
- Seattle Children’s Hospital Research Institute, Seattle, WA, USA
One of the most striking examples of dysfunctional hypothalamic signaling of energy homeostasis is observed in patients with hypothalamic lesions leading to hypothalamic obesity (HO). This drastic condition is frequently seen in patients with craniopharyngioma (CP), an embryological tumor located in the hypothalamic and/or pituitary region, frequently causing not only hypopituitarism, but also leading to damage of medial hypothalamic nuclei due to the tumor and its treatment. HO syndrome in CP patients is characterized by fatigue, decreased physical activity, uncontrolled appetite, and morbid obesity, and is associated with insulin and leptin resistance. Mechanisms leading to the profoundly disturbed energy homeostasis are complex. This review summarizes different aspects of important clinical studies as well as data obtained in rodent studies. In addition a model is provided describing how medial hypothalamic lesion can interact simultaneously with several weight-regulating circuitries.
Hypothalamic Obesity in Patients with Craniopharyngioma: Summary of Key Clinical Findings
Excessive weight gain is one of the most distressing manifestations of hypothalamic injury associated with CP. In 30% of all patients with hypothalamic tumors, BMI is increased at the time of diagnosis (Muller et al., 2004). After surgery, hyperphagia and obesity occur on average in about 50% of all CP patients (Curtis et al., 1994; Muller et al., 2001; Muller, 2006), although study results vary from 6% (Galatzer et al., 1981) to 91% (Imura et al., 1987; Muller et al., 2004), with higher rates in patients with hypothalamic damage. In children and adolescents, the incidence of severe obesity following CP resection surgery is between 22 and 62% (Brauner et al., 1987; Sorva, 1988; Muller, 2008). Structural defects in parts of the medial hypothalamus due to surgery, irradiation, or the tumor itself often cause treatment resistant uncontrolled appetite and rapid weight gain. Tumor resection with and without radiotherapy represents the therapeutic standard of care. But following treatment, patients often suffer from partial hypopituitarism or panhypopituitarism. Despite optimal endocrine management, uncontrolled appetite and rapid weight gain are common, typically most pronounced during the first months after surgery (Sorva, 1988; Muller et al., 2004), making severe obesity a major risk factor for CP related morbidity. Risk factors for developing obesity in CP patients include large hypothalamic lesions affecting several medial hypothalamic nuclei such as the arcuate nucleus (ARC), ventromedial nucleus (VMN), and the dorsomedial nucleus (DMN), and tumors that reach the floor of the third ventricle; hydrocephalus; transcranial surgery, which often causes greater damage to the hypothalamus compared to transnasal surgical tumor removal; aggressiveness of resection; reoperation for tumor recurrence; and hypothalamic irradiation (Muller et al., 2007; Gardner et al., 2008; Vinchon et al., 2009; Roth et al., 2011c; Figure 1).
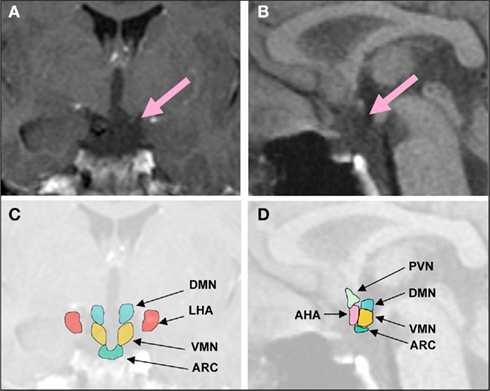
Figure 1. Representative coronal (A), midsagittal brain MRI (B), and schematic overview (C,D) of a patient with hypothalamic obesity and hyperphagia following surgery for CP. Lesion affects the ARC, VMN, DMN, and the posterior part of the AHA, but not the LHA. DMN, dorsomedial nucleus; LHA, lateral hypothalamus; VMN, ventromedial nucleus; PVN, paraventricular nucleus; ARC, arcuate nucleus; AHA, anterior hypothalamic area.
