 A. E. MacKenzie1
A. E. MacKenzie1

- 1 Molecular Pharmacology Group, Institute of Neuroscience and Psychology, College of Medical, Veterinary and Life Sciences, University of Glasgow, Glasgow, UK
- 2 Institute of Cardiovascular and Medical Sciences, College of Medical, Veterinary and Life Sciences, University of Glasgow, Glasgow, UK
- 3 Centre for Therapeutics Discovery, MRC Technology, London, UK
G protein-coupled receptors (GPCRs) remain the best studied class of cell surface receptors and the most tractable family of proteins for novel small molecule drug discovery. Despite this, a considerable number of GPCRs remain poorly characterized and in a significant number of cases, endogenous ligand(s) that activate them remain undefined or are of questionable physiological relevance. GPR35 was initially discovered over a decade ago but has remained an “orphan” receptor. Recent publications have highlighted novel ligands, both endogenously produced and synthetic, which demonstrate significant potency at this receptor. Furthermore, evidence is accumulating which highlights potential roles for GPR35 in disease and therefore, efforts to characterize GPR35 more fully and develop it as a novel therapeutic target in conditions that range from diabetes and hypertension to asthma are increasing. Recently identified ligands have shown marked species selective properties, indicating major challenges for future drug development. As we begin to understand these issues, the continuing efforts to identify novel agonist and antagonist ligands for GPR35 will help to decipher its true physiological relevance; translating multiple assay systems in vitro, to animal disease systems in vivo and finally to man.
Introduction
Since the discovery of GPR35 as an “orphan” GPCR in 1998, limited information on this receptor has appeared. In part, this reflects, until recently, the lack of available ligands that regulate this receptor. This has begun to change with the identification of both potential endogenous agonists and the screening of small scale chemical libraries to identify synthetic ligands with agonist action at GPR35. These advances, in concert with genetic links between GPR35 and disease, have stimulated research into the pharmacology and pathophysiology of GPR35. Evidence now suggests possible links between GPR35 and a range of pathological conditions including inflammation, asthma, hypertension, and diabetes; consequently highlighting GPR35 as a novel potential therapeutic target (Milligan, 2011).
GPR35 was initially discovered as an open reading frame (ORF), situated on chromosome 2q37.3 in human (O’Dowd et al., 1998) which produces two alternatively spliced variants: GPR35a and GPR35b. Human GPR35a encodes an ORF of 309 amino acids, while GPR35b contains an N-terminal extension of 31 amino acids (Okumura et al., 2004). GPR35b was found to be highly expressed in gastric cancer tissue, and suggested to have transforming capabilities (Okumura et al., 2004). However, the exact relevance of the N-terminal extension has yet to be elucidated, with GPR35a and GPR35b generating similar pharmacological responses in vitro (Guo et al., 2008).
Efforts to characterize novel G protein-coupled receptors (GPCRs) such as GPR35 have been assisted by sequence and phylogenetic analysis; these place GPR35 in the rhodopsin-like family of GPCRs. GPR35 shares closest homology with GPR55, the lysophosphatidic acid receptors LPAR4, LPAR5, and LPAR6, and the nicotinic acid receptor HM74 (Fredriksson et al., 2003; Vassilatis et al., 2003). GPR55 was suggested initially as an atypical cannabinoid receptor; based on relatedness of sequence to GPR35 it has been used in ligand screens alongside GPR35 in a bid to identify or to eliminate common ligands (Heynen-Genel et al., 2010). However, to date these receptors appear to share no overlap in terms of ligand interaction or function (Oka et al., 2010; Brown et al., 2011). Despite this, the endogenous agonist of LPAR4, lysophosphatidic acid, has been shown to display agonist action at GPR35 (Oka et al., 2010).
GPR35 Expression Pattern
In addition to identification of GPR35, O’Dowd et al. (1998) also reported GPR35 transcripts in endogenous tissue, with significant expression in the rat small intestine. Subsequent studies have shown a high level of human GPR35 in the pancreas and the small intestine (Wang et al., 2006; Leonard et al., 2007); with significant expression in the human colon, spleen, and immune cells (monocytes, neutrophils, T cells, and dendritic cells); and with lower levels of GPR35 reported in the stomach, skeletal muscle, and adipose tissue, kidney, liver, and thymus (Taniguchi et al., 2006; Wang et al., 2006). In mouse, extremely high levels of GPR35 were detected in the spleen, with similar expression levels to human detected in the small intestine, colon, stomach, thymus, and adipose tissue (Wang et al., 2006). In rat there is high expression of GPR35 in the spleen and colon, dorsal root ganglion, and uterus (Taniguchi et al., 2006; Ohshiro et al., 2008); and moderate expression in cerebrum, heart, liver, bladder, and spinal cord has been reported (Taniguchi et al., 2006; Ohshiro et al., 2008; Cosi et al., 2010).
GPR35 Polymorphisms
Correlation between single nucleotide polymorphisms (SNPs) and disease incidence can hint at links between a gene and disease and may promote interest in the development of novel therapeutics. The coding and intergenic region surrounding GPR35 is substantially polymorphic, with various publications to date reporting around 70 SNPs, six of which have been associated with disease. The first paper to document GPR35 SNPs employed a genome wide association study (GWAS) and focused on genetic variation associated with type 2 diabetes (Horikawa et al., 2000). Whilst sequencing chromosome 2q37.3 in a diabetic Mexican–American population, 19 polymorphisms were identified within GPR35, with an additional 40 located in the intergenic regions situated 5′ and 3′ to GPR35; 4 of which were found to show some association with type 2 diabetes (Horikawa et al., 2000). The majority of SNPs identified by Horikawa and colleagues, were also identified in a GWAS aiming to resolve terminal deletions on chromosome 2q37 associated with Albright hereditary osteodystrophy and mild mental retardation. This study found that patients lacked one copy (either maternal or paternal) of GPR35 (Shrimpton et al., 2004). However, it should be noted that chromosomal deletions at 2q37 were not mapped specifically to GPR35, but to a 3 Mb region containing at least 30 other genes which may also contribute to the Albright phenotype. Additionally, a computerized GWAS reported implications for GPR35 in the pathology of atherosclerotic plaque formation, with a non-synonymous SNP (Ser294Arg) located in the predicted intracellular C-terminal tail, significantly associated with the burden of coronary artery calcification (quantified by computed tomography), a measure of atherosclerotic plaque development and coronary artery disease (CAD) risk in a patient cohort (Sun et al., 2008). These authors postulated that Ser294Arg may have significant implications for receptor–protein interactions (Sun et al., 2008) because this variation might alter the phosphorylation pattern of GPR35. However, this hypothesis is yet to be tested directly. Lastly, a GWAS for early on-set inflammatory bowel disease (IBD) linked an SNP within GPR35 (rs4676410) to ulcerative colitis (Imielinski et al., 2009). However, as with the four SNPs associated with type 2 diabetes, the authors did not comment specifically on the potential contribution of GPR35. Interestingly, however, a clinical study of CAD risk factors highlighted a significant statistical link between the incidence of CAD and irritable bowel syndrome (Yarur et al., 2011). These findings, along with the therapeutically relevant tissue expression pattern of GPR35, suggest potential roles for GPR35 in diabetes, cardiovascular disease, and IBD.
Methods Used to Identify GPR35 Ligands
In order to better characterize GPR35, a number of studies have screened commercially available chemical libraries in a bid to identify potential ligands which may act at GPR35. A number of techniques commonly used to explore the pharmacology and ligand binding of orphan GPCRs have been employed in GPR35 screening and are discussed below.
