindall, D. J., Dedman, J. R., Mena, C. R., and Means, A. R. (1976). Biochemical actions of follicle-stimulating hormone in the Sertoli cell of the rat testis. Endocrinology 98, 392–402.
Fan, H.-Y., Liu, Z., Cahill, N., and Richards, J. S. (2008). Targeted disruption of Pten in ovarian granulosa cells enhances ovulation and extends the life span of luteal cells. Mol. Endocrinol. 22, 2128–2140.
Fan, H.-Y., O’Connor, A., Shitanaka, M., Shimada, M., Liu, Z., and Richards, J. S. (2010). Beta-catenin (CTNNB1) promotes preovulatory follicular development but represses LH-mediated ovulation and luteinization. Mol. Endocrinol. 24, 1529–1542.
Flores, J. A., Veldhuis, J. D., and Leong, D. A. (1990). Follicle-stimulating hormone evokes an increase in intracellular free calcium ion concentrations in single ovarian (granulosa) cells. Endocrinology 127, 3172–3179.
Gonzalez-Robayna, I. J., Falender, A. E., Ochsner, S., Firestone, G. L., and Richards, J. S. (2000). Follicle-Stimulating hormone (FSH) stimulates phosphorylation and activation of protein kinase B (PKB/Akt) and serum and glucocorticoid-lnduced kinase (Sgk): evidence for A kinase-independent signaling by FSH in granulosa cells. Mol. Endocrinol. 14, 1283–1300.
Guan, R., Wu, X., Feng, X., Zhang, M., Hebert, T. E., and Segaloff, D. L. (2010). Structural determinants underlying constitutive dimerization of unoccupied human follitropin receptors. Cell. Signal. 22, 247–256.
Hans, F., and Dimitrov, S. (2001). Histone H3 phosphorylation and cell division. Oncogene 20, 3021–3027.
Huhtaniemi, I., Ahtiainen, P., Pakarainen, T., Rulli, S. B., Zhang, F. P., and Poutanen, M. (2006). Genetically modified mouse models in studies of luteinising hormone action. Mol. Cell. Endocrinol. 252, 126–135.
Jayes, F. C., Day, R. N., Garmey, J. C., Urban, R. J., Zhang, G., and Veldhuis, J. D. (2000). Calcium ions positively modulate follicle-stimulating hormone- and exogenous cyclic 3′,5′-adenosine monophosphate-driven transcription of the P450(scc) gene in porcine granulosa cells. Endocrinology 141, 2377–2384.
Kara, E., Crépieux, P., Gauthier, C., Martinat, N., Piketty, V., Guillou, F., and Reiter, E. (2006). A phosphorylation cluster of five serine and threonine residues in the C-terminus of the follicle-stimulating hormone receptor is important for desensitization but not for beta-arrestin-mediated ERK activation. Mol. Endocrinol. 20, 3014–3026.
Kayampilly, P. P., and Menon, K. M. J. (2007). Follicle-stimulating hormone increases tuberin phosphorylation and mammalian target of rapamycin signaling through an extracellular signal-regulated kinase-dependent pathway in rat granulosa cells. Endocrinology 148, 3950–3957.
Kayampilly, P. P., and Menon, K. M. J. (2009). Follicle-stimulating hormone inhibits adenosine 5′-monophosphate-activated protein kinase activation and promotes cell proliferation of primary granulosa cells in culture through an Akt-dependent pathway. Endocrinology 150, 929–935.
Kenakin, T. (2005). New concepts in drug discovery: collateral efficacy and permissive antagonism. Nat. Rev. Drug Discov. 4, 919–927.
Kishi, H., Krishnamurthy, H., Galet, C., Bhaskaran, R. S., and Ascoli, M. (2002). Identification of a short linear sequence present in the C-terminal tail of the rat follitropin receptor that modulates arrestin-3 binding in a phosphorylation-independent fashion. J. Biol. Chem. 277, 21939–21946.
Kitano, H., Funahashi, A., Matsuoka, Y., and Oda, K. (2005). Using process diagrams for the graphical representation of biological networks. Nat. Biotechnol. 23, 961–966.
Kluetzman, K. S., Thomas, R. M., Nechamen, C. A., and Dias, J. A. (2011). Decreased degradation of internalized follicle-stimulating hormone caused by mutation of aspartic acid 6.30550 in a protein kinase-CK2 consensus sequence in the third intracellular loop of human follicle-stimulating hormone receptor. Biol. Reprod. 84, 1154–1163.