Key Brain Areas and Neuronal Pathways Involved in Energy Homeostasis
Hypothalamic Nuclei and Weight Regulatory Pathways
The hypothalamus is central to regulation of energy homeostasis. Hypothalamic nuclei are the target of circulating peptides, including leptin, insulin, peptide YY3–36 (PYY), and ghrelin, as well as nutrients such as free fatty acids, glucose, and amino acids (Schwartz et al., 2000; Cota et al., 2006; Morton et al., 2006; Parton et al., 2007). Changes in the circulating level of these afferent inputs can trigger potent efferent responses affecting both food intake (FI) and energy expenditure (EE) in the regulation of energy homeostasis. It is well established that insulin and leptin are two peripherally secreted hormones that play critical roles in FI and EE (Schwartz et al., 2000) and that levels of both are proportionate to body fat and interact with their respective receptors expressed in the ARC, a key brain area transmitting peripheral weight-regulating signals to the brain. Many neurons are regulated by leptin and insulin, and central administration of either hormone reduces FI and body weight. The main mediators in the hypothalamic regulation of energy intake and EE appear to be proopiomelanocortin (POMC) and neuropeptide-Y/agouti-related peptide (NPY/AgRP) producing neurons, which are concentrated in the ARC and project mainly to the paraventricular nucleus (PVN) in the hypothalamus but also to the VMN, lateral hypothalamus (LHA), as well as the DMN and the median preoptic area (Bai et al., 1985; Kerkerian and Pelletier, 1986; Schwartz et al., 2000; Schwartz and Gelling, 2002). Alpha-melanocyte stimulating hormone (α-MSH), a posttranslational product of the prohormone POMC, is one of the key weight-regulating neuropeptides (Bai et al., 1985; Kerkerian and Pelletier, 1986; Schwartz et al., 2000; Schwartz and Gelling, 2002). The VMN contains gamma-amino butyric acid- (GABA)ergic and anorexigenic brain-derived neurotrophic factor (BDNF) neurons and is an important nucleus for insulin dependent neurotransmission and is an important site for the regulation of the autonomous nervous system outflow from the brain (Morton et al., 2006; Berthoud and Morrison, 2008). The DMN contains GABAergic neurons but also expresses NPY in particular in its compact region and expresses leptin receptors such as the ARC, VMN, and PVN (Choi et al., 1999; Glavas et al., 2007). NPY in the DMN may play a role in maintaining energy homeostasis (Bi, 2007). The PVN receives projections from NPY-synthesizing neurons located in the ARC, which are also present in the brainstem (Sawchenko et al., 1985; Sahu et al., 1988). Neurons that synthesize the hunger hormones, MCH or orexin A, are located in the LHA and are downstream mediators of feeding responses initiated by ARC neurons (Schwartz, 2006). The PVN contains a variety of appetite suppressing peptides, such as corticotropin-releasing factor (CRF), thyrotropin-releasing hormone (TRH), and oxytocin, that promote negative energy balance by conveying input from the ARC to other key brain areas such as the NTS (Schwartz, 2006; Blevins et al., 2009). Recent evidence also suggests that the adiposity hormones act in brain areas linked with food reward, which mediate motivation and the decision to eat. (Petrovich et al., 2002; Figlewicz and Benoit, 2009). Limbic and reward circuitry are densely innervated by neurocircuits originating in the hypothalamus and are directly responsive to ghrelin, leptin, and insulin (Figlewicz, 2003; Figlewicz et al., 2007). These structures receive significant cortical input as well, sending projections to the nucleus accumbens and pallidum, and from there, via the LHA, to motor regions charged with executing feeding behavior.
Hindbrain Signaling and Peripheral Regulators
Peptide YY3–36, glucagon-like peptide (GLP)-1, pancreatic polypeptide (PP), and cholecystokinin (CCK) are gastrointestinal-derived hormones that are released postprandially in proportion to the amount of calories ingested (Wynne et al., 2005). These anorexigenic hormones inhibit FI through neurons in the ARC, which is frequently damaged in CP patients, and/or neurons located in the hindbrain solitary tract nucleus, which is usually intact in CP patients (Batterham et al., 2002; Small et al., 2005). Additionally, plasma concentrations of orexigenic ghrelin decrease after FI. An intact vagal-brainstem-hypothalamic pathway is required in order for some of these hormones to exert their effects on FI which is controversial for PYY (Batterham et al., 2002; Small et al., 2005). Although there is published evidence that increased insulin secretion is caused by an increased vagal tone secondary to VMN lesions in humans (Lustig et al., 2003; Ahmet et al., 2006) as in rats (Bray and Nishizawa, 1978), it is unclear how the increased vagal tone and decreased sympathetic tone affect secretion of different gut peptides. It has been shown that the anorexigenic effects of peripheral administration of both PYY on FI and activation of ARC neurons are abolished following either bilateral sub-diaphragmatic total truncal vagotomy or brainstem-hypothalamic pathway transectioning in rodents (Abbott et al., 2005). In addition, it has been reported that disturbed adrenergic sympathetic dysregulation in obesity influences anorexigenic gut hormone secretion, in particular PYY release (Lin et al., 2003). Even though vagotomy is currently a therapeutic approach for treating hypothalamic obesity (HO) in CP patients (Lustig et al., 2009), from a mechanistic standpoint it remains unclear if potential benefits, e.g., reduction of hyperinsulinemia, outweigh potential problems, e.g., disturbed post-meal gut hormone release. Long acting GLP-1 agonists, and orally active inhibitors of the incretin-degrading enzyme dipeptidyl peptidase-IV are approved for treating type-2 diabetes (Bailey, 2005; DeFronzo et al., 2005), and it is compelling to test in future studies the responsiveness to peripherally injected gut peptides in HO, as these might offer human obesity treatment opportunities.
