 Roxana I. Albu1,2
Roxana I. Albu1,2 Stefan N. Constantinescu1,2*
Stefan N. Constantinescu1,2*- 1 Ludwig Institute for Cancer Research, Brussels, Belgium
- 2 de Duve Institute, Université catholique de Louvain, Brussels, Belgium
The thrombopoietin receptor (TpoR) is a type I transmembrane protein that mediates the signaling functions of thrombopoietin (Tpo) in regulating megakaryocyte differentiation, platelet formation, and hematopoietic stem cell renewal. We probed the role of each of the four extracellular domain putative N-glycosylation sites for cell surface localization and function of the receptor. Single N-glycosylation mutants at any of the four sites were able to acquire the mature N-glycosylated pattern, but exhibited a decreased Tpo-dependent JAK2–STAT response in stably transduced Ba/F3 or Ba/F3-JAK2 cell lines. The ability of JAK2 to promote cell surface localization and stability of TpoR required the first N-glycosylation site (Asn117). In contrast, the third N-glycosylation site (Asn298) decreased receptor maturation and stability. TpoR mutants lacking three N-glycosylation sites were defective in maturation, but N-glycosylation on the single remaining site could be detected by sensitivity to PNGaseF. The TpoR mutant defective in all four N-glycosylation sites was severely impaired in plasma membrane localization and was degraded by the proteasome. N-glycosylation receptor mutants are not misfolded as, once localized on the cell surface in overexpression conditions, they can bind and respond to Tpo. Our data indicate that extracellular domain N-glycosylation sites regulate in a combinatorial manner cell surface localization of TpoR. We discuss how mutations around TpoR N-glycosylation sites might contribute to inefficient receptor traffic and disease.
Introduction
The thrombopoietin receptor (TpoR) is a major regulator of megakaryopoiesis and of platelet formation, and is required for maintaining the quiescence of hematopoietic stem cells, regulating proliferation of early myeloid progenitors and removal of circulating thrombopoietin (Tpo) ligand from blood by circulating platelets (Kaushansky et al., 1994; Solar et al., 1998). TpoR is an N-glycosylated, single-pass transmembrane protein with the N-terminus in the extracellular space. It belongs to the homodimeric type I cytokine receptor subfamily, which includes the receptors for erythropoietin (Epo), granulocyte-colony stimulating factor (GCSF), growth hormone (GH), and prolactin (Prl; Vigon et al., 1992, 1993; Skoda et al., 1993; Drachman and Kaushansky, 1995). Proper folding and traffic to cell surface are crucial for these cytokine receptors to receive signals from their cognate cytokines. TpoR lacks intrinsic kinase activity and relies on the cytokine-dependent activation of the cytoplasmic non-receptor Janus kinase (JAKs) family proteins that are bound to their intracellular domains (Drachman et al., 1995; Ezumi et al., 1995), with JAK2 being the main kinase required for receptor effects (Drachman et al., 1999). Tpo stimulation of the cell surface localized TpoRs results in trans-phosphorylation of TpoR-bound JAKs and the subsequent activation of several downstream pathways, including the signal transducer and activator of transcription 5 (STAT5), STAT3, Ras/mitogen-activated protein kinase, and phosphatidylinositol-3′-kinase/AKT (Miyakawa et al., 1995, 1996; Onishi et al., 1996).
The type I hematopoietic cytokine receptor family, of which TpoR is a member, consists of more than 10 members that bear one or two cytokine receptor motifs (CRM), an approximately 200 aminoacid module, containing four spatially conserved cysteine residues, 14 beta-sheets and a juxtamembrane Trp-Ser-X-Trp-Ser conserved sequence required for receptor folding and intracellular traffic (Bazan, 1990; Yoshimura et al., 1992). All mammalian TpoRs contain duplications of the CRM domains, when compared to other receptors of the family such as the EpoR, or human prolactin receptor (hPrlR). The two TpoR CRMs can be divided in four sub-domains of approximately 100 aminoacids (namely D1D2 for the distal, N-terminal CRM and D3D4 for the proximal, C-terminal CRM), each showing homologies with the fibronectin type III module (Figure 1B). Structural modeling and ligand binding affinity experiments showed that D1D2 is responsible for the Tpo binding (Deane et al., 1997; Feese et al., 2004; Figure 1B) and possibly for preventing the rest of the receptor to signal in the absence of ligand (Sabath et al., 1999).
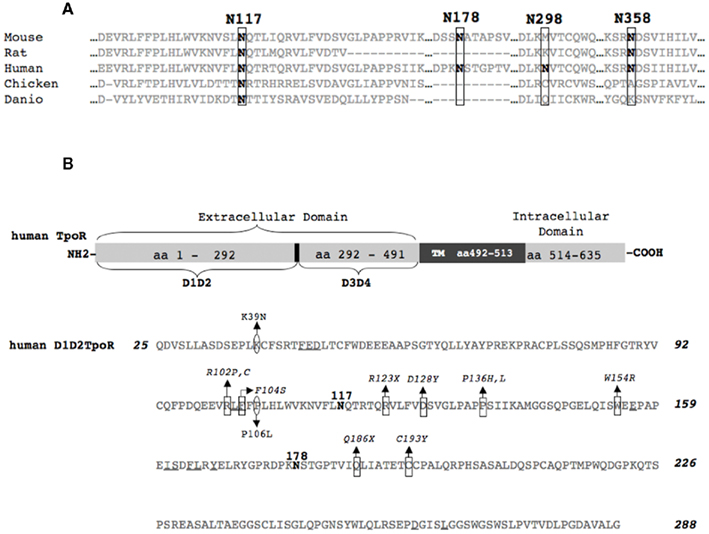
Figure 1. Schematic representation of human TpoR and multiple sequence alignment of the N-glycosylation sites of TpoR from different species. (A) Multiple sequence alignment showing conservation of the first N-glycosylation site from different species. The alignment was obtained with the ClustalW program for the following sequences: Homo sapiens (UniProtKB/Swiss-Prot accession number P40238), Mus musculus (Q08351), Rattus norvegicus (D4A2R0), Gallus gallus (Q6IYE8), Danio rerio (Q6EIY6). The positions of the four putative N-glycosylation sites of human TpoR are depicted. (B) Schematic representation of human TpoR. The bottom panel of (B) depicts the D1D2 cytokine receptor module. The important aminoacids for Tpo binding are underlined (Deane et al., 1997). In square (□) are depicted the aminoacids that are mutated in congenital amegakaryocytic thrombocytopenia (Ballmaier et al., 2001; Germeshausen et al., 2006; Tijssen et al., 2008; Fox et al., 2009) and in round circles (○) the aminoacids that are mutated in familial thrombocytosis (Moliterno et al., 2004; El-Harith El et al., 2009). TM, transmembrane region; aa, aminoacid; D1D2, sub-domains of the distal cytokine receptor module; D3D4, sub-domains of the proximal cytokine receptor module; NH2, N-terminus of the protein; COOH, C-terminus of the protein; X, any aminoacid.
Little is known about regulation of TpoR traffic to the cell surface or about the mechanisms that govern receptor internalization, degradation, and recycling. On the cytosolic side, JAK2 and TYK2 were shown to promote cell surface localization and stability of TpoR (Royer et al., 2005), but the extracellular determinants of endoplasmic reticulum (ER) to cell surface traffic remain unknown.
The role of extracellular domain N-glycosylation for cytokine receptor traffic remains debated. N-glycosylation is apparently not required for induction of gene transcription via PrlR and gp130, but is crucial for receptors’ stability and cell surface localization (Buteau et al., 1998; Bolander, 1999; Waetzig et al., 2010). On the other hand, in the case of leptin and GM-CSF receptors, N-glycans are important for ligand binding (Ding et al., 1995; Niu et al., 2000; Kamikubo et al., 2008). Eliminating individual N-glycans in human β-common subunit of the IL3, IL5, and GM-CSF receptors did not affect ligand binding and receptor activation, but the authors did not exclude the possibility that N-glycans may exert some sort of fine control (Murphy et al., 2008). TpoR presents four putative N-glycosylation sites in its extracellular domain, two in each cytokine receptor module (Figure 1). In this study we aimed to probe the role of each N-glycosylation site in the cell surface expression and function of TpoR.
