 Georges Rammouz1
Georges Rammouz1
- 1 Department of Medicine, The Research Institute of the McGill University Health Centre, McGill University, Montreal, QC, Canada
- 2 Department of Biochemistry, McGill University, Montreal, QC, Canada
- 3 Department of Pharmacology and Therapeutics, McGill University, Montreal, QC, Canada
Neurosteroids are steroids made by brain cells independently of peripheral steroidogenic sources. The biosynthesis of most neurosteroids is mediated by proteins and enzymes similar to those identified in the steroidogenic pathway of adrenal and gonadal cells. Dehydroepiandrosterone (DHEA) is a major neurosteroid identified in the brain. Over the years we have reported that, unlike other neurosteroids, DHEA biosynthesis in rat, bovine, and human brain is mediated by an oxidative stress-mediated mechanism, independent of the cytochrome P450 17α-hydroxylase/17,20-lyase (CYP17A1) enzyme activity found in the periphery. This alternative pathway is induced by pro-oxidant agents, such as Fe2+ and β-amyloid peptide. Neurosteroids are involved in many aspects of brain function, and as such, are involved in various neuropathologies, including Alzheimer’s disease (AD). AD is a progressive, yet irreversible neurodegenerative disease for which there are limited means for ante-mortem diagnosis. Using brain tissue specimens from control and AD patients, we provided evidence that DHEA is formed in the AD brain by the oxidative stress-mediated metabolism of an unidentified precursor, thus depleting levels of the precursor in the blood stream. We tested for the presence of this DHEA precursor in human serum using a Fe2+-based reaction and determined the amounts of DHEA formed. Fe2+ treatment of the serum resulted in a dramatic increase in DHEA levels in control patients, whereas only a moderate or no increase was observed in AD patients. The DHEA variation after oxidation correlated with the patients’ cognitive and mental status. In this review, we present the cumulative evidence for oxidative stress as a natural regulator of DHEA formation and the use of this concept to develop a blood-based diagnostic tool for neurodegenerative diseases linked to oxidative stress, such as AD.
Introduction
The crucial roles of steroid hormones in the development and function of the central nervous system (CNS) have been well established (Compagnone and Mellon, 1998; Baulieu et al., 1999; Karishma and Herbert, 2002; Suzuki et al., 2004; Wang et al., 2005). Depending on their chemical structure and plasma concentrations, steroids can exert either protective or adverse effects on neural tissues (Kimonides et al., 1998; Schumacher et al., 2000; Wise et al., 2001; Charalampopoulos et al., 2004, 2006; Melcangi and Panzica, 2006; Singh, 2006). Whereas many steroids originate from peripheral steroidogenic organs, such as adrenals and gonads, recent research has shown that some steroids are synthesized in the nervous system and display beneficial neuroprotective properties, which may be of particular importance in treating diseases in which neurodegeneration is predominant, including age-dependent dementia, stroke, epilepsy, spinal cord injury, Alzheimer’s disease (AD), Parkinson’s disease (PD), and Niemann–Pick type C disease (NP-C). Neurosteroids, according to the definition proposed by Baulieu (1997), are steroid hormones that are synthesized in the central and peripheral nervous systems, either de novo from cholesterol or by in situ metabolism of blood-borne precursors, and that accumulate in the nervous system independently of the classical steroidogenic gland secretion rates. The term “neuroactive steroids” refers to steroid hormones that exert their effects on neural tissue. Neuroactive steroids may be synthesized in both the nervous system and in endocrine glands.
Neurosteroids exert a wide array of biological activities in the brain (Lapchak and Araujo, 2001; Belelli et al., 2006; Strous et al., 2006), either through conventional genomic action or interaction with membrane receptors. In particular, neurosteroids have been found to act as allosteric modulators of the GABAA/central type benzodiazepine receptor complex (Majewska, 1992; Covey et al., 2001; Lapchak and Araujo, 2001), N-methyl D-aspartate (NMDA) receptors (Jang et al., 2004; Mameli et al., 2005), kainate receptors (Costa et al., 2000; Dubrovsky, 2005), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid AMPA receptors (Rupprecht and Holsboer, 2001; Rupprecht et al., 2001), sigma receptors (Monnet et al., 1995; Maurice et al., 1996, 1999, 2006; Takebayashi et al., 2004), glycine receptors (Maksay et al., 2001; Jiang et al., 2006; Mitchell et al., 2007), serotonin receptors (Kostowski and Bienkowski, 1999; Shannon et al., 2005), nicotinic receptors (Paradiso et al., 2000; Pereira et al., 2002; Arias et al., 2006), and muscarinic receptors (Horishita et al., 2005; Steffensen et al., 2006). More recently, it was reported that neurosteroids may directly activate G-protein-coupled membrane receptors (Ueda et al., 2001; He et al., 2003; Zhu et al., 2003; Schiess and Partridge, 2005; Tasker, 2006) or indirectly modulate binding of neuropeptides to their receptors (Sullivan and Moenter, 2003; Torres and Ortega, 2003; Lipschitz et al., 2005). Finally, neurosteroids have been shown to bind to microtubule-associated protein 2 (MAP2) and to stimulate tubulin polymerization in cultured neurons. In fact, the binding of dehydroepiandrosterone (DHEA) and pregnenolone (PREG) to MAP2 involved polar and hydrophobic interactions, such the hydrogen bonds, and was localized at the N-terminal domain of the protein. This domain is specific to high molecular weight MAP2 isoforms, and absent in other MAPs, notably Tau, a component of AD pathology. In AD and other tauopathies the Tau protein forms fibrillar deposits in the brain. The difference between the N-terminal sequence of MAP2 and Tau could account for the difference in their aggregation properties. The direct interaction of DHEA and PREG with MAP2 raises the possibility that, in addition to the well-known steroid hormone–receptor interactions, direct interaction of neurosteroids with the cytoskeleton may participate in brain plasticity (Murakami et al., 2000; Laurine et al., 2003; Iwata et al., 2005).
In vivo studies also indicate that neurosteroids are involved in regulating various neurophysiological and behavioral processes, including cognition, stress, depression, anxiety, and sleep, as well as in sexual- and feeding-related behaviors and locomotion (Vallee et al., 1997, 2001; Engel and Grant, 2001; Mayo et al., 2003; Schumacher et al., 2004; Dubrovsky, 2005, 2006; Mellon, 2007; Mitchell et al., 2008). Paradoxically, although steroids play major roles as signaling molecules within the brain, to date, little is known regarding the neural mechanisms regulating neurosteroid biosynthesis in the CNS.
In this review, we present evidence for oxidative stress as a natural regulator of specific neurosteroid formation. This alternative steroid biosynthesis pathway was used to develop a blood-based diagnostic tool for neurodegenerative diseases linked to oxidative stress, like AD, with the goal of monitoring the onset and progression of the disease as well as its response to existing and experimental therapies.
Pathways of Neurosteroid Biosynthesis
It has long been thought that steroidogenic glands, including the adrenal cortex, gonads, and placenta, were the only sources of steroids that could act on the brain. However, seminal observations made by the Baulieu and Robel group have shown that this view is incorrect. First, these authors discovered that the concentrations of several steroids, such as PREG, DHEA, and their sulfate esters are much higher in the brain than in the plasma (Baulieu, 1981; Corpechot et al., 1981, 1983). Second, they showed that the levels of these steroids in brain tissue remain elevated long after adrenalectomy and castration (Cheney et al., 1995). Third, they found that the circadian variations of steroid concentrations in brain tissue are not synchronized with those of circulating steroids (Robel et al., 1986). These observations led them to propose that the brain can actually synthesize biologically active steroids, or “neurosteroids” (Robel and Baulieu, 1985, 1994; Baulieu, 1997, 1998).
Steroid biosynthesis begins with the transfer of free cholesterol from intracellular stores into mitochondria. Two proteins appear to play a crucial role in intramitochondrial cholesterol transport: the peripheral-type benzodiazepine receptor (Papadopoulos, 1993), renamed translocator protein 18 kDa (TSPO; Papadopoulos et al., 2006; Rone et al., 2009), and the steroidogenic acute regulatory protein (STAR; Stocco and Clark, 1996). TSPO serves as a gatekeeper in protein and cholesterol import into mitochondria, and STAR serves the role of the hormone-induced activator. Thus, both proteins work in concert to bring cholesterol into mitochondria (Hauet et al., 2005).
The first step in the biosynthesis of neurosteroids is the conversion of cholesterol to PREG. This reaction is catalyzed by the cytochrome P450 cholesterol side-chain cleavage (P450scc; CYP11A1) in three successive chemical reactions: 20α-hydroxylation, 22-hydroxylation, and scission of the C20–C22 carbon bond of cholesterol. The products of this reaction are PREG and isocaproic acid. PREG can be converted to DHEA via cytochrome P450c17 (CYP17A1). Both PREG and DHEA are 3β-hydroxy-Δ5-steroids, which are present in neural tissue in the free forms and their sulfate ester forms. 17β-hydroxysteroid dehydrogenase (17β-HSD) and 3β-hydroxysteroid dehydrogenase-isomerase (3β-HSD) mediate the conversion of DHEA into androgens. The cytochrome P450 aromatase (CYP19) converts testosterone to estradiol, whereas 5α-reductase converts testosterone to dihydrotestosterone. PREG can be oxidized to active 3-oxo-Δ4-steroids, such as progesterone (PROG), by 3β-HSD. PROG is a substrate for 5α/β-reductase enzymes and is converted into 5α/β-dihydroprogesterone (5α/β-DH PROG). Further reduction of 5α/β-DH PROG at the C3 position, by the 3α-hydroxysteroid oxidoreductase (3α-HSOR), yields 3α-hydroxy-5α/β-pregnan-20-one (3α,5α/β-tetrahydroprogesterone; 3α,5α/β-TH PROG; allopregnanolone; Mellon et al., 2001; Plassart-Schiess and Baulieu, 2001).