Hypothalamic Obesity: What Can be Learned from Electrolytic and Chemical Lesion Models in Rodents?
Patients with CP often complain about uncontrolled hunger. As early as, Brobeck et al. (1943) described rats with lesions in the VMN as “voracious,” whereas lesions of the LHA led to anorexia and cachexia. Electrolytic or surgical lesions in the PVN produced extreme overeating and obesity, suggesting an important role for this nucleus in energy regulation. However, conflicting results have been published. PVN lesions lead to marked stimulation of FI, but variable frequencies of obesity (Tokunaga et al., 1991, 1993). VMN-lesioned rats showed lower activity levels and disturbed circadian rhythms, whereas rats with PVN lesions did not (Tokunaga et al., 1991). Rats treated at neonatal age with monosodium L-glutamate (MSG) develop neuroendocrine and metabolic abnormalities, resulting in a phenotype of adiposity characterized by GH deficiency (Kubota et al., 1994), hyperinsulinemia (de Souza et al., 2003), and hyperleptinemia, due to leptin resistance (Perello et al., 2004). Vagotomy decreased high insulin secretion induced in MSG-obese rats (Balbo et al., 2002). Neonatal administration of MSG to rodents and monkeys destroyed 80–90% of the ARC, showing cell rarification and cell death most significantly in the rostromedial ARC, but also in the adjacent VMN where cell density was not distinctly reduced (Schoelch et al., 2002), resulting in several neuroendocrine and metabolic abnormalities and decreased thermogenesis. In hamster studies, MSG decreased numbers of hypothalamic neuropeptide-Y (NPY)- and agouti-related peptide (AgRP) fibers in the PVN, because the ARC has projections to the PVN, including α-MSH projections (Leitner and Bartness, 2008). MSG-treated animals develop an obesity syndrome in the absence of hyperphagia (Dawson et al., 1989; Elfers et al., 2011; Roth et al., 2011b), characterized by excess fat deposition and reduced lean body mass, accompanied by reduced metabolic rates, due to decreased sympathetic activation of brown adipose tissue (Poon and Cameron, 1978). Recently we developed a rat model of combined medial hypothalamic lesion (CMHL) affecting the VMN, DMN, and the ARC. Only the CMHL model exhibited all key features observed in patients with HO induced by CP (Elfers et al., 2011; Roth et al., 2011b). These features included excessive weight gain due to increased adiposity, increased FI, and pronounced hyperinsulinemia and hyperleptinemia. Similar to findings in CP patients (Roth et al., 2010), CMHL animals exhibited reduced plasma levels of alpha-melanocyte stimulating hormone and reduced ambulatory activity compared to weight-matched controls. Ablation of the ARC/VMN can destroy the leptin sensitive melanocortin ARC–PVN circuit affecting satiety signaling not only in the hypothalamus but also in the hindbrain (see model Figure 2). The CMHL model best mimics the complex metabolic abnormalities observed in obese CP patients compared to lesions to other hypothalamic areas and provides a foundation for future pharmacological approaches to treat obesity in children with hypothalamic damage.
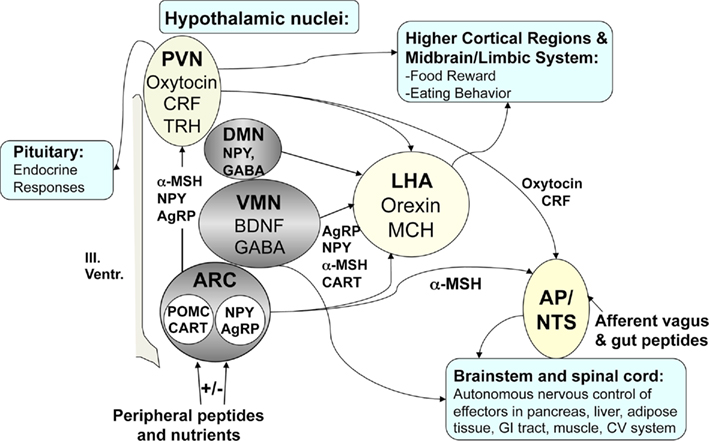
Figure 2. Key regulators of energy homeostasis in the brain. Model of weight regulatory circuits and hypothalamic nuclei potentially affected by craniopharyngioma and surgery (gray area). The lesion of medial hypothalamic nuclei (ARC, VMN, DMN) typically does not involve the lateral hypothalamus (LHA) potentially leading to unbalanced expression and secretion of orexigens (MCH, orexigens A and B) by the LHA. Additionally the communication to (1) higher cortical brain areas and limbic system involved in food reward and eating behavior, (2) pituitary, and (3) hindbrain nuclei (AP, area postrema, NTS: solitary tract nucleus) affecting the autonomous nervous control of effectors in pancreas, liver, adipose tissue, GI tract, muscle, CV system might be affected as well. CRF, corticotrophin relasing factor; TRH, thyrotropin-releasing hormone; NPY, neuropeptide-Y; GABA, gamma aminobutyric acid; BDNF, brain-derived neurotrophic factor; POMC, proopiomelanocortin; AgRP, agouti-related peptide; CART, cocaine amphetamine regulated transcript; α-MSH, alpha-melanocyte stimulating hormone.