Materials and Methods
Plasmids and Generation of Human TpoR N-glycosylation Defective Variants
The human TpoR wild-type (WT) was cloned into the pMX-IRES-GFP bicistronic retroviral vector upstream of the IRES as described (Liu et al., 2000). Several mutagenesis reactions with specific primers were carried out on human TpoR WT (Swiss-Prot accession number P40238) with the QuickChange Site-Directed Mutagenesis Kit (Stratagene). Human TpoR variants lacking single N-glycosylation sites were obtained by changing the codon for Asn (AAC or AAT) to the codon for Gln (CAA) in each of the four potential N-glycosylation sequons. The resulting proteins were named (Δ (1), Δ (2), Δ (3), and Δ (4), respectively, according to the particular N-glycosylation site that was removed (Table 1). Other mutants lacking more than one N-glycosylation site were obtained leaving only one N-glycosylated site: (Δ (123), (Δ (134), (Δ (124), (Δ (234) (Table 1). All constructs were verified by sequencing. All TpoR constructs contain an HA tag at their N-terminus, after the signal peptide cleavage site. The murine JAK2 WT was subcloned into the pREX-IRES-CD4 vector.
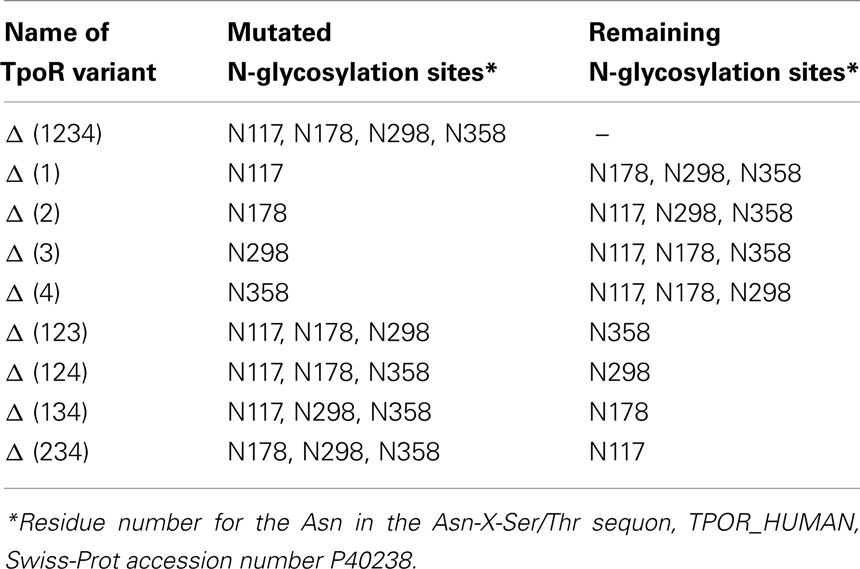
Table 1. Name and description of the engineered human TpoR variants.
Generation of Cell Lines and Retroviral Transductions
HEK293-derived BOSC cells are ecotropic retrovirus packaging cells (Pear et al., 1993). γ2A are JAK2-deficient human fibrosarcoma cells (Pellegrini et al., 1989). Ba/F3 parental and Ba/F3-JAK2 cells are IL3-dependent murine pro-B cells (Palacios and Steinmetz, 1985). Ba/F3-JAK2 is a stably transduced cell line with murine JAK2 WT (Royer et al., 2005) in order to have increased levels of JAK2 (approximately fivefold higher JAK2 levels than the endogenous JAK2 levels of Ba/F3 cells).
Stably transduced Ba/F3 or Ba/F3-JAK2 cell lines were obtained for each TpoR variant. Typically, high titer replication-defective retroviral supernatants were generated by Lipofectamine transient transfection of BOSC packaging cells using standard protocols (Seubert et al., 2003). The viruses were used to stably transduce 2 × 106 cells by centrifugation at 1,300 rpm, at 37°C for 3 h in the presence of 4 μg/ml Polybrene (Sigma). Although the efficiency of infection was around 30–50%, populations of cells expressing the same level of marker, GFP were isolated by fluorescence-activated cell sorter (FACS) 72 h after infection and cultured in IL3.
Dual Luciferase Assays
The STAT5 and STAT3 transcriptional activities of the various mutants were measured in γ2A or HEK293 cells by dual luciferase assays with the STAT reporters, pGRR5-luc or pLHRE-luc (Dumoutier et al., 2000; Gerland et al., 2000). pGRR5-luc contains five copies of the STAT-binding site of the FcγRI gene inserted upstream of a luciferase gene under the transcriptional control of the thymidine kinase (TK) promoter. pLHRE-luc harbors tandem copies of the STAT5-inducible lactogenic hormone response element (LHRE) of the rat β-casein gene promoter, inserted upstream a luciferase gene. As an internal control, we used pRLTK-luc vector that contains the Renilla luciferase gene under the control of the TK promoter. Briefly, cells were seeded in 24-well plates with 24 h before being transfected. The transfection was performed using Lipofectamine (Invitrogen), with pGRR5-luc, pRLTK-luc, and the cDNA coding for each individual TpoR mutant, if not mentioned otherwise (in some cases the cDNA of human JAK2 WT was added). Empty vector was added in order to normalize the total transfected cDNA. Four hours after transfection the cells were stimulated with recombinant human Tpo (R&D System) or left unstimulated for 20 h. Twenty-four hours after transfection, cells were lysed in 100 μl 1× passive lysis (Promega) and luminescence was recorded on a Glomax 96-well plate luminometer. When performing the assay on Ba/F3 or Ba/F3-JAK2 cells (also stably expressing each human TpoR mutant), cells were starved 3–4 h in RPMI medium with 1 mg/ml BSA, and electroporated (250 V, 75 Ω, 1,500 μF) with the pGRR5 and pRLTK luciferase reporters. The cells were subsequently cultured for 2 h, stimulated with recombinant human Tpo (rhTpo, R&D System) or IL3 or left unstimulated. After 2 h, the cells were lysed in 100 μl 1× passive lysis buffer and their luminescence was recorded. All the luciferase assays were performed using the dual luciferase reporter kit (Promega).
Tpo-dependent Proliferation Assay
Three thousand stably transduced Ba/F3 cells were seeded in 96-well plates and stimulated with different concentrations of rhTpo, or mock. Control for proliferation was performed using 10 ng IL3/ml for each cell line. After 72 h, tritiated thymidine was added to the cells for 4 h. Cells were then collected on microfiltered plates, and thymidine incorporation was measured with Top Count microplate scintillation counter (Canberra-Packard, Meriden, CT, USA).
Surface Expression of HA Tagged TpoR Mutants
Surface expression of the receptors was measured in 10 × 106 Ba/F3, Ba/F3-JAK2, or HEK293 cells by flow cytometry using 10 μg/ml monoclonal anti-HA antibody (HA.11, Covance) and 5 μg/ml R-phycoerythrin-conjugated donkey F(ab′) anti-mouse IgG secondary antibody (Jackson ImmunoResearch; Huang et al., 2001).
Western Blotting, Endoglycosidase-H (EndoH), and N-glycosidase F (PNGaseF) Digestion
Ba/F3, Ba/F3-JAK2, or HEK293 cells expressing stably or transiently each human TpoR mutant were washed in cold PBS, lysed in 200 μl Nonidet P-40 buffer with sodium orthovanadate, sodium fluoride, phenylmethanesulfonyl fluoride, and complete protease inhibitor cocktail (Roche Applied Science). Cell lysates were digested with EndoH or PNGaseF (both from New England BioLabs) or left untreated for 16 h at 37°C. The lysate supernatants/products of the digestions were then mixed with Laemmli Blue 4×, boiled for 10 min, and centrifuged for 1 min at 14,000 rpm. A volume equivalent to 30 μg protein/well was loaded on 4–12% BisTris precast gels (Invitrogen). After transfer to nitrocellulose membranes and blocking in 20% milk/TBS–Tween, immunoblotting was performed overnight at 4°C with mouse anti-HA tag (HA.11, Covance), mouse anti-β-actin (Sigma), or rabbit anti-JAK2 (Cell Signaling) antibodies in a solution of 3% milk/TBS–Tween with a 1:1,000 antibody dilution. Secondary mouse or rabbit-horseradish peroxidase antibodies (GE Healthcare, UK) were used in a 1:10,000 dilution in 3% milk/TBS–Tween.
Measurements of TpoR Mutant Half-Life and Degradation
For measuring the contribution of the proteasome- or lysosome-mediated degradation of the TpoR N-glycosylation defective mutant Δ (1234) versus WT, stably transduced Ba/F3 or Ba/F3-JAK2 cells were incubated 3 h at 37°C with 50 μg/ml cycloheximide (Sigma; to block protein synthesis) and proteasome inhibitor MG132 (10 μM; Sigma) or with 50 μg/ml cycloheximide and lysosome inhibitors (200 μM chloroquine or 10 μg/ml leupeptin; Roche). Control cells were kept untreated to have 100% protein expression. Cells were lysed and analyzed by Western blotting with antibodies anti-HA for TpoR levels.