Krishnamurthy, H., Galet, C., and Ascoli, M. (2003a). The association of arrestin-3 with the follitropin receptor depends on receptor activation and phosphorylation. Mol. Cell. Endocrinol. 204, 127–140.
Krishnamurthy, H., Kishi, H., Shi, M., Galet, C., Bhaskaran, R. S., Hirakawa, T., and Ascoli, M. (2003b). Postendocytotic trafficking of the follicle-stimulating hormone (FSH)-FSH receptor complex. Mol. Endocrinol. 17, 2162–2176.
Kumar, T. R., Wang, Y., Lu, N., and Matzuk, M. M. (1997). Follicle stimulating hormone is required for ovarian follicle maturation but not male fertility. Nat. Genet. 15, 201–204.
Lai, T.-H., Lin, Y.-F., Wu, F.-C., and Tsai, Y.-H. (2008). Follicle-stimulating hormone-induced Galphah/phospholipase C-delta1 signaling mediating a noncapacitative Ca2+ influx through T-type Ca2+ channels in rat sertoli cells. Endocrinology 149, 1031–1037.
Landmark, B. F., Fauske, B., Eskild, W., Skalhegg, B., Lohmann, S. M., Hansson, V., Jahnsen, T., and Beebe, S. J. (1991). Identification, characterization, and hormonal regulation of 3′, 5′-cyclic adenosine monophosphate-dependent protein kinases in rat Sertoli cells. Endocrinology 129, 2345–2354.
Layman, L. C., Lee, E. J., Peak, D. B., Namnoum, A. B., Vu, K. V., Van Lingen, B. L., Gray, M. R., Mcdonough, P. G., Reindollar, R. H., and Jameson, J. L. (1997). Delayed puberty and hypogonadism caused by mutations in the follicle-stimulating hormone beta-subunit gene. N. Engl. J. Med. 337, 607–611.
Lazari, M. F., Liu, X., Nakamura, K., Benovic, J. L., and Ascoli, M. (1999). Role of G protein-coupled receptor kinases on the agonist-induced phosphorylation and internalization of the follitropin receptor. Mol. Endocrinol. 13, 866–878.
Lécureuil, C., Tesseraud, S., Kara, E., Martinat, N., Sow, A., Fontaine, I., Gauthier, C., Reiter, E., Guillou, F., and Crépieux, P. (2005). Follicle-stimulating hormone activates p70 ribosomal protein S6 kinase by protein kinase A-mediated dephosphorylation of Thr 421/Ser 424 in primary Sertoli cells. Mol. Endocrinol. 19, 1812–1820.
Lefkowitz, R. J., and Shenoy, S. K. (2005). Transduction of receptor signals by beta-arrestins. Science 308, 512–517.
Lin, Y. F., Tseng, M. J., Hsu, H. L., Wu, Y. W., Lee, Y. H., and Tsai, Y. H. (2006). A novel follicle-stimulating hormone-induced G alpha h/phospholipase C-delta1 signaling pathway mediating rat sertoli cell Ca2+-influx. Mol. Endocrinol. 20, 2514–2527.
Loutradis, D., Drakakis, P., Milingos, S., Stefanidis, K., and Michalas, S. (2003). Alternative approaches in the management of poor response in controlled ovarian hyperstimulation (COH). Ann. N. Y. Acad. Sci. 997, 112–119.
Loutradis, D., Elsheikh, A., Kallianidis, K., Drakakis, P., Stefanidis, K., Milingos, S., and Michalas, S. (2004). Results of controlled ovarian stimulation for ART in poor responders according to the short protocol using different gonadotrophins combinations. Arch. Gynecol. Obstet. 270, 223–226.
Lunenfeld, B. (2004). Historical perspectives in gonadotrophin therapy. Hum. Reprod. Update 10, 453–467.
Macklon, N. S., Stouffer, R. L., Giudice, L. C., and Fauser, B. C. (2006). The science behind 25 years of ovarian stimulation for in vitro fertilization. Endocr. Rev. 27, 170–207.
Maizels, E. T., Cottom, J., Jones, J. C., and Hunzicker-Dunn, M. (1998). Follicle stimulating hormone (FSH) activates the p38 mitogen-activated protein kinase pathway, inducing small heat shock protein phosphorylation and cell rounding in immature rat ovarian granulosa cells. Endocrinology 139, 3353–3356.