β-Arrestin-2 Recruitment
Measurement of β-arrestin-2 recruitment to an agonist ligand–receptor complex is a useful and efficient method of gathering substantial data regarding ligand efficacy and potency for many GPCRs. Publications have reported using various β-arrestin-2 based assays and GPR35 generally produces strong signal to background in such assays. For example, Jenkins et al. (2010) screened the Prestwick Chemical Library to identify ligands with agonism at GPCR using a bioluminescence resonance transfer (BRET) assay. In this approach human and rat GPR35 were modified by introducing both an N-terminal epitope-tag and C-terminal in-frame fusion of enhanced yellow fluorescent protein (eYFP). These were then co-transfected into HEK293 cells with Renilla luciferase-tagged β-arrestin-2. Following addition of the luciferase substrate coelenterazine-h, agonist activation of GPR35 resulted in interaction between GPR35 and β-arrestin-2 and substantial BRET signal that was dependent on agonist concentration. An alternative GPR35-β-arrestin-2 assay to explore GPR35 pharmacology was recently reported using the “Tango™” system. This method utilizes a protease-tagged β-arrestin-2 which cleaves GPR35 at a modified c-terminal encoding a β-lactamase transcription site. β-lactamase then becomes the reporter gene for which FRET technology can measure a fluorescent signal (Deng et al., 2011). Two further variants of the β-arrestin-2 interaction technology have also been used to identify and characterize GPR35 agonists. The first of these is based on high content analysis of the translocation of a GFP-tagged form of β-arrestin-2 (Zhao et al., 2010), whilst the “PathHunter®” assay kit produced by DiscoveRx has also been employed to measure both ligand potency and species selectivity of zaprinast and kynurenic acid at human and rat orthologs of GPR35 (Jenkins et al., 2011). This assay is based on a complementation system of fragments of β-galactosidase linked to either GPR35 or β-arrestin-2. Importantly, the nature of β-arrestin-2 recruitment to the cell membrane upon ligand activation contributes to both desensitization and internalization of the receptor protein complex, therefore, the signals generated from this assay are essentially independent of G-protein activation (Hamdan et al., 2005; DeWire et al., 2007). This may be a limitation in assessing certain aspects of receptor function; however, publications have primarily used this method to identify initial ligand hits and subsequently employed downstream signaling assays to provide secondary confirmation of ligand receptor interactions (Zhao et al., 2010; Jenkins et al., 2011).
Guanine Nucleotide Binding Assays
[35S]guanosine-5′-O-(3-thio)triphosphate ([35S]GTPγS) assays measure the guanine nucleotide exchange process which occurs on Gα subunits upon GPCR activation and by using an analog of GTP which is resistant to the GTPase activity of the Gα subunit, trap the active complex. [35S]GTPγS assays are popular as they allow monitoring of GPCR activation at an early stage of the signal transduction pathway and can be performed in cell membrane preparations. [35S]GTPγS binding assays have been most widely used in the measurement of GPCRs that couple to the Gαi/o family of G-proteins – a result of higher Gαi/o expression in cell membranes and a higher guanine nucleotide exchange rate compared with other G-proteins. Additionally, GPCR–G-protein fusions and immunoprecipitation of [35S]GTPγS labeled Gα subunits have enabled measurement of activation of Gαq, Gα12/13, or Gαs via modifications of the standard [35S]GTPγS assay (Milligan, 2003). Both the standard [35S]GTPγS binding assay and the immunoprecipitation [35S]GTPγS assay have been used to demonstrate GPR35 activation by kynurenic acid (Wang et al., 2006) or ligands from the Prestwick Chemical Library (Jenkins et al., 2010; Figure 1).
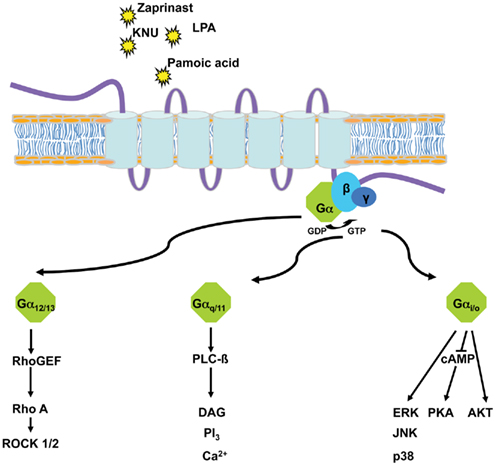
Figure 1. G-protein-dependent pathways either used endogenously by GPR35 (G12/13, Gi/o) or subverted (Gq/11) to allow screening of potential agonists of GPR35 via chimeric and/or promiscuous G-proteins.
Calcium Mobilization Assays
Further downstream, GPCR activation can be monitored by measuring ligand induced changes in second messenger levels and pathways. These include assays which detect modulation of intracellular cAMP, inositol phosphates, and Ca2+ levels. Calcium mobilization assays are among the most widely used functional assays for GPCR screening. Historically, Ca2+ assays have been used to measure Gαq mediated calcium release from the endoplasmic reticulum; measured by the addition of a Ca2+ sensitive dye which alters fluorescence upon binding of Ca2+. More recently, this assay has been adapted to allow measurement of Gαs, Gαi/o, and Gα12/13 responses through the Ca2+ pathway, via the use of G-protein chimeras and promiscuous G-proteins (Kostenis et al., 2005; Milligan and Kostenis, 2006). Such Gαq chimeras are generated by substitution of, usually, the last five, but sometimes more, amino acids of the Gαq carboxyl terminus with the corresponding residues of Gαs, Gαi/o, or Gα12/13, to create G-proteins that alter their GPCR-coupling specificity but that still link to phospholipase C-mediated regulation of intracellular Ca2+ levels. Expression of so called “promiscuous” G-proteins (Gα15 and Gα16; Kostenis et al., 2005) which interact with a broad range of GPCRs can also result in activation of the Ca2+ pathway. Such Ca2+ mobilization assays have been particularly useful in searches for ligands which act upon “orphan” GPCRs, for which there has often been no information available regarding the subsets of G-proteins or the signal pathways usually regulated upon receptor activation. Indeed, two of the earliest studies to identify GPR35 agonists used calcium mobilization assays to show agonist activated GPR35 mediated changes in [Ca2+]i via addition of zaprinast (Taniguchi et al., 2006), and kynurenic acid (Wang et al., 2006).
Yeast-Based Assays
The chimeric G-protein approach has also been applied to yeast model systems, most usually via manipulation of the mating response pathway in Saccharomyces cerevisiae. Deletion of the single GPCR in haploid yeast can allow heterologous expression and measurement of functional mammalian GPCRs without interference from other receptors and signaling pathways (Dowell and Brown, 2009). Chimeric yeast transplants are generated in much the same way as with the Gαq chimeras: by altering the last five C-terminal amino acids of the endogenous Gα to those of a mammalian G-protein of interest. Such a yeast assay has been used to confirm GPR35 activity and Gα13 G-protein coupling of Prestwick Chemical Library hits identified initially using β-arrestin-2 assays (Jenkins et al., 2010). However, caution must be applied when analyzing data obtained from assays employing chimeric G-proteins; as the chimeras only contain a small fragment of the original G-protein: independent assays must be employed to confirm findings using full length, unmodified G-proteins.
ERK1/2 MAP Kinase Phosphorylation
One of the more popular signaling pathways to be measured following GPCR activation is the extracellular signal-regulated kinase 1/2 (ERK1/2) mitogen-activated kinase pathway. This reflects that most GPCRs generate signals that intersect with and alter the activity of proteins within this cascade. GPCRs can influence ERK1/2 signaling through a variety of means including modulation of protein kinase A (via Gαs), protein kinase C (via Gαq and Gαo), the Gαi subunit βγ dimers, and β-arrestins (Luttrell, 2003; Tohgo et al., 2003). Thus, ERK1/2 assays incorporate multiple G-protein signals that converge within the same cell, generating an experimental system that can measure GPCR activation without prior knowledge of G-protein coupling. Phospho-ERK1/2 immunoblotting has been used to demonstrate ligand activation of GPR35, whereby an upstream kinase phosphorylates specific threonine and tyrosine residues, which can be measured using phospho-specific antibodies that recognize these modifications. For example, 2-oleoyl lysophosphatidic acid (Oka et al., 2010) and tyrophostin-51 (Deng et al., 2011) were identified as GPR35 agonists using such assays. Furthermore, ERK1/2 activation assays have also been used to illustrate GPCR–G-protein coupling specificity: by use of pertussis toxin, which abolishes Gαi/o mediated responses, pamoic acid, and zaprinast were shown to activate human GPR35 in a Gαi/o-dependent manner (Zhao et al., 2010).
