
- Department of Pediatrics, University of California San Francisco, San Francisco, CA, USA
Obesity is a common complication after craniopharyngioma therapy, occurring in up to 75% of survivors. Its weight gain is unlike that of normal obesity, in that it occurs even with caloric restriction, and attempts at lifestyle modification are useless to prevent or treat the obesity. The pathogenesis of this condition involves the inability to transduce afferent hormonal signals of adiposity, in effect mimicking a state of CNS starvation. Efferent sympathetic activity drops, resulting in malaise and reduced energy expenditure, and vagal activity increases, resulting in increased insulin secretion and adipogenesis. Lifestyle intervention is essentially useless in this syndrome, termed “hypothalamic obesity.” Pharmacologic treatment is also difficult, consisting of adrenergics to mimic sympathetic activity, or suppression of insulin secretion with octreotide, or both. Recently, bariatric surgery (Roux-en-Y gastric bypass, laparoscopic gastric banding, truncal vagotomy) have also been attempted with variable results. Early and intensive management is required to mitigate the obesity and its negative consequences.
Introduction
When it comes to brain tumors, the three laws of New York real estate prevail: “Location, location, location.” Craniopharyngiomas are problematic less for what they are than for where they are. The hypothalamus, as is true for most hormonal systems, is the anatomic seat of peripheral energy regulation. When the hypothalamus is damaged, a syndrome of intractable weight gain ensues. This syndrome, termed “hypothalamic obesity,” originally described by Babinski (1900) and Frohlich (1901) at the turn of the twentieth century, documents the “organicity” of obesity. Hypothalamic obesity can occur due to the tumor itself, the surgery to extirpate it, or due to subsequent radiation therapy (Bray, 1984; Lustig, 2002). Although this co-morbidity usually manifests in children due to their increased incidence of tumors localized to the posterior fossa (Stahnke et al., 1984; Sorva, 1988; Pinto et al., 2000), adults can also exhibit similar weight gain after completion of therapy (Daousi et al., 2005). Craniopharyngioma accounts for half of the reported cases, with other posterior fossa tumors each contributing smaller numbers. However, the syndrome has also been reported in cases of pseudotumor cerebri, trauma, and infiltrative or inflammatory diseases of the hypothalamus (Bray, 1984).
Incidence and Risk Factors
Hypothalamic obesity can occur in response to any hypothalamic damage. Most studies have been performed in the acute lymphoblastic leukemia (ALL) survivor population (Lustig, 2002; Rogers et al., 2005), in which obesity may be due to several factors, including glucocorticoids and alterations in activity. Nonetheless, the majority of these studies document an abnormal increase in weight for height long after tumor therapy has been discontinued, and many of these studies demonstrate that cranial radiation is an important risk factor (Lustig, 2002).
An extremely high frequency of hypothalamic obesity of 30–77% has been documented after craniopharyngioma treatment (Stahnke et al., 1984; Sorva, 1988; Pinto et al., 2000; Muller, 2008; Vinchon et al., 2009). We analyzed the BMI curves of 148 children with brain tumors who survived longer than 5 years post-therapy, in order to determine risk factors for the development of obesity (Lustig et al., 2003a). We identified four parameters as being predictive. First, those with tumors localized to the hypothalamus or thalamus, along with those originating in the temporal lobe (due to stereoscopic position of the hypothalamus during radiation for this area) gained weight much more rapidly as did those with tumors in the posterior fossa or other hemispheric areas. Secondly, those with tumor histologies prominent in the diencephalon (craniopharyngioma, germinoma, optic glioma, prolactinoma, hypothalamic astrocytoma) also gained weight more rapidly. Third, those with quantitative direct radiation exposure of the hypothalamus of greater than 51 Gy gained excessive weight twice as rapidly after the completion of tumor therapy, even when those with hypothalamic or thalamic locations were removed from the analysis. Lastly, those with some other form of hypothalamic endocrinopathy (i.e., GH deficiency, hypothyroidism, precocious or delayed puberty, ACTH deficiency, diabetes insipidus) exhibited a BMI curve with a steeper upward slope. Thus, each significant risk factor was either linked to hypothalamic location, damage, or dysfunction. Factors not associated with obesity after tumor therapy included hydrocephalus, initial high-dose glucocorticoids, and peripheral or intrathecal chemotherapy.
More recently, Müller et al. (2011) respectively evaluated the long-term outcome data on the Kraniopharyngeom database in Germany. In this analysis, pre-operative hypothalamic involvement was specifically implicated in the development of post-operative hypothalamic obesity, suggesting again that tumor location is the most important risk factor for obesity.
The Energy Balance Negative Feedback Pathway
Animal studies elaborating the negative feedback energy balance pathway have predicted the pathogenesis and symptomatology of hypothalamic obesity. This can best be described as “organic leptin resistance”; that is, a failure in leptin signaling in the afferent arm, due to hypothalamic damage; leading to autonomic dysfunction in the efferent arm, promoting inadequate energy expenditure, and excessive energy storage.
The Afferent Arm
Circulating leptin (derived from peripheral adipocytes) crosses the blood–brain barrier, and synapses on receptors located on neurons within the ventromedial hypothalamus [VMH; which consists of the ventromedial nucleus (VMN) and arcuate nucleus (AN)]. In the energy replete state, both insulin and leptin levels are increased, which acts on a set of “anorexigenic” neurons to increase the synthesis and processing of proopiomelanocortin (POMC) in the VMH to its component peptides, including α-melanocyte stimulating hormone (α-MSH) and its co-localized neuromodulator cocaine–amphetamine regulated transcript (CART), both of which act at the lateral hypothalamic area (LHA) and paraventricular nucleus (PVN) to alter melanocortin receptor-4 (MC4R) occupancy, which decreases appetite and food intake (Elmquist et al., 1999; Kalra et al., 1999; Schwartz et al., 2000; Balthasar et al., 2005). The stomach hormone ghrelin stimulates, while insulin and leptin inhibit a set of “orexigenic” neurons to inhibit the release of neuropeptide Y (NPY) and agouti-related protein (AgRP), further limiting feeding and providing for unantagonized MC4R occupancy (Elmquist et al., 1998). Immediately after a meal, ghrelin levels are low, which prevents orexigenic neuronal activation and NPY neurotransmission (Kamegai et al., 2000), keeping hunger at a minimum; furthermore, PYY levels increase after a meal; this hormone binds to the Y2 receptor on orexigenic neurons, activating gamma-amino butyric acid (GABA), which inhibits signal transduction of NPY to inhibit further food intake (Small and Bloom, 2004).