The half-life measurements of different TpoR variants were determined by treating the Ba/F3 stably transduced cell lines with 50 μg/ml cycloheximide for different time lengths. Cells were then lysed and analyzed by Western blotting with antibodies anti-HA for the levels of immature and mature TpoR. Anti-β-actin was used as a control.
Results
Human TpoR Cell Surface Levels and Activity are Not Critically Dependent on Individual N-glycosylation
The first question of our study was whether there is a single critical N-glycosylation site of human TpoR that is responsible for the efficiency of cell surface localization. Taking into account that JAK2 is able to promote cell surface localization and to increase TpoR protein stability in the Ba/F3 cell line (Royer et al., 2005), we assessed cell surface levels of single N-glycosylation mutant TpoR variants in Ba/F3 cells with endogenous or elevated JAK2 levels.
Thrombopoietin receptor mutants lacking one out of four putative N-glycosylation sites (Figure 1; Table 1) were obtained by using site-directed mutagenesis on a human TpoR WT template, and then cloned into the bicistronic vector, pMX-IRES-GFP. The constructs were then used to produce retroviral supernatants in HEK293-derived BOSC cells. The retroviral supernatants were subsequently utilized to infect murine Ba/F3 cells expressing or not increased levels of murine JAK2 WT (cloned in bicistronic pREX-IRES-CD4 vector). To avoid clone-specific artifacts in expression or signal transduction, stably transduced Ba/F3 cell pools, with the same level of TpoR construct expression were used. More precisely, the cells were sorted for similar GFP levels 72 h later (correlated with the same mRNA level for each TpoR variant). Sorted cell lines were lysed and the protein for each TpoR variant was analyzed for the correct molecular size using anti-HA in Western blotting (data not shown).
Flow cytometry using HA.11 antibody performed in Ba/F3 cell lines exhibiting equivalent GFP expression consistently showed that, compared with the WT receptor, Δ (1) cell surface localization was decreased, while Δ (3) cell surface localization was increased (Figure 2A). In the Ba/F3-JAK2 cell line, the four variants lacking single N-glycosylation site are expressed at the same level [Δ (2), (Δ (3), Δ (4)] or lower, as is the case for Δ (1) (Figure 2A). We also transiently expressed these variants in HEK293 cells, achieving overexpression. Also in these conditions Δ (1) cell surface localization was decreased, while Δ (3) cell surface localization was increased (data not shown).
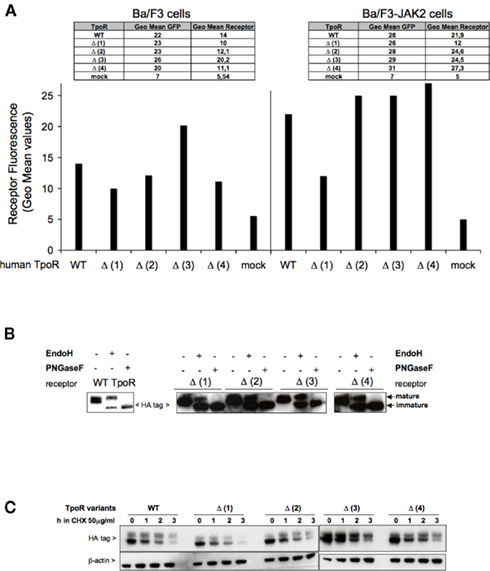
Figure 2. Effect of single N-glycosylation site removal on the cell surface levels and stability of human TpoR. (A) Sorted Ba/F3 parental or Ba/F3-JAK2 cells, expressing TpoR variants defective in one N-glycosylation site [Δ (1), Δ (2), Δ (3), Δ (4)] were tested for cell surface levels using anti-HA antibody based flow cytometry. Similar results were obtained in four independent experiments, on two different cell lines obtained for each TpoR variant. The differences between the cell surface levels of Δ (1) TpoR in Ba/F3 and Ba/F3-JAK2 cells were statistically significant when compared with the levels of WT TpoR (p = 0.006 and p = 0.0086, respectively). (B) HEK293 cells were plated in 6-well plates and transiently transfected with 3 μg of each of the indicated TpoR variants. Twenty-four hours post-transfection cells were lysed and treated with EndoH or PNGaseF or left untreated and subjected to Western blot analysis. Membranes were probed with anti-HA. Similar results were obtained in three different experiments. (C) A pool of the cell lines used in (A) were treated at 37°C with 50 μg/ml cycloheximide (CHX) over different time periods then lysed. The lysates were analyzed for TpoR variants stability using anti-HA and normalizing by anti-β-actin Western blotting.
The flow cytometry results were verified by Western blots experiments, where we enzymatically analyzed the N-glycosylation pattern. The fact that all the TpoR variants defective in single N-glycosylation site are able to localize at the cell surface is confirmed in Figure 2B. We showed in the HEK293 cell line that these variants presented a strong EndoH resistant band, which correlated with the mature form of each variant. When the first N-glycosylation site is removed, the level of the mature receptor band seems to be weaker compared with the mature band of the WT, correlating very well with the decreased cell surface localization for this mutant.
To test whether the cell surface receptors defective in single N-glycosylation site are as stable as the WT TpoR, we used Ba/F3 cells stably transduced with each of the mutants. The cells were treated with CHX (cycloheximide) over different time periods, in order to block protein synthesis and determine the half-life of each TpoR variant by using anti-HA and anti-β-actin Western blotting. Compared with the WT, Δ (3) TpoR showed higher and Δ (1) TpoR lower stability (Figure 2C), which correlated well with the cell surface level differences.
We were interested to determine the relevance of each of the N-glycans for the TpoR function. For each of the human TpoR variants lacking one N-glycosylation site, we assessed the STAT3 and STAT5 transcriptional activities using luciferase assays in stably transduced Ba/F3 or Ba/F3-JAK2 cell models. These TpoR variants presented a decreased transcriptional activity in the Tpo-stimulated condition, compared with the WT TpoR (Figure 3A). Between the TpoR variants the levels of Tpo-dependent transcriptional activity correlated well with the cell surface levels of the TpoR variants, with Δ (1) having the lowest and Δ (3) the highest Tpo-dependent transcriptional activity.
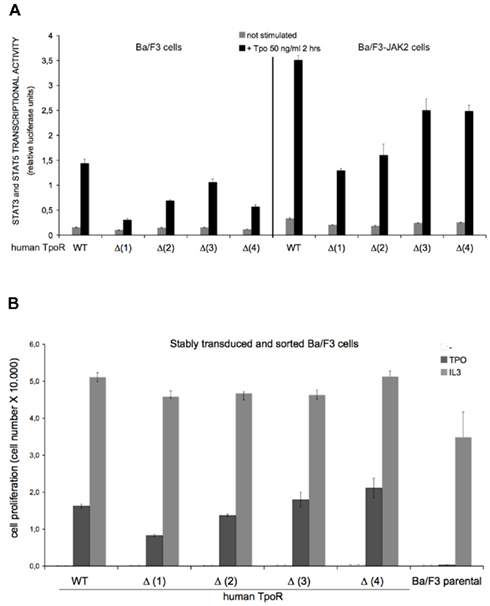
Figure 3. Effect of single N-glycosylation site removal on the Tpo-dependent transcriptional activation of human TpoR. (A) Ba/F3 or Ba/F3-JAK2 cells expressing stably expressing each human TpoR defective in single N-glycosylation site [Δ (1), Δ (2), Δ (3), Δ (4)] were starved 3 h in RPMI medium +1 mg/ml BSA and electroporated with the luciferase reporters pGRR5-luc and pRLTK-luc. The cells were stimulated with 50 ng Tpo/ml or left unstimulated for 2 h. Upon treatment the cells were lysed and their luminescence recorded. Results are the mean ± variation of triplicate samples. One of three independent experiments is depicted. The t-test showed statistically significant differences for the Tpo-dependent transcriptional levels of the TpoR mutants when compared with the WT TpoR (p < 0.05 for all the TpoR variants). Of importance, when Tpo-dependent Δ (1) TpoR transcriptional activity in the stimulated condition was compared with the transcriptional activity of the stimulated WT TpoR, the p value equaled 0.0086. (B) Ba/F3 stably transduced cells for each of the TpoR variants [Δ (1), Δ (2), Δ (3), and Δ (4)] were washed three times and seeded in 96-well plates at a density of 3,000 cells/well. The cells were stimulated with 5 ng Tpo/ml, or with 5 ng IL3/ml or left unstimulated during 72 h. After 72 h, the proliferation of the cells was determined by the DNA synthesis assay reflected by the measurements of the thymidine incorporation as described in Experimental procedures. Results are the mean ± variation of triplicate samples. One of three independent experiments is depicted. The t-test showed significant differences between the Tpo-dependent proliferations level of Δ (1) TpoR (p = 0.0001) and Δ (2) TpoR (p = 0.0019) when compared with WT TpoR.