Receptor Internalization
Receptor internalization studies have also been used to visualize and quantify the process of endocytosis which often ensues in response to occupancy of a GPCR with high efficacy ligands (Marchese et al., 2008). By modifying GPR35 to encode an external N-terminal epitope-tag, it is possible to carry out fluorescence imaging via immunostaining whilst simultaneously performing “on-cell” quantification analysis. Studies which have benefited from these techniques include those which have identified kynurenic acid as an endogenous ligand at GPR35 (Wang et al., 2006), pamoic acid (Zhao et al., 2010), and most recently tyrophostin-51 (Deng et al., 2011). These assays all demonstrated receptor internalization in response to appropriate ligand activation at GPR35.
Endogenous GPR35 Ligands
Kynurenic Acid
To explore the mechanisms by which GPR35 mediates signaling endogenously, successful ligand identification is imperative. The tryptophan metabolite kynurenic acid (4-oxo-1H-quinoline-2-carboxylic acid; Table 1) was the first reported agonist ligand for GPR35 that is generated endogenously and is present in many tissues including the brain, pancreas, colon, kidney, lung, intestine, spleen, and muscle. In addition to acting as a potential GPR35 agonist kynurenic acid also acts as a glycine and allosteric site antagonist of the glutamate N-methyl-D-aspartate (NMDA)-receptor ion channel complex; and as a non-competitive antagonist at the α7 nicotinic acetylcholine receptor (Hilmas et al., 2001; Stone, 2001; Prescott et al., 2006). The ability of kynurenic acid to activate and internalize human, rat, and mouse orthologs of GPR35 at micromolar concentrations was initially demonstrated by Wang et al. (2006). Using a Ca2+/aequorin assay, this group also demonstrated that kynurenic acid could increase intracellular calcium levels whilst coupling to the chimeric G-proteins Gqo5, Gqi9, and also the pertussis toxin-sensitive Gi/o pathway by employing a [35S]GTPγS binding assay (Wang et al., 2006). However, it may be important to consider the concentrations of kynurenic acid required to activate GPR35 in vitro and compare these with endogenous levels in vivo. Although plasma kynurenic acid levels are often reported in nanomolar concentrations in humans, kynurenic acid can be increased to micromolar concentrations in inflammatory states. Interestingly, a recent publication demonstrated that kynurenic acid levels are elevated during inflammation in colon cancer patients (Walczak et al., 2011). Furthermore, levels of kynurenic acid have also been reported to increase in the kidney, lung, intestine, spleen, and muscles in rats suffering chronic renal failure (Pawlak et al., 2002), in spontaneously hypertensive rats (Mizutani et al., 2002) and in the urine of type 2 diabetic primates (Patterson et al., 2011). Additionally, it is argued that the measurement of endogenous concentrations can be underestimated by dilution factors occurring during the measurement process, whereby concentrations may be misjudged at the receptor complex, and may actually be potentially more potent than recorded (Wang et al., 2006). A subsequent publication assessing endogenous concentrations in rodent small intestine also reported endogenous kynurenic acid at micromolar levels (Kuc et al., 2008); suggesting that kynurenic acid could feasibly be an endogenous agonist at rodent GPR35. Meanwhile, evidence relating to the activation of GPR35 by kynurenic acid in humans remains inconsistent across the literature. These discrepancies may be a result of variation in the techniques which are employed to quantify the ligand potency. For example, Oka et al. (2010) were unable to generate a response at human GPR35 to kynurenic acid when measuring intracellular [Ca2+], nor were they able to demonstrate GPR35 internalization following transfection of a fluorescently tagged form of human GPR35. Further contrasting data has emerged from Jenkins et al. (2011) who reported via a BRET-based β-arrestin-2 interaction assay that although kynurenic acid can activate human GPR35, potency is low and at least 100 fold lower than at the rat ortholog.
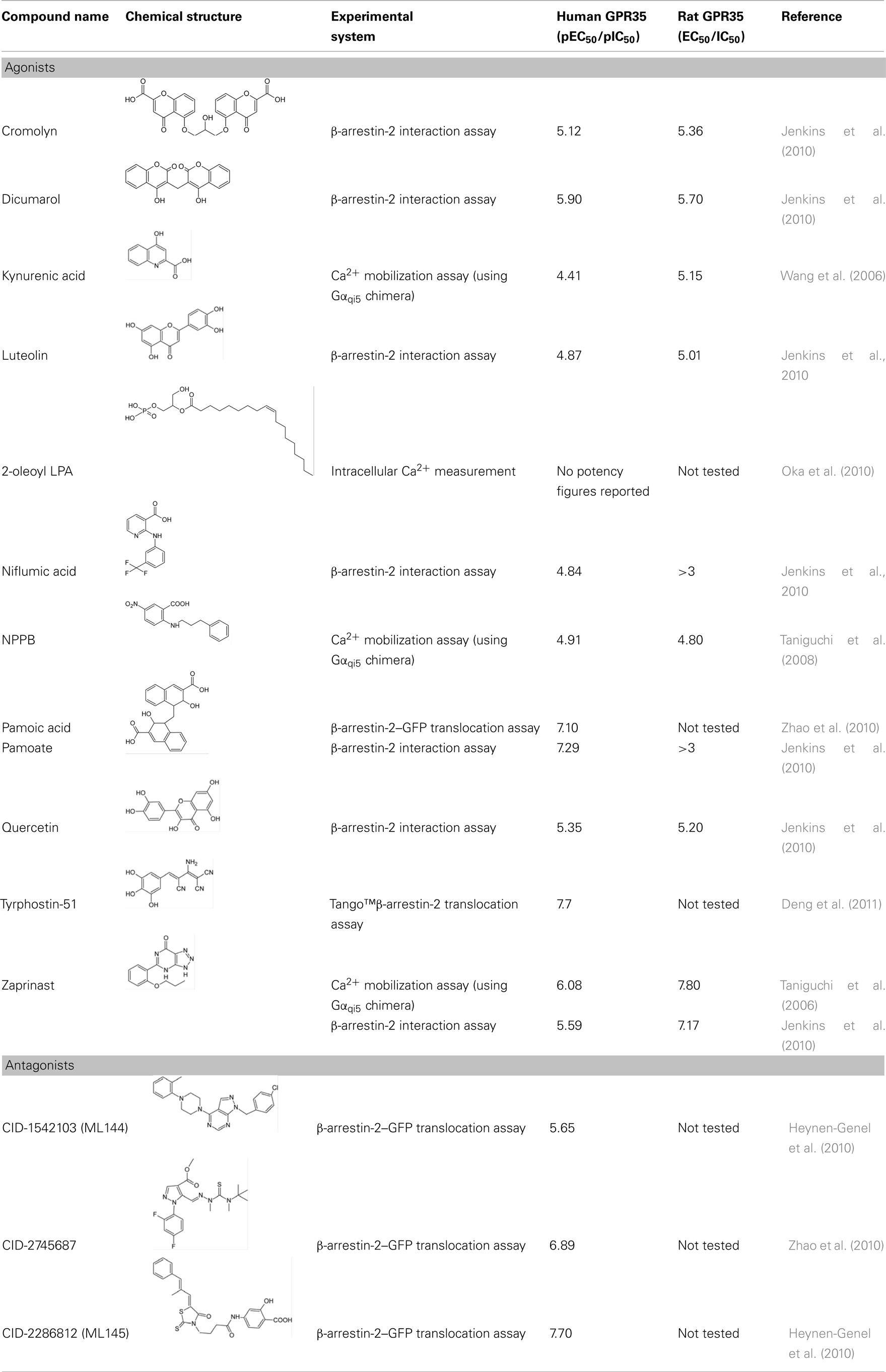
Table 1. GPR35 ligands, their chemical structures and reported potency values.