Hypothalamic Obesity: Results from Studies in Humans
Imaging Studies and Food Reward
Risk factors for developing obesity in CP patients include: large hypothalamic lesions and tumors that reach the floor of the third ventricle; hydrocephalus; aggressive resection; and hypothalamic irradiation (Muller et al., 2007; Gardner et al., 2008; Vinchon et al., 2009). Extensive hypothalamic infiltration and a completely deficient third ventricle have been associated with the highest risk of obesity (DeVile et al., 1996; Pascual et al., 2008). however, results are not entirely consistent (Daousi et al., 2005). Disturbance of the hypothalamus by the tumor itself (Vinchon et al., 2009) and/or its radiation/surgical treatment has been discussed as a major cause of hyperphagia and obesity. In our own clinical series we found in patients with morbid obesity lesions affecting several medial hypothalamic nuclei such as ARC, VMN, and DMN (representative picture is shown in Figure 1). Functional magnetic resonance imaging (fMRI) is a powerful tool for observing the human brain’s in vivo responses to stimuli. The question is whether damage to homeostatic centers, such as medial hypothalamic nuclei by CP, also alters eating behavior by alteration of neuronal activity in food reward brain areas (see for schematic overview Figure 2) and whether central processing of satiety is affected. In a pilot study, we utilized fMRI to examine food-related activity in brain centers that control appetite. We hypothesized that hypothalamic damage due to CP and its treatment results in enhanced perception of food reward. Participants viewed blocked photographs of food and mean z-scores for regions of interest (ROI; bilateral N. accumbens, insula, striatum, and medial orbitofrontal cortex) were calculated for the contrast of fattening > non-fattening food. Subjects underwent the first fMRI scan, received a high-calorie test meal to suppress appetite, and had a second fMRI scan 30 min after the test meal. Before and after the scans, hunger ratings were performed. Following the test meal, controls showed suppression of activation in ROIs while CP patients showed trends toward higher activation in ROIs (Roth et al., 2011a). These preliminary data support our hypothesis that perception of food cues may be altered in HO, especially after eating. The fMRI approach may be applicable for performing future mechanistic studies of the brains’ response to food in patients with HO.
Central and Peripheral Peptides Involved in Energy Homeostasis
There is good evidence for alterations in peripheral hormone signaling in CP. Previous clinical studies showed that hypothalamic tumor involvement leads to greater leptin resistance in patients with CP than in obese controls (Roth et al., 1998). CP patients are reported to be less physically active than controls with comparable BMIs (Harz et al., 2003) and catecholamine levels are lower in CP patients, indicating low sympathetic tone (Roth et al., 2007). Plasma α-MSH levels were significantly reduced in CP, similar to findings in our CMHL rat model, which might explain lower EE in peripheral tissues via reduced fat and muscle fatty acid oxidation (Roth et al., 2010). In an ancillary study to the German multicenter study Kraniopharyngeom 2000, plasma insulin, and gut hormone responses, such as ghrelin and PYY, in response to meal tests were assessed in lean and obese CP patients and BMI matched controls. Obese CP subjects had significantly higher baseline and post-meal insulin and weaker post-meal changes for PYY compared to obese controls. Patients with hypothalamic CP had significantly weaker post-meal reduction of ghrelin levels than CP without hypothalamic localization (Roth et al., 2011c). In sum, prior studies of HO have shown impaired function of peripherally released hormones that act centrally to modulate responses to food (see Figure 3).
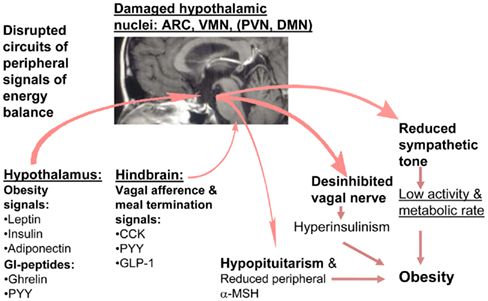
Figure 3. Hypothetical model of disturbed energy homeostasis in hypothalamic obesity.