Our preliminary observation was that Tpo binding to TpoR is not critically dependent on individual N-glycosylation sites. To test this observation in a second set of experiments, we measured Tpo-dependent cell growth of Ba/F3 stably transduced with each of the TpoR variant. Cells were washed to remove IL3, incubated 72 h in Tpo or IL3 or left untreated. As a negative control, we used Ba/F3 cells without any TpoR variant. As a positive control for cell growth, we used IL3 treatment of each cell line. After 72 h, tritiated thymidine was added to the cells for 4 h. The proliferation of the cells was quantified according to the level of the incorporated thymidine (Figure 3B). The order of the cell proliferation level was Δ4 > Δ3 = WT > (Δ2 > Δ1 and the result is consistent with the result obtained in the luciferase test of Figure 3A.
Removal of Three N-glycosylation Sites Decreased Cell Surface Levels and Activity of Human TpoR
The Ba/F3 and Ba/F3-JAK2 cells were stably transduced with the variants missing different combinations of N-glycosylation sites (Table 1). The cells were sorted for the same protein expression level for each TpoR variant and tested for cell surface expression of the receptors. All of the variants, with only one N-glycosylation site left [Δ (123), Δ (124), (Δ (134), Δ (234)] were located at a lower level at the cell surface, compared with the WT TpoR (Figure 4A). JAK2 overexpression did not increase the cell surface level of these variants. We also tested the cell surface levels of these variants in transiently transfected HEK293 cells and found the levels are higher than in Ba/F3 but still lower than the WT or the single N-glycosylation site defective variants.
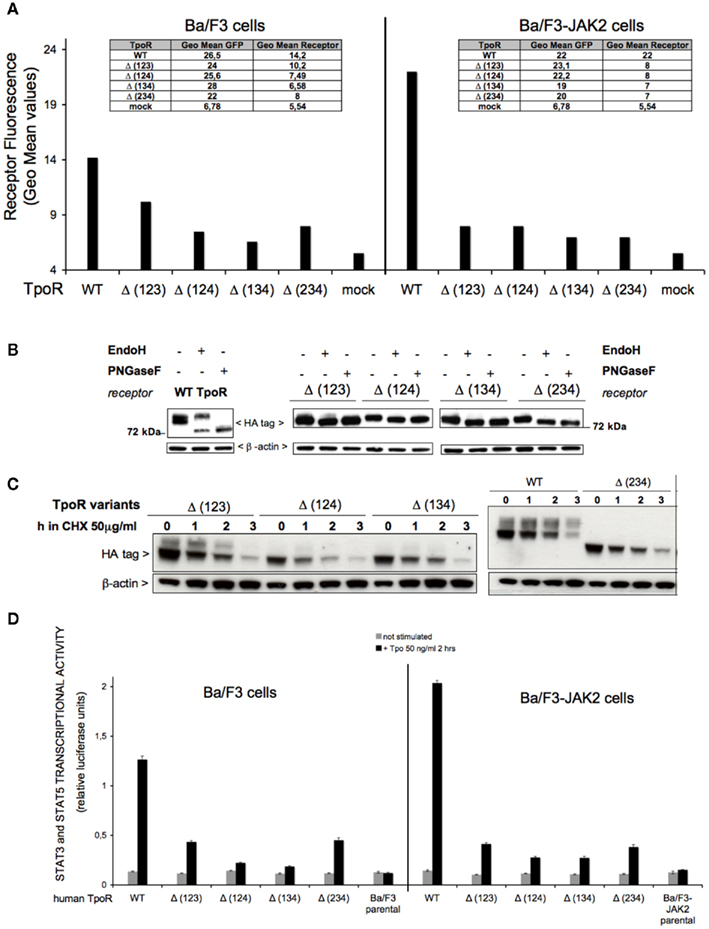
Figure 4. Effect of multiple N-glycosylation sites removal on the cell surface levels and function of human TpoR. (A) Sorted Ba/F3 parental or Ba/F3-JAK2 cells, expressing TpoR variants defective in three N-glycosylation sites [Δ (123), Δ (124), Δ (134), Δ (234)] were tested for cell surface levels using anti-HA antibody based flow cytometry. Similar results were obtained in two independent experiments, on two different cell lines obtained for each TpoR variant. For the Ba/F3 cells, the t-test showed statistically significant differences between Δ (123) TpoR (p = 0.0019), Δ (124) TpoR (p = 0.0036), Δ (134) TpoR (p = 0.0016), and Δ (234) TpoR (p = 0.0086) when compared with WT TpoR. (B) HEK293 cells were seeded in 6-well plates and transiently transfected with 3 μg of each of the indicated TpoR variants. Twenty-four hours post-transfection cells were lysed and treated with EndoH or PNGaseF or left untreated during 16 h at 37°C. The digestion products were subjected to Western blot analysis. Membranes were probed with anti-HA and anti-β-actin. Similar results were obtained in two different experiments. (C) A pool of each cell line used in (A) was treated at 37°C with 50 μg/ml cycloheximide (CHX) over different time periods then lysed. The lysates were analyzed for TpoR variants stability using anti-HA and normalized by anti-β-actin Western blotting. (D) Ba/F3 or Ba/F3-JAK2 cells stably expressing each human TpoR defective in three N-glycosylation sites were starved 3 h in RPMI medium +1 mg/ml BSA and electroporated with the pGRR5-luc and pRLTK-luc reporters. The cells were stimulated with 50 ng Tpo/ml or left unstimulated 2 h at 37°C. Upon treatment, the cells were lysed and their luminescence recorded. Results are the mean ± variation of triplicate samples. One of three independent experiments is depicted. The t-test showed statistically significant differences for Tpo-dependent transcriptional activity levels of the all the TpoR variants, when compared with the WT TpoR (all the p values < 0.0001).
We next sought to determine the carbohydrate occupancy of the four putative N-glycosylation sites. We performed EndoH (to detect shifts in the case of high mannose glycosylated immature receptors that are localized in ER or early Golgi) or PNGaseF (to remove the N-glycans from the immature and mature proteins) treatments on the transiently transfected HEK293 cell lysates. We analyzed the enzymatic products in electrophoresis under reducing conditions, and Western blotting for HA immunodetection. Because all these TpoR variants have only one putative N-glycosylation site left and all of them showed a faster migration in the presence of EndoH or PNGaseF (Figure 4B) compared with the untreated condition, we concluded that all of the possible sites could be N-glycosylated. Thus sites 1, 2, 3, and 4 at Asn residues 117, 178, 298, and 358, respectively, are fully occupied.
The results from the enzymatic assessment of these TpoR variants on transiently transfected HEK293 cell lysates showed that there is very low or absent EndoH resistant band (mature receptor, cell surface localized receptor) when compared with the WT TpoR (Figure 4B), supporting the observation of the flow cytometry results of Figure 4A.
To test the stability of TpoRs defective in multiple N-glycosylation sites, we used stably transduced Ba/F3 cells with each of the mutants and treated the cells with CHX over different time periods. A very unstable pattern was observed for the three N-glycosylation mutated variants (Figure 4C), indicating that N-glycosylation exerts a protective action on TpoR stability.
We next tested in a more sensitive assay, the ability of these TpoR variants to localize at the cell surface and to bind Tpo. Compared with the WT, a dramatic decrease of JAK2–STAT transcriptional activity for these mutants was observed in stably transduced Ba/F3 or Ba/F3-JAK2 cell models (Figure 4D). In the case of Δ (123) and Δ (234) the Tpo-dependent JAK2–STAT transcriptional activity was decreased compared with the WT but higher than in the case of Δ (124) and Δ (134) variants. We further tested the same mutants in transiently transfected HEK293 cells (see below Figure 6), but this difference between the mutants defective in three glycosylation sites was not statistically significant, suggesting that it might either reflect cell type specific differences, or be relevant at the lower stable expression levels achieved in the Ba/F3 system after sorting. These results pointed to the first (Asn117) or the last N-glycosylation site (Asn358) being important, for cell surface localization (Figure 4A) or stability (Figure 4C) in Ba/F3 cells.
