Abstract
Neurosteroids were initially defined as steroid hormones locally synthesized within the nervous tissue. Subsequently, they were described as steroid hormone derivatives that devoid hormonal action but still affect neuronal excitability through modulation of ionotropic receptors. Neurosteroids are further subdivided into natural (produced in the brain) and synthetic. Some authors distinguish between hormonal and regular neurosteroids in the group of natural ones. The latter group, including hormone metabolites like allopregnanolone or tetrahydrodeoxycorticosterone, is devoid of hormonal activity. Both hormones and their derivatives share, however, most of the physiological functions. It is usually very difficult to distinguish the effects of hormones and their metabolites. All these substances may influence seizure phenomena and exhibit neuroprotective effects. Neuroprotection offered by steroid hormones may be realized in both genomic and non-genomic mechanisms and involve regulation of the pro- and anti-apoptotic factors expression, intracellular signaling pathways, neurotransmission, oxidative, and inflammatory processes. Since regular neurosteroids show no affinity for steroid receptors, they may act only in a non-genomic mode. Multiple studies have been conducted so far to show efficacy of neurosteroids in the treatment of the central and peripheral nervous system injury, ischemia, neurodegenerative diseases, or seizures. In this review we focused primarily on neurosteroid mechanisms of action and their role in the process of neurodegeneration. Most of the data refers to results obtained in experimental studies. However, it should be realized that knowledge about neuroactive steroids remains still incomplete and requires confirmation in clinical conditions.
Introduction
In the century of spectacular medical therapies, there is still a lack of substance that may protect the brain cells against neurodegeneration. Since the discovery of neurosteroids, scientists have made a great effort to evaluate their function, including possible neuroprotective properties. According to most commonly used definition neurosteroids cover both steroid hormones, which are synthesized de novo in neurons and glial cells, and their metabolites acting within the central nervous system (CNS) (Compagnone and Mellon, 2000) (Figure 1). Hormones may regulate brain function in an autocrine and paracrine manner via membrane or nuclear receptors (Baulieu, 1998; Baulieu et al., 2001; Plassart-Schiess and Baulieu, 2001; Singh, 2006). Regular neurosteroids act as positive or negative modulators of neurotransmitter receptors: gamma-amino butyric acid A (GABAA) receptor, N-methyl-d-aspartate (NMDA) receptor, α-amino-3-hydroxy-5-methylisoxazole propionate (AMPA) receptor, sigma-1 (σ1), nicotinic, muscarinic, kainic, serotoninergic and glycine receptors, regulate gene expression, or signaling cascades (Wojtal et al., 2006; Borowicz et al., 2008). In this article we review the main neurosteroids, including pregnenolone (PREG) and pregnenolone sulfate (PREGS), progesterone (PROG), allopregnanolone (ALLO), dehydroepiandrosterone (DHEA), DHEA sulfate (DHEAS), deoxycorticosterone (DOC), and tetrahydrodeoxycorticosterone (THDOC).
Figure 1
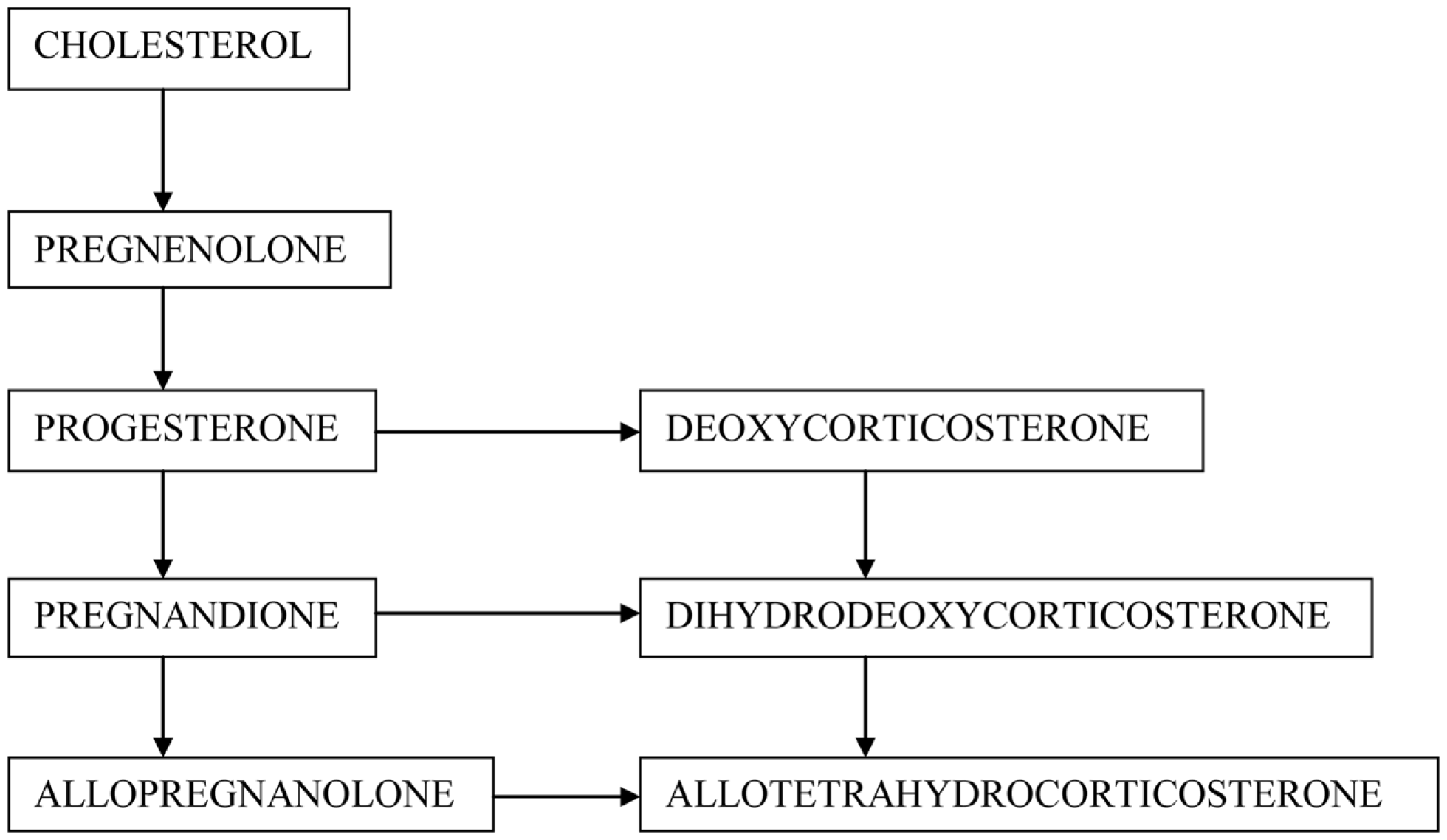
Schematic synthesis of neurosteroids in the CNS.
From the biochemical point of view, PREG is a hormone precursor, PROG, DOC, and DHEA represent hormones, while ALLO (a PROG metabolite) and THDOC (a DOC derivative) belong to neurosteroids, which are entirely devoid of hormonal action. However, their ketone metabolites may activate hormonal receptors. An additional advantage of regular neurosteroids is better bioavailability and longer half-life. Synthetic neurosteroids have even longer half-time, and may be administered orally, in contrast to natural steroids (Wojtal et al., 2006; Borowicz et al., 2008).
Neuroprotection involves mechanisms protecting against neuronal injury or degeneration in the central and peripheral nervous system. All these processes may be a consequence of acute disorders (e.g., stroke or nervous system injury) or chronic neurodegenerative processes (e.g., in the course of Alzheimer’s, Parkinson’s, and Huntington’s disease). The overriding aim is, of course, to prevent the development of a disease, but it is equally important to limit neuronal dysfunction after the CNS injury and maintain undisturbed neural function of the brain with the highest possible cellular integrity. At present, a wide range of neuroprotective products is available or under investigation. Many of them can potentially be used in more than one disorder, because most of the underlying mechanisms of neurodegeneration (in both acute and chronic disorders) seems to be quite similar.
Until recently two mechanisms of cell death were known: the programmed cell death (apoptosis) and unregulated process of necrosis. Necrosis and apoptosis differ with numerous morphological and biochemical features. In necrosis, extreme physiological conditions (e.g., hypoxia) lead to damage to the plasma membrane. The whole cell and intracellular organelles, particularly mitochondria, swell, and rapture. As a consequence of loss of membrane integrity, the cytoplasmic contents, including lysosomal enzymes, are released into the extracellular fluid. This process ends with cell lysis and is often associated with a vast tissue damage resulting from an intense inflammatory response (Majno and Joris, 1995; Kanduc et al., 2002; Van Cruchten and Van Den Broeck, 2002). Nevertheless, it has been proved that non-apoptotic forms of programmed cell death may also exist. In fact, three main types of the programmed death has been distinguished: 1. nuclear (apoptotic); 2. autophagic (lysosomal); and 3. cytoplasmic. Autophagic cell death can be activated by Ras genes, while type 3 (cytoplasmic) was shown to be triggered by stimulation of the insulin-like growth factor I receptor (IGFIR) or the binding of substance P to its neurokinin-1. This form, characterized by cytoplasmic vacuolization, lack of apoptotic morphology, and lack of caspase activation, was named paraptosis (Sperandio et al., 2000, 2004, 2010; Leist and Jäättelä, 2001; Yakovlev and Faden, 2004; Kroemer et al., 2009).
However, accumulating evidences indicate that necrotic cell death can also be a regulated process. In 2005 the term necroptosis was introduced to describe a little known yet alternative form of cell death – the programmed necrosis (Teng et al., 2005; Vandenabeele et al., 2010). Necroptosis is defined as a programmed but caspase-independent cell death that activates autophagy and morphologically resembles necrosis (Degterev et al., 2005; Teng et al., 2005). Whether this form of cell death should be equated with the programmed autophagic form remains uncertain. This process, in fact intermediate between necrosis and apoptosis, develops often as an answer to hypoxia, reperfusion, and excitotoxicity. These findings open a possibility to develop new therapeutic strategies that may extend the window for neuroprotection in such disorders as stroke, trauma and neurodegenerative diseases. Interestingly, necrostatin-1 and 5, lately identified specific and potent small-molecule inhibitors of necroptosis, were shown to block a critical step of the cell death process (Yuan et al., 2003; Degterev et al., 2005; Mehta et al., 2007; Wang et al., 2007). In fact, research on neuronal death mechanisms may help to develop an effective neuroprotective treatment and contribute to a significant improvement in patient quality of life (Charalampopoulos et al., 2008).