Lysophosphatidic Acid
Although Oka et al. (2010) were unable to record a response at GPR35 via kynurenic acid, they did observe that lysophosphatidic acid (2-hydroxy-3-phosphonooxypropyl) (Z)-octadec-9-enoate), particularly 2-oleoyl LPA, acted as an agonist at GPR35. LPA is a simple bioactive phospholipid, present in biological fluids such as serum and plasma, with notable tissue levels in the brain (Sugiura et al., 1999; Noguchi et al., 2009). LPA has many cellular effects, including cellular proliferation, prevention of apoptosis, cell migration, cytokine and chemokine secretion, platelet aggregation, smooth muscle contraction, and neurite retraction (Aoki et al., 2008). A number of GPCRs are reported to be responsive to LPA including LPA1 (EDG2), LPA2 (EDG4), LPA3 (EDG7), LPA4 (GPR23), LPA5 (GPR92), LPA6 (P2Y5), and GPR82. Oka et al. (2010) identified LPA as a GPR35 agonist whilst screening endogenous and structurally related GPR55 ligands in HEK293 cells stably expressing GPR35. While the GPR55 ligands were unable to induce [Ca2+]i mobilization, the structurally related analogs 2-oleoyl lysophosphatidic acid (LPA), 2-arachidonoyl LPA, 1-oleoyl LPA, and 1-palmtoyl LPA did demonstrate an ability to mobilize [Ca2+]i in these cells. In addition, a number of in vitro assays including ERK1/2 phosphorylation, GTP-bound RhoA protein quantification and internalization assays were also employed. Overall these provide a robust data set, which could shift the focus of the true functional endogenous ligand from kynurenic acid to LPA, at least at the human receptor. Interestingly, LPA has been associated with peripheral nociception in mouse (Renback et al., 1999), the regulation of blood pressure and atherosclerotic plaque formation (Smyth et al., 2008), and cancer cell invasion (Mills and Moolenaar, 2003).
Synthetic GPR35 Ligands
Zaprinast
Synthetic ligands have proved invaluable in the characterization of GPR35, with moderately potent agonists, and more recently, antagonists, becoming available to facilitate receptor characterization efforts and a small number of in vivo research efforts (Table 1). The synthetic chemical zaprinast (5-(2-propoxyphenyl)-1H-[1,2,3]triazolo[4,5-d]pyrimidin-7(4H)-one) is currently the standard GPR35 agonist. Zaprinast was initially shown to allow GPR35 coupling to the Gi/o and G16 pathways using a calcium mobilization assay, in a manner that is distinct from the well-known effects of this ligand as an inhibitor of cyclic GMP phosphodiesterases (Taniguchi et al., 2006). Jenkins et al. (2010) have since demonstrated that zaprinast activates both human and rat orthologs of GPR35 at micromolar concentrations but, like kynurenic acid, zaprinast is significantly less potent at human than rat GPR35 with reported pEC50 values in BRET-based β-arrestin-2 assays of 5.4 and 7.1, respectively. Zaprinast is better known as an inhibitor of cyclic GMP phosphodiesterases, specifically PDE5 and PDE6. This creates difficulties in disentangling the separate effects of this ligand which reflect activation of GPR35, from effects that are produced via elevated levels of cyclic GMP. However, given its robust potency at both species orthologs within multiple assay systems in vitro, zaprinast can be utilized as an excellent reference compound in the identification process of other possible ligands.
Pamoic Acid and Other Prestwick Chemical Library® Hits
The majority of other currently described GPR35 synthetic ligands (Table 1) have been identified via screens of the Prestwick Chemical Library®, which contains 1120 small molecule drugs with known bioavailability and safety characteristics. This approach identified pamoic acid (4-[(3-carboxy-2-hydroxynaphthalen-1-yl)methyl]-3-hydroxynaphthalene-2-carboxylic acid) as a potent activator of human GPR35 (Jenkins et al., 2010; Zhao et al., 2010). Interestingly, pamoate is a compound previously considered by the Food and Drug Administration as an inactive “salt” component, used to prolong the life of formulations in a wide range of drugs which are currently available on the market (Neubig, 2010). Pamoic acid was elegantly shown to activate human GPR35 using a wide array of in vitro techniques, including ERK1/2 phosphorylation, β-arrestin-2 recruitment and fluorescence internalization assays. Importantly, pamoic acid is significantly more potent than kynurenic acid and zaprinast (Jenkins et al., 2010; Zhao et al., 2010). However, although Zhao et al. (2010) have also reported pamoic acid to be a useful agonist for mouse GPR35, Jenkins et al. (2010) noted that pamoic acid displayed very weak potency at rat GPR35 and demonstrated it to be a partial agonist when compared to zaprinast at human GPR35 in both G-protein-dependent and independent assays. Jenkins et al. (2010) also identified the anti-asthma medicine cromolyn disodium, the flavonoids luteolin and quercetin, the anti-coagulant dicumarol, and the anti-inflammatory agent niflumic acid as GPR35 agonists (Jenkins et al., 2010). Interestingly, as with the ligands noted above a number of these compounds also showed marked species selectivity. So, although cromolyn disodium and dicumarol activated human and rat receptors with similar potency, niflumic acid was highly selective for human GPR35 whilst zaprinast and luteolin were selective at rat GPR35 (Jenkins et al., 2010).
Antagonists of GPR35
To date, the only GPR35 antagonist studied significantly is CID2745687 (methyl-5-[(tert-butylcarbamothioylhydrazinylidene)methyl]-1-(2,4-difluorophenyl)pyrazole-4-carboxylate; Zhao et al., 2010; Table 1). This is a nanomolar, potentially competitive antagonist of human GPR35 and also reported to inhibit the effects of both pamoic acid and zaprinast at mouse GPR35 (Zhao et al., 2010). Two further structural classes including, ML145 (CID-2286812) and ML144 (CID-1542103) as representative examples, have been reported as GPR35 antagonists (Table 1) via a high content screen of nearly 300,000 compounds with ML145 in particular being reported as a high affinity (20 nM) ligand with good selectivity over GPR55 (Heynen-Genel et al., 2010). The availability of such ligands offers potential to further increase knowledge of the pharmacology and functional roles of GPR35.
Implications of Species Selectivity of GPR35 Agonists
From the information relating to reported variation in ligand potencies at multiple species orthologs of GPR35 it is apparent that species selectivity and possible ligand bias may exist (Milligan, 2011). This may have significant implications for the development of GPR35 as a therapeutic target in disease. It will be vital to improve understanding of the basis of ligand selectivity and to document this fully in order to promote translation of information from multiple in vivo animal models to man. To date, little is known on these issues or even if the various reported ligands bind to the same region of GPR35. Homology modeling and ligand docking studies can provide insight. Based on models of human and rat GPR35 Jenkins et al. (2011) hypothesized that the carboxylate moiety of kynurenic acid might form an ionic interaction with the arginine located in transmembrane domain III of GPR35 at position 3.36, because a number of other GPCRs with acidic ligands, including the lactate receptor GPR81, also have an arginine residue at this position. Following alteration of this arginine to alanine in both rat and human GPR35, both kynurenic acid and zaprinast lost function (Jenkins et al., 2011). The same was also true with mutation of the tyrosine residue located at position 3.32, one turn of the alpha helix higher than the arginine residue (Jenkins et al., 2011). These results highlight these amino acids as key residues of the binding pocket. Much further work is required to both define the nature and extent of the binding pocket and to explain ligand species selectivity. With the application of further mutagenesis and analysis of the effects of a wider range of ligands at such mutants, such studies will truly begin to dissect the nature of species selectivity at GPR35. Further investigation may also be required to assess whether selective ligands also lead to distinct signaling pathways, thus inducing a potential signal bias (Milligan, 2011). The G-protein coupling profile of GPR35 has been reported to span Gi/o, and Gα12/13 families but no coherent attempt has yet been made to assess if ligands activate these pathways differentially (Milligan, 2011).