Major Impairment of TpoR Cell Surface Localization and Activity by Removal of All N-glycosylation Sites
Although the removal of three N-glycosylation sites dramatically affected the cell surface localization and Tpo-dependent cell signaling in Ba/F3 cells, we also wanted to test the TpoR variant lacking all of the four putative N-glycosylation sites, Δ (1234). This variant was completely absent from the cell surface by flow cytometry both in Ba/F3 or Ba/F3 overexpressing JAK2 (Figure 5A). Our preliminary observation is that, for a cell surface level detectable by flow cytometry, at least one N-glycosylation site needs to be glycosylated (Figures 4A,D).
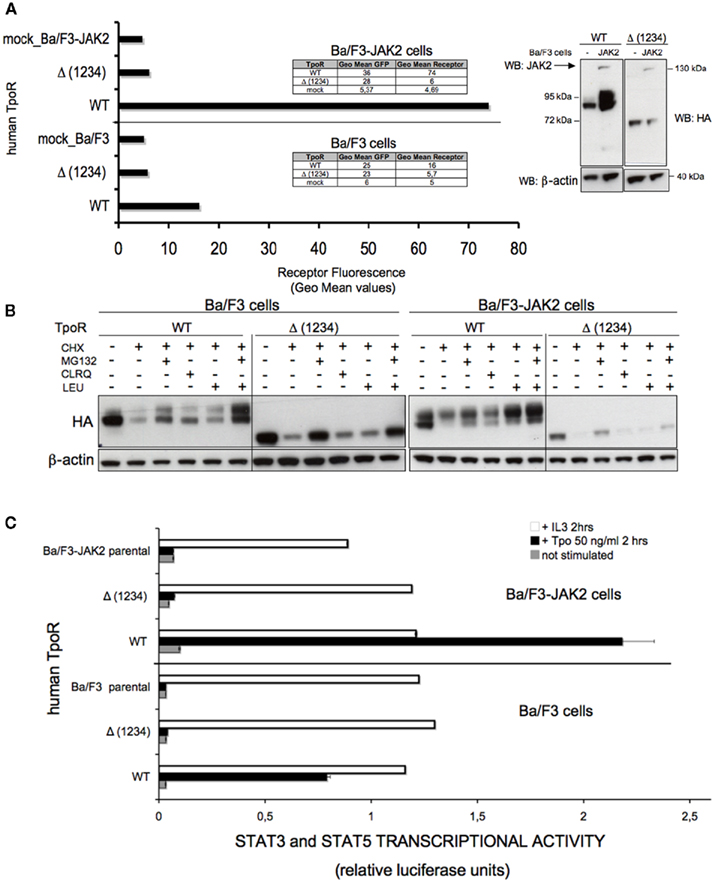
Figure 5. Removal of all four N-glycosylation sites dramatically affects the cell surface localization of human TpoR in Ba/F3 cells. (A) Ba/F3 or Ba/F3-JAK2 cells stably expressing the WT TpoR or the variant of the receptor defective in all four N-glycosylation sites [Δ (1234)] were tested for cell surface levels using anti-HA antibody based flow cytometry. Similar results were obtained in five independent experiments, on two different cell lines obtained for the Δ (1234) variant (p < 0.0001). Right panel of the (A) shows a Western blot of the cells used in the experiment, probing the receptor with anti-HA antibodies, the murine JAK2 WT with anti-JAK2 antibodies and using anti-β-actin as a control. (B) Ba/F3 or Ba/F3-JAK2 cells used in (A) were tested for the contribution of the proteasome- or lysosome-mediated degradation of the TpoR N-glycosylation defective mutant Δ (1234) versus WT. The cells were incubated 3 h at 37°C with 50 μg/ml cycloheximide and proteasome inhibitor MG132 (10 μM) or with 50 μg/ml cycloheximide and lysosome inhibitors (200 μM chloroquine or 10 μg/ml leupeptin). Control cells were kept untreated to have 100% protein expression. Cells were lysed and analyzed by Western blotting with antibodies against HA for the levels of TpoR and anti-β-actin as a loading control. (C) Pools from the same cell lines used in (A,B) were starved 3 h in RPMI medium +1 mg/ml BSA and electroporated with pGRR5-luc and pRLTK-luc reporters. The cells were also stimulated with Tpo or left unstimulated during 2 h at 37°C. Upon treatment the cells were lysed and their luminescence recorded. Results are the mean ± variation of triplicate samples. One of three independent experiments is depicted. The t-test showed statistically significant differences between the Tpo-dependent transcriptional activities level of the Δ (1234) TpoR and that of WT TpoR (p < 0.0001 for both Ba/F3 and Ba/F3-JAK2 cell lines).
Due to the fact the Δ (1234) TpoR was not detected at the cell surface, we tested its degradation features compared with the WT. We performed Western blot on the lysates of Ba/F3 and Ba/F3-JAK2 cell lines stably transfected with this mutant or the WT TpoR. Before lysis, these cells were incubated 3 h at 37°C with 50 μg/ml cycloheximide (to block protein synthesis) and proteasome inhibitor MG132 (10 μM) or with 50 μg/ml cycloheximide and lysosome inhibitors (200 μM chloroquine or 10 μg/ml leupeptin). We observed a stabilization effect due to the blocking of the proteasomal degradation (Figure 5B). Thus, the TpoR variant lacking all four putative N-glycosylation sites [Δ (1234)] is a very unstable protein that can be stabilized by blocking proteasome-mediated degradation. It is very likely that this mutant is an ER-associated degradation (ERAD) substrate, but future analysis will be needed to demonstrate this hypothesis.
The removal of all the N-glycosylation sites [Δ (1234) TpoR] dramatically affected the STAT transcriptional activity in stably transfected Ba/F3 and Ba/F3-JAK2 cell lines, compared with WT TpoR (Figure 5C). This result was expected, because the cell surface level of this mutant was undetectable by flow cytometry in Ba/F3 cells. We also tested the Tpo-dependent cell growth of these stable cell lines in a more sensitive assay (thymidine incorporation), confirming the data obtained in the luciferase assay. We could not detect any Tpo-dependent growth even in the presence of high Tpo levels (10 ng Tpo/ml; Figure A1 in Appendix).
The Defective N-glycosylation Variants are Able to Activate Tpo-Dependent STAT Activity in Overexpression Conditions in HEK293 and γ2A Cells
In order to assess whether the TpoR variants that exhibit defective localization and stability [Δ (123), Δ (124), (Δ (134), Δ (234)] or [Δ (1234)] are able to bind Tpo and activate Tpo-dependent signaling pathways, we tested all the TpoR variants in an overexpression cell system.
In transiently transfected γ2A or HEK293 cells, in the case of single N-glycosylation site [Δ (1), (Δ (2), (Δ (3), Δ (4)] defective variants, there was no difference in the Tpo-dependent JAK2–STAT transcriptional activity when compared with the WT receptor (Figure 6). In the case of mutation of multiple N-glycosylation sites [Δ (124), Δ (134), Δ (234)], their Tpo-dependent JAK2–STAT transcriptional activity in transiently transfected was lower than that of WT TpoR (Figure 6). Mutant Δ (124) was similar to Δ (134) with respect to transcriptional activity in γ2A cells (data not shown). For the mutant Δ (1234) TpoR, removal of all N-glycosylation sites led to a significantly decreased, but still detectable, level of transcriptional activity in response to Tpo (Figure 6). These data suggest that overexpression conditions allow for some cell surface localization, which is sufficient to support a response to Tpo ligand in the very sensitive luciferase assays. Importantly, these data indicate that the receptor mutants are not misfolded and, once localized on the surface, they can bind Tpo.