Marion, S., Kara, E., Crepieux, P., Piketty, V., Martinat, N., Guillou, F., and Reiter, E. (2006). G protein-coupled receptor kinase 2 and beta-arrestins are recruited to FSH receptor in stimulated rat primary Sertoli cells. J. Endocrinol. 190, 341–350.
Marion, S., Robert, F., Crepieux, P., Martinat, N., Troispoux, C., Guillou, F., and Reiter, E. (2002). G protein-coupled receptor kinases and beta arrestins are relocalized and attenuate cyclic 3′,5′-adenosine monophosphate response to follicle-stimulating hormone in rat primary Sertoli cells. Biol. Reprod. 66, 70–76.
Matthews, C. H., Borgato, S., Beck-Peccoz, P., Adams, M., Tone, Y., Gambino, G., Casagrande, S., Tedeschini, G., Benedetti, A., and Chatterjee, V. K. (1993). Primary amenorrhoea and infertility due to a mutation in the beta-subunit of follicle-stimulating hormone. Nat. Genet. 5, 83–86.
McDonald, C. A., Millena, A. C., Reddy, S., Finlay, S., Vizcarra, J., Khan, S. A., and Davis, J. S. (2006). Follicle-stimulating hormone-induced aromatase in immature rat Sertoli cells requires an active phosphatidylinositol 3-kinase pathway and is inhibited via the mitogen-activated protein kinase signaling pathway. Mol. Endocrinol. 20, 608–618.
Means, A. R., Macdougall, E., Soderling, T. R., and Corbin, J. D. (1974). Testicular adenosine 3′:5′-monophosphate-dependent protein kinase. Regulation by follicle-stimulating hormone. J. Biol. Chem. 249, 1231–1238.
Meroni, S. B., Riera, M. F., Pellizzari, E. H., and Cigorraga, S. B. (2002). Regulation of rat Sertoli cell function by FSH: possible role of phosphatidylinositol 3-kinase/protein kinase B pathway. J. Endocrinol. 174, 195–204.
Meroni, S. B., Riera, M. F., Pellizzari, E. H., Galardo, M. N., and Cigorraga, S. B. (2004). FSH activates phosphatidylinositol 3-kinase/protein kinase B signaling pathway in 20-day-old Sertoli cells independently of IGF-I. J. Endocrinol. 180, 257–265.
Musnier, A., Heitzler, D., Boulo, T., Tesseraud, S., Durand, G., Lécureuil, C., Guillou, H., Poupon, A., Reiter, E., and Crépieux, P. (2009). Developmental regulation of p70 S6 kinase by a G protein-coupled receptor dynamically modelized in primary cells. Cell. Mol. Life Sci. 66, 3487–3503.
Nakamura, K., Krupnick, J. G., Benovic, J. L., and Ascoli, M. (1998). Signaling and phosphorylation-impaired mutants of the rat follitropin receptor reveal an activation- and phosphorylation-independent but arrestin-dependent pathway for internalization. J. Biol. Chem. 273, 24346–24354.
Naz, R. K., Gupta, S. K., Gupta, J. C., Vyas, H. K., and Talwar, G. P. (2005). Recent advances in contraceptive vaccine development: a mini-review. Hum. Reprod. 20, 3271–3283.
Nechamen, C. A., Thomas, R. M., Cohen, B. D., Acevedo, G., Poulikakos, P. I., Testa, J. R., and Dias, J. A. (2004). Human follicle-stimulating hormone (FSH) receptor interacts with the adaptor protein APPL1 in HEK 293 cells: potential involvement of the PI3K pathway in FSH signaling. Biol. Reprod. 71, 629–636.
Nechamen, C. A., Thomas, R. M., and Dias, J. A. (2007). APPL1, APPL2, Akt2 and FOXO1a interact with FSHR in a potential signaling complex. Mol. Cell. Endocrinol. 260–262, 93–99.
Northup, J. K., Sternweis, P. C., Smigel, M. D., Schleifer, L. S., Ross, E. M., and Gilman, A. G. (1980). Purification of the regulatory component of adenylate cyclase. Proc. Natl. Acad. Sci. U.S.A. 77, 6516–6520.