Neurosteroid Action in Neurobehavioral Disorders
Considering the role identified for TSPO in steroid biosynthesis in peripheral tissues, Papadopoulos et al. (1992, 2006) investigated whether TSPO ligands affect PREG formation in glioma cell mitochondria. We showed that at nanomolar levels, high-affinity TSPO drug ligands increase PREG production (Papadopoulos et al., 1992, 2006; Rupprecht et al., 2010). These results were confirmed in situ in rat and human glioma cell cultures upon addition of the precursor mevalonactone (Guarneri et al., 1992; Brown et al., 2000). In addition, TSPO drug ligands were found to increase allopregnanolone levels in vivo in rat forebrain (Korneyev et al., 1993; Costa et al., 1994; Trapani et al., 2005) and hippocampus (Bitran et al., 2000), and to induce antineophobic, anticonflict, and anxiolytic actions via their TSPO-mediated steroidogenic effect and the subsequent action of the synthesized neuroactive steroids on the GABAA receptor (Costa et al., 1994; Bitran et al., 2000). Thus, if TSPO levels in the CNS are reduced, neurosteroid synthesis also will be reduced. This decrease will act at the beginning of the pathway, controlling the supply of cholesterol to support neurosteroid biosynthesis that may be reduced under various pathological conditions, such as anxiety disorders.
To determine the function of increased TSPO levels in AD, the Papadopoulos lab investigated the levels of steroids present in various brain areas in specimens obtained from postmortem AD and age-matched controls, with a focus on DHEA (Brown et al., 2003).
DHEA Properties
DHEA and its sulfate ester dehydroepiandrosterone sulfate (DHEA-S) were identified in the brain by Baulieu et al. (1965). DHEA is a major neuroactive steroid that exerts a broad range of biological effects and constitutes the majority of neurosteroids found in the brain (Baulieu and Robel, 1996). Various studies showed that DHEA also has functions on modulating membrane receptors, such as NMDA, GABA, sigma receptors, and on Ca2+ channels (Compagnone and Mellon, 1998). These effects may explain, at least in part, its ability to protect hippocampal (Kimonides et al., 1998) and brain cells (Roberts et al., 1987), to regulate neuronal activity (Meyer and Gruol, 1994), and to enhance or restore memory and learning processes (Maurice et al., 1998). Finally, the sulfate ester DHEA-S has been shown to enhance the in vitro release of hippocampal acetylcholine (Rhodes et al., 1997), a neurotransmitter likely involved in memory processes and impairment in AD (Kasa et al., 1997). It is well-known that DHEA also displays antioxidant properties, because it can reduce lipid peroxidation in the rat brain (Aragno et al., 1997; Boccuzzi et al., 1997) and in the periphery in older humans (Araghiniknam et al., 1996).
DHEA peripheral levels peak early in adulthood and gradually decline with age (Baulieu, 1996; Parker, 1999). The role of DHEA in the periphery is not well established, but DHEA may serve as a precursor for androgens and estrogens. The levels of DHEA in the brain were found to exceed those seen in the periphery, and these levels were maintained after removal of the peripheral steroidogenic endocrine glands (Corpechot et al., 1981; Akwa et al., 1991).
The neurotrophic effects of DHEA were first reported in mouse brain cell cultures (Roberts et al., 1987) and DHEA was subsequently found to protect rat and human hippocampal neuronal cells against oxidative stress-induced cellular damage (Bastianetto et al., 1999). Given neuroprotective and cognitive-enhancing properties of DHEA, it has been hypothesized that elevated DHEA levels observed in AD brain tissue potentially represent an adaptive or compensatory process (Alhaj et al., 2006).
CYP17A1-Independent Pathway for DHEA Formation
Baulieu and Robel (1990) demonstrated that PREG and DHEA accumulate in the brain independently of the supply by peripheral endocrine organs (Baulieu and Robel, 1990). Despite these initial findings and numerous subsequent studies, the data available to date on the synthesis of DHEA do not account for the mechanisms responsible for its synthesis. First, the levels of CYP17A1 enzymatic activity, immunoreactivity, and mRNA are not consistent with each other (Mellon and Deschepper, 1993). Second, neither CYP17A1 protein nor its activity has been detected in the brain (Le Goascogne et al., 1995). Only transient expression of the mRNA for this enzyme during embryonic life was reported (Compagnone et al., 1995) and contradictory data on the presence of its mRNA in the adult have been presented (Sanne and Krueger, 1995; Stromstedt and Waterman, 1995). Thus, the pathway by which DHEA is synthesized in the adult brain is unknown, and brain steroid synthesis seemingly may not fit in at all with the steps of the well-accepted scheme for adrenal and gonadal steroidogenesis, suggesting that alternate pathways for the synthesis of some steroids, such as DHEA, may exist.
Prasad et al. (1994) proposed the existence of a few alternative precursors present in rat brain extracts that can react with compounds unrelated to peripheral steroid biosynthesis and produce neurosteroids. These authors demonstrated that treatment of organic extracts of rat brain with different oxidizing and reducing compounds not known to cleave lipoidal or sulfate conjugates resulted in the liberation of DHEA. The results were based on experiments that compared the concentrations of these steroids found in the treated aliquots with those measured in untreated samples estimated by mass spectrometric analysis. Cascio et al. (1998) subsequently reported that rat tumor glioma cells, which do not contain the enzyme CYP17A1, are still able to produce DHEA through an alternative pathway. The same pathway was also found in MA-10 Leydig tumor cells. However, in Leydig tumor cells, this process accounts for only a small portion of the steroids produced, suggesting that in this steroidogenic tissue, the principal pathway by which DHEA is biosynthesized involves CYP17A1. Because CYP17A1 enzyme, protein, and activity have not yet been found in rat and guinea pig brain (Mellon and Deschepper, 1993; Baulieu, 1996) where high levels of DHEA have been measured, it may be possible that this DHEA arises from a similar alternative process.
Other Alternative Pathways for Steroid Biosynthesis
Several studies have shown that steroids may be formed via alternative biosynthesis pathways. Some pathways are described as microbial aerobic pathways, and many steroidal components have been detected in axillary secretions, such as the unsaturated steroids of the androstane (C19) family, where the precursors and the biochemical routes of production remain matters for debate (Austin and Ellis, 2003). Other studies have proposed the existence of a “sodium status factor” that regulates aldosterone biosynthesis whereby during severe sodium deficiency, there is a switch in the aldosterone pathway to a pathway using 18-hydroxy-deoxycorticosterone rather than corticosterone as an intermediate (Boon et al., 1998). Moreover, some studies have shown that biotransformation of lithocholic acid by Pseudomonas sp. strain NCIB 10590 under anaerobic conditions leads to a wide range of steroidal products, such as androsta-1,4-diene-3,17-dione 17β-hydroxyandrost-4-ene-3-one, 17β-hydroxyandrosta-1,4-diene-3-one, 3-oxo-5β-cholan-24-oic acid, 3-oxochola-1,4-diene-24-oic acid, 3-oxopregn-4-ene-20-carboxylic acid, and 3-oxopregna-1,4-diene-20 carboxylic acid (Owen and Bilton, 1984).
Oxidative Stress, β-Amyloid (Aβ) Peptide, Iron, and DHEA Levels
The generation of oxygen radicals has been implicated in all types of neurodegenerative diseases (Coyle and Puttfarcken, 1993). Evidence of increased oxidative stress has been shown in the AD brain (Subbarao et al., 1990; Mecocci et al., 1994; Smith et al., 1995), where it contributes to the formation of amyloid plaques and neurofibrillary tangles (Dyrks et al., 1992; Smith et al., 1994). A possible source of oxidative stress in the AD brain is the Aβ peptide. Aβ is a component of AD plaques (Joachim and Selkoe, 1992) and can cause increases in reactive oxygen species (ROS) via several mechanisms. Aβ can acquire a free radical state on its own (Hensley et al., 1994) or activate microglia to produce free radicals (Klegeris et al., 1994; Klegeris and McGeer, 1997). Studies have shown that addition of Aβ to PC12 cells results in increased intracellular calcium levels and oxygen radical production, leading to mitochondrial dysfunction and apoptosis (Guo et al., 1998). These events can be prevented using antioxidants such as vitamin E (Guo et al., 1996).
These findings led us to hypothesize that if the alternative pathway of DHEA formation is present in human brain tissue, DHEA formation should be elevated in AD brain tissue due to the presence of increased Aβ and oxidative stress levels. Brown et al. (2003) measured the levels of DHEA in AD and age-matched control brains and indeed found that DHEA levels are significantly higher in the AD brain in all three areas examined, and are maximal in AD hippocampus. This may reflect increased oxidative stress in the AD brain, potentially due to the actions of Aβ. Aβ, a major component of AD neuritic plaques (Joachim and Selkoe, 1992), increases free radicals in neurons (Subbarao et al., 1990; Mecocci et al., 1994; Nunomura et al., 2000) and glia (Brown et al., 2000), and directly produces hydrogen peroxide through metal ion reduction (Huang et al., 1999). Other studies have shown increased levels of carbonyls in neuronal cytoplasm and in nuclei of neurons and glia from AD brains (Smith et al., 1995), as well as increases in lipid peroxidation, protein peroxidation, disruption of mitochondria energy metabolism in AD, and increased RNA oxidation, suggesting a role for ROS in the development of AD (Subbarao et al., 1990; Mecocci et al., 1994; Markesbery, 1997; Nunomura et al., 2000). Indeed, it is now well established that Aβ and oxidative stress play major roles in the pathogenesis of AD (Markesbery, 1997).
In addition to AD pathology, head trauma and stroke-related massive bleeding may also significantly produce iron-mediated oxidative stress and neurodegeneration. In AD, iron accumulation in AD plaques and neurofibrillary tangles can act as a source of redox-generated free radicals (Smith et al., 1997). If our hypothesis is correct, we expect DHEA levels to be elevated in patients with AD, a disease that involves increased iron and oxidative stress levels. Indeed, as stated above, Brown et al. (2003) reported that DHEA levels are significantly higher in all regions of the AD brain compared to age-matched control brain (Brown et al., 2003). Furthermore, increased oxidative stress-induced by either positive ions (ferrous, copper, zinc, etc.) or Aβ was shown to induce DHEA formation by human brain cells in vitro (Brown et al., 2000).