Summary and Etiological Theories
Damage of medial hypothalamic nuclei due to the tumor and its treatment is frequently seen in patients with CP leading to HO characterized by fatigue, decreased physical activity, uncontrolled appetite, and morbid obesity, and is associated with insulin and leptin resistance. Disruption of feeding circuits by damage of medial hypothalamic nuclei has the potential to increase hunger, by (i) enhancing the response to orexigenic signals such as ghrelin and by stimulating the orexigenic signaling through NPY/AgRP or (ii) blocking the response to adiposity signals such as leptin and inhibiting the anorexigenic POMC signaling in the hypothalamus (Figure 2). In either case, the result can involve unopposed activation of the LHA which increases FI while decreasing sympathetic nervous system (SNS)-mediated stimulation of EE. VMN damage can lead to disinhibition of the vagal tone, resulting in excess stimulation of pancreatic β-cells, hyperinsulinemia, and obesity (Lustig et al., 2003). Alternatively, the sympathetic nervous output might be reduced, leading to decreased physical activity (Harz et al., 2003; Roth et al., 2007). As the hypothalamic damage in CP frequently includes the ARC, which is the main hypothalamic binding site for hormones from the periphery (Figures 2 and 3), the hypothalamus might be unable to respond normally to adiposity and satiety signals such as insulin, leptin, and gut hormones, including PYY and ghrelin (Stellar, 1954; Blundell, 1982, 1990; Inge et al., 2007). In addition, excessive weight gain could be the result of deficient hypothalamic neural circuits communicating with corticolimbic and other brain regions to regulate appetite and thus leading to unbalanced eating (see summary Figure 4).
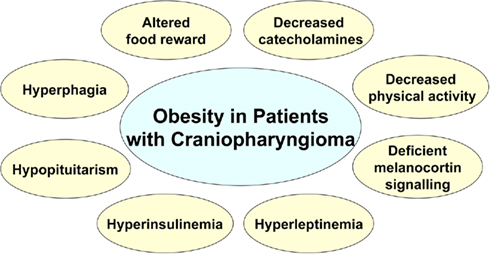
Figure 4. Summary of metabolic and clinical findings involved in dysregulated energy balance in hypothalamic obesity.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author thanks Michael W. Schwartz, and Gregory Morton, Diabetes and Obesity Center of Excellence, Seattle, as well as James Blevins, Department of Veterans Affairs Medical Center, Seattle, WA, USA, for their constructive ideas and comments while developing the CMHL rat model. In addition the author is grateful to Ellen Schur, Department of Medicine, University of Washington, for her support in functional neuroimaging studies, as well as to Clinton Elfers, for his expertise in performing rat studies.
References
Abbott, C. R., Monteiro, M., Small, C. J., Sajedi, A., Smith, K. L., Parkinson, J. R., Ghatei, M. A., and Bloom, S. R. (2005). The inhibitory effects of peripheral administration of peptide YY(3-36) and glucagon-like peptide-1 on food intake are attenuated by ablation of the vagal-brainstem-hypothalamic pathway. Brain Res. 1044, 127–131.
Ahmet, A., Blaser, S., Stephens, D., Guger, S., Rutkas, J. T., and Hamilton, J. (2006). Weight gain in craniopharyngioma – a model for hypothalamic obesity. J. Pediatr. Endocrinol. Metab. 19, 121–127.
Bai, F. L., Yamano, M., Shiotani, Y., Emson, P. C., Smith, A. D., Powell, J. F., and Tohyama, M. (1985). An arcuato-paraventricular and -dorsomedial hypothalamic neuropeptide Y-containing system which lacks noradrenaline in the rat. Brain Res. 331, 172–175.
Balbo, S. L., Bonfleur, M. L., Carneiro, E. M., Amaral, M. E., Filiputti, E., and Mathias, P. C. (2002). Parasympathetic activity changes insulin response to glucose and neurotransmitters. Diabetes Metab. 28, 3S13–3S17; discussion 3S108–3S112.
Batterham, R. L., Cowley, M. A., Small, C. J., Herzog, H., Cohen, M. A., Dakin, C. L., Wren, A. M., Brynes, A. E., Low, M. J., Ghatei, M. A., Cone, R. D., and Bloom, S. R. (2002). Gut hormone PYY(3-36) physiologically inhibits food intake. Nature 418, 650–654.
Berthoud, H. R., and Morrison, C. (2008). The brain, appetite, and obesity. Annu. Rev. Psychol. 59, 55–92.
Bi, S. (2007). Role of dorsomedial hypothalamic neuropeptide Y in energy homeostasis. Peptides 28, 352–356.
Blevins, J. E., Morton, G. J., Williams, D. L., Caldwell, D. W., Bastian, L. S., Wisse, B. E., Schwartz, M. W., and Baskin, D. G. (2009). Forebrain melanocortin signaling enhances the hindbrain satiety response to CCK-8. Am. J. Physiol. Regul. Integr. Comp. Physiol. 296, R476–R484.
Blundell, J. E. (1982). Neuroregulators and feeding: implications for the pharmacological manipulation of hunger and appetite. Rev. Pure Appl. Pharmacol. Sci. 3, 381–462.