Conversely, in the fasting state, gastric secretion of ghrelin is increased (Kamegai et al., 2000; Tschöp et al., 2000), while leptin, insulin, and PYY levels are low, which leads to stimulation of the orexigenic pathway (NPY/AgRP), and antagonism of the anorexigenic pathway (α-MSH/CART). The resultant lack of anorexigenic pressure on the MC4R results in increased feeding behavior and energy efficiency (with reduced fat oxidation), in order to store energy substrate as fat. This is accomplished through signal transduction within the efferent pathway, consisting of the sympathetic nervous system (SNS) and the vagus (see below).
The Efferent Arm
From the PVN and LHA, efferent projections synapse in the locus coeruleus (LC), which controls the SNS; and in the dorsal motor nucleus of the vagus (DMV), which controls the vagus nerve, the chief output of the parasympathetic nervous system.
In the energy replete state, elevated leptin and insulin levels cause the anorexigenic arm to activate the SNS (Muntzel et al., 1994; Vollenweider et al., 1995; Rahmouni et al., 2003). Stimulation of β2-adrenergic receptors by the SNS (Blaak et al., 1993) increase the expression of numerous genes in skeletal muscle (Viguerie et al., 2004), which promote mitochondrial biogenesis, glycogenolysis, thermogenesis, and increased movement (Boss et al., 1999; Lowell and Spiegelman, 2000), all associated with increased energy expenditure (Collins et al., 1996). The SNS also activates α2a- and α2c-adrenoreceptors on the β-cell, which stimulate Gi and inhibit adenyl cyclase, lower cAMP, and maintain potassium channels in an open configuration with a negative resting membrane potential (Sharp, 1996), in order to reduce pancreatic insulin secretion, and thus reduce energy deposition into adipose tissue. Lastly, SNS activation stimulates the β3-adrenergic receptor on the adipocyte to promote lipolysis (Susulic et al., 1995). These coordinate sympathetic events serve to reduce adipose tissue leptin expression and secretion; thus this forms a negative feedback loop with the afferent system (Figure 1).
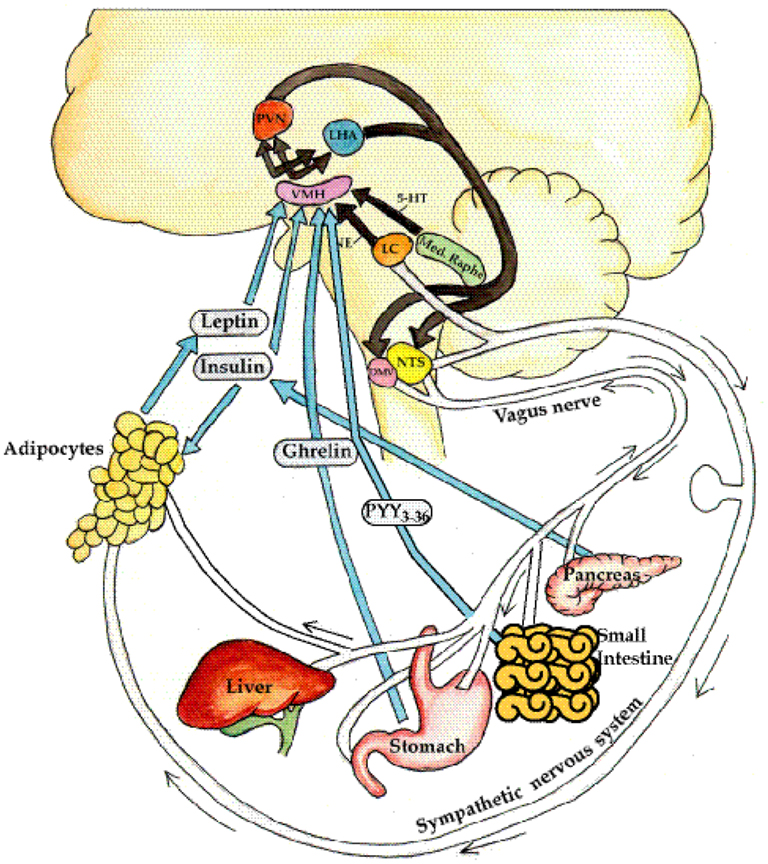
Figure 1. Neuroendocrine regulation of energy balance. The afferent system: neural (e.g., vagal) and hormonal (ghrelin, insulin, leptin) signals are generated from the liver, gut, pancreas, and adipose. In addition, norepinephrine from the locus cœruleus and serotonin (5-HT) from the median raphe are elaborated. These signals of satiety vs. hunger, and thinness vs. fatness are interpreted in the ventromedial hypothalamus (VMH). These signals are then integrated in the paraventricular nucleus (PVN) and lateral hypothalamus (LHA). The efferent system: efferent signals from these areas in turn stimulate the sympathetic nervous system (SNS) to expend energy by activating β3-adrenergic receptors and uncoupling proteins in the adipocyte, to release energy the form of lipolysis, heat, or physical activity. Conversely, the parasympathetic nervous system (efferent vagal) increases insulin secretion, with resultant adipogenesis and energy storage, and also increases insulin sensitivity through direct effects on the adipose tissue (Lustig, 2006). From Nature Publishing Group, with permission.