Pregnenolone
Pregnenolone, usually considered as a neurosteroid precursor, exhibited neuroprotective effects against glutamate- and amyloid β protein-induced neurotoxicity in clonal mouse hippocampal cell line (HT-22; Gursoy et al., 2001). It also stabilized microtubules (Hsu et al., 2006), enhanced its polymerization and activated neurite outgrowth in nerve growth factor-pretreated clonal rat pheochromocytoma cell line of neuronal crest origin, PC12 (Fontaine-Lenoir et al., 2006), improves myelination (Koenig et al., 1995). According to Leskiewicz et al. (2008) PREG reduced the staurosporine- and glutamate-induced LDH release and diminished the number of apoptotic cells in primary neuronal cortical cultures. Additionally, PREG exerted neuroprotective effects against kainate-induced cell death in the hippocampus of gonadectomized rats. The exact mechanism of PREG action is not clear, however, it may be mediated by estradiol, the main product of aromatase action. Fadrozole, an aromatase inhibitor, prevented the neuroprotective effects of both neurosteroids (Veiga et al., 2003). In patients with Alzheimer’s disease, PREG concentrations were reduced in the striatum and cerebellum. Interestingly, the negative correlation was shown between PREG and the level of cortical β-amyloid (Weill-Engerer et al., 2002; Luchetti et al., 2011). In rat hippocampal slices, PREG inhibited, or enhanced NMDA response. The former effect appeared in the presence of haloperidol and indicates an indirect σ receptor modulation. The latter one seems to be dependent on a direct NMDA receptor response (Monnet et al., 1995) (Table 1).
Table 1
| Neurosteroid | Seizures | Excitotoxicity | ND | TBI | IBI |
|---|---|---|---|---|---|
| PREG | + | + | |||
| PROG | + | + | + | + | |
| ALLO | + | + | + | + | |
| DHEA/DHEAS | + | + | |||
| DOC/THDOC | + | ||||
| Synthetic neurosteroids | + |
Leading actions of chosen neurosteroids.
PREG, pregnenolone; PROG, progesterone; ALLO, allopregnanolone; DHEA, dehydroepiandrosterone; DHEAS, dehydroepiandrosterone sulfate ester; DOC, deoxycorticosterone; THDOC, allotetrahydrodeoxycorticosterone.
Progesterone and Derivatives
Progesterone is produced primarily in females in the ovarian corpus luteum and placenta, while in males it is mostly found in testes and adrenal glands. However, this hormone may also be synthesized de novo within the nervous system. Initially, it was observed that brain concentrations of PROG maintained even in gonadectomized rats (Corpéchot et al., 1983). Then, glial cell cultures were reported to synthesize PROG and express PROG receptors (Jung-Testas et al., 1989, 1999). The hormone and its derivatives are positive modulators of GABAA receptors (Borowicz et al., 2008). PROG is synthesized in human brain at the similar level in both sexes (Stein and Wright, 2010). Identification of neuroprotective properties of this steroid began with the observation of a better recovery of female pseudopregnant rats after the traumatic brain injury (TBI; Roof et al., 1993). The authors observed the inverse correlation between serum PROG concentration and degree of brain edema after injury (Wright et al., 2001). In male and normal cycling female rats, administration of PROG (in the 1st, 6th, 24th, and 48th h after TBI) significantly protected animals against the development of brain edema and cognitive impairment (Roof et al., 1996; Stein, 2001; Wright et al., 2001). It could be concluded that the positive effect of PROG treatment could be achieved when the steroid is administered within 24 h after injury, although the sooner onset of the treatment means the better outcome. The optimal dose of PROG ranged from 8 to 16 mg/kg (Roof et al., 1996; Stein, 2008). Furthermore, Shear et al. (2002) tested 3- and 5-days regimen of PROG administration after TBI and observed a significant reduction size of injury-induced necrosis and diminished cell loss in the dorsomedial nucleus of the thalamus. The latter process facilitated behavioral recovery of animals. PROG, used at low physiological doses in ovariectomized female rats after TBI, reduced alterations in mitochondrial respiration and hippocampal cell loss (Robertson et al., 2006). In another study, treatment with PROG following TBI diminished production of inflammatory proteins in rats (Pettus et al., 2005). Roof et al. (1997) confirmed antioxidant effect of PROG. This hormone administered after a TBI episode significantly lowered the brain concentration of isoprostaglandin, an established marker of lipid peroxidation.
Advantageous properties of PROG were also observed in models of the spinal cord and peripheral nerve injury. The hormone administered for 3 days following the spinal cord injury improved myelination, increased the level of brain derived neurotrophic factor (BDNF) mRNA and reduced chromatolysis (De Nicola et al., 2006), whereas 5-day administration diminished the size of lesions and prevented secondary neuronal loss (Thomas et al., 1999). According to Labombarda et al. (2010) PROG significantly enhanced BDNF neuronal expression, up-regulated growth-associated protein 43 (GAP-43) necessary for axonal regeneration, prevented the injury-induced chromatolytic changes of spinal neurons and increased activity of enzymes crucial for normal neuronal metabolism and neurotransmission, and restored impaired expression of the Na,K-ATPase subunits and choline acetyltransferase. On the other hand, Coughlan et al. (2009) found no evidence for neuroprotective relationship between BDNF and PROG. Administration of PROG following acute spinal cord injury in rats attenuated the loss of microtubule-associated protein 2, a major component of the cytoskeleton (González et al., 2009). In addition, this steroid acts as a promyelinating factor by stimulating synthesis of myelin proteins and proliferation/differentiation of oligodendrocyte progenitors (Gonzalez et al., 2005; Labombarda et al., 2010).
Yu (1989) observed that PROG treatment following motor neuron axotomy attenuated loss of neurons in rats. According to Koenig et al. (1995) PROG induced myelin repair after cryolesion of peripheral nerves in mice. The same hormone increased expression of genes coding crucial myelin proteins in rat Schwann cells (Désarnaud et al., 1998). In rat peripheral nerves PROG increased glial cell growth and differentiation, induced synthesis of myelin-specific proteins in oligodendrocytes, and potentiated the formation of new myelin sheaths by Schwann cells (Jung-Testas et al., 1999; Baulieu and Schumacher, 2000).
Progesterone administration following the cortical contusion injury protected against secondary neuronal loss and facilitated cognitive recovery in rats (Roof et al., 1994; Asbury et al., 1998). In the diffused TBI model, administration of physiological doses of PROG reduced cell loss, axonal injury, decreased caspase-3 immunoreactivity, and facilitated the motor and cognitive performance recovery in male and ovariectomized female rats (O’Connor et al., 2007). In the model of ischemic injury, treatment with PROG reduced brain cell damage, improved neurological outcome in rats (Jiang et al., 1996; Chen et al., 1999; Kumon et al., 2000), and increased survival rate in mice (Gibson and Murphy, 2004). In the study of Jiang et al. (1996) the effects were comparable either when the hormone was given immediately before or 2 h after reperfusion. Post-injury administration of PROG in rats with the brain global ischemia resulted in a significant reduction of cortical shrinkage and cell loss in the CA1 and CA2 hippocampal subfields. Additionally, the hormone prevented ventricular dilatation compared with control animals (Moralí et al., 2005). In another study, application of PROG and ALLO after severe transient forebrain ischemia in rats resulted in better functional outcome and preserved dimensions of hippocampal formation despite pyramidal neurons loss (Moralí et al., 2011). Similarly, in cats after acute global ischemia PROG reduced cell loss, especially in the CA1/CA2 areas, and caudate nucleus regions (González-Vidal et al., 1998; Cervantes et al., 2002). In permanent middle cerebral artery occlusion model in rats, this steroid diminished cortical and optionally subcortical infarct volumes (Alkayed et al., 2000). Both PROG and ALLO improved behavioral outcome of animals (Sayeed et al., 2007). Recently, Cai et al. (2008) found that acute neuroprotective effects of PROG in ischemic brain injury may depend on its antagonistic action toward the σ1 receptor and subsequent inhibition of the NMDA-induced Ca influx. Finally, PROG was investigated in the ProTECT clinical study. It was observed that patients with acute TBI, treated with PROG, had a lower 30-day mortality rate than control individuals (Wright et al., 2006). Similarly, lower mortality and better functional outcomes were found in the PROG group of patients by Xiao et al. (2008) and Stein and Wright (2010). At least two third-phase clinical trials are currently run to confirm neuroprotective properties of this hormone in patients after brain injury (Stein and Wright, 2010).
Neuroprotective effects of PROG were also shown in animal models of spinal cord degeneration. Wobbler mice receiving the hormon presented less pronounced neuropathology, reduced number of vacuolated cells, preserved mitochondrial ultrastructure (Gonzalez Deniselle et al., 2002), attenuated motoneuron degeneration and boosted myelination in Schwann cells and oligodendrocytes in the CNS (Schumacher et al., 2004). According to Martini et al. (2003) PROG and its derivatives: dihydroprogesterone (DHP) and tetrahydroprogesterone (THP) may control the expression of the glycoprotein Po peripheral myelin and protein 22 (PMP22) via activation of the PROG receptor or GABAA receptor, respectively. Neuroprotective properties of PROG and its two metabolites were shown by Leonelli et al. (2007) in streptozotocin-induced diabetic neuropathy model in rats. Chronic (1 month lasting) treatment with these neurosteroids preserved the nerve conduction velocity, thermal threshold, intra-epidermal nerve fiber density, Na(+)K(+)-ATPase activity, and mRNA levels of myelin proteins (Leonelli et al., 2007).