Therapeutic Potential for GPR35
Cardiovascular Disease
Two recent publications have suggested a role for GPR35 in hypertension and the pathology of both heart failure and atherosclerosis. Firstly, a Ser294Arg SNP within GPR35, discussed previously, was shown to have significant association with coronary artery calcification in a patient cohort (Sun et al., 2008). In addition, a link between GPR35, heart failure and hypertension has been implied (Min et al., 2010). This study explored 12 heart failure patient samples (significantly ranging in severity) and two healthy “controls.” A selection of genes up-regulated in the heart failure patients was identified via global expression microarray analysis. These investigators then went on to correlate highly expressed genes with traditional heart failure biomarkers such as plasma brain natriuretic peptide, ejection fraction, and pulmonary arterial pressure; thereby facilitating a degree of adjustment for disease severity. Within the up-regulated genes was GPR35. Subsequently, adenovirus-mediated over-expression of GPR35 in primary neonatal cardiomyocytes resulted in hypertrophy and, most interestingly, these studies also demonstrated a significant 37.5 mmHg increase in blood pressure in a transgenic strain of GPR35 knock-out mice over wild type littermates (Min et al., 2010). It is also clear that Gα13 plays significant regulatory roles in embryonic development, cell migration, proliferation, and contraction (Suzuki et al., 2009), thus establishing Gα13 as an important mediator in pathways relating to the pathology of CVD. Thus, evidence that GPR35 couples selectively to Gα13 (Jenkins et al., 2011) may provide a further link to hypertension and CVD. Ruppel et al. (2005) demonstrated a loss of vascular development and fatality in transgenic mouse neonates via generation of embryonic vascular endothelial cell specific transgenic knockouts of Gα13. Additionally, neonatal fatality and abnormal vascular development can be rescued in transgenic models with a Gα13 “floxed” allele, in which Gα13 is flanked by loxP recombination sites, elegantly demonstrating that transgenically targeting vascular endothelial cells is not harmful in itself and therefore, abnormal vascular development can be entirely attributable to the knock down of Gα13 (Ruppel et al., 2005). These reports are consistent with previous studies which have demonstrated both vascular defects and embryonic lethality in response to Gα13 deficiency (Offermanns et al., 1997) and others which associate Gα13 with the regulation of morphogenesis and capillary assembly via Rho GTPases in angiogenesis (Connolly et al., 2002).
Subsequent activation of the Rho A pathway following activation of Gα13 has been reported in a number of studies. Most recently, evidence has emerged regarding the role of subsequent effectors of the Rho A pathway, for example the Rho associated kinases ROCK1/2 and c-Jun NH2 terminal kinase, have been implicated in mediating angiogenesis and cardiac hypertrophic responses via the Rho pathway, respectively (Maruyama et al., 2002; Bryan et al., 2010). In relation to GPR35, it may be worthwhile to employ techniques to assess JNK activation in neonatal cardiomyocytes given the indirect, yet measurable relation to Gα13 reported here and the hypertrophic response previously reported by Min et al. (2010) may have significant implications relating to GPR35. Although activation of Gα13 is promising regarding CVD pathology, it is important to note that GPR35 also couples to Gαi/o which has been demonstrated to be up-regulated in the on-set of hypertension in spontaneous hypertensive rats (Anand-Srivastava et al., 1991; Li and Anand-Srivastava, 2002). Therefore, it might be relevant to suggest that multiple G-protein pathways contribute toward the hypertensive phenotype associated with GPR35.
Anti-Nociception
GPR35 has also been suggested as a potential therapeutic target in the treatment of pain. Rat dorsal root ganglion neurons endogenously expressing GPR35 were found to inhibit adenylate cyclase in a pertussis toxin-sensitive manner in response to kynurenic acid and zaprinast, and to co-localize with TRPV1 receptors (Ohshiro et al., 2008). Neurons expressing TRPV1 are involved in nociception, mediation of hyper-analgesia, neurogenic inflammation and neuropathic pain. Therefore, Ohshiro et al. (2008) hypothesized that GPR35 may also be involved in nociceptive signaling. This initial hypothesis has since been supported in a number of in vivo studies using various GPR35 agonist compounds. Subcutaneous injection of zaprinast or kynurenic acid prior to acetic acid administration significantly reduced the number of writhes in Swiss male mice (Cosi et al., 2010). Interestingly, the co-administration of kynurenic acid and zaprinast did not induce an additive effect on anti-nociception, suggesting that the two ligands were acting via the same pathway (Cosi et al., 2010). However, since kynurenic acid is associated with anti-nociception via antagonism of NMDA receptors (Chen et al., 2009), and zaprinast is also an effective cGMP phosphodiesterase inhibitor, it may be that these anti-nociceptive effects are not solely attributable to activation of GPR35. Nevertheless, zaprinast has also been shown to induce anti-nociceptive properties in a manner distinct from phosphodiesterase inhibition via the formalin test in rats (Yoon et al., 2006). Additionally, pamoic acid has been demonstrated to induce a dose-dependent anti-nociceptive response in mice (Zhao et al., 2010).
Inflammation
A number of studies have linked GPR35 to inflammatory regulation, either by the presence of the receptor at the surface of immune-specific cells, or by agonist activation leading to changes in immune response. One of the earliest studies aiming to identify GPR35 agonists showed that treatment with kynurenic acid induced an anti-inflammatory response in GPR35 expressing human peripheral blood mononuclear cells via a reduction in lipopolysaccharide-induced tumor necrosis factor-α (TNF-α) secretion in a dose-dependent manner (Wang et al., 2006). TNF-α is a cytokine which acts as part of the innate immune response, principally produced by macrophages in response to infection, trauma, and ischemia. Moreover, large scale gene expression studies have also demonstrated significant up-regulation of GPR35 in macrophages upon response to the polycyclic aromatic hydrocarbon benzo[a]pyrene, an environmental contaminant associated with carcinogenic and inflammatory responses (Sparfel et al., 2010); and lipopolysaccharide, which caused up-regulation of GPR35 in inflammatory macrophages but not non-inflammatory macrophages (Lattin et al., 2008).
Further links between GPR35 and pro-inflammatory effects have been demonstrated via treatment of human invariant natural killer like T (iNKT) cells with kynurenic acid or zaprinast (Fallarini et al., 2010). Activated iNKT cells, which are involved in the maturation of dendric cells, can counter-regulate autoimmunity by secreting cytokines IL-4, IL-5 and TGFβ; or exacerbate autoimmunity through secretion of the pro-inflammatory cytokine, INFγ. In this study iNKT cells stimulated with super-agonist α-Gal-Cer (a chemically synthesized α-galactosylceramide) and kynurenic acid or zaprinast, reduced IL-4 release in a pertussis toxin, dose–responsive manner, without significantly reducing levels of INFγ (Fallarini et al., 2010). Although this study did not speculate on the outcome of GPR35 activation on iNKT cells, previous data show that stimulation of iNKT cells with synthetic glycolipids prevent spontaneous type 1 diabetes in non-obese diabetic mice (Naumov et al., 2001; Sharif et al., 2001), with repeated administration of α-Gal-Cer suppressing type 1 diabetes by promoting differentiation and recruiting tolerogenic dendritic cells in draining lymph nodes (Gillessen et al., 2003). However, an inability to produce IL-4 and an iNKT T helper type 1 bias has also been associated with human type 1 diabetes (Wilson et al., 1998; Kent et al., 2005). It will be interesting to determine the role of GPR35 in iNKT cells, and whether modulation of GPR35 has an affect on the development or protection from type 1 diabetes.
Lastly, one of the most prominent studies to associate GPR35 with immune regulation shows that treatment of monocytes and neutrophils with kynurenic acid can cause β2 integrin mediated firm arrest to intracellular adhesion molecule 1-expressing monolayers of human umbilical vein endothelial cells (Barth et al., 2009). Kynurenic acid triggered adhesion of monocytic cells was diminished by pertussis toxin treatment, and reduced by small hairpin-RNA delivery targeting GPR35, thereby implicating GPR35 as a direct mediator of leukocyte adhesion (Barth et al., 2009). Taken together, these data associate GPR35 with immune regulation; although the concentrations of kynurenic acid used in these studies were substantially lower than consistent with the known potency of this ligand at GPR35. Clearly there is a need for further in vivo analysis to fully understand the role of GPR35 in immunity.
Metabolic Syndrome
In 2007, a patent application regarding “GPR35 and modulators thereof as treatment for metabolic-related disorders” was the first to implicate GPR35 in the pathology of metabolic disorders (Leonard et al., 2007). A variety of compounds with agonist action at GPR35 including “compound 1” [(Z)-5-(3-ethoxy-4-hydoxy-5-nitro-benzylidene)-thiazolidine-2,4-dione] and “compound 16” [(Z)-5-(4-hydoxy-3-nitro-5-propoxy-benzylidene)-thiazolidine-2,4-dione] were able to reduce blood glucose levels in oral glucose tolerance tests, stimulate glucose uptake in differentiated 3T3-L1 adipocytes, and reduce free fatty acid plasma levels in both fasted wild type (C57bl/6) and diabetic (db/db) mice (Leonard et al., 2007). Additionally, and rather interestingly, GPR35 was identified in the pancreas of db/db mice but not obese (ob/ob) diabetic mice using quantitative polymerase chain reaction, whereas expression levels in adipose, liver, spleen, and colon remained similar between these two transgenic lines. Altogether, these data suggests GPR35 may play a role in glucose uptake, storage, and transport. However, as these findings are yet to be confirmed in subsequent studies, further analysis will be required to probe the role of GPR35 in the mediation of glucose homeostasis and diabetes.