Conversely, in the fasting state, leptin and insulin are low, leading to reduced SNS tone, and reduced skeletal muscle thermogenesis, and reduced adipose tissue lipolysis. In addition, the LHA and PVN send efferent projections residing in the medial longitudinal fasciculus to the DMV nerve (Powley and Laughton, 1981). By slowing the heart rate, the vagus reduces myocardial oxygen consumption. Through its effects on the alimentary tract, the vagus promotes peristalsis, and energy substrate absorption. Through its effects on the adipocyte, the vagus promotes increased lipoprotein lipase activity to increase the clearance of energy substrate into adipose tissue (Boden and Hoeldtke, 2003). Lastly, through effects on the β-cell (D’Alessio et al., 2001), the vagus accentuates postprandial insulin hypersecretion in response to a meal, which promotes energy deposition into the adipocyte (Rohner-Jeanrenaud and Jeanrenaud, 1985; Marin et al., 1988; Peles et al., 1995; Lustig, 2003). Overactive vagal neurotransmission increases insulin secretion through three distinct but overlapping mechanisms (Gilon and Henquin, 2001; Figure 2):
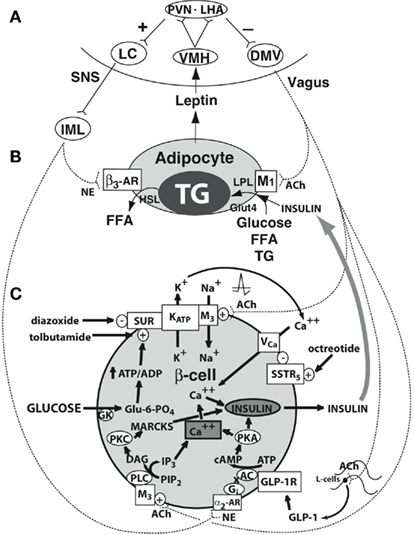
Figure 2. Central regulation of leptin signaling, autonomic innervation of the adipocyte and β-cell, and the starvation response. (A) The arcuate nucleus transduces the peripheral leptin signal as one of sufficiency or deficiency. In leptin sufficiency, efferents from the hypothalamus synapse in the locus coeruleus, which stimulates the sympathetic nervous system. In leptin deficiency, efferents from the hypothalamus stimulate the dorsal motor nucleus of the vagus. (B) Autonomic innervation and hormonal stimulation of white adipose tissue. In leptin sufficiency, norepinephrine binds to the β3-adrenergic receptor, which stimulates hormone-sensitive lipase, promoting lipolysis of stored triglyceride into free fatty acids. In leptin deficiency, vagal acetylcholine increases adipose tissue insulin sensitivity (documented only in rats to date), promotes uptake of glucose and free fatty acids for lipogenesis, and promotes triglyceride uptake through activation of lipoprotein lipase. (C) Autonomic innervation and hormonal stimulation of the β-cell. Glucose entering the cell is converted to glucose-6-phosphate by the enzyme glucokinase, generating ATP, which closes an ATP-dependent potassium channel, resulting in cell depolarization. A voltage-gated calcium channel opens, allowing for intracellular calcium influx, which activates neurosecretory mechanisms leading to insulin vesicular exocytosis. In leptin sufficiency, norepinephrine binds to α2-adrenoceptors on the β-cell membrane to stimulate inhibitory G proteins, decrease adenyl cyclase and its product cAMP, and thereby reduce protein kinase A levels and insulin release. In leptin deficiency, the vagus stimulates insulin secretion through three mechanisms. First, acetylcholine binds to a M3 muscarinic receptor, opening a sodium channel, which augments the ATP-dependent cell depolarization, increasing the calcium influx, and insulin exocytosis. Secondly, acetylcholine activates a pathway that increases protein kinase C, which also promotes insulin secretion. Thirdly, the vagus innervates L-cells of the small intestine, which secrete glucagon-like peptide-1, which activates protein kinase A, contributing to insulin exocytosis. Octreotide binds to a somatostatin receptor on the β-cell, which is coupled to the voltage-gated calcium channel, limiting calcium influx and the amount of insulin released in response to glucose. (Lustig, 2007; reprinted with kind permission of Humana, Totowa, NJ, USA). α2-AR, α2-adrenergic receptor; β3-AR, β3-adrenergic receptor; AC, adenyl cyclase; ACh, acetylcholine; DAG, diacylglycerol; DMV, dorsal motor nucleus of the vagus; FFA, free fatty acids; Gi, inhibitory G protein; GK, glucokinase; GLP-1, glucagon-like peptide-1; GLP-1R, GLP-1 receptor; Glu-6-PO4, glucose-6-phosphate; Glut4, glucose transporter-4; HSL, hormone-sensitive lipase; IML, intermediolateral cell column; IP3, inositol triphosphate; LC, locus coeruleus; LHA, lateral hypothalamic area; LPL, lipoprotein lipase; MARCKS, myristoylated alanine-rich protein kinase C substrate; NE, norepinephrine; PIP2, phosphatidylinositol; PKA, protein kinase A; PKC, protein kinase C; PLC, phospholipase C; PVN, paraventricular nucleus; SSTR5, somatostatin-5 receptor; TG, triglyceride; VCa, voltage-gated calcium channel; VMH, ventromedial hypothalamus; SUR, sufonylurea receptor (Lustig, 2006). From Nature Publishing Group, with permission.
1. Vagal firing increases acetylcholine availability and binding to the M3 muscarinic receptor on the β-cell, which is coupled to a sodium channel within the pancreatic β-cell membrane (Miura et al., 1996). As glucose enters the β-cell after ingestion of a meal, the enzyme glucokinase phosphorylates glucose to form glucose-6-phosphate. This increases the generation of intracellular ATP, which induces closure of the β-cell’s ATP-dependent potassium channel. Upon channel closure, the β-cell experiences an ATP concentration-dependent β-cell depolarization (Nishi et al., 1987; Zawalich et al., 1989), and the opening of a separate voltage-gated calcium channel within the membrane. Intracellular calcium influx increases acutely, which results in rapid insulin vesicular exocytosis. Concomitant opening of the sodium channel by vagally derived acetylcholine augments the β-cell depolarization, which augments the intracellular calcium influx, and results in insulin hypersecretion (Berthoud and Jeanrenaud, 1979; Komeda et al., 1980; Rohner-Jeanrenaud and Jeanrenaud, 1980).
2. Vagally mediated acetylcholine increases phospholipases A2, C, and D, within the β-cell, which hydrolyze intracellular phosphatidylinositol to diacylglycerol (DAG) and inositol triphosphate (IP3; Gilon and Henquin, 2001). DAG is a potent stimulator of protein kinase C (PKC; Tian et al., 1996) which phosphorylates myristoylated alanine-rich Protein Kinase C substrate (MARCKS), which then binds actin and calcium–calmodulin, and induces insulin vesicular exocytosis (Arbuzova et al., 1998). IP3 potentiates release of calcium within β-cells from intracellular stores, which also promotes insulin secretion (Blondel et al., 1994).
3. The vagus also stimulates the release of glucagon-like peptide-1 (GLP-1) from intestinal L-cells, which circulates and binds to the β-cell GLP-1 receptor. Activation of this receptor induces a calcium–calmodulin-sensitive adenyl cyclase, with generation of cAMP, which activates protein kinase A (PKA), stimulating phosphorylation of vesicular proteins, with resultant insulin exocytosis (Kiefer and Habener, 1999).
In this way, the afferent system is entrained with the efferent system by an intricate servo-mechanism to coordinate central and peripheral signals either for appetite and energy storage, or satiety and energy expenditure.