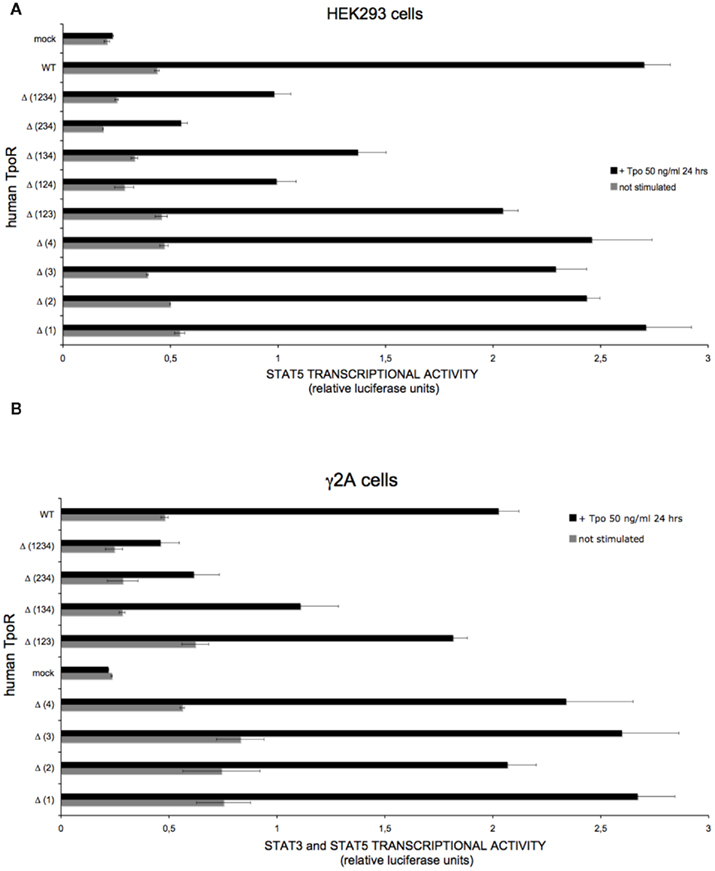
Figure 6. Human TpoR mutants defective in N-glycosylation sites are able, in overexpression conditions, to support Tpo-dependent STAT3 and STAT5 transcriptional activities. (A) HEK293 cells were plated in 24-well plates the day before transfection. The cells were co-transfected with plasmids encoding for each of the TpoR variants, the pLHRE-luc and pRLTK-luc reporters, and JAK2 WT. Cells were lysed 24 h after treatment in 100 μl 1× passive lysis buffer and luminescence values were recorded. The experiment was independently performed three times and the presented result represents ± variation of triplicate samples. The t-test showed that the unstimulated condition is significantly different from the stimulated condition for the Δ (1234) TpoR (p = 0.0058). (B), JAK2-deficient γ2A cells were plated in 24-well plates a day before transfection. The cells were co-transfected with plasmids encoding for each of the TpoR variants, the pGRR5-luc and pRLTK-luc reporters, and JAK2 WT. Four hours post-transfection cells were stimulated or left unstimulated. Cells were lysed 24 h after treatment in 100 μl 1× passive lysis buffer and luminescence values were recorded. The experiment was independently performed two times and the result presented represents ± variation of triplicate samples.
Discussion
Our major finding is that N-glycosylation sites on the extracellular TpoR domain exert essential regulatory roles on TpoR cell surface localization and function. Using a combination of mutagenesis and biochemistry approaches in stably transduced hematopoietic cells and in several other cell lines, we show that the four sites exert positive and negative roles on receptor traffic, that at least one glycosylation site is essential for cell surface transport, and that exit from ER and stability are regulated by N-glycosylation. When three of the four N-glycosylation sites were mutated, we could demonstrate that the remaining N-glycosylation site was occupied.
Previous data presented a differential role of the N-glycans for different cytokine receptor traffic, ligand binding, and function (Ding et al., 1995; Buteau et al., 1998; Bolander, 1999; Niu et al., 2000; Kamikubo et al., 2008; Waetzig et al., 2010). There are no available studies on the importance of the carbohydrate groups on the cell surface expression and function of human TpoR. In order to perform our study, we constructed nine human TpoR variants, defective in one, three, or four out of four putative N-glycosylation sites located of the extracellular domain. Taking into account that TpoR is very important for the cells of hematopoietic lineage, most of the results of this study were obtained in Ba/F3 and Ba/F3-JAK2 cell lines.
The first question we asked was whether the physiologic human TpoR cell surface localization requires the occupancy of a specific N-glycosylation site from the four putative ones (Figure 1A). None of the four single sites was critical for cell surface localization, for Tpo binding or for Tpo-dependent activation of the JAK2–STAT signal pathway. Nevertheless, the TpoR mutant lacking the first N-glycosylation site (Asn117) is the most interesting and it constantly presents striking differences compared with the WT TpoR. The cell surface level of Δ (1) TpoR was decreased in Ba/F3 and Ba/F3-JAK2 cells (Figure 2A). Compared with the WT, the JAK–STAT transcriptional activity of this variant was also decreased in Ba/F3 and Ba/F3-JAK2 (Figure 3A), and we consequently observed a decrease in the Tpo-dependent cell proliferation of Ba/F3 stably transduced with this receptor, compared with Ba/F3 stably transduced with WT TpoR (Figure 3B). Alignments of TpoR from different species (Figure 1A) pointed out that this first N-glycosylation site of human TpoR is highly conserved between species. We hypothesize that occupancy of this site is important but not crucial for TpoR cell surface expression. Rather, this carbohydrate group might exert a fine control on the cell surface level of TpoR. Consistent with our results, previous observations found that cell surface levels of TpoR are important for setting the correct blood platelet production. Insufficient expression of TpoR at the surface of late megakaryocytes and of platelets can lead to the unexpected phenotype of thrombocytosis, since Tpo ligand was not properly removed from circulation and it could still stimulate early megakaryocyte proliferation and differentiation (Lannutti et al., 2009; Tiedt et al., 2009). Moreover, TpoR defective N-glycosylation and impaired cell surface localization was detected in the megakaryocytes and platelets of Polycythemia Vera patients (Moliterno et al., 1998; Moliterno and Spivak, 1999).
In contrast to the results with Δ (1) TpoR, the Δ (3) TpoR mutant was localized at the cell surface to higher extents than the WT TpoR and showed enhanced maturation and activity. This glycosylation site is not conserved in rodents. Interestingly, its presence limits receptor function in human TpoR. It is interesting to note that normal platelet levels are higher in mice than in humans. One possibility is that absence of this site allows higher levels of platelet production in mice. Further studies using a knock-in approach of a murine TpoR containing this N-glycosylation site could answer this question.
Several mutations in the first five exons coding for the first cytokine receptor homology domain have been described to affect the cell surface display of TpoR. Mutations located around residue Asn117, possibly affecting the N-glycosylation pattern, are at the basis of two types of hematological diseases, with opposite pathologies: congenital amegakaryocytic thrombocytopenia (CAMT; Ballmaier et al., 2001; Germeshausen et al., 2006; Tijssen et al., 2008; Fox et al., 2009) and familial thrombocytosis (FT; Moliterno et al., 2004; El-Harith El et al., 2009; Teofili and Larocca, 2011; Figure 1B). Extracellular domain mutations in CAMT abolish cell surface receptor localization, while FT extracellular domain mutations severely impair, though do not abolish, cell surface localization. For the latter, since TpoR on platelets performs clearance of Tpo from the circulation, low platelet TpoR levels are associated with increased levels of Tpo in the blood and in the bone marrow, a situation similar to the one reported when TpoR expression levels are decreased at the late megakaryocyte and platelet levels (Lannutti et al., 2009; Tiedt et al., 2009). Our results show that Asn117 occupancy is able to control the cell surface expression of human TpoR and consequently its availability for Tpo. We hypothesize that this first N-glycan is important for efficient cell surface localization and that mutations around Asn117 might either impact the efficiency of glycosylation of this site, or its interaction with ER chaperone proteins. More studies are required in order to determine which scenario is true for each mutation detected in humans.
A major finding of our study is that removal of three out of four N-glycosylation sites did not abolish, but significantly decreased, the cell surface localization, and activity of TpoR (Figure 4). In Ba/F3 cell lines, at least one N-glycosylation site of human TpoR needs to be occupied in order to have detectable cell surface levels and Tpo-dependent gene transcription. Neither JAK2 nor TYK2 were able to restore to normal the cell surface level of these variants (Figure 4). JAK2 levels differ between the cell types of the hematopoietic lineage (Dalal et al., 1998) and JAK2 was shown to promote WT TpoR traffic to the cell surface (Royer et al., 2005). Interestingly, in this study we show that the ability of JAK2 to promote cell surface localization of TpoR requires a normal N-glycosylation, with more than one site occupied. It is possible that JAK2 is not able to rescue the cell surface localization of TpoRs that do not acquire a specific N-glycosylation pattern. In any case, our data suggest that JAK2 binding to the cytosolic juxtamembrane region induces an inside-out conformational change that must be sensed by the extracellular domain Asn117. More studies are required in order to determine whether N-glycosylation places TpoR in a particular region of the ER, or facilitates interaction with export proteins. Such studies are currently underway.