Ongeri, E. M., Verderame, M. F., and Hammond, J. M. (2007). The TATA binding protein associated factor 4b (TAF4b) mediates FSH stimulation of the IGFBP-3 promoter in cultured porcine ovarian granulosa cells. Mol. Cell. Endocrinol. 278, 29–35.
Papkoff, H., and Ekblad, M. (1970). Ovine follicle stimulating hormone: preparation and characterization of its subunits. Biochem. Biophys. Res. Commun. 40, 614–621.
Park, Y., Maizels, E. T., Feiger, Z. J., Alam, H., Peters, C. A., Woodruff, T. K., Unterman, T. G., Lee, E. J., Jameson, J. L., and Hunzicker-Dunn, M. (2005). Induction of cyclin D2 in rat granulosa cells requires FSH-dependent relief from FOXO1 repression coupled with positive signals from Smad. J. Biol. Chem. 280, 9135–9148.
Piketty, V., Kara, E., Guillou, F., Reiter, E., and Crepieux, P. (2006). Follicle-stimulating hormone (FSH) activates extracellular signal-regulated kinase phosphorylation independently of beta-arrestin- and dynamin-mediated FSH receptor internalization. Reprod. Biol. Endocrinol. 4, 33–44.
Quintana, J., Hipkin, R. W., Sanchez-Yague, J., and Ascoli, M. (1994). Follitropin (FSH) and a phorbol ester stimulate the phosphorylation of the FSH receptor in intact cells. J. Biol. Chem. 269, 8772–8779.
Radu, A., Pichon, C., Camparo, P., Antoine, M., Allory, Y., Couvelard, A., Fromont, G., Hai, M. T., and Ghinea, N. (2010). Expression of follicle-stimulating hormone receptor in tumor blood vessels. N. Engl. J. Med. 363, 1621–1630.
Rajagopal, S., Rajagopal, K., and Lefkowitz, R. J. (2010). Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat. Rev. Drug Discov. 9, 373–386.
Rangel-Aldao, R., and Rosen, O. M. (1976). Dissociation and reassociation of the phosphorylated and nonphosphorylated forms of adenosine 3′:5′-monophosphate-dependent protein kinase from bovine cardiac muscle. J. Biol. Chem. 251, 3375–3380.
Reiter, E., and Lefkowitz, R. J. (2006). GRKs and beta-arrestins: roles in receptor silencing, trafficking and signaling. Trends Endocrinol. Metab. 17, 159–165.
Reiter, E., Marion, S., Robert, F., Troispoux, C., Boulay, F., Guillou, F., and Crepieux, P. (2001). Kinase-inactive G-protein-coupled receptor kinases are able to attenuate follicle-stimulating hormone-induced signaling. Biochem. Biophys. Res. Commun. 282, 71–78.
Salvador, L. M., Park, Y., Cottom, J., Maizels, E. T., Jones, J. C., Schillace, R. V., Carr, D. W., Cheung, P., Allis, C. D., Jameson, J. L., and Hunzicker-Dunn, M. (2001). Follicle-stimulating hormone stimulates protein kinase A-mediated histone H3 phosphorylation and acetylation leading to select gene activation in ovarian granulosa cells. J. Biol. Chem. 276, 40146–40155.
Santos, N. C., and Kim, K. H. (2010). Activity of retinoic acid receptor-alpha is directly regulated at its protein kinase A sites in response to follicle-stimulating hormone signaling. Endocrinology 151, 2361–2372.
Shimada, M., Hernandez-Gonzalez, I., Gonzalez-Robayna, I., and Richards, J. S. (2006). Paracrine and autocrine regulation of epidermal growth factor-like factors in cumulus oocyte complexes and granulosa cells: key roles for prostaglandin synthase 2 and progesterone receptor. Mol. Endocrinol. 20, 1352–1365.
Shupe, J., Cheng, J., Puri, P., Kostereva, N., and Walker, W. H. (2011). Regulation of sertoli-germ cell adhesion and sperm release by FSH and nonclassical testosterone signaling. Mol. Endocrinol. 25, 238–252.
Simoni, M., Gromoll, J., and Nieschlag, E. (1997). The follicle-stimulating hormone receptor: biochemistry, molecular biology, physiology, and pathophysiology. Endocr. Rev. 18, 739–773.
Themmen, A. P. N., and Huhtaniemi, I. T. (2000). Mutations of gonadotropins and gonadotropin receptors: elucidating the physiology and pathophysiology of pituitary-gonadal function. Endocr. Rev. 21, 551–583.