Pathogenesis of Hypothalamic Obesity
Rat models of hypothalamic damage, either due to bilateral electrolytic lesions or deafferentation of the VMH, lead to intractable weight gain (Berthoud and Jeanrenaud, 1979; Rohner-Jeanrenaud and Jeanrenaud, 1980; Bray et al., 1981; Jeanrenaud, 1985; Satoh et al., 1997), even upon food restriction (Bray and Nishizawa, 1978). Similarly, children with hypothalamic obesity exhibit weight gain, even in response to forced caloric restriction (Bray and Gallagher, 1975). This seems paradoxical, as one would expect that if hyperphagia were the reason for the obesity, then caloric restriction would be effective in preventing further weight gain. In fact, analysis of energy intake in children with hypothalamic obesity demonstrates no difference vs. control patients with simple obesity (Harz et al., 2003). Instead, both resting energy expenditure (Shaikh et al., 2008) and voluntary energy expenditure (Harz et al., 2003) is severely compromised in these patients. Indeed, the most prominent and concerning complaint in patients with hypothalamic obesity is the persistent fatigue, lack of energy, and lack of physical activity. This generalized malaise is not due to hypopituitarism, as it persists even after full hormonal replacement.
The decrease in energy expenditure is mediated through suppression of SNS activity by the hypothalamic damage. Recent reports demonstrate an impaired ability of such patients to mount an epinephrine response to insulin-induced hypoglycemia (Schofl et al., 2002; Coutant et al., 2003), and document decreased 24-h epinephrine excretion (Coutant et al., 2003), along with decreased urinary homovanillic acid and vanillylmandelic acid (Roth et al., 2007); all pointing to decreased sympathetic tone. It is thought that this malaise and decrease in sympathetic tone may account for decreased rates of lipolysis through the adipocyte β3-adrenergic receptor (al-Adsani et al., 1997), which results in decreased resting and voluntary energy expenditure.
In addition to “organic leptin resistance,” it is possible that such patients also manifest “organic ghrelin resistance,” in that ghrelin’s suppression of hunger may be attenuated in children with hypothalamic obesity (O’Gorman et al., 2911). This may increase total food intake; although alterations in total food intake in these patients is not different from otherwise healthy obese controls (Harz et al., 2003).
Diagnosis
A retrospective analysis of growth records of children with craniopharyngioma (Muller et al., 2004) indicates that increased weight and BMI gain is evident even before the diagnosis of the tumor. However, after surgery or radiotherapy, the weight gain is immediate, rapid, and highly exaggerated. Evidence of aberrant energy deposition is obvious within the first month. Physicians sometimes confuse this weight gain with glucocorticoid effect, and reduce the dose of maintenance hydrocortisone, which does not impact the obesity, and renders the patient with even more fatigue and malaise.
Children with hypothalamic obesity frequently have normal fasting insulin levels, especially during the rapid weight gain phase. It is important that such metabolic testing be dynamic, as the phenomenon that distinguishes hypothalamic obesity is insulin hypersecretion, not insulin resistance, and so may not be obvious with a fasting insulin level. In addition, stimulation of the alimentary tract so as to activate the vagal efferent component of insulin secretion is required to document the effect. Two sets of studies on insulin dynamics demonstrate insulin hypersecretion (as measured by an increased Corrected Insulin Response, or CIR; Sluiter et al., 1976) on oral glucose tolerance testing (OGTT); and surprisingly these patients also have insulin sensitivity (as measured by an increased Composite Insulin Sensitivity Index (Matsuda and DeFronzo, 1999) within the normal range, and certainly better than BMI-matched healthy obese children (Preeyasombat et al., 2005; Simoneau-Roy et al., 2010; Figure 3). However, other studies suggest that some hypothalamic obesity patients may also manifest signs of metabolic syndrome (Tiosano et al., 2003; Srinivasan et al., 2004). These patients may also have an increased incidence of obstructive sleep apnea, which may predispose them to the co-morbidities of the metabolic syndrome (O’Gorman et al., 2010). It is not clear whether those patients with both hypothalamic obesity and metabolic syndrome represent a subgroup, or a different pathogenetic phenomenon entirely, or just the late evolution of their morbid obesity. A retrospective evaluation suggests that the degree of hypothalamic involvement of the tumor at its presentation predicts the degree of metabolic disturbance (Müller et al., 2011), though mechanisms for the metabolic alteration are still unclear.
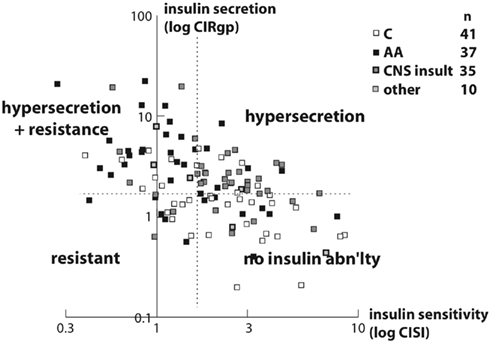
Figure 3. Scatterplot of insulin secretion (Corrected Insulin Response, or CIRgp) vs. sensitivity (Composite Insulin Sensitivity Index, or CISI) plotted logarithmically in 113 obese non-diabetic children. A negative linear correlation was noted (r = −0.54, p < 0.001). Different racial and etiopathogenic groups tended to plot in different areas. Arbitrary cutoffs (dashed lines) for CIRgp (1.5) and CISI (1.7) divide the plot into four quadrants. The majority of Caucasian children (open squares) plotted in the lower right quadrant, with a CIRgp less than 1.5 and a CISI greater than 1.7, indicating lower insulin secretion and better insulin sensitivity. The preponderance of children with hypothalamic obesity (gray squares) plotted in the upper right quadrant, with a CIRgp of greater than 1.5, and with a CISI of greater than 1.7, indicating insulin hypersecretion with better insulin sensitivity. Finally the majority of African American children (filled squares) plotted in the upper left quadrant, with a CIRgp of greater than 1.5 and a CISI of less than 1.7, indicating both insulin hypersecretion and resistance (Preeyasombat et al., 2005). From Elsevier, with permission.