Neuroprotective action of PROG seem to be partially due to reduction of cerebral edema that may be achieved via different mechanisms: protecting or rebuilding the blood–brain barrier (Duvdevani et al., 1995; Roof et al., 1997), modulating aquaporins (Amiry-Moghaddam et al., 2003), up-regulation of membrane PROG binding protein 25-Dx, which is co-expressed with vasopressin and regulates brain water homeostasis, and reduction of the cytotoxic phase of edema (Guennoun et al., 2008; Stein et al., 2008). Other possible actions of PROG include: down-regulation of the inflammatory cascade by decelerating cytokine (IL-1; IL-6; TNFα)-induced reactions, slowing the immune cell activation, and migration (Arvin et al., 1996; Grossman et al., 2004; Stein, 2008). This neurosteroid limits cellular necrosis and apoptosis by: 1. lowering the concentration of nuclear factor κβ and expression of its target genes such as IL-1β, C3, iNOS, COX2 (Pettus et al., 2005; Stein, 2008), 2. reducing excitotoxicity by inhibition of glutamate receptors, 3. boosting the effects of GABAA, 4 depression of spontaneous firing of neurons, a possible causative factor of posttraumatic seizures (Bergeron et al., 1996; Pierson et al., 2005; Stein, 2008), 5. stimulation of Schwann’s cells via nuclear receptor to produce myelin, and reducing glial scarring in the CNS (Koenig et al., 1995; Plassart-Schiess and Baulieu, 2001), 6. decreasing injury-induced lipid peroxidation and oxidative stress via inhibition of TNF-α production or up-regulation of antioxidant enzymes (Roof et al., 1997; Stein, 2008; Stein and Wright, 2010).
Progesterone activates signaling pathways associated with neuroprotection like mitogen activated protein kinase, extracellular signal regulated kinase (MAPK/ERK), or serine/threonine protein kinase Akt (Brinton et al., 2008). Nilsen and Brinton (2002) showed that PROG increased expression of the Bcl-2 anti-apoptotic gene, which prevented cell death in rat hippocampal neuronal cultures. In the TBI model in rats, administration of PROG increased mRNA levels of the Bcl-2 and Bcl-x(L) anti-apoptotic genes, and their protein derivatives. On the contrary, the hormone reduced mRNA levels of pro-apoptotic bax and bad genes, as well as their protein products (Yao et al., 2005). Additionally, the neurosteroid attenuates release of the proapoptotic cytochrome c (Stein and Wright, 2010). In cultured hippocampal neurons, PROG attenuated lipid peroxidation induced by FeSO4 and amyloid β-peptide, as well as lightened elevation of intracellular Ca2+ concentration following β-amyloid- or glutamate-induced toxicity (Goodman et al., 1996).
In rats after TBI, administration of ALLO decreased apoptotic DNA fragmentation and expression of caspase-3 and Bax pro-apoptotic proteins. Moreover, the neurosteroid improved behavioral outcome of animals similarly to PROG (Djebaili et al., 2004, 2005). ALLO and PROG protected the rat brain from post-injury edema. Both neurosteroids reduced production of pro-inflammatory cytokines (IL-1, TNF-α; He et al., 2004) and enhanced production of the CD55 protein, a complement convertase inhibitor, which was reported to limit inflammatory processes after contusion of the cerebral cortex in rats (VanLandingham et al., 2007). In patients with Alzheimer’s disease, the level of ALLO in the temporal cortex was significantly lower than in controls, in contrast to PREG and DHEA which concentrations were increased. This may be explained by altered regulation of neurosteroid biosynthetic pathway, blocking ALLO formation (Naylor et al., 2010). Similarly, the ALLO level was found to be decreased in Niemann-Pick type. The neurosteroid administration doubled the animal lifespan and delayed the onset of neurological symptoms. Furthermore, ALLO enhanced myelination and reduced inflammatory processes in brains of Npc1(−/−) mice, a genetic model of Niemann-Pick type C disease (Griffin et al., 2004; Ahmad et al., 2005; Mellon et al., 2008; Liao et al., 2009). Callier et al. (2001) revealed that PROG protected dopaminergic neurons against MPTP-induced degeneration in rats. Moreover, in MPTP mice, PROG did not reverse the protective effect of estrogens on dopamine neurons. In methamphetamine model of Parkinson’s disease, PROG used at low doses exerted protective action on striatal dopaminergic neurons in gonadectomized male mice, whereas much higher doses were needed to achieve a beneficial effect in ovariectomized mice (Bourque et al., 2009).
Higher concentrations of PROG, ALLO, and 5α-DHP were found in the substantia nigra and nucleus caudate in comparison with other brain areas in female control patients. In patients with Parkinson’s disease, lower concentrations of these neurosteroids than in controls were found in the cerebrospinal fluid (CSF; Luchetti et al., 2011). During further studies the authors observed reduced mRNA expression of one enzyme synthesizing ALLO in the substantia nigra, and increased expression of another one in the caudate nucleus. It seems that disturbed synthesis of ALLO may result in a dysfunction of endogenous neuroprotective effects (Luchetti et al., 2011). Similarly, diminished ALLO concentration and reduced activity of enzymes synthesizing the neurosteroid were found in patients with multiple sclerosis (Luchetti et al., 2011). Such phenomenon as NMDA-induced excitotoxicity in P-19 cell cultures (pluripotent embryonic cells differentiated into neurons) was attenuated by ALLO and DHEA, while DHEAS remained less effective in this respect. ALLO prevented the release of cytochrome c to cytoplasm and Bax protein translocation to mitochondria. All these processes are considered to be signs of apoptotic death. Since both neurosteroids enhanced expression of GABAA receptor subunits, their neuroprotective action seems to be mediated by enhanced GABA-ergic neurotransmission (Xilouri and Papazafiri, 2006).
Allopregnanolone appears to play a significant role in the pathogenesis of Alzheimer’s disease. Reduced levels of this steroid were found in the patient plasma and prefrontal cortex. Additionally, elevated concentrations of enzymes metabolizing pregnane steroids were found in brains from early stages of the disease (Luchetti et al., 2011). In the mouse model of Alzheimer’s disease PROG treatment improved cognitive functions and reduced hyperphosphorylation of tau protein. But on the other hand, PROG antagonized beneficial effects of estrogens in reducing β-amyloid deposits. It is possible that the sequential therapy with estrogen and PROG may bring better results than the treatment with only one hormone (Pike et al., 2009).
Allopregnanolone prevented apoptotic cell death in the human NT2 cell line culture in NMDA-induced excitotoxicity (Lockhart et al., 2002).
Progesterone is widely considered as a pleiotropic drug that can be markedly effective in the treatment of TBI. At present, more than 180 articles showed protective activity of PROG in both experimental animals and humans. Moreover, two independent phase II clinical trials have revealed that PROG group of patients had significantly better survival and functional outcomes than patients given placebo. Currently, phase III clinical trial is conducted in the United States. Nevertheless, in some studies PROG was not effective or even worsen the prognosis of animals after TBI or ischemic injury (Murphy et al., 2000; Toung et al., 2004; Ciriza et al., 2006; Gilmer et al., 2008; Stein, 2008, 2011; Stein and Wright, 2010). The probable reason of PROG treatment failure may be advanced age of used animals. Anyway, PROG proved efficacy and safety in most models of TBI. Surprisingly, medroxyprogesterone acetate, a PROG synthetic derivative, may exhibit quite opposite properties in the CNS than the parent compound. Medroxyprogesterone did not bring better functional outcome after TBI, moreover, it blocked the expression of Bcl-2 gene and estrogen-dependent neuroprotection, increased calcium-induced toxicity (Stein, 2008).
Anticonvulsive and antiexcitotoxic properties of PROG were widely examined in animal models and clinical studies. It seems that antiseizure action of this hormone is not mediated by steroid receptor since it occurs quickly (within minutes) and is not blocked by the respective receptor antagonist – RU486. GABAA receptor activation induces neuronal membrane hyperpolarization and reduces its excitability.
In kainic acid-induced seizures in rats the hormone significantly reduced seizures, mortality, and neuronal death in hippocampus (Hoffman et al., 2003). In the same model in mice, ALLO protected against seizures and reduced neuronal loss in hippocampal fields, while other neurosteroids – negative modulators of GABAA receptor (DHEAS, PREGS) – remained ineffective or even toxic (Leskiewicz et al., 1997). Holmes and Weber (1984) tested PROG in amygdala-kindled rats and observed kindling inhibition. Secondary generalization of seizures was prevented in immature, but not adult animals. In the same model, PROG and ALLO protected against secondarily generalized seizures, being ineffective or effective only at toxic doses against focal seizures. On the other hand, 5α-DHP inhibited both components of seizures (Lonsdale et al., 2006). Growing evidence suggests that anticonvulsive and neuroprotective properties of PROG are mediated by its metabolites 5α-DHP and ALLO, positive modulators of GABAA receptors (Reddy, 2004; Ciriza et al., 2006; Verrotti et al., 2010; Stevens and Harden, 2011). ALLO itself protected against picrotoxin-induced neuronal cell death (Brinton, 1994). Rhodes and Frye (2005) showed that inhibition of 5α-reductase enzyme (finasteride) attenuated antiseizure activity of PROG and reduced concentration of ALLO. However, according to Leskiewicz et al. (2003) ALLO showed no significant anticonvulsive action against cocaine-induced kindling in mice, in contrast to isopregnanolon and a representative of negative GABAA modulators – DHEAS. The authors suggest that antiseizure properties may not depend exclusively on GABAA receptor modulation. Some other neurosteroids were reported to inhibit seizures.
Both PROG derivatives: DHP and ALLO dose-dependently protected the kainate-induced loss of hilar neurons and the induction of vimentin expression in reactive astrocytes (a sign of neural damage) in ovariectomized rats (Ciriza et al., 2004) (Table 1).
Dehydroepiandrosterone and Dehydroepiandrosterone Sulfate Ester
Dehydroepiandrosterone and its sulfate ester (DHEAS) were reported to act as neurotropic or neuroprotective factors, defending neurons against many harmful events, including excitotoxicity. Protective properties of DHEA may be related to inhibition of the NMDA-induced nitric oxide (NO) production in hippocampal cells or modulation of the calcium/NO signaling pathway. The action of this hormone can be also mediated by sigma receptors. Additionally, DHEAS was shown to attenuate the destructive action of glutamate and NMDA on cerebellar granule cells in rats. Protective effects of both DHEA and DHEAS can be realized through preventing the mitochondria against intracellular Ca2+ overload (Mao and Barger, 1998). Time- and dose-dependent neuroprotective action of DHEA was documented in the in vivo model of global cerebral ischemia in rats. Administration of DHEA at the dose of 20 mg/kg during 3–48 h after ischemia reduced neuronal death in the hippocampal CA1 region and ameliorated ischemia-induced deficits in spatial learning. Whereas, the treatment of DHEA (20 mg/kg, 1 h before or after ischemia) may intensify the ischemia-induced neuronal damage and learning failure. Moreover, treatment with doses 25–100 mg/kg (for 15 days before induction of ischemia) led to more pronounced neuroprotective effects and reduced hippocampal CA1 neuronal injury (Li et al., 2001). These study may suggest that DHEA, dependently on time of treatment, exhibits a dual action – it may be either neurotoxic or neuroprotective, depending on the time of treatment initiation and duration (Li et al., 2009).