Thomas, R. M., Nechamen, C. A., Mazurkiewicz, J. E., Muda, M., Palmer, S., and Dias, J. A. (2007). Follice-stimulating hormone receptor forms oligomers and shows evidence of carboxyl-terminal proteolytic processing. Endocrinology 148, 1987–1995.
Thomas, R. M., Nechamen, C. A., Mazurkiewicz, J. E., Ulloa-Aguirre, A., and Dias, J. A. (2011). The Adapter Protein APPL1 Links FSH Receptor to inositol 1,4,5-trisphosphate production and is implicated in intracellular Ca2+ mobilization. Endocrinology 152, 1691–1701.
Tranchant, T., Durand, G., Gauthier, C., Crépieux, P., Ulloa-Aguirre, A., Royère, D., and Reiter, E. (2011). Preferential β-arrestin signalling at low receptor density revealed by functional characterization of the human FSH receptor A189 V mutation. Mol. Cell. Endocrinol. 331, 109–118.
Troispoux, C., Guillou, F., Elalouf, J. M., Firsov, D., Iacovelli, L., Blasi, A. D., Combarnous, Y., and Reiter, E. (1999). Involvement of G protein-coupled receptor kinases and arrestins in desensitization to follicle-stimulating hormone action. Mol. Endocrinol. 13, 1599–1614.
Ulloa-Aguirre, A., Crépieux, P., Poupon, A., Maurel, M.-C., and Reiter, E. (2011). Novel pathways in gonadotropin receptor signaling and biased agonism. Rev. Endocr. Metab. Disord. doi: 10.1007/s11154-011-9176-2. [Epub ahead of print].
Vloeberghs, V., Peeraer, K., Pexsters, A., and D’Hooghe, T. (2009). Ovarian hyperstimulation syndrome and complications of ART. Best Pract. Res. Clin. Obstet. Gynaecol. 23, 691–709.
Wang, W., Chen, X., Li, X., Wang, L., Zhang, H., He, Y., Wang, J., Zhao, Y., Zhang, B., and Xu, Y. (2011). Interference RNA (RNAi)-based silencing of endogenous Smad4 in porcine granulosa cells resulted in decreased FSH-mediated GCs proliferation and steroidogenesis. Reproduction 141, 643–651.
Wang, Y., Chan, S., and Tsang, B. K. (2002). Involvement of inhibitory nuclear factor-kappaB (NFkappaB)-independent NFkappaB activation in the gonadotropic regulation of X-linked inhibitor of apoptosis expression during ovarian follicular development in vitro. Endocrinology 143, 2732–2740.
Wayne, C. M., Fan, H.-Y., Cheng, X., and Richards, J. S. (2007). Follicle-stimulating hormone induces multiple signaling cascades: evidence that activation of Rous sarcoma oncogene, RAS, and the epidermal growth factor receptor are critical for granulosa cell differentiation. Mol. Endocrinol. 21, 1940–1957.
Wehbi, V., Decourtye, J., Piketty, V., Durand, G., Reiter, E., and Maurel, M.-C. (2010a). Selective modulation of follicle-stimulating hormone signaling pathways with enhancing equine chorionic gonadotropin/antibody immune complexes. Endocrinology 151, 2788–2799.
Wehbi, V., Tranchant, T., Durand, G., Musnier, A., Decourtye, J., Piketty, V., Butnev, V. Y., Bousfield, G. R., Crépieux, P., Maurel, M.-C., and Reiter, E. (2010b). Partially deglycosylated equine LH preferentially activates beta-arrestin-dependent signaling at the follicle-stimulating hormone receptor. Mol. Endocrinol. 24, 561–573.
Whalen, E. J., Rajagopal, S., and Lefkowitz, R. J. (2011). Therapeutic potential of beta-arrestin- and G protein-biased agonists. Trends. Mol. Med. 17, 126–139.
Xiao, K., Mcclatchy, D. B., Shukla, A. K., Zhao, Y., Chen, M., Shenoy, S. K., Yates, J. R. III, and Lefkowitz, R. J. (2007). Functional specialization of beta-arrestin interactions revealed by proteomic analysis. Proc. Natl. Acad. Sci. U.S.A. 104, 12011–12016.