Treatment
The best treatment is prevention. The hypothalamus is extremely sensitive to both surgical intervention and/or external beam radiation (Lustig et al., 2003a). Rather than employing gross total or subtotal resection as a primary therapy for some posterior fossa tumors, newer strategies have been developed which treat them more conservatively, using stereotactic biopsy and conformal irradiation (Karavitaki et al., 2006; Spoudeas et al., 2006). A retrospective single-institutional followup of craniopharyngioma subjected either to gross total resection or stereotactic surgery and conformal radiation demonstrates equal survival and residual rates of hypopituitarism; however those treated with gross total resection exhibit higher incidences of obesity and neurological complications (Merchant et al., 2002).
Bray demonstrated the futility of lifestyle intervention by noting weight gain even with severe caloric restriction (Bray and Gallagher, 1975). Thus, treatment needs to be early and intensive to have any chance at success. A recent report suggests that intensive lifestyle can reduce the rate of BMI gain by half (from 8.4 kg/m2/year to 4.5 kg/m2/day), but the rate of increase is still quite unacceptable to rely on (Rakhshani et al., 2010).
Pharmacotherapy
Since the hypothalamus is not amenable to therapy, and aberrant afferent hormonal signal transduction cannot be corrected, pharmacotherapy must instead address the alterations in the efferent pathways. Several attempts to use serotonin or norepinephrine reuptake inhibitors (e.g., phen–fen, fluoxetine, sibutramine) have been met with only salutary efficacy (Molloy et al., 1998). One study assessed the effects of sibutramine 10–15 mg PO qd, with a small but reproducible effect in BMI (Danielsson et al., 2007); however, sibutramine has been withdrawn from the market. These medications work centrally to reduce food intake, but do not work peripherally to stimulate skeletal muscle to increase energy expenditure, and thus have limited value. Mason et al. (2002) used dextroamphetamine 5 mg PO bid, which acts both centrally and peripherally, and achieved weight stability for an interval of 6 months. We have also seen improvement in affect and alertness, which is a major benefit of dextroamphetamine.
In an attempt to reduce hyperinsulinemia, Hamilton have attempted to treat patients with a combination of diazoxide and metformin (Hamilton et al., 2011). Weight gain over 6 months was reduced as compared to pre-treatment; however, side-effects were significant, including edema, and there were some discontinuations.
In an attempt to reduce hyperinsulinemia and simultaneously enhancing insulin action, we examined the effects of the somatostatin analog octreotide (an agonist of the somatostatin-5 receptor on the β-cell, which inhibits the voltage-gated calcium channel; Figure 2). A pilot, open-label trial of octreotide 15 μg/kg/day subcutaneously for 6 months in eight subjects (Lustig et al., 1999) demonstrated BMI loss commensurate with the degree of insulin suppression, along with decrease in caloric intake, and subjective improvements in spontaneous physical activity and quality of life. A double-blind, placebo-controlled trial of 20 subjects (Lustig et al., 2003b) resulted in insulin suppression and stabilization of BMI, decreased leptin, decreased caloric intake, increased spontaneous physical activity, and improvement in quality of life commensurate with the degree of insulin suppression. A retrospective analysis demonstrated that octreotide was most effective in those patients who exhibited both insulin hypersecretion with continued insulin sensitivity (Preeyasombat et al., 2005).
Surgery
The severity and morbidity of obesity in these patients, and the relative lack of alternatives, have led to attempts at bariatric surgery. Inge et al. (2007) reported a 25-kg weight loss after Roux-en-Y gastric bypass in one subject, but whose weight stabilized at an unacceptable level. Recently, Müller et al. (2007) reported in abstract form his experience with four subjects who underwent laparoscopic adjustable gastric banding, with reductions in food intake and slow reductions in BMI. Lastly, vagotomy may be effective in this syndrome (Smith et al., 1983), by reducing the efferent output to both beta-cells and adipocytes. We have recently performed laparoscopic truncal vagotomy in four subjects with hypothalamic obesity, with early results being supportive of this procedure, and with relatively few complications or side-effects (Lustig et al., 2009).
Summary
The hypothalamus interprets afferent signals for energy balance, and transduces them into autonomic efferent signals to either expend or store energy. When this negative feedback system breaks down, as after craniopharyngioma therapy, the phenomenon of hypothalamic obesity ensues. While this disorder is a defect in the afferent pathway, treatment focuses on the efferent pathway, as it is modulable with drugs and surgical techniques that are currently available. Physicians need to explain the risks of this disorder to patients prior to tumor therapy, and must be willing to act quickly and decisively once the intractable weight gain begins, in order to provide intensive management so that the obesity will not get worse.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
al-Adsani, H., Hoffer, L. J., and Silva, J. E. (1997). Resting energy expenditure is sensitive to small dose changes in patients on chronic thyroid hormone replacement. J. Clin. Endocrinol. Metab. 82, 1118–1125.
Arbuzova, A., Murray, D., and McLaughlin, S. (1998). MARCKS, membranes, and calmodulin: kinetics of their interaction. Biochim. Biophys. Acta 1376, 369–379.
Babinski, M. J. (1900). Tumeur du corps pituitaire san acromegalie et avec arret de developpement des organes genitaux. Rev. Neurol. 8, 531–533.
Balthasar, N., Dalgaard, L. T., Lee, C. E., Yu, J., Funahashi, H., Williams, T., Ferreira, M., Tang, V., McGovern, R. A., Kenny, C. D., Christiansen, L. M., Edelstein, E., Choi, B., Boss, O., Aschkenasi, C., Zhang, C. Y., Mountjoy, K., Kishi, T., Elmquist, J. K., and Lowell, B. B. (2005). Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell 123, 493–505.
Berthoud, H. R., and Jeanrenaud, B. (1979). Acute hyperinsulinemia and its reversal by vagotomy following lesions of the ventromedial hypothalamus in anesthetized rats. Endocrinology 105, 146–151.
Blaak, E. E., Saris, W. H., and van Baak, M. A. (1993). Adrenoceptor subtypes mediating catecholamine-induced thermogenesis in man. Int. J. Obes. 17, S78–S81.
Blondel, O., Bell, G. I., Moody, M., Miller, R. J., and Gibbons, S. J. (1994). Creation of an inositol 1,4,5-triphosphate-sensitive Ca2+ store in secretory granules of insulin-producing cells. J. Biol. Chem. 269, 27167–27170.
Boden, G., and Hoeldtke, R. D. (2003). Nerves, fat, and insulin resistance. N. Engl. J. Med. 349, 1966–1967.
Boss, O., Bachman, E., Vidal-Puig, A., Zhang, C. Y., Peroni, O., and Lowell, B. B. (1999). Role of the β3-adrenergic receptor and/or a putative β3-adrenergic receptor on the expression of uncoupling proteins and peroxisome proliferator-activated receptor-g coactivator-1. Biochem. Biophys. Res. Commun. 261, 870–876.