The analysis of the cell surface localization of TpoRs with different N-glycosylation patterns is very important, as one of the essential functions of the N-glycans is to secure efficient protein production. The N-glycosylation sites are crucial for the interaction of the receptor with the lectin-like chaperones from ER, in the process of proper folding. The calnexin–calreticulin quality control cycle was previously described, in which these two chaperones are acting in the initial steps of glycoprotein folding. Prolonged interaction with calnexin, due to inefficient folding, results in the substrate being directed to the proteasome for degradation. We hypothesize that the TpoR mutants reported in different hematological diseases and that are defective in cell surface localization, could be retained in early folding compartments due to this prolonged interactions with the lectin-like chaperones. In this model, N-glycans are a limiting event for proper cell surface localization. The nature and expression levels of chaperones differ between cell types or stages of differentiation, thus possibly explaining why a particular mutant can be localized at the surface of some progenitors and not others. Future experiments will have to address the importance of N-glycans for human TpoR interactions with the ER chaperones. Besides its key role in Tpo-dependent signaling, JAK2 acts as a cytosolic chaperone that increases the receptor traffic to cell surface (Royer et al., 2005), but it is possible that the main determinants that govern the ER to plasma membrane traffic of human TpoR to be located in ER and to be influenced by receptor N-glycosylation pattern.
We also observed in our study that the removal of all four putative N-glycosylation sites dramatically affects cell surface localization of TpoR and Tpo-dependent cell proliferation in the Ba/F3 cell model (Figure 5 and data not shown). This observation supports the notion that N-glycosylation is crucial for the ER to cell surface traffic of TpoR. In this model, the presence of N-glycans promotes traffic of the receptor to the cell surface. JAK2 was not able to promote cell surface localization of the mutant defective in all N-glycosylation sites. Along with the results obtained with single and multiple defective glycosylation sites, this result again points to an inside-out conformational change induced by JAK2 binding to the cytosolic domain that is sensed by the extracellular domain via N-glycosylation sites.
Finally, our results showed that all the receptors defective in different combinations of N-glycosylation sites were able to support Tpo-dependent transcriptional activity in an overexpressed system (Figure 6), thus indicating that defective N-glycosylation does not simply induce misfolding and that N-glycosylation defective TpoR mutants are able to bind Tpo, once they reach the cell surface. This is quite important and it can serve as a starting point for targeting ER-associated folding machinery in diseases with TpoR mutants blocked in this compartment, as receptors could be functional once they escape from the ER and localize at the cell surface.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank to Dr Alexandra Dusa for critical reading of this manuscript. We thank to André Tonon for support with flow cytometry. Roxana I. Albu currently holds a F.N.R.S Télévie PhD fellowship and was also funded by the Marie Curie ReceptEUR Network PhD fellowship. Stefan N. Constantinescu is a Senior Research Associate of the F.N.R.S., Belgium. We acknowledge support from the Fondation contre le Cancer, the Salus Sanguinis Foundation, the Action de Recherche Concertée MEXP31C1 of the Université catholique de Louvain, the PAI Program BCHM61B5, Belgium and the Atlantic Philanthropies, New York.
References
Ballmaier, M., Germeshausen, M., Schulze, H., Cherkaoui, K., Lang, S., Gaudig, A., Krukemeier, S., Eilers, M., Strauss, G., and Welte, K. (2001). c-mpl Mutations are the cause of congenital amegakaryocytic thrombocytopenia. Blood 97, 139–146.
Bazan, J. F. (1990). Structural design and molecular evolution of a cytokine receptor superfamily. Proc. Natl. Acad. Sci. U.S.A. 87, 6934–6938.
Bolander, F. F. Jr. (1999). Regulation of prolactin receptor glycosylation and its role in receptor location. Mol. Cell. Endocrinol. 149, 85–92.
Buteau, H., Pezet, A., Ferrag, F., Perrot-Applanat, M., Kelly, P. A., and Edery, M. (1998). N-glycosylation of the prolactin receptor is not required for activation of gene transcription but is crucial for its cell surface targeting. Mol. Endocrinol. 12, 544–555.
Dalal, I., Arpaia, E., Dadi, H., Kulkarni, S., Squire, J., and Roifman, C. M. (1998). Cloning and characterization of the human homolog of mouse Jak2. Blood 91, 844–851.
Deane, C. M., Kroemer, R. T., and Richards, W. G. (1997). A structural model of the human thrombopoietin receptor complex. J. Mol. Graph. Model. 15, 170–178, 185–178.
Ding, D. X., Vera, J. C., Heaney, M. L., and Golde, D. W. (1995). N-glycosylation of the human granulocyte-macrophage colony-stimulating factor receptor alpha subunit is essential for ligand binding and signal transduction. J. Biol. Chem. 270, 24580–24584.
Drachman, J. G., Griffin, J. D., and Kaushansky, K. (1995). The c-Mpl ligand (thrombopoietin) stimulates tyrosine phosphorylation of Jak2, Shc, and c-Mpl. J. Biol. Chem. 270, 4979–4982.
Drachman, J. G., and Kaushansky, K. (1995). Structure and function of the cytokine receptor superfamily. Curr. Opin. Hematol. 2, 22–28.
Drachman, J. G., Millett, K. M., and Kaushansky, K. (1999). Thrombopoietin signal transduction requires functional JAK2, not TYK2. J. Biol. Chem. 274, 13480–13484.
Dumoutier, L., Van Roost, E., Colau, D., and Renauld, J. C. (2000). Human interleukin-10-related T cell-derived inducible factor: molecular cloning and functional characterization as an hepatocyte-stimulating factor. Proc. Natl. Acad. Sci. U.S.A. 97, 10144–10149.
El-Harith El, H. A., Roesl, C., Ballmaier, M., Germeshausen, M., Frye-Boukhriss, H., Von Neuhoff, N., Becker, C., Nurnberg, G., Nurnberg, P., Ahmed, M. A., Hubener, J., Schmidtke, J., Welte, K., and Stuhrmann, M. (2009). Familial thrombocytosis caused by the novel germ-line mutation p.Pro106Leu in the MPL gene. Br. J. Haematol. 144, 185–194.
Ezumi, Y., Takayama, H., and Okuma, M. (1995). Thrombopoietin, c-Mpl ligand, induces tyrosine phosphorylation of Tyk2, JAK2, and STAT3, and enhances agonists-induced aggregation in platelets in vitro. FEBS Lett. 374, 48–52.
Feese, M. D., Tamada, T., Kato, Y., Maeda, Y., Hirose, M., Matsukura, Y., Shigematsu, H., Muto, T., Matsumoto, A., Watarai, H., Ogami, K., Tahara, T., Kato, T., Miyazaki, H., and Kuroki, R. (2004). Structure of the receptor-binding domain of human thrombopoietin determined by complexation with a neutralizing antibody fragment. Proc. Natl. Acad. Sci. U.S.A. 101, 1816–1821.
Fox, N. E., Chen, R., Hitchcock, I., Keates-Baleeiro, J., Frangoul, H., and Geddis, A. E. (2009). Compound heterozygous c-Mpl mutations in a child with congenital amegakaryocytic thrombocytopenia: functional characterization and a review of the literature. Exp. Hematol. 37, 495–503.
Gerland, K., Bataille-Simoneau, N., Basle, M., Fourcin, M., Gascan, H., and Mercier, L. (2000). Activation of the Jak/Stat signal transduction pathway in GH-treated rat osteoblast-like cells in culture. Mol. Cell. Endocrinol. 168, 1–9.
Germeshausen, M., Ballmaier, M., and Welte, K. (2006). MPL mutations in 23 patients suffering from congenital amegakaryocytic thrombocytopenia: the type of mutation predicts the course of the disease. Hum. Mutat. 27, 296.
Huang, L. J., Constantinescu, S. N., and Lodish, H. F. (2001). The N-terminal domain of Janus kinase 2 is required for Golgi processing and cell surface expression of erythropoietin receptor. Mol. Cell 8, 1327–1338.
Kamikubo, Y., Dellas, C., Loskutoff, D. J., Quigley, J. P., and Ruggeri, Z. M. (2008). Contribution of leptin receptor N-linked glycans to leptin binding. Biochem. J. 410, 595–604.
