

 V. V. Smith3
V. V. Smith3
- 1 Institute of Biomedical and Clinical Science, Peninsula Medical School, University of Exeter, Exeter, UK
- 2 Clinical and Molecular Genetics Unit, University College London Institute of Child Health and Great Ormond Street Hospital for Children, London, UK
- 3 Paediatric Surgery Unit, University College London Institute of Child Health, London, UK
Background: Loss of function mutations in the genes encoding the pancreatic β-cell ATP-sensitive potassium (KATP) channel are identified in approximately 80% of patients with diazoxide unresponsive hyperinsulinemic hypoglycemia (HH). For a small number of patients HH can occur as part of a multisystem disease such as Beckwith–Wiedemann syndrome (BWS). In approximately 20% of patients, BWS results from chromosome 11 paternal uniparental disomy (UPD), which causes dysregulation of imprinted growth regulation genes at 11p15.5. There is a considerable range in the clinical features and phenotypic severity associated with BWS which is likely to be due to somatic mosaicism. The cause of HH in these patients is not known. Research Design and Methods: We undertook microsatellite analysis of 12 markers spanning chromosome 11p in two patients with severe HH and diffuse disease requiring a pancreatectomy. In both patients mutations in the KATP channel genes had not been identified. Results: We identified segmental paternal UPD in DNA extracted from pancreatic tissue in both patients. UPD was not observed in DNA extracted from the patient’s leukocytes or buccal samples. In both cases the UPD encompassed the differentially methylated region at chromosome 11p15.5. Despite this neither patient had any further features of BWS. Conclusion: Paternal UPD of the chromosome 11p15.5 differentially methylated region limited to the pancreatic tissue may represent a novel cause of isolated diazoxide unresponsive HH. Loss of heterozygosity studies should therefore be considered in all patients with severe HH who have undergone pancreatic surgery when KATP channel mutation(s) have not been identified.
Introduction
Hyperinsulinemic hypoglycemia (HH) is a heterogeneous disorder characterized by dysregulated insulin secretion. A range in clinical severity is observed with some patients presenting with mild transient hypoglycemia whilst others have persistent HH that requires medical management. In the most severe cases where euglycemia is not achieved despite intensive therapy, pancreatic surgery may be required.
For approximately 80% of patients who require pancreatic surgery, HH is due to inactivating mutation(s) in genes encoding the SUR1 (ABCC8) and Kir6.2 (KCNJ11) subunits of the pancreatic β-cell ATP-sensitive potassium (KATP) channel (Thomas et al., 1995; Thomas et al., 1996; Bellanne-Chantelot et al., 2010). A genetic diagnosis is clinically important for these patients as the mode of inheritance provides information on the histological subtype. Typically, diffuse disease affects all the β-cells and is most commonly due to recessive mutations in KCNJ11 or ABCC8. Patients with diazoxide unresponsive diffuse disease often require a near total pancreatectomy. In contrast, in focal disease, the affected area is localized to a single region of the pancreas.
The genetic etiology of focal HH is distinct from that of the diffuse form and involves a paternally inherited ABCC8/KCNJ11 mutation and paternal uniparental isodisomy (UPD) within the focal lesion (de Lonlay et al., 1997; Damaj et al., 2008). The UPD unmasks the recessive mutation and causes a dysregulation of imprinted genes at chromosome 11p15.5. For patients with a paternally inherited mutation a 18FDOPA-PET CT scan is required to confirm and localize the focal lesion following which the patient may undergo a curative lesionectomy (de Lonlay et al., 1997; Otonkoski et al., 2006).