DHEA sulfate significantly inhibited the amplitude of persistent sodium currents, and this result was canceled by the Gi protein inhibitor, protein kinase C inhibitor, sigma-1 receptor blockers, but not by the protein kinase A inhibitor. These results suggest that DHEAS may protect neurons against ischemia by activation of sigma-1 receptor (Cheng et al., 2008). DHEAS has been also investigated in the reversible spinal cord ischemia model. The neurosteroid exhibited a preventive effect when it was administered 5 min, but not 30 min after ischemia. The beneficial effect of DHEA was reversed by bicuculline, the GABAA antagonist, which indicates GABA-ergic mechanism of the neurosteroid action (Lapchak et al., 2000). Aforementioned results show that DHEA and DHEAS may provide neuroprotection against ischemia. However, it should be remembered that DHEA administered during ischemia may be also neurotoxic.
According Kimonides et al. (1998), DHEA showed a dose-dependent neuroprotective effect against glutamate excitotoxicity mediated by NMDA receptors (in vivo and in vitro) or evoked by AMPA/KA receptor agonists (in vitro). For instance, the neurosteroid prevented hippocampal CA1/2 neurons against toxicity induced by NMDA infusions. Thus, decreased level of DHEA in aging people may increase vulnerability of the brain to such a damage. Age-related declines of neurosteroids have been postulated to play a role in the pathogenesis of neurodegenerative diseases (Hillen et al., 2000; Brown et al., 2003; Naylor et al., 2008). Interestingly, DHEA-induced protection against KA-induced excitotoxicity in hippocampal hilar neurons was attenuated by letrozole, an aromatase inhibitor (Veiga et al., 2003). Taking this into consideration, neuroprotective effects of DHEA in the hippocampus can be mediated by estradiol (Azcoitia et al., 1998; Garcia-Segura et al., 2003). It should be stressed that expression of aromatase in reactive glial cells is induced after brain injury (Garcia-Segura et al., 2003).
Some results suggest that DHEAS may be a negative modulator of GABAA receptors. In the study of Czlonkowska et al. (2000), intracerebroventricularly administered DHEAS induced seizures in a dose-dependent manner. But on the other hand, DHEAS (25 mg/kg, i.p.) significantly increased the dose of NMDA necessary to induce clonic convulsions in mice, indicating that the neurosteroids can protect mice against NMDA-induced seizures and mortality (Budziszewska et al., 1998). The antiseizure effect of DHEA was also proved in the iron-induced model of posttraumatic epilepsy in rats. DHEA at the dose of 30 mg/kg/day prevented epileptiform electrophysiological activity and attenuated cognitive defects produced by epileptic activity (Mishra et al., 2010). It is worth mentioning that neurosteroids may serve as regulators of epileptogenesis. This raises the possibility that exogenously administered neurosteroids or their synthetic analogs could be clinically effective as antiepileptogenic drugs, e.g., after TBI (Biagini et al., 2006). In the study of Fiore et al. (2004), DHEA provided better locomotor recovery, left–right coordination, and fine motor control in mice after contusive spinal cord injury. Possibly, these drugs may create a promising novel therapeutic avenue for the treatment of this dysfunction in humans. Further, DHEA was reported to reduce the extent of denervation atrophy. The neurosteroid accelerated axonal regeneration (Ayhan et al., 2003) and increased the number of myelinated fibers and fiber diameters after sciatic nerve transection in rats (Gudemez et al., 2002).
According to Frye and Reed (1998), a 3α-Diol metabolite of another androgen – testosterone – exhibited anticonvulsive effect against seizures induced in rats by kainate or perforant pathway stimulation. Moreover, the neurosteroid reduced hippocampal cell death and improved animal performance in the Morris water maze. Interestingly, this action was inhibited by GABAA receptor blockade, therefore it is not clear whether the action of 3α-Diol is due to enhanced GABA-ergic neurotransmission or to increased estrogen concentration (Table 1).
Deoxycorticosterone and Allotetrahydrodeoxycorticosterone
Although there is no literature data on neuroprotective properties of deoxycorticosterone DOC) and its derivative allotetrahydrodeoxycorticosterone (THDOC), the two neurosteroids exhibited anticonvulsive effects in some experimental studies. For instance, THDOC protected against convulsions induced by the GABAA receptor antagonists, like picrotoxin, pentylenetetrazole, or bicuculline, and the glutamate receptor agonists, such as kainic acid or NMDA. The neurosteroid, administered at relatively high doses, showed also the anticonvulsive action in the rat model of amygdala kindling and the maximal electroshock test in mice. In human studies, decreased serum level of THDOC was found in women with catamenial epilepsy, which may indicate a role of this neurosteroid in the pathophysiology of perimenstrual seizures. The antiseizure effect of THDOC seems to depend on positive allosteric modulation of GABAA receptor (Reddy, 2009) (Table 1).
Synthetic Neurosteroids
The main disadvantage of natural neurosteroids is their short biological half-life. Therefore some modified derivatives were synthesized in order to select drugs, which are not so rapidly metabolized.
Neurosteroid analogs offered mainly a broad spectrum of anticonvulsive activity in different experimental seizure models. Therefore, this group of neurosteroids are mostly considered as a novel class of antiepileptic agents. The overriding example is ganaxolone, a representative of III generation antiepileptic drugs.
Ganaxolone (GNX, 3α-hydroxy-3β-methyl-5α-prengan-20-one) is the 3β-methylated analog of ALLO. As the parent compound, GNX is a positive allosteric modulator of GABAA receptors and increases chloride channel permeability within the GABAA–benzodiazepine receptor–chloride ionophore complex (Carter et al., 1997; Monaghan et al., 1997; Nohria and Giller, 2007). The neurosteroid exhibited effectiveness in a broad range of animal models of epilepsy, including convulsions induced by pentylenetetrazole, bicuculline, fluorothyl, cocaine, aminophylline, as well as in pentylenetetrazole kindling, amygdala kindling, and 6 Hz electrical stimulation (Gasior et al., 2000; Ungard et al., 2000; Reddy and Rogawski, 2000a,b; Kaminski et al., 2004). Powerful antiseizure effects of GNX in the amygdala-kindled model in mice strongly support the utility of this substance in the treatment of temporal lobe epilepsy (Reddy and Rogawski, 2010). However, most data on this neurosteroid was obtained in preclinical models of catamenial epilepsy. According to Reddy and Rogawski (2009), GNX treatment (7 mg/kg, sc) significantly reduced the frequency of spontaneous seizures in rats. These results raise the possibility that GNX might provide a specific treatment for catamenial epilepsy in humans. On the other hand, this ALLO analog proved to be ineffective against NMDA- and strychnine-induced seizures in mice (Carter et al., 1997; Gasior et al., 1997). In clinical studies, GNX was shown to be well tolerated in adults and children. In early phase II studies, GNX was found to be effective in adult patients with partial-onset seizures and in epileptic children with history of infantile spasms. Currently, further investigations are conducted in infants with newly diagnosed infantile spasms, women with catamenial epilepsy, and adults with refractory partial-onset seizures (Rogawski and Reddy, 2004).
It was recently reported that the pretreatment with a new synthesized analog of ALLO, 3α-hydroxy-21,22-oxido-21-homo-5α-pregnan-20-on (HOHP), suppressed generalized tonic–clonic seizures in pentylenetetrazol-induced seizures in mice, and efficacy of this analog resembled that of GNX (Mareš et al., 2010).
Two synthetic neurosteroids, minaxolone (2β,3α,5α,11α)-11-(dimethylamino)-2-(ethoxy-3-hydroxypregnan-20-one), and alfaxalone (5α-pregnan-3α-ol-11,20-dione) seem to be effective positive allosteric modulators of the α1 glycine receptor (Maksay and Biro, 2002; Weir et al., 2004). Both analogs proved efficacy against pentylenetetrazole- and bicuculline-induced convulsions in animals. Alphaxalone showed, moreover, a moderate anticonvulsive activity in the rat model of kindling and maximal electroshock in mice (Rogawski and Reddy, 2004).
Another synthetic neurosteroid, Co 2-1068 (3β-(4acetylphenyl)ethynyl-3α,21-dihydroxy-5β-20-one-21-hemisuccinate) was re-ported to be effective against pentylenetetrazole-, NMDA-, and cocaine-induced seizures in mice (Gasior et al., 1997).
So far, only fluasterone (DHEF), a novel DHEA analog, was considered as a candidate for a drug used in the treatment of TBI in humans. This steroid was reported to improve functional recovery in the rat TBI model. Possible DHEF mechanisms of action are still unclear and discussed. According to Malik et al. (2003), this neurosteroid markedly inhibited synthesis of prostaglandin (PGE2) and interleukin-1βa-induced cyclooxygenase-2 (COX2) in cultures of rat mesangial, but not cortical or hippocampal cells (Malik et al., 2003) (Table 1).
Conclusion
Neurosteroids may exhibit neuroprotective effects in both central and peripheral nervous system. These natural and synthetic compounds attenuate the excitotoxicity, brain edema, inflammatory processes, oxidative stress, and neural degeneration. Additionally, neurosteroids accelerate and improve regeneration and myelination. Promising results of preclinical studies directed scientist attention toward possible profits of neurosteroid treatment in a wide range of diseases, especially the brain and spinal cord injury, stroke, Parkinson’s and Alzheimer’s diseases, or epilepsy. Unfortunately, therapeutic application of natural neurosteroids was significantly limited by its rapid biotransformation. Only synthetic analogs of these substances, particularly ganaxolone or 3α-ol-5β-pregnan-20-one, can be considered as potential anticonvulsive drugs. In fact, ganaxolone has been already classified to antiepileptic drugs of III generation. This gives a real hope that neurosteroids can be also used as effective neuroprotective drugs.