Fe2+ Triggers the Alternative Pathway
As noted above, iron (Fe) is a significant component of senile plaques, and iron inlays of blood vessels are common in AD. Fe2+ levels are elevated and iron mobility decreased in AD brains (Beard et al., 1993). Fe2+ used to trigger the alternative pathway causes an increase in ROS, both by the formation of ferric iron and superoxide and by reacting with hydrogen peroxide to form the hydroxyl radical by Fenton reaction (Figure 1; Galey, 1997). Considering the effect of Fe2+ on ROS and DHEA formation, Cascio et al. (2000) showed that Aβ causes an increase in oxygen free radicals in cells, and this rise in free radicals affects DHEA levels (Cascio et al., 2000).
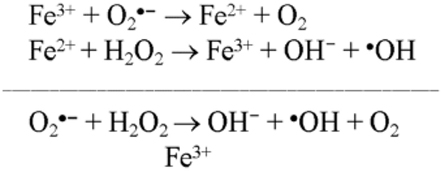
Figure 1. Fenton’s reaction occurs between hydrogen peroxide and Ferrous Iron(II). Ferrous Iron(II) oxidized by hydrogen peroxide to ferric iron(III), a hydroxyl radical, and a hydroxyl anion. Iron(III) is then reduced back to iron(II), a peroxide radical, and a proton by the same hydrogen peroxide.
The structure of at least one form of a putative DHEA precursor of the CYP17A1-independent pathway was suggested by the results obtained with Fe2+ (Prasad et al., 1994). This ion, considered as a reducing agent, is known to produce carbonyl-containing compounds from hydroperoxides (Hawkins, 1949; Hawkins and Young, 1950). This reaction occurs by one-electron reduction of the O-O bond of the peroxide, with subsequent fragmentation of the resulting alkoxy radical (Figure 2). Therefore, the lability of such molecules to heat, air, light, and other environmental conditions may make a delusive contribution to the concentrations of the free neurosteroids estimated by other analytical methods (Mathur et al., 1993). Hawkins and Young (1950) have shown, treatment of tertiary hydroperoxides with aqueous solutions of FeSO4 leads to several products, only one of which may be a ketone.
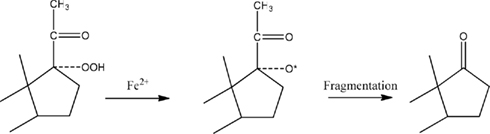
Figure 2. Mechanism of ketone formation from hydroperoxides. This scheme illustrates how the addition of FeSO4 could reduce the 17-hydroperoxide of PREG to an intermediate alkoxy radical. Cleavage of the two-carbon side-chain by b-fragmentation results in the formation of DHEA.
Fe2+ ions affect many cellular processes, including those that stimulate oxygenases and hydroxylases. It is also conceivable that Fe2+ forms complexes with constituents within glioma cells that are able to mimic the catalytic oxidative behavior of a P450 enzyme (Brown et al., 2003). It should be noted that although Fe2+ is a pleiotropic agent in the CNS, the specificity of its reaction with endogenous precursors to form DHEA is characterized by the following observations: (i) it is specific for the formation of DHEA because no effect on PROG production is seen; (ii) it appears to be tissue-specific; (iii) its action is found in the microsomal fraction; (iv) it is dose dependent, but not in a stoichiometric manner; and (v) the effect of FeSO4 could not be replicated to the same extent when using FeCl3 or H2O2.
Nature of the DHEA Precursors
The suggestion that an alternate pathway involving a hydroperoxide could exist for the biosynthesis of steroid hormones was first proposed by Van Lier and Smith (1970). They showed that the 20-hydroperoxide derivative of cholesterol could be converted into PREG by incubation with adrenal cortex mitochondrial enzymes. Thus, a pathway involving sterol peroxide as the precursor of PREG or DHEA could differ from the traditional one.
Cascio et al. (1998) reported that addition of FeSO4 directly to glial cells in culture resulted in a 5- to 10-fold increase in DHEA. This is probably due to the fragmentation of an in situ-formed tertiary hydroperoxide initiated by Fe2+. Evidence already exists that mammalian tissues contain enzymes that catalyze the fragmentation of peroxy constituents to steroid ketones. Larroque and Van Lier (1986) found that when 20-hydroperoxycholesterol is incubated with purified P450scc for only 30 s at 0°C, it is readily converted to PREG. Moreover, Tan and Rousseau (1975) showed that in the rat testis, microsomal fraction can convert 17-hydroperoxyprogesterone to androstenedione when incubated in the presence of oxygen and nicotinamide adenine dinucleotide phosphate. The cofactor was essential, but oxygen was not, because they showed that oxygen can be substituted with argon. These authors also reported that the 17-hydroperoxide can be trapped when PROG is incubated with adrenal homogenates in the presence of the hydroxylase inhibitor p-hydroxymercuribenzoate (Tan and Rousseau, 1975). Adding exogenous PREG along with FeSO4 to C6 microsomes results in a large increase in the amount of DHEA formed. This example, perhaps the first, of a mammalian brain cell converting PREG to DHEA indicates that Fe2+ can activate the conversion process, which may, in fact, involve hydroperoxylation at C17 of the added PREG.
Further studies showed that the peroxy-precursor of DHEA is not soluble in organic solvents, suggesting the potential requirement for a cellular component for activity. Thus, the effect of Fe2+ on the formation of DHEA may be mediated by a protein microsomal component associated with iron reduction. However, since the peroxy-precursor(s) of DHEA made in C6 cells was not soluble in organic solvents, it appears that the process for making DHEA in C6 cells is more complicated than that which simply involves fragmentation of peroxy compounds (Cascio et al., 1998).
Previous reports (Parton et al., 1994; Sawyer et al., 1994) suggested that the level of activation of oxygen is similar to that for Fenton reagents and P450 hydroxylases, and thus it can affect oxygen insertion at the C17 bond. Therefore, we cannot be certain of the mechanism(s) by which Fe2+ acts and increases DHEA production in C6 glioma cells. However, the absence of CYP17A1 activity, protein, and mRNA in these glial cells indicates that, whatever process leads to DHEA formation in these cells, it differs from that involving this steroidogenic enzyme. Even if Fe2+ stimulates an existing enzyme to produce DHEA, the precursor of that DHEA also appears to differ from the precursor customarily assumed to be used in peripheral steroidogenic tissues.
Indeed, addition of exogenous PREG is an artificial way to demonstrate the physiological role and activity of CYP17A1. Therefore, it is important to perform experiments on alternative pathways of steroid synthesis in the absence of the precursor to identify alternative activities. Cascio et al. (2000) proposed that the final answer to the question of how DHEA is made in the brain will depend on the isolation and characterization of brain CYP17A1 or the Fe2+-dependent activity (Cascio et al., 2000). However, in the human brain, all cell types (neurons, oligodendrocytes, and astrocytes) express message and protein CYP17A1, but show no CYP17A1 activity. Nevertheless and as mentioned before, addition of Fe2+ or Aβ increases cellular ROS and results in the formation of DHEA (Brown et al., 2000).
Brown et al. (2000) showed a ROS-induced DHEA synthesis, which may vary between oligodendrocytes and astrocytes. Astrocytes seem to produce more ROS in response to Fe2+ than glioma cells, which may indicate an increased sensitivity to FeSO4 and potentially a higher activity for ROS regulation of DHEA synthesis in normal human brain cells. These data suggest that the oxidative environment of the brain under different conditions may influence DHEA formation by glia. Neurons do not have the alternative pathway for DHEA synthesis, suggesting that the alternative pathway is specific to glia. Oligodendrocytes that do not respond to Fe2+ can synthesize DHEA de novo in a CYP17A1-independent manner, potentially due to high endogenous levels of ROS (Brown et al., 2000).
In support of these findings, the CYP17A1 inhibitor SU10603 (LaCagnin et al., 1989) was found to not affect PREG levels in any of the cell lines used, indicating that this drug is not altering CYP11A1 activity or PREG metabolism. Considering that SU10603 did not inhibit DHEA formation in any human brain cell system tested, a conclusion could be made that CYP17A1 does not mediate DHEA formation in human brain cells.
AD and a CYP17A1-Independent Pathway
Brown et al. (2003) examined the potential for alternative pathway activity in AD and age-matched control brain specimens (Figure 3). Treatment with the reducing reagent FeSO4 caused a significant increase in DHEA levels in the hippocampus and cortex of control patients, possibly indicating the presence of an alternative precursor in these areas. AD patients showed a significant increase in DHEA levels only in the frontal cortex, indicating the presence of an alternative precursor is in this area, but not in the AD hypothalamus or hippocampus. The higher levels of DHEA present in the hypothalamus and hippocampus of the AD brain before FeSO4 treatment suggest that the precursor of the alternative pathway has already been converted to DHEA by endogenous oxidative stress due to the disease process (Brown et al., 2003).
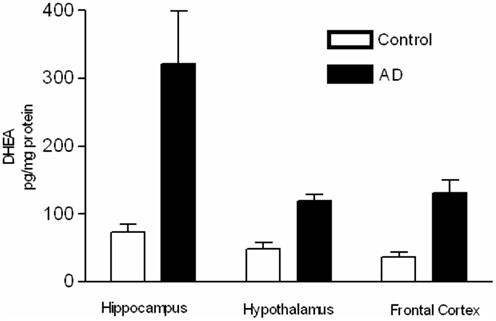
Figure 3. Samples were extracted, and DHEA was purified by HPLC and measured using a specific radioimmunoassay. DHEA levels (pg/mg protein) in brain samples from AD and age-matched control patients with and without treatment with 30 mM FeSO4 (adapted from Brown et al., 2003).
The role of the neurosteroid DHEA in aging is unknown, although a number of attempts have been made to link low serum DHEA with dementia and memory disorders, particularly AD (Berr et al., 1996; Guazzo et al., 1996; Baulieu, 1997; Wolf et al., 1997a,b). Although DHEA levels are lower in AD serum compared to controls, the differences seen may not be as significant due to high variability among specimens (Lacroix et al., 1987; Rammouz et al., 2011). Thus, although DHEA is known to affect NMDA receptors (Bergeron et al., 1996) and to potentiate both memory formation (Flood et al., 1992) and hippocampal long-term potentiation (Yoo et al., 1996), there is no in vivo evidence for its role in memory and dementia.