Medically unresponsive HH has also been reported in patients with Beckwith–Wiedemann syndrome (BWS; MIM #130650; Debaun et al., 2000; Munns and Batch, 2001). In these patients BWS can result from multiple genetic mechanisms, including mosaic paternal UPD, which causes a dysregulation of imprinted genes at chromosome 11p15.5 (reviewed in Weksberg et al., 2005). A range of clinical features and severity of phenotype is observed in BWS with typical features including macrosomia, macroglossia, abdominal wall defects, hemihyperplasia, embryonal tumors, adrenocortical cytomegaly, ear anomalies, visceromegaly, renal abnormalities, cleft palate, and neonatal HH. Hypoglycemia occurs in approximately 50% of cases and is highly variable in terms of duration and clinical severity. In some patients HH may be transient, resolving within the first few days of life, whilst in approximately 5% of children the HH is persistent and may require pancreatic surgery (Debaun et al., 2000; Munns and Batch, 2001). Although the underlying mechanism leading to HH in these patients is unclear, the variability in clinical phenotype has been attributed to the level of mosaicism for the somatic mutation within the various affected tissues (Smith et al., 2007).
We report two cases with severe, medically unresponsive HH and diffuse histological disease in whom paternal UPD of the chromosome 11p15.5 imprinted region was found within pancreatic tissue. Neither patient has any other feature associated with BWS.
Materials and Methods
Clinical Studies
The study was conducted in accordance with the Declaration of Helsinki 2000 and informed consent was obtained from the parents on behalf of their children.
Molecular Genetics
Genomic DNA was extracted from peripheral leukocytes of the patients and parents and from resected pancreatic tissue and buccal cells (when available) from the patients using standard procedures. The coding regions, intron exon/boundaries, and the minimal promoter regions of the ABCC8 and KCNJ11 genes were amplified and sequenced as previously described (Flanagan et al., 2007; primers for minimal promoter regions available on request). Sequences were compared to published sequences (NM_000525.3 and NM_000352.2) using Mutation Surveyor software (SoftGenetics, State College, PA, USA). When no coding mutation was identified dosage analysis of the ABCC8 gene was undertaken using mulitplex ligation-dependent probe amplification (MLPA) as per the manufacturer’s instructions (Kit P117, MRC Holland, Amsterdam, Netherlands). Products were analyzed using GeneMarker software v1.85 (SoftGenetics, State College, PA, USA).
Loss of heterozygosity of chromosome 11 was investigated by the analysis of 12 microsatellite makers on chromosome 11p. Details of the microsatellite markers are provided in Figure 1. The 12 markers were amplified by PCR using DNA extracted from the patient and parental leukocytes and DNA from the patients resected pancreatic tissue and when available buccal cells (details of primers available on request). The fluorescently tagged PCR products were run on an ABI 3100 (Applied Biosystems, Warrington, UK) and the allele peak heights were compared using GeneMarker software v1.85 (SoftGenetics, State College, PA, USA).
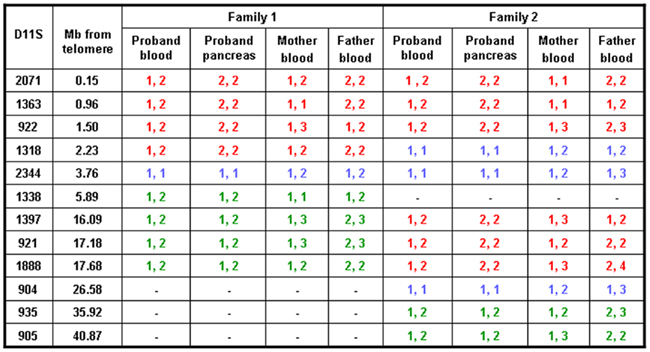
Figure 1. Results of microsatellite analysis of markers on chromosome 11p amplified from leukocyte DNA of the probands and their parents and from the pancreatic DNA from the probands. Red text represents markers which are informative for paternal UPD, green text represents markers which show no evidence of UPD, and blue texts represents markers which are uninformative. The distance between the marker and the telomere at 11p is provided.