Statements
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
AhmadI.Lope-PiedrafitaS.BiX.HicksC.YaoY.YuC.ChaitkinE.HowisonC. M.WebergL.TrouardT. P.EricksonR. P. (2005). Allopregnanolone treatment, both as a single injection or repetitively, delays demyelination and enhances survival of Niemann-Pick C mice. J. Neurosci. Res.82, 811–821.10.1002/jnr.20685
2
AlkayedN. J.MurphyS. J.TraystmanR. J.HurnP. D.MillerV. M. (2000). Neuroprotective effects of female gonadal steroids in reproductively senescent female rats. Stroke31, 161–168.10.1161/01.STR.31.1.161
3
Amiry-MoghaddamM.OtsukaT.HurnP. D.TraystmanR. J.HaugF. M.FroehnerS. C.AdamsM. E.NeelyJ. D.AgreP.OttersenO. P.BhardwajA. (2003). An alpha-syntrophin-dependent pool of AQP4 in astroglial end-feet confers bidirectional water flow between blood and brain. Proc. Natl. Acad. Sci. U.S.A.100, 2106–2111.10.1073/pnas.0437946100
4
ArvinB.NevilleL. F.BaroneF. C.FeuersteinG. Z. (1996). The role of inflammation and cytokines in brain injury. Neurosci. Biobehav. Rev.20, 445–452.10.1016/0149-7634(95)00026-7
5
AsburyE. T.FrittsM. E.HortonJ. E.IsaacW. L. (1998). Progesterone facilitates the acquisition of avoidance learning and protects against subcortical neuronal death following prefrontal cortex ablation in the rat. Behav. Brain Res.97, 99–106.10.1016/S0166-4328(98)00031-X
6
AyhanS.MarkalN.SiemionowK.AraneoB.SiemionowM. (2003). Effect of subepineurial dehydroepiandrosterone treatment on healing of transected nerves repaired with the epineurial sleeve technique. Microsurgery23, 49–53.10.1002/micr.10088
7
AzcoitiaI.SierraA.Garcia-SeguraL. M. (1998). Estradiol prevents kainic acid-induced neuronal loss in the rat dentate gyrus. Neuroreport14, 3075–3079.10.1097/00001756-199809140-00029
8
BaulieuE. E. (1998). Neurosteroids: a novel function of the brain. Psychoneuroendocrinology23, 963–987.10.1016/S0306-4530(98)00071-7
9
BaulieuE. E.RobelP.SchumacherM. (2001). Neurosteroids: beginning of the story. Int. Rev. Neurobiol.46, 1–32.
10
BaulieuE. E.SchumacherM. (2000). Progesterone as a neuroactive neurosteroid, with special reference to the effect of progesterone on myelination. Steroids65, 605–612.10.1016/S0039-128X(00)00173-2
11
BergeronR.de MontignyC.DebonnelG. (1996). Potentiation of neuronal NMDA response induced by dehydroepiandrosterone and its suppression by progesterone: effects mediated via sigma receptors. J. Neurosci.16, 1193–1202.
12
BiaginiG.BaldelliE.LongoD.PradelliL.ZiniI.RogawskiM. A.AvoliM. (2006). Endogenous neurosteroids modulate epileptogenesis in a model of temporal lobe epilepsy. Exp. Neurol.201, 519–524.10.1016/j.expneurol.2006.04.029
13
BorowiczK. K.GolyskaD.MorawskaM.Furmanek-KarwowskaK. (2008). Neuroprotekcyjne wlasciwosci neurosteroidów. Epileptologia16, 59–68.
14
BourqueM.DluzenD. E.Di PaoloT. (2009). Neuroprotective actions of sex steroids in Parkinson’s disease. Front. Neuroendocrinol.30, 142–157.10.1016/j.yfrne.2009.04.014
15
BrintonR. D. (1994). The neurosteroid 3 alpha-hydroxy-5 alpha-pregnan-20-one induces cytoarchitectural regression in cultured fetal hippocampal neurons. J. Neurosci.14, 2763–2774.
16
BrintonR. D.ThompsonR. F.FoyM. R.BaudryM.WangJ.FinchC. E.MorganT. E.PikeC. J.MackW. J.StanczykF. Z.NilsenJ. (2008). Progesterone receptors: form and function in brain. Front. Neuroendocrinol.29, 313–339.10.1016/j.yfrne.2008.02.001
17
BrownR. C.HanZ.CascioC.PapadopoulosV. (2003). Oxidative stress-mediated DHEA formation in Alzheimer’s disease pathology. Neurobiol. Aging24, 57–65.10.1016/S0197-4580(02)00048-9
18
BudziszewskaB.SiwanowiczJ.Le kiewiczM.Jaworska-FeilL.LasonW. (1998). Protective effects of neurosteroids against NMDA-induced seizures and lethality in mice. Eur. Neuropsychopharmacol.8, 7–12.10.1016/S0924-977X(98)90017-7
19
CaiW.ZhuY.FuruyaK.LiZ.SokabeM.ChenL. (2008). Two different molecular mechanisms underlying progesterone neuroprotection against ischemic brain damage. Neuropharmacology55, 127–138.10.1016/j.neuropharm.2008.04.023
20
CallierS.MorissetteM.GrandboisM.PelapratD.Di PaoloT. (2001). Neuroprotective properties of 17beta-estradiol, progesterone, and raloxifene in MPTP C57Bl/6 mice. Synapse41, 131–138.10.1002/syn.1067
21
CarterR. B.WoodP. L.WielandS.HawkinsonJ. E.BelelliD.LambertJ. J.WhiteH. S.WolfH. H.MirsadeghiS.TahirS. H.BolgerM. B.LanN. C.GeeK. W. (1997). Characterization of the anticonvulsant properties of ganaxolone (CCD 1042; 3alpha-hydroxy-3beta-methyl-5alpha-pregnan-20-one), a selective, high-affinity, steroid modulator of the gamma-aminobutyric acid(A) receptor. J. Pharmacol. Exp. Ther.280, 1284–1295.
22
CervantesM.González-VidalM. D.RuelasR.EscobarA.MoralíG. (2002). Neuroprotective effects of progesterone on damage elicited by acute global cerebral ischemia in neurons of the caudate nucleus. Arch. Med. Res.33, 6–14.10.1016/S0188-4409(01)00347-2
23
CharalampopoulosI.RemboutsikaE.MargiorisA. N.GravanisA. (2008). Neurosteroids as modulators of neurogenesis and neuronal survival. Trends Endocrinol. Metab.19, 300–307.10.1016/j.tem.2008.07.004
24
ChenJ.ChoppM.LiY. (1999). Neuroprotective effects of progesterone after transient middle cerebral artery occlusion in rat. J. Neurol. Sci.171, 24–30.10.1016/S0022-510X(99)00247-6
25
ChengZ. X.LanD. M.WuP. Y.ZhuY. H.DongY.MaL.ZhengP. (2008). Neurosteroid dehydroepiandrosterone sulphate inhibits persistent sodium currents in rat medial prefrontal cortex via activation of sigma-1 receptors. Exp. Neurol.210, 128–136.10.1016/j.expneurol.2007.10.004
26
CirizaI.AzcoitiaI.Garcia-SeguraL. M. (2004). Reduced progesterone metabolites protect rat hippocampal neurons from kainic acid excitotoxicity in vivo. J. Neuroendocrinol.16, 58–63.10.1111/j.1365-2826.2004.01121.x
27
CirizaI.CarreroP.FryeC. A.Garcia-SeguraL. M. (2006). Reduced metabolites mediate neuroprotective effects of progesterone in the adult rat hippocampus. The synthetic progestin medroxyprogesterone acetate (Provera) is not neuroprotective. J. Neurobiol.66, 916–928.10.1002/neu.20293
28
CompagnoneN. A.MellonS. H. (2000). Neurosteroids: biosynthesis and function of these novel neuromodulators. Front. Neuroendocrinol.21, 1–56.10.1006/frne.1999.0188
29
CorpéchotC.SynguelakisM.TalhaS.AxelsonM.SjövallJ.VihkoR.BaulieuE. E.RobelP. (1983). Pregnenolone and its sulfate ester in the rat brain. Brain Res.270, 119–125.10.1016/0006-8993(83)90797-7
30
CoughlanT.GibsonC.MurphyS. (2009). Progesterone, BDNF and neuroprotection in the injured CNS. Int. J. Neurosci.119, 1718–1740.10.1080/00207450903116430
31
CzlonkowskaA. I.KrzascikP.Sienkiewicz-JaroszH.SiemiatkowskiM.SzyndlerJ.BidzinskiA.PlaznikA. (2000). The effects of neurosteroids on picrotoxin-, bicuculline- and NMDA-induced seizures, and a hypnotic effect of ethanol. Pharmacol. Biochem. Behav.67, 345–353.10.1016/S0091-3057(00)00369-5
32
De NicolaA. F.GonzalezS. L.LabombardaF.Gonzalez DeniselleM. C.GarayL.GuennounR.SchumacherM. (2006). Progesterone treatment of spinal cord injury: effects on receptors, neurotrophins, and myelination. J. Mol. Neurosci.28, 3–15.10.1385/JMN:28:1:3
33
DegterevA.HuangZ.BoyceM.LiY.JagtapP.MizushimaN.CunyG. D.MitchisonT. J.MoskowitzM. A.YuanJ. (2005). Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol.1, 112–119.10.1038/nchembio0905-234a
34
DésarnaudF.Do ThiA. N.BrownA. M.LemkeG.SuterU.BaulieuE. E.SchumacherM. (1998). Progesterone stimulates the activity of the promoters of peripheral myelin protein-22 and protein zero genes in Schwann cells. J. Neurochem.71, 1765–1768.10.1046/j.1471-4159.1998.71041765.x
35
DjebailiM.GuoQ.PettusE. H.HoffmanS. W.SteinD. G. (2005). The neurosteroids progesterone and allopregnanolone reduce cell death, gliosis, and functional deficits after traumatic brain injury in rats. J. Neurotrauma22, 106–118.10.1089/neu.2005.22.106
36
DjebailiM.HoffmanS. W.SteinD. G. (2004). Allopregnanolone and progesterone decrease cell death and cognitive deficits after a contusion of the rat pre-frontal cortex. Neuroscience123, 349–359.10.1016/j.neuroscience.2003.09.023
37
DuvdevaniR.RoofR. L.FülöpZ.HoffmanS. W.SteinD. G. (1995). Blood-brain barrier breakdown and edema formation following frontal cortical contusion: does hormonal status play a role?J. Neurotrauma12, 65–75.10.1089/neu.1995.12.65
38
FioreC.InmanD. M.HiroseS.NobleL. J.IgarashiT.CompagnoneN. A. (2004). Treatment with the neurosteroid dehydroepiandrosterone promotes recovery of motor behavior after moderate contusive spinal cord injury in the mouse. J. Neurosci. Res.75, 391–400.10.1002/jnr.10821
39
Fontaine-LenoirV.ChambraudB.FellousA.DavidS.DuchossoyY.BaulieuE. E.RobelP. (2006). Microtubule-associated protein 2 (MAP2) is a neurosteroid receptor. Proc. Natl. Acad. Sci. U.S.A.103, 4711–4716.10.1073/pnas.0600113103
40
FryeC. A.ReedT. A. (1998). Androgenic neurosteroids: anti-seizure effects in an animal model of epilepsy. Psychoneuroendocrinology23, 385–399.10.1016/S0306-4530(98)00009-2
41
Garcia-SeguraL. M.VeigaS.SierraA.MelcangiR. C.AzcoitiaI. (2003). Aromatase: a neuroprotective enzyme. Prog. Neurobiol.71, 31–41.10.1016/j.pneurobio.2003.09.005
42
GasiorM.CarterR. B.GoldbergerS. R.WitkinJ. M. (1997). Anticonvulsant and behavioral effects of neuroactive steroids alone and in conjunction with diazepam. J. Pharmacol. Exp. Ther.282, 543–552.