Liu et al. (2009) reported the first in vivo evidence for the existence of DHEA in brain tissue coming from a CYP17A1-independent pathway present in peripheral steroidogenic tissues using a CYP17A1 chimeric mouse model. In CYP17A1 chimeric mice, the Leydig cell CYP17A1 mRNA and intratesticular and circulating testosterone levels were reduced by 65%, which is in agreement with previous data and consistent with the role of CYP17A1 in peripheral steroidogenesis (Liu et al., 2009). In the same study, the authors observed that although the CYP17A1 protein was undetectable in adult mouse brain extracts, CYP17A1 mRNA was present, but was reduced by 50% in CYP17A1 chimeric mouse brain compared to wild-type brain tissue. Despite this difference in CYP17A1 mRNA levels and lack of CYP17A1 protein, CYP17A1 chimeric mice contained the same endogenous levels of DHEA as wild-type mice. These data raised the question of whether CYP17A1 activity is required for the synthesis of DHEA in adult mouse brain tissue. In support of these findings, Maayan et al. (2005) reported that the increased DHEA synthesis seen in the brain of castrated male mice was completely blocked by the antioxidant N-acetylcysteine amide. The CYP17A1 and 3β-HSD inhibitors SU10693 and trilostane, respectively, did not block the FeSO4-induced DHEA formation, providing additional evidence that CYP17A1 does not mediate the Fe2+-induced DHEA synthesis in the adult mouse brain.
A reduction of DHEA levels throughout development would be expected to affect the cognitive process of the CYP17A1 chimeric mice. However, no spatial memory deficiency was seen in the CYP17A1 chimeric mice in comparison to wild-type mice, suggesting that CYP17A1 gene deletion and the reduction of CYP17A1 mRNA levels in the brain did not affect learning or memory. This finding is supported by clinical studies from patients with CYP17A1 defects (Yanase, 1995). Indeed, there are no reports of cognitive deficiencies in CYP17A1-deficient patients. These results demonstrate the lack of relationship between learning and memory and CYP17A1 expression in brain. Considering the proposed roles of DHEA in neuronal function, DHEA production defects in humans would be expected to have a dramatic impact on brain development and function. The only explanation for the lack of cognitive deficits in CYP17A1-deficient patients and the chimeric mouse model is that DHEA is synthesized by a CYP17A1-independent, Fe2+-sensitive pathway that maintains or compensates for reduced DHEA synthesis.
In further support of a CYP11A1-independent pathway in AD, Brown et al. (2003) measured CYP17A1, and whereas CYP17A1 mRNA was found in the frontal cortex of control and AD patients, there was no CYP17A1 mRNA or immunoreactivity was detected in the hippocampus (Brown et al., 2003). In contrast, the enzyme CYP11A1 was present in all specimens, suggesting that these tissues can synthesize PREG from cholesterol. Thus, the high levels of DHEA found in the AD brain are either derived from peripheral sources and accumulated and stored in specific brain regions, such as the hippocampus, or are derived from local activity of the alternative pathway.
CYP17A1-Independent Pathway in the Diagnostic of Neurodegenerative Diseases
One of the major problems with AD diagnosis and treatment is the inability to determine the onset of the disease. Currently, AD diagnosis is performed using a combination of magnetic resonance imaging scans to measure generalized shrinkage of the brain, and cognitive tests to determine the state of dementia. Unfortunately, symptoms typically do not occur until very late in the disease process. If the changes in DHEA in the cerebral spinal fluid (CSF) are indeed regulated by oxidative stress conditions within the brain, alterations in CSF DHEA levels may occur very early in the progression of the disease. Furthermore, analysis of the data in Figure 3 (Brown et al., 2000) suggests that the serum shows a greater response to FeSO4 treatment than does any brain area tested. Because there was an increase in all control sera after FeSO4 treatment, but no change in AD sera, the serum may be a useful compartment for determining the progression of brain oxidative stress in AD. Therefore, by determining whether the alternative precursor is present in the blood, and measuring CSF DHEA levels, it may be possible to determine the conditions of oxidative stress in the brain. The availability of the alternate precursor for DHEA in serum could thus be used as a diagnostic tool to identify the onset and follow the progression of AD.
It has been long speculated that DHEA is important in the aging process, particularly in modulating memory formation (Parker, 1999). Although serum DHEA levels decrease with age in the human, there are no available studies on the levels of DHEA in the brain. Several groups have tried to correlate serum levels of DHEA and DHEA sulfate with cognitive function (Guazzo et al., 1996) or future development of AD (Berr et al., 1996), but these studies have failed to find a connection between DHEA levels and cognitive ability, or to demonstrate serum DHEA levels as a predictor of future AD development.
Contrary to current hypotheses, we reported that DHEA levels are much higher in the AD brain than in age-matched control specimens (Brown et al., 2003). Additionally, we demonstrated that: (i) in contrast to decreasing serum levels of DHEA in patients with AD, brain DHEA is significantly higher in AD patients than in age-matched controls; and (ii) CYP17A1 protein and mRNA are not found in the hippocampus. In agreement with these observations, DHEA levels in the brain tissue and the CSF of AD patients are significantly higher than in controls. Furthermore, there is evidence for alternative pathway activity in specimens from both AD and control patients, although treatment of control, but not AD, sera with FeSO4 results in increased DHEA levels. Thus, the measurement of CSF DHEA levels, in conjunction with serum DHEA levels and alternative pathway activity examined in the presence of FeSO4, could be used as a predictive diagnostic measurement of AD neuropathology.
Although a larger sampling is required to validate the results of Brown et al. (2003), we speculated that by measuring DHEA levels in the serum and CSF of aging patients, and looking for evidence of alternative pathway activity in these compartments, it may be possible to determine early changes in the levels of oxidative stress in the brain. These changes could reflect early damage in AD or even in other neurodegenerative disorders involving oxidative stress. We investigated the presence of the DHEA precursor in human serum using a Fe2+-based reaction and determined the amounts of DHEA formed in 86 subjects (Rammouz et al., 2011). Our results confirmed the preliminary data of Brown et al. (2003). Indeed, in vitro oxidation of sera from aged-matched control subjects led to a more than 50% increase in DHEA levels compared to respective baseline levels. This increase was significantly higher than that observed in AD patients (14 and 3% increases in the mild and severe AD groups, respectively) assessed using the mini-mental state examination (Figure 4). Interestingly, the correlation between cognitive impairment and percent DHEA increase in the oxidative pathway seemed to be stronger in women than in men. However, more samples must be analyzed before we can reach any conclusions on gender differences in the evolution and outcomes of AD.
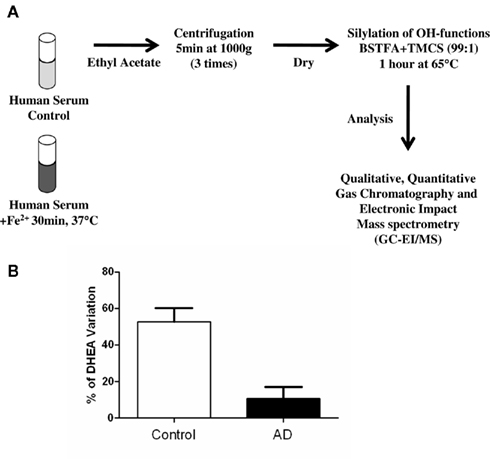
Figure 4. Outline of the methodology used to determine serum DHEA levels and formation in response to FeSO4 treatment (A). Variation of DHEA levels before and after Fe2+ oxidation of human sera from AD and age-matched control patients (adapted from Rammouz et al., 2011).
Mild cognitive impairment (MCI) is currently proposed as a transition state between normal aging and dementia (Mariani et al., 2007). Although MCI is a rather elusive entity for which no consensual definition has been provided, it is commonly divided into two groups: amnestic MCI, which is thought to be an early stage of AD, and non-amnestic MCI, which is associated with cognitive alterations other than memory. Because MCI is considered an early stage of dementia and early diagnosis can lead to clinically relevant treatment, a diagnostic tool for early stage identification of the disease is required to allow clinicians to discriminate MCI from AD. This subset of data demonstrates that monitoring oxidative stress-mediated DHEA formation in sera allows one to discriminate MCI from healthy subjects, and more so from AD patients, regardless of the severity of the disease status.
Many candidate disease-modifying therapies that target the underlying pathogenic mechanism of AD are currently being tested in clinical trials. However, the clinically relevant implementation of any therapy depends on the reliability of the diagnosis. Currently, the diagnosis of AD follows a logical sequence: family history information, mental assessment, and the physical examination which, thus far, focuses on neurological signs (Burns and Iliffe, 2009). An accurate, easy, and specific non-invasive biochemical test that correlates with clinical findings is needed. Our data show that a blood assay based on the existence of an alternative pathway for DHEA biosynthesis in the brain can be successfully used as an early diagnostic tool for AD (Rammouz et al., 2011). The results obtained demonstrate that the DHEA biosynthesis process tightly correlates with cognitive deficits of AD patients and could allow one to discriminate MCI from AD. Such an assay could be used to diagnose AD at a very early stage and to monitor the effect of therapeutic modalities on disease evolution.
Conclusion
We initially reported a cell-specific alternative pathway for DHEA synthesis in the human brain. These results suggested a model for neurosteroid biosynthesis in the CNS. In the case of an oxidative environment and increase in ROS due to ischemia, trauma, or neurodegeneration, glia may also act as reservoirs of DHEA by using the alternative pathway to produce this steroid. Thus, DHEA synthesis via the alternative pathway may be a mechanism by which the brain can protect sensitive areas against oxidative stress or limit the spread of damage. If the cholesterol–cholesterol peroxide-DHEA pathway does exist in the brain, or even in the steroid-producing endocrine glands, then it is undoubtedly associated with its own regulatory system (trophic factors, etc.) and represents a new aspect of steroidogenesis.