Results
Case Reports
Proband 1
The male patient was born at 40 weeks gestation with a birth weight of 3900 g to unaffected parents. He was noted to be hypoglycemic on the first day of life (blood glucose of 1.1 mmol/l) with biochemical evidence of HH. An intravenous glucose infusion of 24 mg/kg/min was required to maintain normoglycemia. He was unresponsive to diazoxide and hence was stabilized on a subcutaneous infusion of octreotide while awaiting surgery. Pancreatic biopsy performed in the 7th week of life revealed enlarged clusters of islet tissue without nuclear enlargement or hyperchromasia and arranged in ribbon-like pattern. Very little exocrine tissue was apparent. He underwent a near total pancreatectomy at 9 weeks of age following which he achieved normoglycemia. The resected pancreas showed the same histological changes as the biopsy. These appearances are those seen in focal HH but involved the entire pancreas. Currently, at 6 years of age, he remains normoglycemic and is neurodevelopmentally appropriate for his age.
Proband 2
The male patient was born at term with a birth weight of 3870 g to healthy parents. Symptomatic hypoglycemia was noted at 7 h of life and HH was confirmed biochemically. He required an intravenous glucose infusion of 25 mg/kg/min to maintain normoglycemia and did not respond to a trial of medical treatment (diazoxide, octreotide, and nifedipine). At 8 weeks of age, he underwent 50% resection of his pancreas, followed by a near total pancreatectomy as hypoglycemia persisted post-operatively. Histology of the resected pancreas revealed excess of enlarged islets with endocrine cells forming ribbons. The amount of exocrine tissue was greatly reduced. These histological appearances are seen in focal HH but the pathology involved the entire pancreas.
Following a near total pancreatectomy, he was managed on oral diazoxide and regular gastrostomy feeds (continuous overnight and daytime bolus feeds) to maintain normoglycemia. The patient is currently 6.8 years of age and is on minimal doses of diazoxide (6 mg/kg/day) and daytime gastrostomy feeds to maintain normoglycemia along with pancreatic enzyme supplementation for exocrine insufficiency. There are no neurodevelopmental concerns.
Genetics
Sequencing analysis of the KCNJ11 and ABCC8 genes did not identify a mutation in leukocyte DNA from either patient and MLPA analysis of the ABCC8 gene confirmed normal gene dosage in leukocyte and pancreatic tissue from both cases.
In proband 1 microsatellite analysis of four markers spanning the chromosome 11p15.5 region (2.23 Mb) demonstrated a reduced peak height for the maternal allele in the pancreatic tissue when compared to the leukocyte and buccal cell DNA (Figures 1 and 2, results from buccal cells not shown). One marker was uninformative and four markers were heterozygous in all three samples. These results confirmed segmental loss of the short arm of the maternal chromosome 11 (maximum size 5.89 Mb) within the pancreatic tissue but not within the buccal or leukocyte DNA (Figure 2).
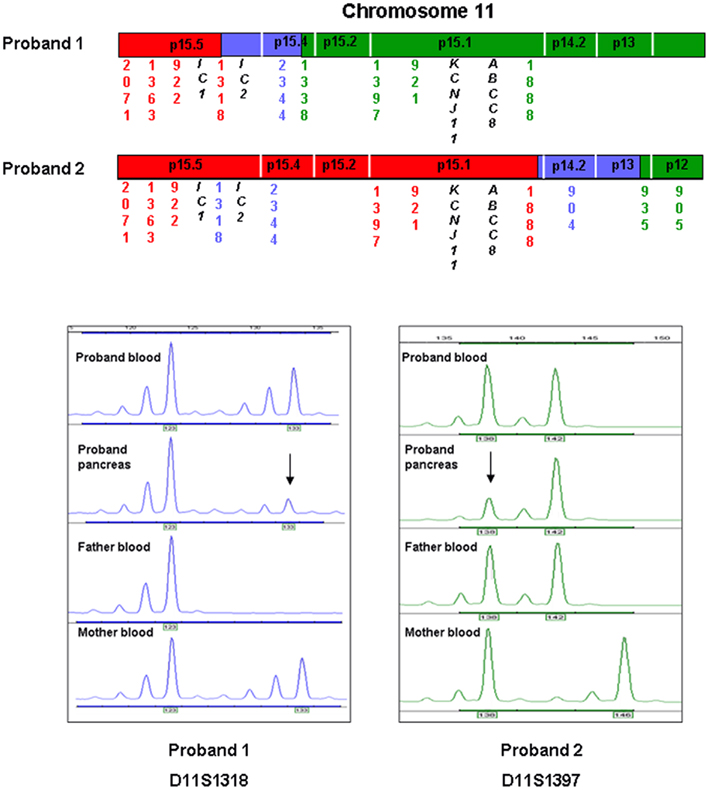