Conclusion
Although GPR35 was identified 13 years ago, initial progress has been slow to elucidate the function and pharmacology of this “orphan” receptor. This is largely due to a lack of potent ligands and species selectivity between orthologs. Nonetheless, a number of groups have begun to elucidate the pharmacology, function, and therapeutic potential of GPR35. As a result, a number of endogenous and synthetic ligands have been identified which act upon GPR35. Additionally, the first antagonist compounds have been described, and this should greatly enhance receptor characterization in the future. Furthermore, GPR35 has demonstrated a therapeutically relevant tissue expression profile, with significant expression in the immune and gastrointestinal tissue. GWAS have also identified polymorphisms which link to disease; both in the coding and intergenic regions surrounding GPR35. Taking these findings together, it is becoming apparent that GPR35 may play a significant role in inflammatory pain, asthma, diabetes, hypertension, cardiovascular disease and irritable bowl disease. Future studies will focus on characterization of the receptor binding pocket, elucidation of receptor specific and potent ligands, and in vivo research to determine the extent of GPR35 regulation and its subsequent contribution to disease.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
These studies are supported by the Biotechnology and Biosciences Research Council (BBSRC) and British Heart Foundation (BHF). A. E. Mackenzie thanks BBSRC for a “CASE” studentship. J. E. Lappin thanks BHF for a studentship.
References
Anand-Srivastava, M. B., Picard, S., and Thibault, C. (1991). Altered expression of inhibitory guanine nucleotide regulatory proteins (Gi alpha) in spontaneously hypertensive rats. Am. J. Hypertens. 4, 840–843.
Aoki, J., Inoue, A., and Okudaira, S. (2008). Two pathways for lysophosphatidic acid production. Biochem. Biophys. Acta 1781, 513–518.
Barth, M. C., Ahluwalia, N., Anderson, T. J., Hardy, G. J., Sinha, S., Alvarez-Cardona, J. A., Pruitt, I. E., Rhee, E. P., Colvin, R. A., and Gerszten, R. E. (2009). Kynurenic acid triggers firm arrest of leukocytes to vascular endothelium under flow conditions. J. Biol. Chem. 284, 19189–19195.
Brown, A. J., Daniels, D. A., Kassim, M., Brown, S., Haslam, C. P., Terrell, V. R., Brown, J., Nichols, P. L., Staton, P. C., Wise, A., and Dowell, S. J. (2011). Pharmacology of GPR55 in yeast and identification of GSK494581A as a mixed-activity glycine transporter subtype 1 inhibitor and GPR55 agonist. J. Pharmacol. Exp. Ther. 337, 236–246.
Bryan, B. A., Dennstedt, E., Mitchell, D. C., Walshe, T. E., Noma, K., Loureiro, R., Saint-Geniez, M., Campaigniac, J. P., Liao, J. K., and D’Amore, P. A. (2010). RhoA/ROCK signaling is essential for multiple aspects of VEGF-mediated angiogenesis. FASEB J. 24, 3186–3195.
Chen, Y., Meininger, V., and Guillemin, G. J. (2009). Recent advances in the treatment of amyotrophic lateral sclerosis. Emphasis on kynurenine pathway inhibitors. Cent. Nerv. Syst. Agents Med. Chem. 9, 32–39.
Connolly, J. O., Simpson, N., Hewlett, L., and Hall, A. (2002). Rac regulates endothelial morphogenesis and capillary assembly. Mol. Biol. Cell 13, 2474–2485.
Cosi, C., Mannaioni, G., Cozzi, A., Carla, V., Sili, M., Cavone, L., Maratea, D., and Moroni, F. (2010). G-protein coupled receptor 35 (GPR35) activation and inflammatory pain: Studies on the antinociceptive effects of kynurenic acid and zaprinast. Neuropharmacology 60, 1227–1231.
Deng, H., Hu, H., and Fang, Y. (2011). Tyrphostin analogs are GPR35 agonists. FEBS Lett. 585, 1957–1962.
DeWire, S. M., Ahn, S., Lefkowitz, R. J., and Shenoy, S. K. (2007). Beta-arrestins and cell signaling. Annu. Rev. Physiol. 69, 483–510.
Dowell, S. J., and Brown, A. J. (2009). Yeast assays for G protein-coupled receptors. Methods Mol. Biol. 552, 213–229.
Fallarini, S., Magliulo, L., Paoletti, T., de Lalla, C., and Lombardi, G. (2010). Expression of functional GPR35 in human iNKT cells. Biochem. Biophys. Res. Commun. 398, 420–425.
Fredriksson, R., Lagerstrom, M. C., Lundin, L. G., and Schioth, H. B. (2003). The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 63, 1256–1272.
Gillessen, S., Naumov, Y. N., Nieuwenhuis, E. E., Exley, M. A., Lee, F. S., Mach, N., Luster, A. D., Blumberg, R. S., Taniguchi, M., Balk, S. P., Strominger, J. L., Dranoff, G., and Wilson, S. B. (2003). CD1d-restricted T cells regulate dendritic cell function and antitumor immunity in a granulocyte-macrophage colony-stimulating factor-dependent fashion. Proc. Natl. Acad. Sci. U.S.A. 100, 8874–8879.
Guo, J., Williams, D. J., Puhl, H. L. III., and Ikeda, S. R. (2008). Inhibition of N-type calcium channels by activation of GPR35, an orphan receptor, heterologously expressed in rat sympathetic neurons. J. Pharmacol. Exp. Ther. 324, 342–351.
Hamdan, F. F., Audet, M., Garneau, P., Pelletier, J., and Bouvier, M. (2005). High-throughput screening of G protein-coupled receptor antagonists using a bioluminescence resonance energy transfer 1-based beta-arrestin2 recruitment assay. J. Biomol. Screen. 10, 463–475.
Heynen-Genel, S., Dahl, R., Shi, S., Sauer, M., Hariharan, S., Sergienko, E., Dad, S., Chung, T., Stonich, D., Su, Y., Caron, M., Abood, M. E., and Barak, L. S. (2010). Antagonists for the Orphan Receptor GPR35. Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda, MD: National Center for Biotechnology Information.
Hilmas, C., Pereira, E. F., Alkondon, M., Rassoulpour, A., Schwarcz, R., and Albuquerque, E. X. (2001). The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increaes non-alpha7 nicotinic receptor expression: physiopathological implications. J. Neurosci. 21, 7463–7473.
Horikawa, Y., Oda, N., Cox, N. J., Li, X., Orho-Melander, M., Hara, M., Hinokio, Y., Lindner, T. H., Mashima, H., Schwarz, P. E., del Bosque-Plata, L., Horikawa, Y., Oda, Y., Yoshiuchi, I., Colilla, S., Polonsky, K. S., Wei, S., Concannon, P., Iwasaki, N., Schulze, J., Baier, L. J., Bogardus, C., Groop, L., Boerwinkle, E., Hanis, C. L., and Bell, G. I. (2000). Genetic variation in the gene encoding calpain-10 is associated with type 2 diabetes mellitus. Nat. Genet. 26, 163–175.