If neither enzyme is specific, the hydroperoxide formed by autoxidation and a non-specific enzyme catalyze the formation of PREG from the autoxidized product, the physiological effects due to the portion of ketosteroids formed through this spurious, non-specific mechanism might be inconsequential. Indeed, a recent study pinpointed that the autoxidation of cholesterol constitutes a potential source of both the lipoidal and the sulfated forms of the DHEA and PREG (Liere et al., 2009). Conversely, considerable amounts of PREG and DHEA were released from unknown precursor(s) present in the lipoidal fraction, distinct from fatty acid ester conjugates. In fact, chromatographic and mass spectrometric studies of the nature of the precursor(s) showed that the autoxidation of brain cholesterol is responsible for the release of PREG and DHEA from the lipoidal fraction (Liere et al., 2009). However, in our recent report (Rammouz et al., 2011), we observed that cholesterol levels do not change during the oxidation process mediated by Fe2+. These results obtained in human cells support the data on the ability of astrocytes and oligodendrocytes to synthesize DHEA. In conclusion, all different brain model systems, including rat glioma cell lines, primary cultures of human and rat oligodendrocytes and astrocytes, microsomes from rat brain, rat brain organic extracts (Prasad et al., 1994; Cascio et al., 1998, 2000; Brown et al., 2000, 2003; Liu et al., 2009), and bovine brain microsomes (data not shown), demonstrate the presence of a Fe2+-dependent, CYP17A1-independent process of DHEA formation. Our recent data suggest that a still unidentified precursor of DHEA in the oxidative pathway is absent from the serum of AD patients and lead us to apply this specific trait in the diagnosis of the disease (Rammouz et al., 2011). A blood test based on in vitro oxidative stress-mediated DHEA increases may be a valuable diagnostic tool for identifying AD at an early stage, monitoring AD progression, differentially diagnosing AD from MCI, and evaluating the efficacy of various AD treatments.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work over several years was supported in part by grants from the National Science Foundation, the National Institutes of Health (1R41 NS048688), and contracts from Samaritan Pharmaceuticals (Las Vegas, NV, USA) and Samaritan Therapeutics (Montreal, QC, Canada). Vassilios Papadopoulos was also supported by a Canada Research Chair in Biochemical Pharmacology. The Research Institute of MUHC is supported in part by a Center grant from Le Fond de la Recherche en Santé du Québec.
References
Akwa, Y., Young, J., Kabbadj, K., Sancho, M. J., Zucman, D., Vourc’h, C., Jung-Testas, I., Hu, Z. Y., Le Goascogne, C., and Jo, D. H. (1991). Neurosteroids: biosynthesis, metabolism and function of pregnenolone and dehydroepiandrosterone in the brain. J. Steroid Biochem. Mol. Biol. 40, 71–81.
Alhaj, H. A., Massey, A. E., and McAllister-Williams, R. H. (2006). Effects of DHEA administration on episodic memory, cortisol and mood in healthy young men: a double-blind, placebo-controlled study. Psychopharmacology (Berl.) 188, 541–551.
Araghiniknam, M., Chung, S., Nelson-White, T., Eskelson, C., and Watson, R. R. (1996). Antioxidant activity of dioscorea and dehydroepiandrosterone (DHEA) in older humans. Life Sci. 59, L147–L157.
Aragno, M., Brignardello, E., Tamagno, E., Gatto, V., Danni, O., and Boccuzzi, G. (1997). Dehydroepiandrosterone administration prevents the oxidative damage induced by acute hyperglycemia in rats. J. Endocrinol. 155, 233–240.
Arias, H. R., Bhumireddy, P., and Bouzat, C. (2006). Molecular mechanisms and binding site locations for noncompetitive antagonists of nicotinic acetylcholine receptors. Int. J. Biochem. Cell Biol. 38, 1254–1276.
Austin, C., and Ellis, J. (2003). Microbial pathways leading to steroidal malodour in the axilla. J. Steroid Biochem. Mol. Biol. 87, 105–110.
Bastianetto, S., Ramassamy, C., Poirier, J., and Quirion, R. (1999). Dehydroepiandrosterone (DHEA) protects hippocampal cells from oxidative stress-induced damage. Brain Res. Mol. Brain Res. 66, 35–41.
Baulieu, E. E. (1981). Steroids Hormones in the Brain: Several Mechanisms? Oxford: Pergamon Press, 3–14.
Baulieu, E. E. (1996). Dehydroepiandrosterone (DHEA): a fountain of youth? J. Clin. Endocrinol. Metab. 81, 3147–3151.
Baulieu, E. E. (1997). Neurosteroids: of the nervous system, by the nervous system, for the nervous system. Recent Prog. Horm. Res. 52, 1–32.
Baulieu, E. E. (1998). Neurosteroids: a novel function of the brain. Psychoneuroendocrinology 23, 963–987.
Baulieu, E. E., Corpechot, C., Dray, F., Emiliozzi, R., Lebeau, M. C., Mauvais Jarvis, P., and Robel, P. (1965). An adrenal-selected “androgen”: dehydroisoandrosterone sulfate. Its metabolism and a tentative generalization on the metabolism of other steroid conjugates in man. Recent Prog. Horm. Res. 21, 411–500.
Baulieu, E. E., and Robel, P. (1990). Neurosteroids: a new brain function? J. Steroid Biochem. Mol. Biol. 37, 395–403.
Baulieu, E. E., and Robel, P. (1996). Dehydroepiandrosterone and dehydroepiandrosterone sulfate as neuroactive neurosteroids. J. Endocrinol. 150(Suppl.), S221–S239.
Baulieu, E. E., Robel, P., and Schumacher, M. (1999). Neurosteroids: A New Regulatory Function in the Nervous System. Totowa, NJ: Humana Press.
Belelli, D., Herd, M. B., Mitchell, E. A., Peden, D. R., Vardy, A. W., Gentet, L., and Lambert, J. J. (2006). Neuroactive steroids and inhibitory neurotransmission: mechanisms of action and physiological relevance. Neuroscience 138, 821–829.
Bergeron, R., de Montigny, C., and Debonnel, G. (1996). Potentiation of neuronal NMDA response induced by dehydroepiandrosterone and its suppression by progesterone: effects mediated via sigma receptors. J. Neurosci. 16, 1193–1202.
Berr, C., Lafont, S., Debuire, B., Dartigues, J. F., and Baulieu, E. E. (1996). Relationships of dehydroepiandrosterone sulfate in the elderly with functional, psychological, and mental status, and short-term mortality: a French community-based study. Proc. Natl. Acad. Sci. U.S.A. 93, 13410–13415.
Bitran, D., Foley, M., Audette, D., Leslie, N., and Frye, C. A. (2000). Activation of peripheral mitochondrial benzodiazepine receptors in the hippocampus stimulates allopregnanolone synthesis and produces anxiolytic-like effects in the rat. Psychopharmacology (Berl.) 151, 64–71.
Boccuzzi, G., Aragno, M., Seccia, M., Brignardello, E., Tamagno, E., Albano, E., Danni, O., and Bellomo, G. (1997). Protective effect of dehydroepiandrosterone against copper-induced lipid peroxidation in the rat. Free Radic. Biol. Med. 22, 1289–1294.
Boon, W. C., McDougall, J. G., and Coghlan, J. P. (1998). Hypothesis: aldosterone is synthesized by an alternative pathway during severe sodium depletion. ‘A new wine in an old bottle.’ Clin. Exp. Pharmacol. Physiol. 25, 369–378.
Brown, R. C., Cascio, C., and Papadopoulos, V. (2000). Pathways of neurosteroid biosynthesis in cell lines from human brain: regulation of dehydroepiandrosterone formation by oxidative stress and beta-amyloid peptide. J. Neurochem. 74, 847–859.
Brown, R. C., Han, Z., Cascio, C., and Papadopoulos, V. (2003). Oxidative stress-mediated DHEA formation in Alzheimer’s disease pathology. Neurobiol. Aging 24, 57–65.
Cascio, C., Brown, R. C., Liu, Y., Han, Z., Hales, D. B., and Papadopoulos, V. (2000). Pathways of dehydroepiandrosterone formation in rat brain glia. J. Steroid Biochem. Mol. Biol. 75, 177–186.
Cascio, C., Prasad, V. V., Lin, Y. Y., Lieberman, S., and Papadopoulos, V. (1998). Detection of P450c17-independent pathways for dehydroepiandrosterone (DHEA) biosynthesis in brain glial tumor cells. Proc. Natl. Acad. Sci. U.S.A. 95, 2862–2867.
Charalampopoulos, I., Alexaki, V. I., Tsatsanis, C., Minas, V., Dermitzaki, E., Lasaridis, I., Vardouli, L., Stournaras, C., Margioris, A. N., Castanas, E., and Gravanis, A. (2006). Neurosteroids as endogenous inhibitors of neuronal cell apoptosis in aging. Ann. N. Y. Acad. Sci. 1088, 139–152.
Charalampopoulos, I., Tsatsanis, C., Dermitzaki, E., Alexaki, V. I., Castanas, E., Margioris, A. N., and Gravanis, A. (2004). Dehydroepiandrosterone and allopregnanolone protect sympathoadrenal medulla cells against apoptosis via antiapoptotic Bcl-2 proteins. Proc. Natl. Acad. Sci. U.S.A. 101, 8209–8214.
Cheney, D. L., Uzunov, D., Costa, E., and Guidotti, A. (1995). Gas chromatographic-mass fragmentographic quantitation of 3 alpha-hydroxy-5 alpha-pregnan-20-one (allopregnanolone) and its precursors in blood and brain of adrenalectomized and castrated rats. J. Neurosci. 15, 4641–4650.
Compagnone, N. A., Bulfone, A., Rubenstein, J. L., and Mellon, S. H. (1995). Steroidogenic enzyme P450c17 is expressed in the embryonic central nervous system. Endocrinology 136, 5212–5223.
Compagnone, N. A., and Mellon, S. H. (1998). Dehydroepiandrosterone: a potential signalling molecule for neocortical organization during development. Proc. Natl. Acad. Sci. U.S.A. 95, 4678–4683.
Corpechot, C., Robel, P., Axelson, M., Sjovall, J., and Baulieu, E. E. (1981). Characterization and measurement of dehydroepiandrosterone sulfate in rat brain. Proc. Natl. Acad. Sci. U.S.A. 78, 4704–4707.