Xiao, K., Sun, J., Kim, J., Rajagopal, S., Zhai, B., Villen, J., Haas, W., Kovacs, J. J., Shukla, A. K., Hara, M. R., Hernandez, M., Lachmann, A., Zhao, S., Lin, Y., Cheng, Y., Mizuno, K., Ma’ayan, A., Gygi, S. P., and Lefkowitz, R. J. (2010). Global phosphorylation analysis of beta-arrestin-mediated signaling downstream of a seven transmembrane receptor (7TMR). Proc. Natl. Acad. Sci. U.S.A. 107, 15299–15304.
Xu, P., and Derynck, R. (2010). Direct activation of TACE-mediated ectodomain shedding by p38 MAP kinase regulates EGF receptor-dependent cell proliferation. Mol. Cell 37, 551–566.
Yamashita, Y., Hishinuma, M., and Shimada, M. (2009). Activation of PKA, p38 MAPK and ERK1/2 by gonadotropins in cumulus cells is critical for induction of EGF-like factor and TACE/ADAM17 gene expression during in vitro maturation of porcine COCs. J. Ovarian Res. 2, 20.
Yamashita, Y., Kawashima, I., Gunji, Y., Hishinuma, M., and Shimada, M. (2010). Progesterone is essential for maintenance of Tace/Adam17 mRNA expression, but not EGF-like factor, in cumulus cells, which enhances the EGF receptor signaling pathway during in vitro maturation of porcine COCs. J. Reprod. Dev. 56, 315–323.
Yamashita, Y., Kawashima, I., Yanai, Y., Nishibori, M., Richards, J. S., and Shimada, M. (2007). Hormone-induced expression of tumor necrosis factor alpha-converting enzyme/A disintegrin and metalloprotease-17 impacts porcine cumulus cell oocyte complex expansion and meiotic maturation via ligand activation of the epidermal growth factor receptor. Endocrinology 148, 6164–6175.
Yang, P., and Roy, S. K. (2006). A novel mechanism of FSH regulation of DNA synthesis in the granulosa cells of hamster preantral follicles: involvement of a protein kinase C-mediated MAP kinase 3/1 self-activation loop. Biol. Reprod. 75, 149–157.
Yang, W., Lu, C.-L., Yu, F.-Q., Liu, T., Hu, Z.-Y., and Liu, Y.-X. (2008). Mitogen-activated protein kinase regulates FSH-induced expression of tissue-type plasminogen activator through an activator protein 1 response element. Endocrine 34, 101–107.
Yu, F.-Q., Han, C.-S., Yang, W., Jin, X., Hu, Z.-Y., and Liu, Y.-X. (2005). Activation of the p38 MAPK pathway by follicle-stimulating hormone regulates steroidogenesis in granulosa cells differentially. J. Endocrinol. 186, 85–96.
Zeleznik, A. J., Saxena, D., and Little-Ihrig, L. (2003). Protein kinase B is obligatory for follicle-stimulating hormone-induced granulosa cell differentiation. Endocrinology 144, 3985–3994.
Zhang, Q., Thomas, S. M., Lui, V. W. Y., Xi, S., Siegfried, J. M., Fan, H., Smithgall, T. E., Mills, G. B., and Grandis, J. R. (2006). Phosphorylation of TNF-alpha converting enzyme by gastrin-releasing peptide induces amphiregulin release and EGF receptor activation. Proc. Natl. Acad. Sci. U.S.A. 103, 6901–6906.
Keywords: follicle-stimulating hormone, receptor, signaling network, topological map, systems biology
Citation: Gloaguen P, Crépieux P, Heitzler D, Poupon A and Reiter E (2011) Mapping the follicle-stimulating hormone-induced signaling networks. Front. Endocrin. 2:45. doi: 10.3389/fendo.2011.00045
Received: 13 July 2011;
Accepted: 14 September 2011;
Published online: 05 October 2011.
Edited by:
Sandhya Srikant Visweswariah, Indian Institute of Science, IndiaReviewed by:
Suraj Unniappan, York University, CanadaRajan R. Dighe, Indian Institute of Science, India
Copyright: © 2011 Gloaguen, Crépieux, Heitzler, Poupon and Reiter. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Eric Reiter, INRA UMR85, CNRS-Université François Rabelais UMR6175, 37380, Nouzilly, France. e-mail:RXJpYy5SZWl0ZXJAdG91cnMuaW5yYS5mcg==