Blundell, J. E. (1990). Appetite disturbance and the problems of overweight. Drugs 39(Suppl. 3), 1–19.
Brauner, R., Malandry, F., Rappaport, R., Pierre-Kahn, A., and Hirsch, J. F. (1987). Craniopharyngioma in children. Endocrine evaluation and treatment. Apropos of 37 cases. Arch. Fr. Pediatr. 44, 765–769.
Bray, G. A., and Nishizawa, Y. (1978). Ventromedial hypothalamus modulates fat mobilisation during fasting. Nature 274, 900–902.
Brobeck, J. R., Tepperman, J., and Long, C. N. H. (1943). Experimental hypothalamic hyperphagia in the albino rat. Yale J. Biol. Med. 15, 831–853.
Choi, S., Sparks, R., Clay, M., and Dallman, M. F. (1999). Rats with hypothalamic obesity are insensitive to central leptin injections. Endocrinology 140, 4426–4433.
Cota, D., Proulx, K., Smith, K. A., Kozma, S. C., Thomas, G., Woods, S. C., and Seeley, R. J. (2006). Hypothalamic mTOR signaling regulates food intake. Science 312, 927–930.
Curtis, J., Daneman, D., Hoffman, H. J., and Ehrlich, R. M. (1994). The endocrine outcome after surgical removal of craniopharyngiomas. Pediatr. Neurosurg. 21(Suppl. 1), 24–27.
Daousi, C., Dunn, A. J., Foy, P. M., MacFarlane, I. A., and Pinkney, J. H. (2005). Endocrine and neuroanatomic features associated with weight gain and obesity in adult patients with hypothalamic damage. Am. J. Med. 118, 45–50.
Dawson, R. Jr., Wallace, D. R., and Gabriel, S. M. (1989). A pharmacological analysis of food intake regulation in rats treated neonatally with monosodium L-glutamate (MSG). Pharmacol. Biochem. Behav. 32, 391–398.
de Souza, C. T., Nunes, W. M., Gobatto, C. A., and de Mello, M. A. (2003). Insulin secretion in monosodium glutamate (MSG) obese rats submitted to aerobic exercise training. Physiol. Chem. Phys. Med. NMR 35, 43–53.
DeFronzo, R. A., Ratner, R. E., Han, J., Kim, D. D., Fineman, M. S., and Baron, A. D. (2005). Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care 28, 1092–1100.
DeVile, C. J., Grant, D. B., Kendall, B. E., Neville, B. G., Stanhope, R., Watkins, K. E., and Hayward, R. D. (1996). Management of childhood craniopharyngioma: can the morbidity of radical surgery be predicted? J. Neurosurg. 85, 73–81.
Elfers, C., Ralston, M., and Roth, C. L. (2011). Studies of different female rat models of hypothalamic obesity. J. Pediatr. Endocrinol. Metab. 24, 131–137.
Figlewicz, D. P., and Benoit, S. C. (2009). Insulin, leptin, and food reward: update 2008. Am. J. Physiol. Regul. Integr. Comp. Physiol. 296, R9–R19.
Figlewicz, D. P., MacDonald Naleid, A., and Sipols, A. J. (2007). Modulation of food reward by adiposity signals. Physiol. Behav. 91, 473–478.
Galatzer, A., Nofar, E., Beit-Halachmi, N., Aran, O., Shalit, M., Roitman, A., and Laron, Z. (1981). Intellectual and psychosocial functions of children, adolescents and young adults before and after operation for craniopharyngioma. Child Care Health Dev. 7, 307–316.
Gardner, P. A., Kassam, A. B., Snyderman, C. H., Carrau, R. L., Mintz, A. H., Grahovac, S., and Stefko, S. (2008). Outcomes following endoscopic, expanded endonasal resection of suprasellar craniopharyngiomas: a case series. J. Neurosurg. 109, 6–16.
Glavas, M. M., Joachim, S. E., Draper, S. J., Smith, M. S., and Grove, K. L. (2007). Melanocortinergic activation by melanotan II inhibits feeding and increases uncoupling protein 1 messenger ribonucleic acid in the developing rat. Endocrinology 148, 3279–3287.
Harz, K. J., Muller, H. L., Waldeck, E., Pudel, V., and Roth, C. (2003). Obesity in patients with craniopharyngioma: assessment of food intake and movement counts indicating physical activity. J. Clin. Endocrinol. Metab. 88, 5227–5231.
Imura, H., Kato, Y., and Nakai, Y. (1987). Endocrine aspects of tumors arising from suprasellar, third ventricular regions. Prog. Exp. Tumor Res. 30, 313–324.