Corpechot, C., Synguelakis, M., Talha, S., Axelson, M., Sjovall, J., Vihko, R., Baulieu, E. E., and Robel, P. (1983). Pregnenolone and its sulfate ester in the rat brain. Brain Res. 270, 119–125.
Costa, E., Auta, J., Guidotti, A., Korneyev, A., and Romeo, E. (1994). The pharmacology of neurosteroidogenesis. J. Steroid Biochem. Mol. Biol. 49, 385–389.
Costa, E. T., Soto, E. E., Cardoso, R. A., Olivera, D. S., and Valenzuela, C. F. (2000). Acute effects of ethanol on kainate receptors in cultured hippocampal neurons. Alcohol. Clin. Exp. Res. 24, 220–225.
Covey, D. F., Evers, A. S., Mennerick, S., Zorumski, C. F., and Purdy, R. H. (2001). Recent developments in structure-activity relationships for steroid modulators of GABA(A) receptors. Brain Res. Brain Res. Rev. 37, 91–97.
Coyle, J. T., and Puttfarcken, P. (1993). Oxidative stress, glutamate, and neurodegenerative diseases. Science 262, 689–695.
Dubrovsky, B. (2006). Neurosteroids, neuroactive steroids, and symptoms of affective disorders. Pharmacol. Biochem. Behav. 84, 644–655.
Dubrovsky, B. O. (2005). Steroids, neuroactive steroids and neurosteroids in psychopathology. Prog. Neuropsychopharmacol. Biol. Psychiatry 29, 169–192.
Dyrks, T., Dyrks, E., Hartmann, T., Masters, C., and Beyreuther, K. (1992). Amyloidogenicity of beta A4 and beta A4-bearing amyloid protein precursor fragments by metal-catalyzed oxidation. J. Biol. Chem. 267, 18210–18217.
Engel, S. R., and Grant, K. A. (2001). Neurosteroids and behavior. Int. Rev. Neurobiol. 46, 321–348.
Flood, J. F., Morley, J. E., and Roberts, E. (1992). Memory-enhancing effects in male mice of pregnenolone and steroids metabolically derived from it. Proc. Natl. Acad. Sci. U.S.A. 89, 1567–1571.
Galey, J. B. (1997). Potential use of iron chelators against oxidative damage. Adv. Pharmacol. 38, 167–203.
Guarneri, P., Papadopoulos, V., Pan, B., and Costa, E. (1992). Regulation of pregnenolone synthesis in C6-2B glioma cells by 4′-chlorodiazepam. Proc. Natl. Acad. Sci. U.S.A. 89, 5118–5122.
Guazzo, E. P., Kirkpatrick, P. J., Goodyer, I. M., Shiers, H. M., and Herbert, J. (1996). Cortisol, dehydroepiandrosterone (DHEA), and DHEA sulfate in the cerebrospinal fluid of man: relation to blood levels and the effects of age. J. Clin. Endocrinol. Metab. 81, 3951–3960.
Guo, Q., Christakos, S., Robinson, N., and Mattson, M. P. (1998). Calbindin D28k blocks the proapoptotic actions of mutant presenilin 1: reduced oxidative stress and preserved mitochondrial function. Proc. Natl. Acad. Sci. U.S.A. 95, 3227–3232.
Guo, Q., Furukawa, K., Sopher, B. L., Pham, D. G., Xie, J., Robinson, N., Martin, G. M., and Mattson, M. P. (1996). Alzheimer’s PS-1 mutation perturbs calcium homeostasis and sensitizes PC12 cells to death induced by amyloid beta-peptide. Neuroreport 8, 379–383.
Hauet, T., Yao, Z. X., Bose, H. S., Wall, C. T., Han, Z., Li, W., Hales, D. B., Miller, W. L., Culty, M., and Papadopoulos, V. (2005). Peripheral-type benzodiazepine receptor-mediated action of steroidogenic acute regulatory protein on cholesterol entry into leydig cell mitochondria. Mol. Endocrinol. 19, 540–554.
Hawkins, E. G. E. (1949). The reactions of organic peroxides. Part I. 2-Phenyl-2-butyl hydroperoxide. J. Chem. Soc. 2076–2077.
Hawkins, E. G. E., and Young, D. P. (1950). Reactions of organic peroxides. Part V. Reaction of ferrous sulphate with methylcyclopentyl and methylcyclohexyl hydroperoxides. J. Chem. Soc. 2804–2807.
He, L. M., Zhang, C. G., Zhou, Z., and Xu, T. (2003). Rapid inhibitory effects of corticosterone on calcium influx in rat dorsal root ganglion neurons. Neuroscience 116, 325–333.
Hensley, K., Carney, J. M., Mattson, M. P., Aksenova, M., Harris, M., Wu, J. F., Floyd, R. A., and Butterfield, D. A. (1994). A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 91, 3270–3274.
Horishita, T., Minami, K., Uezono, Y., Shiraishi, M., Ogata, J., Okamoto, T., Terada, T., and Sata, T. (2005). The effects of the neurosteroids: pregnenolone, progesterone and dehydroepiandrosterone on muscarinic receptor-induced responses in Xenopus oocytes expressing M1 and M3 receptors. Naunyn Schmiedebergs Arch. Pharmacol. 371, 221–228.
Huang, X., Atwood, C. S., Hartshorn, M. A., Multhaup, G., Goldstein, L. E., Scarpa, R. C., Cuajungco, M. P., Gray, D. N., Lim, J., Moir, R. D., Tanzi, R. E., and Bush, A. I. (1999). The A beta peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry 38, 7609–7616.
Iwata, M., Muneoka, K. T., Shirayama, Y., Yamamoto, A., and Kawahara, R. (2005). A study of a dendritic marker, microtubule-associated protein 2 (MAP-2), in rats neonatally treated neurosteroids, pregnenolone and dehydroepiandrosterone (DHEA). Neurosci. Lett. 386, 145–149.
Jang, M. K., Mierke, D. F., Russek, S. J., and Farb, D. H. (2004). A steroid modulatory domain on NR2B controls N-methyl-D-aspartate receptor proton sensitivity. Proc. Natl. Acad. Sci. U.S.A. 101, 8198–8203.
Jiang, P., Yang, C. X., Wang, Y. T., and Xu, T. L. (2006). Mechanisms of modulation of pregnanolone on glycinergic response in cultured spinal dorsal horn neurons of rat. Neuroscience 141, 2041–2050.
Joachim, C. L., and Selkoe, D. J. (1992). The seminal role of beta-amyloid in the pathogenesis of Alzheimer disease. Alzheimer Dis. Assoc. Disord. 6, 7–34.
Karishma, K. K., and Herbert, J. (2002). Dehydroepiandrosterone (DHEA) stimulates neurogenesis in the hippocampus of the rat, promotes survival of newly formed neurons and prevents corticosterone-induced suppression. Eur. J. Neurosci. 16, 445–453.
Kasa, P., Rakonczay, Z., and Gulya, K. (1997). The cholinergic system in Alzheimer’s disease. Prog. Neurobiol. 52, 511–535.
Kimonides, V. G., Khatibi, N. H., Svendsen, C. N., Sofroniew, M. V., and Herbert, J. (1998). Dehydroepiandrosterone (DHEA) and DHEA-sulfate (DHEAS) protect hippocampal neurons against excitatory amino acid-induced neurotoxicity. Proc. Natl. Acad. Sci. U.S.A. 95, 1852–1857.
Klegeris, A., and McGeer, P. L. (1997). Beta-amyloid protein enhances macrophage production of oxygen free radicals and glutamate. J. Neurosci. Res. 49, 229–235.
Klegeris, A., Walker, D. G., and McGeer, P. L. (1994). Activation of macrophages by Alzheimer beta amyloid peptide. Biochem. Biophys. Res. Commun. 199, 984–991.
Korneyev, A., Pan, B. S., Polo, A., Romeo, E., Guidotti, A., and Costa, E. (1993). Stimulation of brain pregnenolone synthesis by mitochondrial diazepam binding inhibitor receptor ligands in vivo. J. Neurochem. 61, 1515–1524.
Kostowski, W., and Bienkowski, P. (1999). Discriminative stimulus effects of ethanol: neuropharmacological characterization. Alcohol 17, 63–80.
LaCagnin, L. B., Levitt, M., Bergstrom, J. M., and Colby, H. D. (1989). Inhibition of adrenocortical, mitochondrial and microsomal monooxygenases by SU-10’603, a steroid 17 alpha-hydroxylase inhibitor. J. Steroid Biochem. 33, 599–604.
Lacroix, C., Fiet, J., Benais, J. P., Gueux, B., Bonete, R., Villette, J. M., Gourmel, B., and Dreux, C. (1987). Simultaneous radioimmunoassay of progesterone, androst-4-enedione, pregnenolone, dehydroepiandrosterone and 17-hydroxyprogesterone in specific regions of human brain. J. Steroid Biochem. 28, 317–325.
Lapchak, P. A., and Araujo, D. M. (2001). Preclinical development of neurosteroids as neuroprotective agents for the treatment of neurodegenerative diseases. Int. Rev. Neurobiol. 46, 379–397.
Larroque, C., and Van Lier, J. E. (1986). Hydroperoxysterols as a probe for the mechanism of cytochrome P-450scc-mediated hydroxylation. Homolytic versus heterolytic oxygen-oxygen bond scission. J. Biol. Chem. 261, 1083–1087.
Laurine, E., Lafitte, D., Gregoire, C., Seree, E., Loret, E., Douillard, S., Michel, B., Briand, C., and Verdier, J. M. (2003). Specific binding of dehydroepiandrosterone to the N terminus of the microtubule-associated protein MAP2. J. Biol. Chem. 278, 29979–29986.
Le Goascogne, C., Sananes, N., Eychenne, B., Gouezou, M., Baulieu, E. E., and Robel, P. (1995). Androgen biosynthesis in the stomach: expression of cytochrome P450 17 alpha-hydroxylase/17,20-lyase messenger ribonucleic acid and protein, and metabolism of pregnenolone and progesterone by parietal cells of the rat gastric mucosa. Endocrinology 136, 1744–1752.