43
GasiorM.UngardJ. T.BeekmanM.CarterR. B.WitkinJ. M. (2000). Acute and chronic effects of the synthetic neuroactive steroid, ganaxolone, against the convulsive and lethal effects of pentylenetrazol in seizure-kindled mice: comparison with diazepam and valproate. Neuropharmacology39, 1184–1196.10.1016/S0028-3908(99)00190-2
44
GibsonC. L.MurphyS. P. (2004). Progesterone enhances functional recovery after middle cerebral artery occlusion in male mice. J. Cereb. Blood Flow Metab.24, 805–813.10.1097/01.WCB.0000125365.83980.00
45
GilmerL. K.RobertsK. N.ScheffS. W. (2008). Efficacy of progesterone following a moderate unilateral cortical contusion injury. J. Neurotrauma25, 593–602.10.1089/neu.2007.0477
46
GonzalezS. L.LabombardaF.Gonzalez DeniselleM. C.MougelA.GuennounR.SchumacherM.De NicolaA. F. (2005). Progesterone neuroprotection in spinal cord trauma involves up-regulation of brain-derived neurotrophic factor in motoneurons. J. Steroid Biochem. Mol. Biol.94, 143–149.10.1016/j.jsbmb.2005.01.016
47
GonzálezS. L.López-CostaJ. J.LabombardaF.DeniselleM. C.GuennounR.SchumacherM.De NicolaA. F. (2009). Progesterone effects on neuronal ultrastructure and expression of microtubule-associated protein 2 (MAP2) in rats with acute spinal cord injury. Cell. Mol. Neurobiol.29, 27–39.10.1007/s10571-008-9291-0
48
Gonzalez DeniselleM. C.Lopez CostaJ. J.GonzalezS. L.LabombardaF.GarayL.GuennounR.SchumacherM.De NicolaA. F. (2002). Basis of progesterone protection in spinal cord neurodegeneration. J. Steroid Biochem. Mol. Biol.83, 199–209.10.1016/S0960-0760(02)00262-5
49
González-VidalM. D.Cervera-GaviriaM.RuelasR.EscobarA.MoralíG.CervantesM. (1998). Progesterone: protective effects on the cat hippocampal neuronal damage due to acute global cerebral ischemia. Arch. Med. Res.29, 117–124.
50
GoodmanY.BruceA. J.ChengB.MattsonM. P. (1996). Estrogens attenuate and corticosterone exacerbates excitotoxicity, oxidative injury, and amyloid beta-peptide toxicity in hippocampal neurons. J. Neurochem.66, 1836–1844.10.1046/j.1471-4159.1996.66051836.x
51
GriffinL. D.GongW.VerotL.MellonS. H. (2004). Niemann-Pick type C disease involves disrupted neurosteroidogenesis and responds to allopregnanolone. Nat. Med.10, 704–711.10.1038/nm1073
52
GrossmanK. J.GossC. W.SteinD. G. (2004). Effects of progesterone on the inflammatory response to brain injury in the rat. Brain Res.1008, 29–39.10.1016/j.brainres.2004.02.022
53
GudemezE.OzerK.CunninghamB.SiemionowK.BrowneE.SiemionowM. (2002). Dehydroepiandrosterone as an enhancer of functional recovery following crush injury to rat sciatic nerve. Microsurgery22, 234–241.10.1002/micr.10039
54
GuennounR.MeffreD.LabombardaF.GonzalezS. L.Gonzalez DeniselleM. C.SteinD. G.De NicolaA. F.SchumacherM. (2008). The membrane-associated progesterone-binding protein 25-Dx: expression, cellular localization and up-regulation after brain and spinal cord injuries. Brain Res. Rev.57, 493–505.10.1016/j.brainresrev.2007.05.009
55
GursoyE.CardounelA.KalimiM. (2001). Pregnenolone protects mouse hippocampal (HT-22) cells against glutamate and amyloid beta protein toxicity. Neurochem. Res.26, 15–21.10.1023/A:1007668213330
56
HeJ.EvansC. O.HoffmanS. W.OyesikuN. M.SteinD. G. (2004). Progesterone and allopregnanolone reduce inflammatory cytokines after traumatic brain injury. Exp. Neurol.189, 404–412.10.1016/j.expneurol.2004.06.008
57
HillenT.LunA.ReischiesF. M.BorcheltM.Steinhagen-ThiessenE.SchaubR. T. (2000). DHEA-S Plasma Levels and Incidence of Alzheimer’s disease. Biol. Psychiatry47, 161–163.10.1016/S0006-3223(00)00800-3
58
HoffmanG. E.MooreN.FiskumG.MurphyA. Z. (2003). Ovarian steroid modulation of seizure severity and hippocampal cell death after kainic acid treatment. Exp. Neurol.182, 124–134.10.1016/S0014-4886(03)00104-3
59
HolmesG. L.WeberD. A. (1984). The effect of progesterone on kindling: a developmental study. Brain Res.318, 45–53.
60
HsuH. J.LiangM. R.ChenC. T.ChungB. C. (2006). Pregnenolone stabilizes microtubules and promotes zebrafish embryonic cell movement. Nature439, 480–483.10.1038/nature04436
61
JiangN.ChoppM.SteinD.FeitH. (1996). Progesterone is neuroprotective after transient middle cerebral artery occlusion in male rats. Brain Res.735, 101–107.10.1016/0006-8993(96)00605-1
62
Jung-TestasI.Do ThiA.KoenigH.DésarnaudF.ShazandK.SchumacherM.BaulieuE. E. (1999). Progesterone as a neurosteroid: synthesis and actions in rat glial cells. J. Steroid Biochem. Mol. Biol.69, 97–107.10.1016/S0960-0760(98)00149-6
63
Jung-TestasI.HuZ. Y.BaulieuE. E.RobelP. (1989). Neurosteroids: biosynthesis of pregnenolone and progesterone in primary cultures of rat glial cells. Endocrinology125, 2083–2091.10.1210/endo-125-4-2083
64
KaminskiR. M.LivingoodM. R.RogawskiM. A. (2004). Allopregnanolone analogs that positively modulate GABA receptors protect against partial seizures induced by 6 Hz electrical stimulation in mice. Epilepsia45, 864–867.10.1111/j.0013-9580.2004.04504.x
65
KanducD.MettelmanA.SerpicoR.SinigagliaE.SinhaA. A.NataleC.SantacroceR.Di CorciaM.LuccheseA.DiniL.PaniP.SantacroseS.SimoneS.BucciR.FarberE. (2002). Cell death: apoptosis versus necrosis. Int. J. Oncol.21, 165–170.
66
KimonidesV. G.KhatibiN. H.SvendsenC. N.SofroniewM. V.HerbertJ. (1998). Dehydroepiandrosterone (DHEA) and DHEA-sulfate (DHEAS) protect hippocampal neurons against excitatory amino acid-induced neurotoxicity. Proc. Natl. Acad. Sci. U.S.A.1795, 1852–1857.10.1073/pnas.95.4.1852
67
KoenigH. L.SchumacherM.FerzazB.ThiA. N.RessouchesA.GuennounR.Jung-TestasI.RobelP.AkwaY.BaulieuE. E. (1995). Progesterone synthesis and myelin formation by Schwann cells. Science268, 1500–1503.10.1126/science.7770777
68
KroemerG.GalluzziL.VandenabeeleP.AbramsJ.AlnemriE. S.BaehreckeE. H.BlagosklonnyM. V.El-DeiryW. S.GolsteinP.GreenD. R.HengartnerM.KnightR. A.KumarS.LiptonS. A.MalorniW.NuñezG.PeterM. E.TschoppJ.YuanJ.PiacentiniM.ZhivotovskyB.MelinoG. (2009). Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ.16, 3–11.10.1038/cdd.2008.150
69
KumonY.KimS. C.TompkinsP.StevensA.SakakiS.LoftusC. M. (2000). Neuroprotective effect of postischemic administration of progesterone in spontaneously hypertensive rats with focal cerebral ischemia. J. Neurosurg.92, 848–852.10.3171/jns.2000.92.5.0848
70
LabombardaF.González DeniselleM. C.De NicolaA. F.GonzálezS. L. (2010). Progesterone and the spinal cord: good friends in bad times. Neuroimmunomodulation17, 146–149.10.1159/000258709
71
LapchakP. A.ChapmanD. F.NunezS. Y.ZivinJ. A. (2000). Dehydroepiandrosterone sulfate is neuroprotective in a reversible spinal cord ischemia model: possible involvement of GABA(A) receptors. Stroke31, 1953–1956.10.1161/01.STR.31.12.3034
72
LeistM.JäätteläM. (2001). Triggering of apoptosis by cathepsins. Cell Death Differ.8, 24–26.10.1038/sj.cdd.4400859
73
LeonelliE.BianchiR.CavalettiG.CarusoD.CrippaD.Garcia-SeguraL. M.LauriaG.MagnaghiV.RoglioI.MelcangiR. C. (2007). Progesterone and its derivatives are neuroprotective agents in experimental diabetic neuropathy: a multimodal analysis. Neuroscience144, 1293–1304.10.1016/j.neuroscience.2006.11.014
74
LeskiewiczM.BudziszewskaB.Jaworska-FeilL.KuberaM.Basta-KaimA.LasonW. (2003). Inhibitory effect of some neuroactive steroids on cocaine-induced kindling in mice. Pol. J. Pharmacol.55, 1131–1136.