Bray, G. A., and Gallagher, T. F. (1975). Manifestations of hypothalamic obesity in man: a comprehensive investigation of eight patients and a review of the literature. Medicine (Baltimore) 54, 301–333.
Bray, G. A., and Nishizawa, Y. (1978). Ventromedial hypothalamus modulates fat mobilization during fasting. Nature 274, 900–902.
Collins, S., Kuhn, C. M., Petro, A. E., Swick, A. G., Chrunyk, B. A., and Surwit, R. S. (1996). Role of leptin in fat regulation. Nature 380, 677.
Coutant, R., Maurey, H., Rouleau, S., Mathieu, E., Mercier, P., Limal, J. M., and Le Bouil, A. (2003). Defect in epinephrine production in children with craniopharyngioma: functional or organic origin? J. Clin. Endocrinol. Metab. 88, 5969–5975.
D’Alessio, D. A., Kieffer, T. J., Taborsky, G. J., and Havel, P. J. (2001). Activation of the parasympathetic nervous system is necessary for normal meal induced-insulin secretion in rhesus macaques. J. Clin. Endocrinol. Metab. 86, 1253–1259.
Danielsson, P., Janson, A., Norgren, S., and Marcus, C. (2007). Impact sibutramine therapy in children with hypothalamic obesity or obesity with aggravating syndromes. J. Clin. Endocrinol. Metab. 92, 4101–4106.
Daousi, C., Dunn, A. J., Foy, P. M., MacFarlane, I. A., and Pinkney, J. H. (2005). Endocrine and neuroanatomic predictors of weight gain and obesity in adult patients with hypothalamic damage. Am. J. Med. 118, 45–50.
Elmquist, J. K., Ahima, R. S., Elias, C. F., Flier, J. S., and Saper, C. B. (1998). Leptin activates distinct projections from the dorsomedial and ventromedial hypothalamic nuclei. Proc. Natl. Acad. Sci. U.S.A. 95, 741–746.
Elmquist, J. K., Elias, C. F., and Saper, C. B. (1999). From lesions to leptin: hypothalamic control of food intake and body weight. Neuron 22, 221–232.
Frohlich, A. (1901). Ein fall von tumor der hypophysis cerebri ohne akromegalie. Weiner Klin. Rundsch. 15, 883–886.
Gilon, P., and Henquin, J. C. (2001). Mechanisms and physiological significance of the cholinergic control of pancreatic β-cell function. Endocr. Rev. 22, 565–604.
Hamilton, J. K., Conwell, L. S., Syme, C., Ahmet, A., Jeffery, A., and Daneman, D. (2011). Hypothalamic obesity following craniopharyngioma surgery: results of a pilot trial of combined diazoxide and metformin therapy. Int. J. Pediatr. Endocrinol. 2011, 417949.
Harz, K. J., Muller, H. L., Waldeck, E., Pudel, V., and Roth, C. (2003). Obesity in patients with craniopharyngioma: assessment of food intake and movement counts indicating physical activity. J. Clin. Endocrinol. Metab. 88, 5227–5231.
Inge, T. H., Pfluger, P., Zeller, M., Rose, S. R., Burget, L., Sundararajan, S., Daniels, S. R., and Tschöp, M. H. (2007). Gastric bypass surgery for treatment of hypothalamic obesity after craniopharyngioma therapy. Nat. Clin. Pract. Endocrinol. Metab. 3, 606–609.
Jeanrenaud, B. (1985). An hypothesis on the aetiology of obesity: dysfunction of the central nervous system as a primary cause. Diabetologia 28, 502–513.
Kalra, S. P., Dube, M. G., Pu, S., Xu, B., Horvath, T. L., and Kalra, P. S. (1999). Interacting appetite-regulating pathways in the hypothalamic regulation of body weight. Endocr. Rev. 20, 68–100.
Kamegai, J., Tamura, H., Shimizu, T., Ishii, S., Sugihara, H., and Wakabayashi, I. (2000). Central effect of ghrelin, an endogenous growth hormone secretagogue, on hypothalamic peptide gene expression. Endocrinology 141, 4797–4800.
Karavitaki, N., Cudlip, S., Adams, C. B., and Wass, J. A. (2006). Craniopharyngiomas. Endocr. Rev. 27, 371–397.
Komeda, K., Yokote, M., and Oki, Y. (1980). Diabetic syndrome in the Chinese hamster induced with monosodium glutamate. Experientia 36, 232–234.
Lowell, B. B., and Spiegelman, B. M. (2000). Towards a molecular understanding of adaptive thermogenesis. Nature 404, 652–660.
Lustig, R. H. (2002). Hypothalamic obesity: the sixth cranial endocrinopathy. Endocrinologist 12, 210–217.
Lustig, R. H. (2003). Autonomic dysfunction of the (ß-cell and the pathogenesis of obesity. Rev. Endocr. Metab. Disord. 4, 23–32.
Lustig, R. H. (2006). Childhood obesity: behavioral aberration or biochemical drive? Reinterpreting the first law of thermodynamics. Nat. Clin. Pract. Endocrinol. Metab. 2, 447–458.
Lustig, R. H. (2007). “The efferent arm of the energy balance regulatory pathway: neuroendocrinology and pathology,” in Obesity and Energy Metabolism: Research and Clinical Applications, ed. P. A. Donahoue (Totowa, NJ: Humana), 69–86.
Lustig, R. H., Post, S. M., Srivannaboon, K., Rose, S. R., Danish, R. K., Burghen, G. A., Xiong, X., Wu, S., and Merchant, T. E. (2003a). Risk factors for the development of obesity in children surviving brain tumors. J. Clin. Endocrinol. Metab. 88, 611–616.
Lustig, R. H., Hinds, P. S., Ringwald-Smith, K., Christensen, R. K., Kaste, S. C., Schreiber, R. E., Rai, S. N., Lensing, S. Y., Wu, S., and Xiong, X. (2003b). Octreotide therapy of pediatric hypothalamic obesity: a double-blind, placebo-controlled trial. J. Clin. Endocrinol. Metab. 88, 2586–2592.
Lustig, R. H., Rose, S. R., Burghen, G. A., Velasquez-Mieyer, P., Broome, D. C., Smith, K., Li, H., Hudson, M. M., Heideman, R. L., and Kun, L. E. (1999). Hypothalamic obesity in children caused by cranial insult: altered glucose and insulin dynamics, and reversal by a somatostatin agonist. J. Pediatr. 135, 162–168.