Figure 2. A region of chromosome 11 has been expanded to demonstrate the area of UPD in relation to the location of the imprinted locus at 11p15.5 and the ABCC8 and KCNJ11 genes. IC denotes the imprinted control regions 1 and 2. Red text represents markers which are informative for paternal UPD, green text represents markers which show no evidence of UPD, and blue texts represents uninformative markers. Directly below are electropherograms that demonstrate the results of microsatellite analysis for one of the markers (D11S1318 and D11S1397) which are informative for UPD in pancreatic tissue for each of the probands. The x-axis indicates the size of the product (base pairs) and the y-axis denotes the product quantity (arbitrary units). In each example mosaic paternal UPD is illustrated by a larger peak for the paternal allele compared to the maternal.
In proband 2, six markers spanning 17.68 Mb on chromosome 11p15.5–11p15.1 demonstrated a reduced peak height for the maternal allele in the pancreatic tissue when compared to the leukocyte DNA (Figures 1 and 2). Three markers were uninformative and four markers were heterozygous in the leukocyte and pancreatic samples defining the maximum region of loss of heterozygosity as 35.92 Mb.
Discussion
Microsatellite analysis revealed a loss of the maternal chromosome 11p15.5 allele in DNA extracted from pancreatic tissue in two patients with diazoxide unresponsive HH and diffuse pancreatic disease. In one patient the region was limited to chromosome 11p15.5 whilst in the second patient the loss of heterozygosity extended over ABCC8 and KCNJ11. In this patient sequencing of the coding regions, intron/exon boundaries, and minimal promoters of the KATP channel genes and ABCC8 gene dosage studies did not identify a mutation, although this does not exclude a mutation in the non-coding regions of these genes.
In both patients the maximum shared region of maternal loss of heterozygosity spanned 5.89 Mb which encompassed the genes IGF2, H19, KCNQ1, KCNQ10T1, CDKN1C, PHLD1, and SLC22A18, and the imprinted control regions which have been implicated in BWS and growth regulation (Figure 2). In proband 2, where the loss of heterozygosity extended over the KATP channel genes, ABCC8 gene dosage studies confirmed normal diploid copy number within the pancreatic tissue which suggests that the loss of heterozygosity is likely to be due to mosaic segmental paternal UPD.
Paternal UPD of chromosome 11p15.5 accounts for approximately 20% of BWS cases and in the vast majority of cases patients exhibit somatic mosaicism (Henry et al., 1991). The presence of mosaicism implies that in these patients UPD arises post-zygotically as a result of somatic recombination and therefore the tissues that will be affected will depend on the timing at which this event occurs during embryogenesis. As severe HH requiring pancreatectomy has been reported in patients with BWS it seems likely that paternal UPD over the BWS locus within the pancreatic tissue is responsible for the severe HH observed in our patients (Debaun et al., 2000; Munns and Batch, 2001; Smith et al., 2007). In addition, the histopathology in these patients is similar to that reported in BWS (Hussain et al., 2005). Further studies are required to elucidate the mechanism of HH in these patients.