Liere, P., Pianos, A., Eychenne, B., Cambourg, A., Bodin, K., Griffiths, W., Schumacher, M., Baulieu, E. E., and Sjovall, J. (2009). Analysis of pregnenolone and dehydroepiandrosterone in rodent brain: cholesterol autoxidation is the key. J. Lipid Res. 50, 2430–2444.
Lipschitz, D. L., Crowley, W. R., Armstrong, W. E., and Bealer, S. L. (2005). Neurochemical bases of plasticity in the magnocellular oxytocin system during gestation. Exp. Neurol. 196, 210–223.
Liu, Y., Pocivavsek, A., and Papadopoulos, V. (2009). Dehydroepiandrosterone formation is independent of cytochrome P450 17alpha-hydroxylase/17, 20 lyase activity in the mouse brain. J. Steroid Biochem. Mol. Biol. 115, 86–90.
Maayan, R., Touati-Werner, D., Ram, E., Galdor, M., and Weizman, A. (2005). Is brain dehydroepiandrosterone synthesis modulated by free radicals in mice? Neurosci. Lett. 377, 130–135.
Majewska, M. D. (1992). Neurosteroids: endogenous bimodal modulators of the GABAA receptor. Mechanism of action and physiological significance. Prog. Neurobiol. 38, 379–395.
Maksay, G., Laube, B., and Betz, H. (2001). Subunit-specific modulation of glycine receptors by neurosteroids. Neuropharmacology 41, 369–376.
Mameli, M., Carta, M., Partridge, L. D., and Valenzuela, C. F. (2005). Neurosteroid-induced plasticity of immature synapses via retrograde modulation of presynaptic NMDA receptors. J. Neurosci. 25, 2285–2294.
Mariani, E., Monastero, R., and Mecocci, P. (2007). Mild cognitive impairment: a systematic review. J. Alzheimers. Dis. 12, 23–35.
Markesbery, W. R. (1997). Oxidative stress hypothesis in Alzheimer’s disease. Free Radic. Biol. Med. 23, 134–147.
Mathur, C., Prasad, V. V., Raju, V. S., Welch, M., and Lieberman, S. (1993). Steroids and their conjugates in the mammalian brain. Proc. Natl. Acad. Sci. U.S.A. 90, 85–88.
Maurice, T., Gregoire, C., and Espallergues, J. (2006). Neuro(active)steroids actions at the neuromodulatory sigma1 (sigma1) receptor: biochemical and physiological evidences, consequences in neuroprotection. Pharmacol. Biochem. Behav. 84, 581–597.
Maurice, T., Phan, V. L., Urani, A., Kamei, H., Noda, Y., and Nabeshima, T. (1999). Neuroactive neurosteroids as endogenous effectors for the sigma1 (sigma1) receptor: pharmacological evidence and therapeutic opportunities. Jpn. J. Pharmacol. 81, 125–155.
Maurice, T., Roman, F. J., and Privat, A. (1996). Modulation by neurosteroids of the in vivo (+)-[3H]SKF-10,047 binding to sigma 1 receptors in the mouse forebrain. J. Neurosci. Res. 46, 734–743.
Maurice, T., Su, T. P., and Privat, A. (1998). Sigma1 (sigma 1) receptor agonists and neurosteroids attenuate B25-35-amyloid peptide-induced amnesia in mice through a common mechanism. Neuroscience 83, 413–428.
Mayo, W., George, O., Darbra, S., Bouyer, J. J., Vallee, M., Darnaudery, M., Pallares, M., Lemaire-Mayo, V., Le Moal, M., Piazza, P. V., and Abrous, N. (2003). Individual differences in cognitive aging: implication of pregnenolone sulfate. Prog. Neurobiol. 71, 43–48.
Mecocci, P., MacGarvey, U., and Beal, M. F. (1994). Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann. Neurol. 36, 747–751.
Melcangi, R. C., and Panzica, G. C. (2006). Neuroactive steroids: old players in a new game. Neuroscience 138, 733–739.
Mellon, S. H. (2007). Neurosteroid regulation of central nervous system development. Pharmacol. Ther. 116, 107–124.
Mellon, S. H., and Deschepper, C. F. (1993). Neurosteroid biosynthesis: genes for adrenal steroidogenic enzymes are expressed in the brain. Brain Res. 629, 283–292.
Mellon, S. H., Griffin, L. D., and Compagnone, N. A. (2001). Biosynthesis and action of neurosteroids. Brain Res. Brain Res. Rev. 37, 3–12.
Meyer, J. H., and Gruol, D. L. (1994). Dehydroepiandrosterone sulfate alters synaptic potentials in area CA1 of the hippocampal slice. Brain Res. 633, 253–261.
Mitchell, E. A., Gentet, L. J., Dempster, J., and Belelli, D. (2007). GABAA and glycine receptor-mediated transmission in rat lamina II neurones: relevance to the analgesic actions of neuroactive steroids. J. Physiol. 583, 1021–1040.
Mitchell, E. A., Herd, M. B., Gunn, B. G., Lambert, J. J., and Belelli, D. (2008). Neurosteroid modulation of GABAA receptors: molecular determinants and significance in health and disease. Neurochem. Int. 52, 588–595.
Monnet, F. P., Mahe, V., Robel, P., and Baulieu, E. E. (1995). Neurosteroids, via sigma receptors, modulate the [3H]norepinephrine release evoked by N-methyl-D-aspartate in the rat hippocampus. Proc. Natl. Acad. Sci. U.S.A. 92, 3774–3778.
Murakami, K., Fellous, A., Baulieu, E. E., and Robel, P. (2000). Pregnenolone binds to microtubule-associated protein 2 and stimulates microtubule assembly. Proc. Natl. Acad. Sci. U.S.A. 97, 3579–3584.
Nunomura, A., Perry, G., Pappolla, M. A., Friedland, R. P., Hirai, K., Chiba, S., and Smith, M. A. (2000). Neuronal oxidative stress precedes amyloid-beta deposition in Down syndrome. J. Neuropathol. Exp. Neurol. 59, 1011–1017.
Owen, R. W., and Bilton, R. F. (1984). Bioconversion of lithocholic acid under anaerobic conditions by Pseudomonas sp. strain NCIB 10590. Appl. Environ. Microbiol. 48, 606–609.
Papadopoulos, V. (1993). Peripheral-type benzodiazepine/diazepam binding inhibitor receptor: biological role in steroidogenic cell function. Endocr. Rev. 14, 222–240.
Papadopoulos, V., Guarneri, P., Kreuger, K. E., Guidotti, A., and Costa, E. (1992). Pregnenolone biosynthesis in C6-2B glioma cell mitochondria: regulation by a mitochondrial diazepam binding inhibitor receptor. Proc. Natl. Acad. Sci. U.S.A. 89, 5113–5117.
Papadopoulos, V., Lecanu, L., Brown, R. C., Han, Z., and Yao, Z. X. (2006). Peripheral-type benzodiazepine receptor in neurosteroid biosynthesis, neuropathology and neurological disorders. Neuroscience 138, 749–756.
Paradiso, K., Sabey, K., Evers, A. S., Zorumski, C. F., Covey, D. F., and Steinbach, J. H. (2000). Steroid inhibition of rat neuronal nicotinic alpha4beta2 receptors expressed in HEK 293 cells. Mol. Pharmacol. 58, 341–351.
Parker, C. R. Jr. (1999). Dehydroepiandrosterone and dehydroepiandrosterone sulfate production in the human adrenal during development and aging. Steroids 64, 640–647.
Parton, R. F., Vankelecom, I. F., Casselman, M. J., Bezoukhanova, C. P., Uytterhoeven, J. B., and Jacobs, P. A. (1994). An efficient mimic of cytochrome P-450 from a zeolite-encaged iron complex in a polymer membrane. Nature 370, 541–544.
Pereira, E. F., Hilmas, C., Santos, M. D., Alkondon, M., Maelicke, A., and Albuquerque, E. X. (2002). Unconventional ligands and modulators of nicotinic receptors. J. Neurobiol. 53, 479–500.
Plassart-Schiess, E., and Baulieu, E. E. (2001). Neurosteroids: recent findings. Brain Res. Brain Res. Rev. 37, 133–140.
Prasad, V. V., Vegesna, S. R., Welch, M., and Lieberman, S. (1994). Precursors of the neurosteroids. Proc. Natl. Acad. Sci. U.S.A. 91, 3220–3223.
Rammouz, G., Lecanu, L., Aisen, P., and Papadopoulos, V. (2011). A lead study on oxidative stress-mediate dehydroepiandrosterone formation in serum: the biochemical basis for a diagnosis of Alzheimer’s disease. J. Alzheimers Dis. 24(Suppl. 2), 5–16.
Rhodes, M. E., Li, P. K., Burke, A. M., and Johnson, D. A. (1997). Enhanced plasma DHEAS, brain acetylcholine and memory mediated by steroid sulfatase inhibition. Brain Res. 773, 28–32.
Robel, P., and Baulieu, E. E. (1985). Neuro-steroids: 3?-hydroxy-?(5)-derivatives in the rodent brain. Neurochem. Int. 7, 953–958.
Robel, P., and Baulieu, E. E. (1994). Neurosteroids biosynthesis and function. Trends Endocrinol. Metab. 5, 1–8.
Robel, P., Synguelakis, M., Halberg, F., and Baulieu, E. E. (1986). Persistence of the circadian rhythm of dehydroepiandrosterone in the brain, but not in the plasma, of castrated and adrenalectomized rats. C. R. Acad. Sci. III Sci. Vie 303, 235–238.
Roberts, E., Bologa, L., Flood, J. F., and Smith, G. E. (1987). Effects of dehydroepiandrosterone and its sulfate on brain tissue in culture and on memory in mice. Brain Res. 406, 357–362.
Rone, M. B., Fan, J., and Papadopoulos, V. (2009). Cholesterol transport in steroid biosynthesis: role of protein-protein interactions and implications in disease states. Biochim. Biophys. Acta 1791, 646–658.