Lustig, R. H., Tsai, P., Hirose, S., and Farmer, D. L. (2009). “Treatment of hypothalamic obesity by laparoscopic truncal vagotomy: early experience,” in Proceedings of the 8th Lawson Wilkins/European Pediatric Endocrine Society meeting. New York, NY.
Marin, P., Russeffé-Scrive, A., Smith, J., and Bjorntorp, P. (1988). Glucose uptake in human adipose tissue. Metab. Clin. Exp. 36, 1154–1164.
Mason, P. W., Krawiecki, N., and Meacham, L. R. (2002). The use of dextroamphetamine to treat obesity and hyperphagia in children treated for craniopharyngioma. Arch. Pediatr. Adolesc. Med. 156, 887–892.
Matsuda, M., and DeFronzo, R. A. (1999). Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care 22, 1462–1470.
Merchant, T. E., Kienha, E. N., Sanford, R. A., Mulhern, R. K., Thompson, S. J., Wilson, M. W., Lustig, R. H., and Kun, L. E. (2002). Craniopharyngioma: the St. Jude Children’s Research Hospital experience 1984–2001. Int. J. Radiat. Oncol. Biol. Phys. 53, 533–542.
Miura, Y., Gilon, P., and Henquin, J. C. (1996). Muscarinic stimulation increases Na+ entry in pancreatic β-cells by a mechanism other than the emptying of intracellular Ca2+ pools. Biochem. Biophys. Res. Commun. 224, 67–73.
Molloy, P. T., Berkowitz, R., Stallings, V. A., Krell, H., Sutton, L. N., Vaughan, S. N., Loeb, N., and Phillips, P. C. (1998). “Pilot study of evaluation and treatment of tumor-related obesity in pediatric patients with hypothalamic/chiasmatic gliomas and craniopharyngiomas,” in Proceedings of the International Pediatric Oncology Meeting, Rome, 156.
Müller, H. L., Gebhardt, U., Wessel, V., Schröder, S., Kolb, R., Sörensen, N., Maroske, J., and Hanisch, E. (2007). First experiences with laparoscopic adjustable gastric banding (LAGB) in the treatment of patients with childhood craniopharyngioma and morbid obesity. Klin. Padiatr. 219, 323–325.
Muller, H. L. (2008). Childhood craniopharyngioma: recent advances in diagnosis, treatment and follow-up. Horm. Res. 69, 193–202.
Muller, H. L., Emser, A., Faldum, A., Bruhnken, G., Etavard-Gorris, N., Gebhardt, U., Oeverink, R., Kolb, R., and Sörensen, N. (2004). Longitudinal study of growth and body mass index before and after diagnosis of childhood craniopharyngioma. J. Clin. Endocrinol. Metab. 89, 3298–3305.
Müller, H. L., Gebhardt, U., Teske, C., Faldum, A., Zwiener, I., Warmuth-Metz, M., Pietsch, T., Pohl, F., Sörensen, N., Calaminus, G., and Study Committee of KRANIOPHARYNGEOM 2000. (2011). Post-operative hypothalamic lesions and obesity in childhood craniopharyngioma: results of the multinational prospective trial KRANIOPHARYNGEOM 2000 after 3-year follow-up. Eur. J. Endocrinol. 165, 17–24.
Muntzel, M., Morgan, D. A., Mark, A. L., and Johnson, A. K. (1994). Intracerebroventricular insulin produces non-uniform regional increases in sympathetic nerve activity. Am. J. Physiol. 267, R1350–R1355.
Nishi, S., Seino, Y., Ishida, H., Seno, M., Taminato, T., Sakurai, H., and Imura, H. (1987). Vagal regulation of insulin, glucagon, and somatostatin secretion in vitro in the rat. J. Clin. Invest. 79, 1191–1196.
O’Gorman, C. S., Simoneau-Roy, J., Pencharz, P., Adeli, K., and Hamilton, J. (2911). Delayed ghrelin suppression following oral glucose tolerance test in children and adolescents with hypothalamic injury secondary to craniopharyngioma compared with obese controls. Int. J. Pediatr. Obes. 6, 285–288.
O’Gorman, C. S., Simoneau-Roy, J., Pencharz, P., MacFarlane, J., MacLusky, I., Narang, I., Adeli, K., Daneman, D., and Hamilton, J. (2010). Sleep-disordered breathing is increased in obese adolescents with craniopharyngioma compared with obese controls. J. Clin. Endocrinol. Metab. 95, 2211–2218.
Peles, E., Goldstein, D. S., Akselrod, S., Nitzan, H., Azaria, M., Almog, S., Dolphin, D., Halkin, H., and Modan, M. (1995). Interrelationships among measures of autonomic activity and cardiovascular risk factors during orthostasis and the oral glucose tolerance test. Clin. Auton. Res. 5, 271–278.
Pinto, G., Bussieres, L., Recasens, C., Souberbielle, J. C., Zerah, M., and Brauner, R. (2000). Hormonal factors influencing weight and growth pattern in craniopharyngioma. Horm. Res. 53, 163–169.
Powley, T. L., and Laughton, W. (1981). Neural pathways involved in the hypothalamic integration of autonomic responses. Diabetologia 20, 378–387.
Preeyasombat, C., Bacchetti, P., Lazar, A. A., and Lustig, R. H. (2005). Racial and etiopathologic dichotomies in insulin secretion and resistance in obese children. J. Pediatr. 146, 474–481.
Rahmouni, K., Haynes, W. G., Morgan, D. A., and Mark, A. L. (2003). Role of melanocortin-4 receptors in mediating renal sympathoactivation to leptin and insulin. J. Neurosci. 23, 5998–6004.
Rakhshani, N., Jeffery, A. S., Schulte, F., Barrera, M., Atenafu, E. G., and Hamilton, J. K. (2010). Evaluation of a comprehensive care clinic model for children with brain tumor and risk for hypothalamic obesity. Obesity (Silver Spring) 18, 1768–1774.
Rogers, P. C., Meacham, L. R., Oeffinger, K. C., Henry, D. W., and Lange, B. J. (2005). Obesity in pediatric oncology. Pediatr. Blood Cancer 45, 881–891.
Rohner-Jeanrenaud, F., and Jeanrenaud, B. (1980). Consequences of ventromedial hypothalamic lesions upon insulin and glucagon secretion by subsequently isolated perfused pancreases in the rat. J. Clin. Invest. 65, 902–910.