Despite extensive clinical examination no further features of BWS were detected in either patient which is likely to reflect the absence of UPD in other tissues. This hypothesis is supported by previous reports which identified chromosome 11p15 UPD in patients with isolated hemihyperplasia with or without embryonal tumors (Grundy et al., 1991; Weksberg et al., 2005). It has been suggested that these patients reflect the milder end of the BWS spectrum (Weksberg et al., 2005). Whilst it would be interesting to have investigated the presence/level of UPD in other “unaffected” tissues in these two cases the only sample in addition to leukocyte and pancreatic DNA that was available for testing was buccal cells from one patient. Microsatellite analysis using DNA extracted from buccal cells demonstrated heterozygosity for all markers tested which is in keeping with the absence of, or low level UPD.
Currently for around 20% of patients with severe HH which requires a pancreatectomy sequencing and dosage analysis of the ABCC8 and KCNJ11 genes does not identify a mutation (Bellanne-Chantelot et al., 2010). Our finding that mosaic paternal UPD of chromosome 11p15.5, limited to pancreatic tissue, can cause HH without features of BWS suggest that loss of heterozygosity studies should be undertaken when pancreatic tissue is available. This genetic diagnosis is clinically important as it provides information regarding recurrence risk and also, in the absence of other tissue available for testing, these patients should be monitored closely as they may be at high risk of embryonal tumors (Henry et al., 1991).
In conclusion, we have demonstrated paternal UPD of the chromosome 11p15.5 differentially methylated region within the pancreatic tissue in two patients with isolated HH. The absence of UPD in other tissues tested is in keeping with the lack of features associated with BWS. This mechanism may represent a novel cause of isolated diazoxide unresponsive HH and should be considered in all patients without a KATP channel mutation where pancreatic tissue is available.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
S. E. Flanagan was the Sir Graham Wilkins Peninsula Medical School Research fellow. R. R. Kapoor is a NIHR-BRC Clinical Research Associate. Sian Ellard is a member of the core staff within the NIHR funded Peninsula Clinical Research Facility. We thank Annet Damhuis for technical assistance. This study was funded by the Wellcome Trust (081188/A/06/Z) and Diabetes UK.
References
Bellanne-Chantelot, C., Saint-Martin, C., Ribeiro, M. J., Vaury, C., Verkarre, V., Arnoux, J. B., Valayannopoulos, V., Gobrecht, S., Sempoux, C., Rahier, J., Fournet, J. C., Jaubert, F., Aigrain, Y., Nihoul-Fekete, C., and De Lonlay, P. (2010). ABCC8 and KCNJ11 molecular spectrum of 109 patients with diazoxide-unresponsive congenital hyperinsulinism. J. Med. Genet. 47, 752–759.
Damaj, L., Le Lorch, M., Verkarre, V., Werl, C., Hubert, L., Nihoul-Fekete, C., Aigrain, Y., De Keyzer, Y., Romana, S. P., Bellanne-Chantelot, C., De Lonlay, P., and Jaubert, F. (2008). Chromosome 11p15 paternal isodisomy in focal forms of neonatal hyperinsulinism. J. Clin. Endocrinol. Metab. 93, 4941–4947.
de Lonlay, P., Fournet, J. C., Rahier, J., Gross-Morand, M. S., Poggi-Travert, F., Foussier, V., Bonnefont, J. P., Brusset, M. C., Brunelle, F., Robert, J. J., Nihoul-Fekete, C., Saudubray, J. M., and Junien, C. (1997). Somatic deletion of the imprinted 11p15 region in sporadic persistent hyperinsulinemic hypoglycemia of infancy is specific of focal adenomatous hyperplasia and endorses partial pancreatectomy. J. Clin. Invest. 100, 802–807.
Debaun, M. R., King, A. A., and White, N. (2000). Hypoglycemia in Beckwith-Wiedemann syndrome. Semin. Perinatol. 24, 164–171.
Flanagan, S. E., Patch, A. M., Mackay, D. J., Edghill, E. L., Gloyn, A. L., Robinson, D., Shield, J. P., Temple, K., Ellard, S., and Hattersley, A. T. (2007). Mutations in ATP-sensitive K+ channel genes cause transient neonatal diabetes and permanent diabetes in childhood or adulthood. Diabetes 56, 1930–1937.