75
LeskiewiczM.BudziszewskaB.Jaworska-FeilL.LasonW. (1997). Effects of neurosteroids on kainate-induced seizures, neurotoxicity and lethality in mice. Pol. J. Pharmacol.49, 411–417.
76
LeskiewiczM.JantasD.BudziszewskaB.LasonW. (2008). Excitatory neurosteroids attenuate apoptotic and excitotoxic cell death in primary cortical neurons. J. Physiol. Pharmacol.59, 457–475.
77
LiH.KleinG.SunP.BuchanA. M. (2001). Dehydroepiandrosterone (DHEA) reduces neuronal injury in a rat model of global cerebral ischemia. Brain Res.888, 263–266.10.1016/S0006-8993(00)03077-8
78
LiZ.CuiS.ZhangZ.ZhouR.GeY.SokabeM.ChenL. (2009). DHEA-neuroprotection and-neurotoxicity after transient cerebral ischemia in rats. J. Cereb. Blood Flow Metab.29, 287–296.10.1038/jcbfm.2009.3
79
LiaoG.CheungS.GaleanoJ.JiA. X.QinQ.BiX. (2009). Allopregnanolone treatment delays cholesterol accumulation and reduces autophagic/lysosomal dysfunction and inflammation in Npc1-/- mouse brain. Brain Res.1270, 140-151.10.1016/j.brainres.2009.03.027
80
LockhartE. M.WarnerD. S.PearlsteinR. D.PenningD. H.MehrabaniS.BoustanyR. M. (2002). Allopregnanolone attenuates N-methyl-D-aspartate-induced excititoxicity and apoptosis in human NT2 cell line in culture. Neurosci. Lett.328, 33–36.10.1016/S0304-3940(02)00448-2
81
LonsdaleD.NylenK.McIntyre BurnhamW. (2006). The anticonvulsant effects of progesterone and its metabolites on amygdala-kindled seizures in male rats. Brain Res.1101, 110–116.10.1016/j.brainres.2006.05.005
82
LuchettiS.HuitingaI.SwaabD. F. (2011). Neurosteroid and GABA-A receptor alterations in Alzheimer’s disease, Parkinson’s disease and multiple sclerosis. Neuroscience191, 6–21.10.1016/j.neuroscience.2011.04.010
83
MajnoG.JorisJ. (1995). Apoptosis, Oncosis and Necrosis. Am. J. Pathol.146, 3–15.
84
MaksayG.BiroT. (2002). Dual cooperative allosteric modulation of vinding to ionotropic glycine receptors. Neuropharmacology43, 597–602.10.1016/S0028-3908(02)00213-7
85
MalikA. S.NarayanR. K.WendlingW. W.ColeR. W.PashkoL. L.SchwartzA. G.StraussK. I. (2003). A novel dehydroepiandrosterone analog improves functional recovery in a rat traumatic brain injury model. J. Neurotrauma20, 463–476.10.1089/089771503765355531
86
MaoX.BargerS. W. (1998). Neuroprotection by dehydroepiandrosterone-sulfate: role of an NF kappa B-like factor. Neuroreport9, 759–763.10.1097/00001756-199803090-00036
87
MarešP.KubovaH.KasalA. (2010). Anticonvulsant action of a new analogue of allopregnanolone in immature rats. Physiol. Res.59, 305–308,
88
MartiniL.MagnaghiV.MelcangiR. C. (2003). Actions of progesterone and its 5alpha-reduced metabolites on the major proteins of the myelin of the peripheral nervous system. Steroids68, 825–829.10.1016/S0039-128X(03)00134-X
89
MehtaS. L.ManhasN.RaghubirR. (2007). Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res. Rev.54, 34–66.10.1016/j.brainresrev.2006.11.003
90
MellonS. H.GongW.SchonemannM. D. (2008). Endogenous and synthetic neurosteroids in treatment of Niemann-Pick Type C disease. Brain Res. Rev.57, 410–420.10.1016/j.brainresrev.2007.05.012
91
MishraM.SinghR.SharmaD. (2010). Antiepileptic action of exogenous dehydroepiandrosterone in iron-induced epilepsy in rat brain. Epilepsy Behav.19, 264–271.10.1016/j.yebeh.2010.06.048
92
MonaghanE. P.NavaltaL. A.ShumL.AshbrookD. W.LeeD. A. (1997). Initial human experience with ganaxolone, a neuroactive steroid with antiepileptic activity. Epilepsia38, 1026–1031.10.1111/j.1528-1157.1997.tb01486.x
93
MonnetF. P.MahéV.RobelP.BaulieuE. E. (1995). Neurosteroids, via sigma receptors, modulate the [3H]norepinephrine release evoked by N-methyl-D-aspartate in the rat hippocampus. Proc. Natl. Acad. Sci. U.S.A.92, 3774–3778.10.1073/pnas.92.9.3774
94
MoralíG.Letechipía-VallejoG.López-LoezaE.MontesP.Hernández-MoralesL.CervantesM. (2005). Post-ischemic administration of progesterone in rats exerts neuroprotective effects on the hippocampus. Neurosci. Lett.382, 286–290.10.1016/j.neulet.2005.03.066
95
MoralíG.MontesP.Hernández-MoralesL.MonfilT.Espinosa-GarcíaC.CervantesM. (2011). Neuroprotective effects of progesterone and allopregnanolone on long-term cognitive outcome after global cerebral ischemia. Restor. Neurol. Neurosci.29, 1–15.
96
MurphyS. J.TraystmanR. J.HurnP. D.DucklesS. P. (2000). Progesterone exacerbates striatal stroke injury in progesterone-deficient female animals. Stroke31, 1173–1178.10.1161/01.STR.31.5.1173
97
NaylorJ. C.HuletteC. M.SteffensD. C.ShampineL. J.ErvinJ. F.PayneV. M.MassingM. W.KiltsJ. D.StraussJ. L.CalhounP. S.CalnaidoR. P.BlazerD. G.LiebermanJ. A.MadisonR. D.MarxC. E. (2008). Cerebrospinal fluid dehydroepiandrosterone levels are correlated with brain dehydroepiandrosterone levels, elevated in Alzheimer’s disease, and related to neuropathological disease stage. J. Clin. Endocrinol. Metab.93, 3173–3178.10.1210/jc.2007-1229
98
NaylorJ. C.KiltsJ. D.HuletteC. M.SteffensD. C.BlazerD. G.ErvinJ. F.StraussJ. L.AllenT. B.MassingM. W.PayneV. M.YoussefN. A.ShampineL. J.MarxC. E. (2010). Allopregnanolone levels are reduced in temporal cortex in patients with Alzheimer’s disease compared to cognitively intact control subjects. Biochim. Biophys. Acta1801, 951–959.
99
NilsenJ.BrintonR. D. (2002). Impact of progestins on estrogen-induced neuroprotection: synergy by progesterone and 19-norprogesterone and antagonism by medroxyprogesterone acetate. Endocrinology143, 205–212.10.1210/en.143.1.205
100
NohriaV.GillerE. (2007). Ganaxolone. Neurotherapeutics4, 102–105.10.1016/j.nurt.2006.11.003
101
O’ConnorC. A.CernakI.JohnsonF.VinkR. (2007). Effects of progesterone on neurologic and morphologic outcome following diffuse traumatic brain injury in rats. Exp. Neurol.205, 145–153.10.1016/j.expneurol.2007.01.034
102
PettusE. H.WrightD. W.SteinD. G.HoffmanS. W. (2005). Progesterone treatment inhibits the inflammatory agents that accompany traumatic brain injury. Brain Res.1049, 112–119.10.1016/j.brainres.2005.05.004
103
PiersonR. C.LyonsA. M.GreenfieldL. J. (2005). Gonadal steroids regulate GABA A receptor subunit mRNA expression in NT2-N neurons. Brain Res. Mol. Brain Res.138, 105–115.10.1016/j.molbrainres.2004.10.047
104
PikeC. J.CarrollJ. C.RosarioE. R.BarronA. M. (2009). Protective actions of sex steroid hormones in Alzheimer’s disease. Front. Neuroendocrinol.30, 239–258.10.1016/j.yfrne.2009.04.015
105
Plassart-SchiessE.BaulieuE. E. (2001). Neurosteroids: recent findings. Brain Res. Rev.37, 133–140.10.1016/S0165-0173(01)00113-8
106
ReddyD. S. (2004). Role of neurosteroids in catamenial epilepsy. Epilepsy Res.62, 99–118.10.1016/j.eplepsyres.2004.09.003
107
ReddyD. S. (2009). The role of neurosteroids of catamenial epilepsy. Epilepsy Res.85, 1–30.10.1016/j.eplepsyres.2009.02.017
108
ReddyD. S.RogawskiM. A. (2000a). Chronic treatment with the neuroactive steroid ganaxolone in the rat induces anticonvulsant tolerance to diazepam but not to itself. J. Pharmacol. Exp. Ther.295, 1241–1248.
109
ReddyD. S.RogawskiM. A. (2000b). Enhanced anticonvulsant activity of ganaxolone after neurosteroid withdrawal in a rat model of catamenial epilepsy. J. Pharmacol. Exp. Ther.294, 909–915.
110
ReddyD. S.RogawskiM. A. (2009). Neurosteroid replacement therapy for catamenial epilepsy. Neurotherapeutics6, 392–401.10.1016/j.nurt.2009.01.006
111
ReddyD. S.RogawskiM. A. (2010). Ganaxolone suppression of behavioral and electrographic seizures in the mause amygdala kindling model. Epilepsy Res.89, 254–260.10.1016/j.eplepsyres.2010.01.009
112
RhodesM. E.FryeC. A. (2005). Attenuating 5alpha-pregnane-3alpha-ol-20-one formation in the hippocampus of female rats increases pentylenetetrazole-induced seizures. Epilepsy Behav.6, 140–146.10.1016/j.yebeh.2005.02.006
113
RobertsonC. L.PuskarA.HoffmanG. E.MurphyA. Z.SaraswatiM.FiskumG. (2006). Physiologic progesterone reduces mitochondrial dysfunction and hippocampal cell loss after traumatic brain injury in female rats. Exp. Neurol.197, 235–243.10.1016/j.expneurol.2005.09.014
114
RogawskiM. A.ReddyD. S. (2004). “Neurosteroids: endogenous modulators of seizures susceptibility,” in Epilepsy: Scientific Foundations of Clinical Practive, eds RhoJ. M.SankarR.CavazosJ. (New York: Marcel Dekker), 319–355.