Imielinski, M., Baldassano, R. N., Griffiths, A., Russell, R. K., Annese, V., Dubinsky, M., Kugathasan, S., Bradfield, J. P., Walters, T. D., Sleiman, P., Kim, C. E., Muise, A., Wang, K., Glessner, J. T., Saeed, S., Zhang, H., Frackelton, E. C., Hou, C., Flory, J. H., Otieno, G., Chiavacci, R. M., Grundmeier, R., Castro, M., Latiano, A., Dallapiccola, B., Stempak, J., Abrams, D. J., Taylor, K., McGovern, D., Silber, G., Wrobel, I., Quiros, A., Barrett, J. C., Hansoul, S., Nicolae, D. L., Cho, J. H., Duerr, R. H., Rioux, J. D., Brant, S. R., Silverberg, M. S., Taylor, K. D., Barmuda, M. M., Bitton, A., Dassopoulos, T., Datta, L. W., Green, T., Griffiths, A. M., Kistner, E. O., Murtha, M. T., Regueiro, M. D., Rotter, J. I., Schumm, L. P., Steinhartm, A. H., Targan, S. R., Xavier, R. J., Libioulle, C., Sandor, C., Lathrop, M., Belaiche, J., Dewit, O., Gut, I., Heath, S., Laukens, D., Mni, M., Rutgeerts, P., Van Gossum, A., Zelenika, D., Franchimont, D., Hugot, J. P., de Vos, M., Vermeire, S., Louis, E., Cardon, L. R., Anderson, C. A., Drummond, H., Nimmo, E., Ahmad, T., Prescott, N. J., Onnie, C. M., Fisher, S. A., Marchini, J., Ghori, J., Bumpstead, S., Gwillam, R., Tremelling, M., Delukas, P., Mansfield, J., Jewell, D., Satsangi, J., Mathew, C. G., Parkes, M., Georges, M., Daly, M. J., Heyman, M. B., Ferry, G. D., Kirschner, B., Lee, J., Essers, J., Grand, R., Stephens, M., Levine, A., Piccoli, D., Van Limbergen, J., Cucchiara, S., Monos, D. S., Guthery, S. L., Denson, L., Wilson, D. C., Grant, S. F., Daly, M., Silverberg, M. S., Satsangi, J., and Hakonarson, H. (2009). Common variants at five new loci associated with early-onset inflammatory bowel disease. Nat. Genet. 41, 1335–1340.
Jenkins, L., Alvarez-Curto, E., Campbell, K., de Munnik, S., Canals, M., Schlyer, S., and Milligan, G. (2011). Agonist activation of the G protein-coupled receptor GPR35 involves transmembrane domain III and is transduced via Galpha and beta-arrestin-2. Br. J. Pharmacol. 162, 733–748.
Jenkins, L., Brea, J., Smith, N. J., Hudson, B. D., Reilly, G., Bryant, N. J., Castro, M., Loza, M. I., and Milligan, G. (2010). Identification of novel species-selective agonists of the G-protein-coupled receptor GPR35 that promote recruitment of beta-arrestin-2 and activate Galpha13. Biochem. J. 432, 451–459.
Kent, S. C., Chen, Y., Clemmings, S. M., Viglietta, V., Kenyon, N. S., Ricordi, C., Hering, B., and Hafler, D. A. (2005). Loss of IL-4 secretion from human type 1a diabetic pancreatic draining lymph node NKT cells. J. Immunol. 175, 4458–4464.
Kostenis, E., Waelbroeck, M., and Milligan, G. (2005). Promiscuous Gα proteins in basic research and drug discovery. Trends Pharmacol. Sci. 26, 595–602.
Kuc, D., Zgrajka, W., Parada-Turska, J., Urbanik-Sypniewska, T., and Turski, W. A. (2008). Micromolar concentration of kynurenic acid in rat small intestine. Amino Acids 35, 503–505.
Lattin, J. E., Schroder, K., Su, A. I., Walker, J. R., Zhang, J., Wiltshire, T., Saijo, K., Glass, C. K., Hume, D. A., Kellie, S., and Sweet, M. J. (2008). Expression analysis of G Protein-Coupled Receptors in mouse macrophages. Immunome Res. 4, 5.
Leonard, J. N., Chu, Z. L., Unett, D. J., Gatlin, J. E., Gaidarov, I., Qiu, J., Skinner, P. J., Boatman, D., Hume, S. A., Kellie, M. J., and Sweet Oatman, P. D. (2007). GPR35 and modulators thereof for the treatment of metabolic-related disorders. United States 20070077602.
Li, Y., and Anand-Srivastava, M. B. (2002). Inactivation of enhanced expression of G(i) proteins by pertussis toxin attenuates the development of high blood pressure in spontaneously hypertensive rats. Circ. Res. 91, 247–254.
Luttrell, L. M. (2003). ‘Location, location, location’: activation and targeting of MAP kinases by G protein-coupled receptors. J. Mol. Endocrinol. 30, 117–126.
Marchese, A., Paing, M. M., Temple, B. R., and Trejo, J. (2008). G Protein-Coupled Receptor Sorting to Endosomes and Lysosomes. Annu. Rev. Pharmacol. Toxicol. 48, 601–629.
Maruyama, Y., Nishida, M., Sugimoto, Y., Tanabe, S., Turner, J. H., Kozasa, T., Wada, T., Nagao, T., and Kurose, H. (2002). Galpha(12/13) mediates alpha(1)-adrenergic receptor-induced cardiac hypertrophy. Circ. Res. 91, 961–969.
Milligan, G. (2003). Principles: extending the utility of [35S]GTP gamma S binding assays. Trends Pharmacol. Sci. 24, 87–90.
Milligan, G. (2011). Orthologue selectivity and ligand bias: translating the pharmacology of GPR35. Trends Pharmacol. Sci. 32, 317–325.
Milligan, G., and Kostenis, E. (2006). Hetero-trimeric G proteins: a short history. Br. J. Pharmacol. 147, S46–S55.
Mills, G. B., and Moolenaar, W. H. (2003). The emerging role of lysophosphatidic acid in cancer. Nat. Rev. Cancer 3, 582–591.
Min, K. D., Asakura, M., Liao, Y., Nakamaru, K., Okazaki, H., Takahashi, T., Fujimoto, K., Ito, S., Takahashi, A., Asanuma, H., Yamazaki, S., Minamino, T., Sanada, S., Seguchi, O., Nakano, A., Ando, Y., Otsuka, T., Furukawa, H., Isomura, T., Takashima, S., Mochizuki, N., and Kitakaze, M. (2010). Identification of genes related to heart failure using global gene expression profiling of human failing myocardium. Biochem. Biophys. Res. Commun. 393, 55–60.
Mizutani, K., Sugimoto, K., Okuda, T., Katsuya, T., Miyata, T., Tanabe, T., Higaki, J., Ogihara, T., Yamori, Y., Tsujita, Y., Tago, N., and Iwai, N. (2002). Kynureninase is a novel candidate gene for hypertension in spontaneously hypertensive rats. Hypertens. Res. 25, 135–140.
Naumov, Y. N., Bahjat, K. S., Gausling, R., Abraham, R., Exley, M. A., Koezuka, Y., Balk, S. B., Strominger, J. L., Clare-Salzer, M., and B. Wilson, S. (2001). Activation of CD1d-restricted T cells protects NOD mice from developing diabetes by regulating dendritic cell subsets. Proc. Natl. Acad. Sci. U.S.A. 98, 13838–13843.
Neubig, R. R. (2010). Mind your salts: when the inactive constituent isn’t. Mol. Pharmacol. 78, 558–559.
Noguchi, K., Herr, D., Mutoh, T., and Chun, J. (2009). Lysophosphatidic acid (LPA) and its receptors. Curr. Opin. Pharmacol. 9, 15–23.
O’Dowd, B. F., Nguyen, T., Marchese, A., Cheng, R., Lynch, K. R., Heng, H. H., Kolakowski, L. F. Jr., and George, S. R. (1998). Discovery of three novel G-protein-coupled receptor genes. Genomics 47, 310–313.
Offermanns, S., Mancino, V., Revel, J. P., and Simon, M. I. (1997). Vascular system defects and impaired cell chemokinesis as a result of Galpha13 deficiency. Science 275, 533–536.
Ohshiro, H., Tonai-Kachi, H., and Ichikawa, K. (2008). GPR35 is a functional receptor in rat dorsal root ganglion neurons. Biochem. Biophys. Res. Commun. 365, 344–348.
Oka, S., Ota, R., Shima, M., Yamashita, A., and Sugiura, T. (2010). GPR35 is a novel lysophosphatidic acid receptor. Biochem. Biophys. Res. Commun. 395, 232–237.