Inge, T. H., Pfluger, P., Zeller, M., Rose, S. R., Burget, L., Sundararajan, S., Daniels, S. R., and Tschop, M. H. (2007). Gastric bypass surgery for treatment of hypothalamic obesity after craniopharyngioma therapy. Nat. Clin. Pract. Endocrinol. Metab. 3, 606–609.
Kerkerian, L., and Pelletier, G. (1986). Effects of monosodium L-glutamate administration on neuropeptide Y-containing neurons in the rat hypothalamus. Brain Res. 369, 388–390.
Kubota, A., Nakagawa, Y., and Igarashi, Y. (1994). Studies of gene expression in liver of insulin-like growth factor (IGF)-I, IGF binding protein-3 and growth hormone (GH) receptor/GH binding protein in rats treated neonatally with monosodium glutamate. Horm. Metab. Res. 26, 497–503.
Leitner, C., and Bartness, T. J. (2008). Food deprivation-induced changes in body fat mobilization after neonatal monosodium glutamate treatment. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, R775–R783.
Lin, H. C., Neevel, C., Chen, P. S., Suh, G., and Chen, J. H. (2003). Slowing of intestinal transit by fat or peptide YY depends on beta-adrenergic pathway. Am. J. Physiol. Gastrointest. Liver Physiol. 285, G1310–G1316.
Lustig, R. H., Post, S. R., Srivannaboon, K., Rose, S. R., Danish, R. K., Burghen, G. A., Xiong, X., Wu, S., and Merchant, T. E. (2003). Risk factors for the development of obesity in children surviving brain tumors. J. Clin. Endocrinol. Metab. 88, 611–616.
Lustig, R. H., Tsai, P., Hirose, S., and Farmer, D. L. (2009). “Treatment of hypothalamic obesity by laparoscopic truncal vagatomy: early experience,” in Abstract LWPES/ESPE 8th Joint Meeting Paediatric Endocrinology, Hormone Research 2009, New York, 72, S3:48.
Morton, G. J., Cummings, D. E., Baskin, D. G., Barsh, G. S., and Schwartz, M. W. (2006). Central nervous system control of food intake and body weight. Nature 443, 289–295.
Muller, H. L. (2006). More or less? Treatment strategies in childhood craniopharyngioma. Childs Nerv. Syst. 22, 156–157.
Muller, H. L. (2008). Childhood craniopharyngioma. Recent advances in diagnosis, treatment and follow-up. Horm. Res. 69, 193–202.
Muller, H. L., Bueb, K., Bartels, U., Roth, C., Harz, K., Graf, N., Korinthenberg, R., Bettendorf, M., Kuhl, J., Gutjahr, P., Sorensen, N., and Calaminus, G. (2001). Obesity after childhood craniopharyngioma – German multicenter study on pre-operative risk factors and quality of life. Clin. Padiatr. 213, 244–249.
Muller, H. L., Emser, A., Faldum, A., Bruhnken, G., Etavard-Gorris, N., Gebhardt, U., Oeverink, R., Kolb, R., and Sorensen, N. (2004). Longitudinal study on growth and body mass index before and after diagnosis of childhood craniopharyngioma. J. Clin. Endocrinol. Metab. 89, 3298–3305.
Muller, H. L., Gebhardt, U., Wessel, V., Schroder, S., Kolb, R., Sorensen, N., Maroske, J., and Hanisch, E. (2007). First experiences with laparoscopic adjustable gastric banding (LAGB) in the treatment of patients with childhood craniopharyngioma and morbid obesity. Klin. Padiatr. 219, 323–325.
Parton, L. E., Ye, C. P., Coppari, R., Enriori, P. J., Choi, B., Zhang, C. Y., Xu, C., Vianna, C. R., Balthasar, N., Lee, C. E., Elmquist, J. K., Cowley, M. A., and Lowell, B. B. (2007). Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature 449, 228–232.
Pascual, J. M., Carrasco, R., Prieto, R., Gonzalez-Llanos, F., Alvarez, F., and Roda, J. M. (2008). Craniopharyngioma classification. J. Neurosurg. 109, 1180–1182; author reply 1182–1183.
Perello, M., Moreno, G., Gaillard, R. C., and Spinedi, E. (2004). Glucocorticoid-dependency of increased adiposity in a model of hypothalamic obesity. Neuro Endocrinol. Lett. 25, 119–126.
Petrovich, G. D., Setlow, B., Holland, P. C., and Gallagher, M. (2002). Amygdalo-hypothalamic circuit allows learned cues to override satiety and promote eating. J. Neurosci. 22, 8748–8753.
Poon, T. K., and Cameron, D. P. (1978). Measurement of oxygen consumption and locomotor activity in monosodium glutamate-induced obesity. Am. J. Physiol. 234, E532–E534.
Roth, C. L., Aylward, E., Liang, O., Pauley, G., Kleinhans, N. M., and Schur, E. (2011a). Functional neuroimaging in craniopharyngioma: useful tool to better understand hypothalamic obesity? Abstract Annual Meeting The Endocrine Society, Boston.