115
RoofR. L.DuvdevaniR.BraswellL.SteinD. G. (1994). Progesterone facilitates cognitive recovery and reduces secondary neuronal loss caused by cortical contusion injury in male rats. Exp. Neurol.129, 64–69.10.1006/exnr.1994.1147
116
RoofR. L.DuvdevaniR.HeyburnJ. W.SteinD. G. (1996). Progesterone rapidly decreases brain edema: treatment delayed up to 24 hours is still effective. Exp. Neurol.138, 246–251.10.1006/exnr.1996.0063
117
RoofR. L.HoffmanS. W.SteinD. G. (1997). Progesterone protects against lipid peroxidation following traumatic brain injury in rats. Mol. Chem. Neuropathol.31, 1–11.10.1007/BF02815156
118
RoofR. L.DuvdevaniR.SteinD. G. (1993). Gender influences outcome of brain injury: progesterone plays a protective role. Brain Res.607, 333–336.10.1016/0006-8993(93)91526-X
119
SayeedI.WaliB.SteinD. G. (2007). Progesterone inhibits ischemic brain injury in a rat model of permanent middle cerebral artery occlusion. Restor. Neurol. Neurosci.25, 151–159.
120
SchumacherM.GuennounR.RobertF.CarelliC.GagoN.GhoumariA.Gonzalez DeniselleM. C.GonzalezS. L.IbanezC.LabombardaF.CoiriniH.BaulieuE. E.De NicolaA. F. (2004). Local synthesis and dual actions of progesterone in the nervous system: neuroprotection and myelination. Growth Horm. IGF Res.14, 18–33.10.1016/j.ghir.2004.03.007
121
ShearD. A.GalaniR.HoffmanS. W.SteinD. G. (2002). Progesterone protects against necrotic damage and behavioral abnormalities caused by traumatic brain injury. Exp. Neurol.178, 59–67.10.1006/exnr.2002.8020
122
SinghM. (2006). Progesterone-induced neuroprotection. Endocrine29, 271–274.10.1385/ENDO:29:2:271
123
SperandioS.de BelleI.BredesenD. E. (2000). An alternative, nonapoptotic form of programmed cell death. Proc. Natl. Acad. Sci. U.S.A.97, 14376–14381.10.1073/pnas.97.26.14376
124
SperandioS.PoksayK.de BelleI.LafuenteM. J.LiuB.NasirJ.BredesenD. E. (2004). Paraptosis: mediation by MAP kinases and inhibition by AIP-1/Alix. Cell Death Differ.11, 1066–1075.10.1038/sj.cdd.4401465
125
SperandioS.PoksayK. S.SchillingB.CrippenD.GibsonB. W.BresedenD. E. (2010). Identification of new modulators and protein alterations in non-apoptotic programmed cell death. J. Cell. Biochem.111, 1401–1412.10.1002/jcb.22870
126
SteinD. G. (2001). Brain damage, sex hormones and recovery: a new role for progesterone and estrogen?Trends Neurosci.24, 386–391.10.1016/S0166-2236(00)01821-X
127
SteinD. G. (2008). Progesterone exerts neuroprotective effects after brain injury. Brain Res. Rev.57, 386–397.10.1016/j.brainresrev.2007.06.012
128
SteinD. G. (2011). Progesterone in the treatment of acute traumatic brain injury: a clinical perspective and update. Neuroscience191, 101–106.10.1016/j.neuroscience.2011.04.013
129
SteinD. G.WrightD. W. (2010). Progesterone in the clinical treatment of acute traumatic brain injury. Expert Opin. Investig. Drugs19, 847–857.10.1517/13543784.2010.489549
130
SteinD. G.WrightD. W.KellermanA. L. (2008). Does progesterone have neuroprotective properties?Ann. Emerg. Med.51, 164–172.10.1016/j.annemergmed.2007.05.001
131
StevensS. J.HardenC. L. (2011). Hormonal therapy for epilepsy. Curr. Neurol. Neurosci. Rep.11, 435–442.10.1007/s11910-011-0196-9
132
TengX.DegterevA.JagtapP.XingX.ChoiS.DenuR.YuanJ.CunyG. D. (2005). Structure-activity relationship study of novel necroptosis inhibitors. Bioorg. Med. Chem. Lett.15, 5039-5044.10.1016/j.bmcl.2005.07.077
133
ThomasA. J.NockelsR. P.PanH. Q.ShaffreyC. I.ChoppM. (1999). Progesterone is neuroprotective after acute experimental spinal cord trauma in rats. Spine24, 2134–2138.10.1097/00007632-199910150-00013
134
ToungT. J.ChenT. Y.Littleton-KearneyM. T.HurnP. D.MurphyS. J. (2004). Effects of combined estrogen and progesterone on brain infarction in reproductively senescent female rats. J. Cereb. Blood Flow Metab.24, 1160–1166.
135
UngardJ. T.BeekmanM.GasiorM.CarterR. B.DijkstraD.WitkinJ. M. (2000). Modification of behavioral effects of drugs in mice by neuroactive steroids. Psychopharmacology (Berl.)148, 336–343.10.1007/s002130050060
136
Van CruchtenS.Van Den BroeckW. (2002). Morphological and biochemical aspects of apoptosis, oncosis and necrosis. Anat. Histol. Embryol.31, 214–223.10.1046/j.1439-0264.2002.00398.x
137
VandenabeeleP.GalluzziL.Vanden BergheT.KroemerG. (2010). Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol.11, 700–714.10.1038/nrm2970
138
VanLandinghamJ. W.CekicM.CutlerS.HoffmanS. W.SteinD. G. (2007). Neurosteroids reduce inflammation after TBI through CD55 induction. Neurosci. Lett.425, 94–98.10.1016/j.neulet.2007.08.045
139
VeigaS.Garcia-SeguraL. M.AzcoitiaI. (2003). Neuroprotection by the steroids pregnenolone and dehydroepiandrosterone is mediated by the enzyme aromatase. J. Neurobiol.56, 398–406.10.1002/neu.10249
140
VerrottiA.LausM.CoppolaG.ParisiP.MohnA.ChiarelliF. (2010). Catamenial epilepsy: hormonal aspects. Gynecol. Endocrinol.26, 783–790.10.3109/09513590.2010.490606
141
WangK.LiJ.DegterevA.HsuE.YuanJ.YuanC. (2007) Structure-activity relationship analysis of a novel necroptosis inhibitor, Necrostatin-5. Bioorg. Med. Chem. Lett.17, 1455–1465.10.1016/j.bmcl.2007.03.022
142
Weill-EngererS.DavidJ. P.SazdovitchV.LiereP.EychenneB.PianosA.SchumacherM.DelacourteA.BaulieuE. E.AkwaY. (2002). Neurosteroid quantification in human brain regions: comparison between Alzheimer’s and nondemented patients. J. Clin. Endocrinol. Metab.87, 5138–5143.10.1210/jc.2002-020878
143
WeirC. J.LingA. T.BelelliD.WildsmithJ. A.PetersJ. A.LambertJ. J. (2004). The interaction of anaesthetic steroids with recombinant glycine and GABAA receptors. Br. J. Anaesth.92, 704–711.10.1093/bja/aeh125
144
WojtalK.TrojnarM. K.CzuczwarS. J. (2006). Endogenous neuroprotective factors: neurosteroids. Pharmacol. Rep.58, 335–340.
145
WrightD. W.BauerM. E.HoffmanS. W.SteinD. G. (2001). Serum progesterone levels correlate with decreased cerebral edema after traumatic brain injury in male rats. J. Neurotrauma18, 901–909.10.1089/089771501750451820
146
WrightD. W.KellermannA. L.HertzbergV. S.ClarkP. L.FrankelM.GoldsteinF. C.SalomoneJ. P.DentL. L.HarrisO. A.AnderD. S.LoweryD. W.PatelM. M.DensonD. D.GordonA. B.WaldM. M.GuptaS.HoffmanS. W.SteinD. G. (2006). ProTECT: a randomized clinical trial of progesterone for acute traumatic brain injury. Ann. Emerg. Med.49, 391–402.10.1016/j.annemergmed.2006.07.932
147
XiaoG.WeiJ.YanW.WangW.LuZ. (2008). Improved outcomes from the administration of progesterone for patients with acute severe traumatic brain injury: a randomized controlled trial. Crit. Care12, R61.10.1186/cc6282
148
XilouriM.PapazafiriP. (2006). Anti-apoptotic effects of allopregnanolone on P19 neurons. Eur. J. Neurosci.23, 43–54.10.1111/j.1460-9568.2005.04548.x
149
YakovlevA. G.FadenA. I. (2004). Mechanisms of neural cell death: implications for development of neuroprotective treatment strategies. NeuroRx1, 5–16.10.1602/neurorx.1.1.5
150
YaoX. L.LiuJ.LeeE.LingG. S.McCabeJ. T. (2005). Progesterone differentially regulates pro- and anti-apoptotic gene expression in cerebral cortex following traumatic brain injury in rats. J. Neurotrauma22, 656–668.10.1089/neu.2005.22.656
151
YuW. H. (1989). Survival of motoneurons following axotomy is enhanced by lactation or by progesterone treatment. Brain Res.491, 379–382.10.1016/0006-8993(89)90075-9
152
YuanJ.LipinskiM.DegterevA. (2003). Diversity in the mechanisms of neuronal cell death. Neuron40, 401–413.10.1016/S0896-6273(03)00601-9
Summary
Keywords
neurosteroids, neurodegeneration, neuroprotection
Citation
Borowicz KK, Piskorska B, Banach M and Czuczwar SJ (2011) Neuroprotective Actions of Neurosteroids. Front. Endocrin. 2:50. doi: 10.3389/fendo.2011.00050
Received
04 July 2011
Accepted
22 September 2011
Published
11 October 2011
Volume
2 - 2011
Edited by
Hubert Vaudry, University of Rouen, France
Reviewed by
Denise D. Belsham, University of Toronto, Canada; Alejandro F. D. Nicola, Instituto de Biologia y Medicina Experimental, Argentina
Copyright
© 2011 Borowicz, Piskorska, Banach and Czuczwar.
This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Stanislaw J. Czuczwar, Department of Pathophysiology, Medical University, Jaczewskiego 8, PL-20-090 Lublin, Poland. e-mail: czuczwarsj@yahoo.com
This article was submitted to Frontiers in Neuroendocrine Science, a specialty of Frontiers in Endocrinology.
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.