Rohner-Jeanrenaud, F., and Jeanrenaud, B. (1985). Involvement of the cholinergic system in insulin and glucagon oversecretion of genetic preobesity. Endocrinology 116, 830–834.
Roth, C. L., Hunneman, D. H., Gebhardt, U., Stoffel-Wagner, B., Reinehr, T., and Müller, H. L. (2007). Reduced sympathetic metabolites in urine of obese patients with craniopharyngioma. Pediatr. Res. 61, 496–501.
Satoh, N., Ogawa, Y., Katsura, G., Tsuji, T., Masuzaki, H., Hiraoka, J., Okazaki, T., Tamaki, M., Hayase, M., Yoshimasa, Y., Nishi, S., Hosoda, K., and Nakao, K. (1997). Pathophysiological significance of the obese gene product, leptin in ventromedial hypothalamus (VMH)-lesioned rats: evidence for loss of its satiety effect in VMH-lesioned rats. Endocrinology 138, 947–954.
Schofl, C., Schleth, A., Berger, D., Terkamp, C., Von Zur Muhlen, A., and Brabant, G. (2002). Sympathoadrenal counterregulation in patients with hypothalamic craniopharyngioma. J. Clin. Endocrinol. Metab. 87, 624–629.
Schwartz, M. W., Woods, S. C., Porte, D., Seeley, R. J., and Baskin, D. G. (2000). Central nervous system control of food intake. Nature 404, 661–671.
Shaikh, M. G., Grundy, R. G., and Kirk, J. M. (2008). Reductions in basal metabolic rate and physical activity contribute to hypothalamic obesity. J. Clin. Endocrinol. Metab. 93, 2588–2593.
Simoneau-Roy, J., O’Gorman, C., Pencharz, P., Adeli, K., Daneman, D., and Hamilton, J. (2010). Insulin sensitivity and secretion in children and adolescents with hypothalamic obesity following treatment for craniopharyngioma. Clin. Endocrinol. (Oxf) 72, 364–370.
Sluiter, W. J., Erkelens, D. W., Terpstra, P., Reitsma, W. D., and Doorendos, H. (1976). Glucose intolerance and insulin release, a mathematical approach.1. Assay of the beta cell response after glucose loading. Diabetes 25, 241–244.
Small, C. J., and Bloom, S. R. (2004). Gut hormones and the control of appetite. Trends Endocrinol. Metab. 15, 259–263.
Smith, D. K., Sarfeh, J., and Howard, L. (1983). Truncal vagotomy in hypothalmic obesity. Lancet 1, 1330–1331.
Sorva, R. (1988). Children with craniopharyngioma: early growth failure and rapid post-operative weight gain. Acta Paediatr. Scand. 77, 587–592.
Spoudeas, H. A., Saran, F., and Pizer, B. (2006). A multimodality approach to the treatment of craniopharyngiomas avoiding hypothalamic morbidity: a UK perspective. J. Pediatr. Endocrinol. Metab. 19, 447–451.
Srinivasan, S., Ogle, G. D., Garnett, S. P., Briody, J. N., Lee, J. W., and Cowell, C. T. (2004). Features of the metabolic syndrome after craniopharyngioma. J. Clin. Endocrinol. Metab. 89, 81–86.
Stahnke, N., Grubel, G., Lagenstein, I., and Willig, R. P. (1984). Long-term follow-up of children with craniopharyngioma. Eur. J. Pediatr. 142, 179–185.
Susulic, V. S., Frederich, R. C., Lawitts, J., Tozzo, E., Kahn, B. B., Harper, M. E., Himms-Hagen, J., Flier, J. S., and Lowell, B. B. (1995). Targeted disruption of the beta 3-adrenergic receptor gene. J. Biol. Chem. 270, 29483–29492.
Tian, Y. M., Urquidi, V., and Ashcroft, S. J. H. (1996). Protein kinase C in β-cells: expression of multiple isoforms and involvement in cholinergic stimulation of insulin secretion. Mol. Cell. Endocrinol. 119, 185–193.
Tiosano, D., Eisenstein, I., Militianu, D., Chrousos, G. P., and Hochberg, Z. (2003). 11β-hydroxysteroid dehydrogenase activity in hypothalamic obesity. J. Clin. Endocrinol. Metab. 88, 384.
Tschöp, M., Smiley, D. L., and Heiman, M. L. (2000). Ghrelin induces adiposity in rodents. Nature 407, 908–913.
Viguerie, N., Clement, K., Barbe, P., Courtine, M., Benis, A., Larrouy, D., Hanczar, B., Pelloux, V., Poitou, C., Khalfallah, Y., Barsh, G. S., Thalamas, C., Zucker, J. D., and Langin, D. (2004). In vivo epinephrine-mediated regulation of gene express in human skeletal muscle. J. Clin. Endocrinol. Metab. 89, 2000–2014.
Vinchon, M., Weill, J., Delestret, I., and Dhellemmes, P. (2009). Craniopharyngioma and hypothalamic obesity in children. Childs Nerv. Syst. 25, 347–352.
Vollenweider, L., Tappy, L., Owlya, R., Jequier, E., Nicod, P., and Scherrer, U. (1995). Insulin-induced sympathetic activation and vasodilation in skeletal muscle. Effects of insulin resistance in lean subjects. Diabetes 44, 641–645.
Keywords: craniopharyngioma, hypothalamic obesity, leptin resistance, insulin, octreotide, vagus nerve, symapthetic nervous system, ghrelin
Citation: Lustig RH (2011) Hypothalamic obesity after craniopharyngioma: mechanisms, diagnosis, and treatment. Front. Endocrin. 2:60. doi: 10.3389/fendo.2011.00060
Received: 29 July 2011;
Paper pending published: 29 August 2011;
Accepted: 06 October 2011;
Published online: 03 November 2011.
Edited by:
Hermann Lothar Mueller, Klinikum Oldenburg gGmbH, GermanyReviewed by:
Fahrettin Kelestimur, Erciyes University, TurkeyVera Popovic-Brkic, University Belgrade, Serbia
Copyright: © 2011 Lustig. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Robert H. Lustig, Division of Pediatric Endocrinology, University of California San Francisco, Box 0434, 500 Parnassus Avenue, San Francisco, CA 94143-0434, USA. e-mail:cmx1c3RpZ0BwZWRzLnVjc2YuZWR1