Rupprecht, R., di Michele, F., Hermann, B., Strohle, A., Lancel, M., Romeo, E., and Holsboer, F. (2001). Neuroactive steroids: molecular mechanisms of action and implications for neuropsychopharmacology. Brain Res. Brain Res. Rev. 37, 59–67.
Rupprecht, R., and Holsboer, F. (2001). Neuroactive steroids in neuropsychopharmacology. Int. Rev. Neurobiol. 46, 461–477.
Rupprecht, R., Papadopoulos, V., Rammes, G., Baghai, T. C., Fan, J., Akula, N., Groyer, G., Adams, D., and Schumacher, M. (2010). Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat. Rev. Drug Discov. 9, 971–988.
Sanne, J. L., and Krueger, K. E. (1995). Aberrant splicing of rat steroid 17 alpha-hydroxylase transcripts. Gene 165, 327–328.
Sawyer, D. T., Liu, X., Redman, C., and Chong, B. (1994). Iron(II)/reductant(DH2)-induced activation of dioxygen for the hydroxylation and ketonization of hydrocarbons; mimics for the cytochrome P-450 hydroxylase/reductase system. Bioorg. Med. Chem. 2, 1385–1395.
Schiess, A. R., and Partridge, L. D. (2005). Pregnenolone sulfate acts through a G-protein-coupled sigma1-like receptor to enhance short term facilitation in adult hippocampal neurons. Eur. J. Pharmacol. 518, 22–29.
Schumacher, M., Akwa, Y., Guennoun, R., Robert, F., Labombarda, F., Desarnaud, F., Robel, P., De Nicola, A. F., and Baulieu, E. E. (2000). Steroid synthesis and metabolism in the nervous system: trophic and protective effects. J. Neurocytol. 29, 307–326.
Schumacher, M., Guennoun, R., Robert, F., Carelli, C., Gago, N., Ghoumari, A., Gonzalez Deniselle, M. C., Gonzalez, S. L., Ibanez, C., Labombarda, F., Coirini, H., Baulieu, E. E., and De Nicola, A. F. (2004). Local synthesis and dual actions of progesterone in the nervous system: neuroprotection and myelination. Growth Horm. IGF. Res. 14(Suppl. A), S18–S33.
Shannon, E. E., Porcu, P., Purdy, R. H., and Grant, K. A. (2005). Characterization of the discriminative stimulus effects of the neuroactive steroid pregnanolone in DBA/2J and C57BL/6J inbred mice. J. Pharmacol. Exp. Ther. 314, 675–685.
Smith, M. A., Harris, P. L., Sayre, L. M., and Perry, G. (1997). Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc. Natl. Acad. Sci. U.S.A. 94, 9866–9868.
Smith, M. A., Rudnicka-Nawrot, M., Richey, P. L., Praprotnik, D., Mulvihill, P., Miller, C. A., Sayre, L. M., and Perry, G. (1995). Carbonyl-related posttranslational modification of neurofilament protein in the neurofibrillary pathology of Alzheimer’s disease. J. Neurochem. 64, 2660–2666.
Smith, M. A., Taneda, S., Richey, P. L., Miyata, S., Yan, S. D., Stern, D., Sayre, L. M., Monnier, V. M., and Perry, G. (1994). Advanced Maillard reaction end products are associated with Alzheimer disease pathology. Proc. Natl. Acad. Sci. U.S.A. 91, 5710–5714.
Steffensen, S. C., Jones, M. D., Hales, K., and Allison, D. W. (2006). Dehydroepiandrosterone sulfate and estrone sulfate reduce GABA-recurrent inhibition in the hippocampus via muscarinic acetylcholine receptors. Hippocampus 16, 1080–1090.
Stocco, D. M., and Clark, B. J. (1996). Regulation of the acute production of steroids in steroidogenic cells. Endocr. Rev. 17, 221–244.
Stromstedt, M., and Waterman, M. R. (1995). Messenger RNAs encoding steroidogenic enzymes are expressed in rodent brain. Brain Res. Mol. Brain Res. 34, 75–88.
Strous, R. D., Maayan, R., and Weizman, A. (2006). The relevance of neurosteroids to clinical psychiatry: from the laboratory to the bedside. Eur. Neuropsychopharmacol. 16, 155–169.
Subbarao, K. V., Richardson, J. S., and Ang, L. C. (1990). Autopsy samples of Alzheimer’s cortex show increased peroxidation in vitro. J. Neurochem. 55, 342–345.
Sullivan, S. D., and Moenter, S. M. (2003). Neurosteroids alter gamma-aminobutyric acid postsynaptic currents in gonadotropin-releasing hormone neurons: a possible mechanism for direct steroidal control. Endocrinology 144, 4366–4375.
Suzuki, M., Wright, L. S., Marwah, P., Lardy, H. A., and Svendsen, C. N. (2004). Mitotic and neurogenic effects of dehydroepiandrosterone (DHEA) on human neural stem cell cultures derived from the fetal cortex. Proc. Natl. Acad. Sci. U.S.A. 101, 3202–3207.
Takebayashi, M., Hayashi, T., and Su, T. P. (2004). A perspective on the new mechanism of antidepressants: neuritogenesis through sigma-1 receptors. Pharmacopsychiatry 37(Suppl. 3), S208–S213.
Tan, L., and Rousseau, J. (1975). Anaerobic degradation of the progesterone side chain: a possible new pathway for the biosynthesis of androgen hormones. Biochem. Biophys. Res. Commun. 65, 1320–1326.
Tasker, J. G. (2006). Rapid glucocorticoid actions in the hypothalamus as a mechanism of homeostatic integration. Obesity (Silver Spring) 14(Suppl. 5), 259S–265S.
Torres, J. M., and Ortega, E. (2003). DHEA, PREG and their sulphate derivatives on plasma and brain after CRH and ACTH administration. Neurochem. Res. 28, 1187–1191.
Trapani, G., Laquintana, V., Denora, N., Trapani, A., Lopedota, A., Latrofa, A., Franco, M., Serra, M., Pisu, M. G., Floris, I., Sanna, E., Biggio, G., and Liso, G. (2005). Structure-activity relationships and effects on neuroactive steroid synthesis in a series of 2-phenylimidazo[1,2-a]pyridineacetamide peripheral benzodiazepine receptors ligands. J. Med. Chem. 48, 292–305.
Ueda, H., Inoue, M., Yoshida, A., Mizuno, K., Yamamoto, H., Maruo, J., Matsuno, K., and Mita, S. (2001). Metabotropic neurosteroid/sigma-receptor involved in stimulation of nociceptor endings of mice. J. Pharmacol. Exp. Ther. 298, 703–710.
Vallee, M., Mayo, W., Darnaudery, M., Corpechot, C., Young, J., Koehl, M., Le Moal, M., Baulieu, E. E., Robel, P., and Simon, H. (1997). Neurosteroids: deficient cognitive performance in aged rats depends on low pregnenolone sulfate levels in the hippocampus. Proc. Natl. Acad. Sci. U.S.A. 94, 14865–14870.
Vallee, M., Mayo, W., and Le Moal, M. (2001). Role of pregnenolone, dehydroepiandrosterone and their sulfate esters on learning and memory in cognitive aging. Brain Res. Brain Res. Rev. 37, 301–312.
Van Lier, J. E., and Smith, L. L. (1970). Sterol metabolism. XVI. Cholesterol 20-alpha-hydroperoxide as an intermediate in pregnenolone biosynthesis from cholesterol. Biochem. Biophys. Res. Commun. 40, 510–516.
Wang, J. M., Johnston, P. B., Ball, B. G., and Brinton, R. D. (2005). The neurosteroid allopregnanolone promotes proliferation of rodent and human neural progenitor cells and regulates cell-cycle gene and protein expression. J. Neurosci. 25, 4706–4718.
Wise, P. M., Dubal, D. B., Wilson, M. E., Rau, S. W., and Liu, Y. (2001). Estrogens: trophic and protective factors in the adult brain. Front. Neuroendocrinol. 22, 33–66.
Wolf, O. T., Koster, B., Kirschbaum, C., Pietrowsky, R., Kern, W., Hellhammer, D. H., Born, J., and Fehm, H. L. (1997a). A single administration of dehydroepiandrosterone does not enhance memory performance in young healthy adults, but immediately reduces cortisol levels. Biol. Psychiatry 42, 845–848.
Wolf, O. T., Neumann, O., Hellhammer, D. H., Geiben, A. C., Strasburger, C. J., Dressendorfer, R. A., Pirke, K. M., and Kirschbaum, C. (1997b). Effects of a two-week physiological dehydroepiandrosterone substitution on cognitive performance and well-being in healthy elderly women and men. J. Clin. Endocrinol. Metab. 82, 2363–2367.
Yanase, T. (1995). 17 alpha-Hydroxylase/17,20-lyase defects. J. Steroid Biochem. Mol. Biol. 53, 153–157.
Yoo, A., Harris, J., and Dubrovsky, B. (1996). Dose-response study of dehydroepiandrosterone sulfate on dentate gyrus long-term potentiation. Exp. Neurol. 137, 151–156.
Keywords: Alzheimer’s disease, dehydroepiandrosterone, diagnostic tool, neurosteroids
Citation: Rammouz G, Lecanu L and Papadopoulos V (2011) Oxidative stress-mediated brain dehydroepiandrosterone (DHEA) formation in Alzheimer’s disease diagnosis. Front. Endocrin. 2:69. doi: 10.3389/fendo.2011.00069
Received: 15 July 2011; Paper pending published: 28 September 2011;
Accepted: 19 October 2011; Published online: 08 November 2011.
Edited by:
Hubert Vaudry, University of Rouen, FranceReviewed by:
Rafael Vazquez-Martinez, University of Cordoba, SpainRoberto Cosimo Melcangi, Università degli Studi di Milano, Italy
Copyright: © 2011 Rammouz, Lecanu and Papadopoulos. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Vassilios Papadopoulos, The Research Institute of the McGill University Health Center, Montreal General Hospital, 1650 Cedar Avenue, C10-148, Montreal, QC, Canada H3G 1A4. e-mail:dmFzc2lsaW9zLnBhcGFkb3BvdWxvc0BtY2dpbGwuY2E=