Grundy, P., Telzerow, P., Paterson, M. C., Haber, D., Berman, B., Li, F., and Garber, J. (1991). Chromosome 11 uniparental isodisomy predisposing to embryonal neoplasms. Lancet 338, 1079–1080.
Henry, I., Bonaiti-Pellie, C., Chehensse, V., Beldjord, C., Schwartz, C., Utermann, G., and Junien, C. (1991). Uniparental paternal disomy in a genetic cancer-predisposing syndrome. Nature 351, 665–667.
Hussain, K., Cosgrove, K. E., Shepherd, R. M., Luharia, A., Smith, V. V., Kassem, S., Gregory, J. W., Sivaprasadarao, A., Christesen, H. T., Jacobsen, B. B., Brusgaard, K., Glaser, B., Maher, E. A., Lindley, K. J., Hindmarsh, P., Dattani, M., and Dunne, M. J. (2005). Hyperinsulinemic hypoglycemia in Beckwith-Wiedemann syndrome due to defects in the function of pancreatic beta-cell adenosine triphosphate-sensitive potassium channels. J. Clin. Endocrinol. Metab. 90, 4376–4382.
Munns, C. F., and Batch, J. A. (2001). Hyperinsulinism and Beckwith-Wiedemann syndrome. Arch. Dis. Child. Fetal Neonatal Ed. 84, F67–F69.
Otonkoski, T., Nanto-Salonen, K., Seppanen, M., Veijola, R., Huopio, H., Hussain, K., Tapanainen, P., Eskola, O., Parkkola, R., Ekstrom, K., Guiot, Y., Rahier, J., Laakso, M., Rintala, R., Nuutila, P., and Minn, H. (2006). Noninvasive diagnosis of focal hyperinsulinism of infancy with [18F]-DOPA positron emission tomography. Diabetes 55, 13–18.
Smith, A. C., Shuman, C., Chitayat, D., Steele, L., Ray, P. N., Bourgeois, J., and Weksberg, R. (2007). Severe presentation of Beckwith-Wiedemann syndrome associated with high levels of constitutional paternal uniparental disomy for chromosome 11p15. Am. J. Med. Genet. A 143A, 3010–3015.
Thomas, P., Ye, Y., and Lightner, E. (1996). Mutation of the pancreatic islet inward rectifier Kir6.2 also leads to familial persistent hyperinsulinemic hypoglycemia of infancy. Hum. Mol. Genet. 5, 1809–1812.
Thomas, P. M., Cote, G. J., Wohllk, N., Haddad, B., Mathew, P. M., Rabl, W., Aguilar-Bryan, L., Gagel, R. F., and Bryan, J. (1995). Mutations in the sulfonylurea receptor gene in familial persistent hyperinsulinemic hypoglycemia of infancy. Science 268, 426–429.
Keywords: uniparental disomy, hyperinsulinemic hypoglycemia, Beckwith–Wiedemann syndrome
Citation: Flanagan SE, Kapoor RR, Smith VV, Hussain K and Ellard S (2011) Paternal uniparental isodisomy of chromosome 11p15.5 within the pancreas causes isolated hyperinsulinemic hypoglycemia. Front. Endocrin. 2:66. doi: 10.3389/fendo.2011.00066
Received: 22 September 2011;
Accepted: 14 October 2011;
Published online: 02 November 2011.
Edited by:
Ken Ong, Medical Research Council, UKReviewed by:
Giles Yeo, University of Cambridge, UKIoannis E. Messinis, University of Thessalia, Greece
Copyright: © 2011 Flanagan, Kapoor, Smith, Hussain and Ellard. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Sian Ellard, Peninsula Medical School, Barrack Road, Exeter EX2 5DW, Devon, UK. e-mail:c2lhbi5lbGxhcmRAcmRlZnQubmhzLnVr