Okumura, S., Baba, H., Kumada, T., Nanmoku, K., Nakajima, H., Nakane, Y., Hioki, K., and Ikenaka, K. (2004). Cloning of a G-protein-coupled receptor that shows an activity to transform NIH3T3 cells and is expressed in gastric cancer cells. Cancer Sci. 95, 131–135.
Patterson, A. D., Bonzo, J. A., Li, F., Krausz, K. W., Eichler, G. S., Aslam, S., Tigno, X., Weinstein, J. N., Hansen, B. C., Idle, J. R., and Gonzalez, F. J. (2011). Metabolomics reveals attenuation of the SLC6A20 kidney transporter in nonhuman primate and mouse models of type 2 diabetes mellitus. J. Biol. Chem. 286, 19511–19522.
Pawlak, D., Tankiewicz, A., Mysliwiec, P., and Buczko, W. (2002). Tryptophan metabolism via the kynurenine pathway in experimental chronic renal failure. Nephron 90, 328–335.
Prescott, C., Weeks, A. M., Staley, K. J., and Partin, K. M. (2006). Kynurenic acid has a dual action on AMPA receptor responses. Neurosci. Lett. 402, 108–112.
Renback, K., Inoue, M., and Ueda, H. (1999). Lysophosphatidic acid-induced, pertussis toxin-sensitive nociception through a substance P release from peripheral nerve endings in mice. Neurosci. Lett. 270, 59–61.
Ruppel, K. M., Willison, D., Kataoka, H., Wang, A., Zheng, Y. W., Cornelissen, I., Yin, L., Xu, S. M., and Coughlin, S. R. (2005). Essential role for Galpha13 in endothelial cells during embryonic development. Proc. Natl. Acad. Sci. U.S.A. 102, 8281–8286.
Sharif, I., Crockett, T. R., Kane, K. A., and Wainwright, C. L. (2001). The effects of endothelin-1 on ischaemia-induced ventricular arrhythmias in rat isolated hearts. Eur. J. Pharmacol. 427, 235–242.
Shrimpton, A. E., Braddock, B. R., Thomson, L. L., Stein, C. K., and Hoo, J. J. (2004). Molecular delineation of deletions on 2q37.3 in three cases with an Albright hereditary osteodystrophy-like phenotype. Clin. Genet. 66, 537–544.
Smyth, S. S., Cheng, H. Y., Miriyala, S., Panchatcharam, M., and Morris, A. J. (2008). Roles of lysophosphatidic acid in cardiovascular physiology and disease. Biochim. Biophys. Acta 1781, 563–570.
Sparfel, L., Pinel-Marie, M. L., Boize, M., Koscielny, S., Desmots, S., Pery, A., and Fardel, O. (2010). Transcriptional signature of human macrophages exposed to the environmental contaminant benzo(a)pyrene. Toxicol. Sci. 114, 247–259.
Stone, T. W. (2001). Kynurenic acid antagonists and kynurenine pathway inhibitors. Expert Opin. Investig. Drugs 10, 633–645.
Sugiura, T., Nakane, S., Kishimoto, S., Waku, K., Yoshioka, Y., Tokumura, A., and Hanahan, D. J. (1999). Occurrence of lysophosphatidic acid and its alkyl ether-linked analog in rat brain and comparison of their biological activities toward cultured neural cells. Biochem. Biophys. Acta 1440, 194–204.
Sun, Y. V., Bielak, L. F., Peyser, P. A., Turner, S. T., Sheedy, P. F. II., Boerwinkle, E., and Kardia, S. L. (2008). Application of machine learning algorithms to predict coronary artery calcification with a sibship-based design. Genet. Epidemiol. 32, 350–360.
Suzuki, N., Hajicek, N., and Kozasa, T. (2009). Regulation and physiological functions of G12/13-mediated signaling pathways. Neurosignals 17, 55–70.
Taniguchi, Y., Tonai-Kachi, H., and Shinjo, K. (2006). Zaprinast, a well-known cyclic guanosine monophosphate-specific phosphodiesterase inhibitor, is an agonist for GPR35. FEBS Lett. 580, 5003–5008.
Taniguchi, Y., Tonai-Kachi, H., and Shinjo, K. (2008). 5-Nitro-2-(3-phenylpropylamino)benzoic acid is a GPR35 agonist. Pharmacology 82, 245–249.
Tohgo, A., Choy, E. W., Gesty-Palmer, D., Pierce, K. L., Laporte, S., Oakley, R. H., Caron, M. G., Lefkowitz, R. J., and Luttell, L. M. (2003). The stability of the G protein-coupled receptor-beta-arrestin interaction determines the mechanism and functional consequence of ERK activation. J. Biol. Chem. 278, 6258–6267.
Vassilatis, D. K., Hohmann, J. G., Zeng, H., Li, F., Ranchalis, J. E., Mortrud, M. T., Brown, A., Rodriguez, S. S., Weller, J. R., Wright, A. C., Bergmann, J. E., and Gaitanaris, G. A. (2003). The G protein-coupled receptor repertoires of human and mouse. Proc. Natl. Acad. Sci. U.S.A. 100, 4903–4908.
Walczak, K. D., Browski, W., Langner, E., Zgrajka, W., Pi At, J., Kocki, T., Rzeski, W., and Turski, W. A. (2011). Kynurenic acid synthesis and kynurenine aminotransferases expression in colon derived normal and cancer cells. Scand. J. Gastroenterol. 46, 903–912.
Wang, J., Simonavicius, N., Wu, X., Swaminath, G., Reagan, J., Tian, H., and Ling, L. (2006). Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J. Biol. Chem. 281, 22021–22028.
Wilson, S. B., Kent, S. C., Patton, K. T., Orban, T., Jackson, R. A., Exley, M., Porcelli, S., Schatz, D. A., Atkinson, M. A., Balk, S. P., Strominger, J. L., and Hafler, D. A. (1998). Extreme Th1 bias of invariant Valpha24JalphaQ T cells in type 1 diabetes. Nature 391, 177–181.
Yarur, A. J., Deshpande, A. R., Pechman, D. M., Tamariz, L., Abreu, M. T., and Sussman, D. A. (2011). Inflammatory Bowel Disease is associated with an increased incidence of cardiovascular events. Am. J. Gastroenterol. 106, 741–747.
Yoon, M. H., Choi, J., Bae, H. B., Kim, S. J., Chung, S. T., Jeong, S. W., Chung, S. S., Yoo, K. Y., and Jeong, C. Y. (2006). Antinociceptive effects and synergistic interaction with morphine of intrathecal metabotropic glutamate receptor 2/3 antagonist in the formalin test of rats. Neurosci. Lett. 394, 222–226.
Zhao, P., Sharir, H., Kapur, A., Cowan, A., Geller, E. B., Adler, M. W., H. Seltzman, H., Reggio, P. H., Heynen-Genel, S., Sauer, M., Chung, T. D., Bai, Y., Chen, W., Caron, M. G., Barak, L. S., and Abood, M. E. (2010). Targeting of the orphan receptor GPR35 by pamoic acid: a potent activator of extracellular signal-regulated kinase and beta-arrestin2 with antinociceptive activity. Mol. Pharmacol. 78, 560–568.
Keywords: GPR35, diabetes, cardiovascular disease, G-proteins
Citation: MacKenzie AE, Lappin JE, Taylor DL, Nicklin SA and Milligan G (2011) GPR35 as a novel therapeutic target. Front. Endocrin. 2:68. doi: 10.3389/fendo.2011.00068
Received: 11 August 2011;
Paper pending published: 22 August 2011;
Accepted: 16 October 2011;
Published online: 09 November 2011.
Edited by:
Nicola J. Smith, Victor Chang Cardiac Research Institute, AustraliaReviewed by:
Nicola J. Smith, Victor Chang Cardiac Research Institute, AustraliaMeritxell Canals, Monash University, Australia
Copyright: © 2011 MacKenzie, Lappin, Taylor, Nicklin and Milligan. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: G. Milligan, Molecular Pharmacology Group, Institute of Neuroscience and Psychology, College of Medical, Veterinary and Life Sciences, University of Glasgow, Glasgow, Scotland G12 8QQ, UK. e-mail:Z3JhZW1lLm1pbGxpZ2FuQGdsYXNnb3cuYWMudWs=