Roth, C. L., Blevins, J. E., Ralston, M., Elfers, C., Ogimoto, K., Kaiyala, K. J., and Morton, G. J. (2011b). A novel rodent model that mimics the metabolic sequelae of obese craniopharyngioma patients. Pediatr. Res. 69, 230–236.
Roth, C. L., Gebhardt, U., and Muller, H. L. (2011c). Appetite-regulating hormone changes in patients with craniopharyngioma. Obesity (Silver Spring) 19, 36–42.
Roth, C. L., Enriori, P. J., Gebhardt, U., Hinney, A., Muller, H. L., Hebebrand, J., Reinehr, T., and Cowley, M. A. (2010). Changes of peripheral alpha-melanocyte-stimulating hormone in childhood obesity. Metab. Clin. Exp. 59, 186–194.
Roth, C. L., Hunneman, D. H., Gebhardt, U., Stoffel-Wagner, B., Reinehr, T., and Muller, H. L. (2007). Reduced sympathetic metabolites in urine of obese patients with craniopharyngioma. Pediatr. Res. 61, 496–501.
Roth, C. L., Wilken, B., Hanefeld, F., Schroter, W., and Leonhardt, U. (1998). Hyperphagia in children with craniopharyngioma is associated with hyperleptinaemia and a failure in the downregulation of appetite. Eur. J. Endocrinol. 138, 89–91.
Sahu, A., Kalra, S. P., Crowley, W. R., and Kalra, P. S. (1988). Evidence that NPY-containing neurons in the brainstem project into selected hypothalamic nuclei: implication in feeding behavior. Brain Res. 457, 376–378.
Sawchenko, P. E., Swanson, L. W., Grzanna, R., Howe, P. R., Bloom, S. R., and Polak, J. M. (1985). Colocalization of neuropeptide Y immunoreactivity in brainstem catecholaminergic neurons that project to the paraventricular nucleus of the hypothalamus. J. Comp. Neurol. 241, 138–153.
Schoelch, C., Hubschle, T., Schmidt, I., and Nuesslein-Hildesheim, B. (2002). MSG lesions decrease body mass of suckling-age rats by attenuating circadian decreases of energy expenditure. Am. J. Physiol. Endocrinol. Metab. 283, E604–E611.
Schwartz, M. W. (2006). Central nervous system regulation of food intake. Obesity (Silver Spring) 14(Suppl. 1), 1S–8S.
Schwartz, M. W., and Gelling, R. W. (2002). Rats lighten up with MCH antagonist. Nat. Med. 8, 779–781.
Schwartz, M. W., Woods, S. C., Porte, D. Jr., Seeley, R. J., and Baskin, D. G. (2000). Central nervous system control of food intake. Nature 404, 661–671.
Small, C. J., Parkinson, J. R., and Bloom, S. R. (2005). Novel therapeutic targets for appetite regulation. Curr. Opin. Investig. Drugs 6, 369–372.
Sorva, R. (1988). Children with craniopharyngioma. Early growth failure and rapid postoperative weight gain. Acta Paediatr. Scand. 77, 587–592.
Tokunaga, K., Bray, G. A., and Matsuzawa, Y. (1993). Improved yield of obese rats using a double coordinate system to locate the ventromedial or paraventricular nucleus. Brain Res. Bull. 32, 191–194.
Tokunaga, K., Matsuzawa, Y., Fujioka, S., Kobatake, T., Keno, Y., Odaka, H., Matsuo, T., and Tarui, S. (1991). PVN-lesioned obese rats maintain ambulatory activity and its circadian rhythm. Brain Res. Bull. 26, 393–396.
Vinchon, M., Weill, J., Delestret, I., and Dhellemmes, P. (2009). Craniopharyngioma and hypothalamic obesity in children. Childs Nerv. Syst. 25, 347–352.
Keywords: hypothalamic lesion, hyperphagia, morbid obesity, neuropeptides, gut hormones, autonomous nervous system
Citation: Roth CL (2011) Hypothalamic obesity in patients with craniopharyngioma: profound changes of several weight regulatory circuits. Front. Endocrin. 2:49. doi: 10.3389/fendo.2011.00049
Received: 25 August 2011;
Paper pending published: 15 September 2011;
Accepted: 19 September 2011;
Published online: 14 October 2011.
Edited by:
Hermann Lothar Mueller, Klinikum Oldenburg gGmbH, GermanyCopyright: © 2011 Roth. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Christian L. Roth, Division of Endocrinology, Seattle Children’s Hospital Research Institute, 1900 Ninth Avenue, Seattle, WA 98101, USA. e-mail:Y2hyaXN0aWFuLnJvdGhAc2VhdHRsZWNoaWxkcmVucy5vcmc=