Kaushansky, K., Lok, S., Holly, R. D., Broudy, V. C., Lin, N., Bailey, M. C., Forstrom, J. W., Buddle, M. M., Oort, P. J., Hagen, F. S., Roth, G. J., Papayannopoulou, T., and Foster, D. C. (1994). Promotion of megakaryocyte progenitor expansion and differentiation by the c-Mpl ligand thrombopoietin. Nature 369, 568–571.
Lannutti, B. J., Epp, A., Roy, J., Chen, J., and Josephson, N. C. (2009). Incomplete restoration of Mpl expression in the mpl-/- mouse produces partial correction of the stem cell-repopulating defect and paradoxical thrombocytosis. Blood 113, 1778–1785.
Liu, X., Constantinescu, S. N., Sun, Y., Bogan, J. S., Hirsch, D., Weinberg, R. A., and Lodish, H. F. (2000). Generation of mammalian cells stably expressing multiple genes at predetermined levels. Anal. Biochem. 280, 20–28.
Miyakawa, Y., Oda, A., Druker, B. J., Kato, T., Miyazaki, H., Handa, M., and Ikeda, Y. (1995). Recombinant thrombopoietin induces rapid protein tyrosine phosphorylation of Janus kinase 2 and Shc in human blood platelets. Blood 86, 23–27.
Miyakawa, Y., Oda, A., Druker, B. J., Miyazaki, H., Handa, M., Ohashi, H., and Ikeda, Y. (1996). Thrombopoietin induces tyrosine phosphorylation of Stat3 and Stat5 in human blood platelets. Blood 87, 439–446.
Moliterno, A. R., Hankins, W. D., and Spivak, J. L. (1998). Impaired expression of the thrombopoietin receptor by platelets from patients with polycythemia vera. N. Engl. J. Med. 338, 572–580.
Moliterno, A. R., and Spivak, J. L. (1999). Posttranslational processing of the thrombopoietin receptor is impaired in polycythemia vera. Blood 94, 2555–2561.
Moliterno, A. R., Williams, D. M., Gutierrez-Alamillo, L. I., Salvatori, R., Ingersoll, R. G., and Spivak, J. L. (2004). Mpl Baltimore: a thrombopoietin receptor polymorphism associated with thrombocytosis. Proc. Natl. Acad. Sci. U.S.A. 101, 11444–11447.
Murphy, J. M., Soboleva, T. A., Mirza, S., Ford, S. C., Olsen, J. E., Chen, J., and Young, I. G. (2008). Clarification of the role of N-glycans on the common beta-subunit of the human IL-3, IL-5 and GM-CSF receptors and the murine IL-3 beta-receptor in ligand-binding and receptor activation. Cytokine 42, 234–242.
Niu, L., Heaney, M. L., Vera, J. C., and Golde, D. W. (2000). High-affinity binding to the GM-CSF receptor requires intact N-glycosylation sites in the extracellular domain of the beta subunit. Blood 95, 3357–3362.
Onishi, M., Mui, A. L., Morikawa, Y., Cho, L., Kinoshita, S., Nolan, G. P., Gorman, D. M., Miyajima, A., and Kitamura, T. (1996). Identification of an oncogenic form of the thrombopoietin receptor MPL using retrovirus-mediated gene transfer. Blood 88, 1399–1406.
Palacios, R., and Steinmetz, M. (1985). Il-3-dependent mouse clones that express B-220 surface antigen, contain Ig genes in germ-line configuration, and generate B lymphocytes in vivo. Cell 41, 727–734.
Pear, W. S., Nolan, G. P., Scott, M. L., and Baltimore, D. (1993). Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. U.S.A. 90, 8392–8396.
Pellegrini, S., John, J., Shearer, M., Kerr, I. M., and Stark, G. R. (1989). Use of a selectable marker regulated by alpha interferon to obtain mutations in the signaling pathway. Mol. Cell. Biol. 9, 4605–4612.
Royer, Y., Staerk, J., Costuleanu, M., Courtoy, P. J., and Constantinescu, S. N. (2005). Janus kinases affect thrombopoietin receptor cell surface localization and stability. J. Biol. Chem. 280, 27251–27261.
Sabath, D. F., Kaushansky, K., and Broudy, V. C. (1999). Deletion of the extracellular membrane-distal cytokine receptor homology module of Mpl results in constitutive cell growth and loss of thrombopoietin binding. Blood 94, 365–367.
Seubert, N., Royer, Y., Staerk, J., Kubatzky, K. F., Moucadel, V., Krishnakumar, S., Smith, S. O., and Constantinescu, S. N. (2003). Active and inactive orientations of the transmembrane and cytosolic domains of the erythropoietin receptor dimer. Mol. Cell 12, 1239–1250.
Skoda, R. C., Seldin, D. C., Chiang, M. K., Peichel, C. L., Vogt, T. F., and Leder, P. (1993). Murine c-mpl: a member of the hematopoietic growth factor receptor superfamily that transduces a proliferative signal. EMBO J. 12, 2645–2653.
Solar, G. P., Kerr, W. G., Zeigler, F. C., Hess, D., Donahue, C., De Sauvage, F. J., and Eaton, D. L. (1998). Role of c-mpl in early hematopoiesis. Blood 92, 4–10.
Teofili, L., and Larocca, L. M. (2011). Advances in understanding the pathogenesis of familial thrombocythaemia. Br. J. Haematol. 152, 701–712.
Tiedt, R., Coers, J., Ziegler, S., Wiestner, A., Hao-Shen, H., Bornmann, C., Schenkel, J., Karakhanova, S., De Sauvage, F. J., Jackson, C. W., and Skoda, R. C. (2009). Pronounced thrombocytosis in transgenic mice expressing reduced levels of Mpl in platelets and terminally differentiated megakaryocytes. Blood 113, 1768–1777.
Tijssen, M. R., Di Summa, F., Van Den Oudenrijn, S., Zwaginga, J. J., Van Der Schoot, C. E., Voermans, C., and De Haas, M. (2008). Functional analysis of single amino-acid mutations in the thrombopoietin-receptor Mpl underlying congenital amegakaryocytic thrombocytopenia. Br. J. Haematol. 141, 808–813.
Vigon, I., Florindo, C., Fichelson, S., Guenet, J. L., Mattei, M. G., Souyri, M., Cosman, D., and Gisselbrecht, S. (1993). Characterization of the murine Mpl proto-oncogene, a member of the hematopoietic cytokine receptor family: molecular cloning, chromosomal location and evidence for a function in cell growth. Oncogene 8, 2607–2615.
Vigon, I., Mornon, J. P., Cocault, L., Mitjavila, M. T., Tambourin, P., Gisselbrecht, S., and Souyri, M. (1992). Molecular cloning and characterization of MPL, the human homolog of the v-mpl oncogene: identification of a member of the hematopoietic growth factor receptor superfamily. Proc. Natl. Acad. Sci. U.S.A. 89, 5640–5644.
Waetzig, G. H., Chalaris, A., Rosenstiel, P., Suthaus, J., Holland, C., Karl, N., Valles Uriarte, L., Till, A., Scheller, J., Grotzinger, J., Schreiber, S., Rose-John, S., and Seegert, D. (2010). N-linked glycosylation is essential for the stability but not the signaling function of the interleukin-6 signal transducer glycoprotein 130. J. Biol. Chem. 285, 1781–1789.
Yoshimura, A., Zimmers, T., Neumann, D., Longmore, G., Yoshimura, Y., and Lodish, H. F. (1992). Mutations in the Trp-Ser-X-Trp-Ser motif of the erythropoietin receptor abolish processing, ligand binding, and activation of the receptor. J. Biol. Chem. 267, 11619–11625.
Appendix
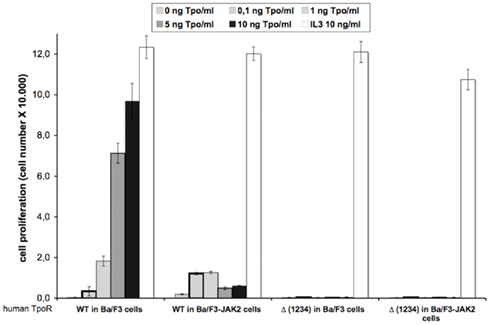
Figure A1. Removal of all four N-glycosylation sites dramatically affects the Tpo-dependent cell proliferation of human TpoR in Ba/F3 and Ba/F3-JAK2 cells. Ba/F3 and Ba/F3-JAK2 cells stably expressing WT or Δ (1234) human TpoR were washed 3 times and seeded in 96-well plates at a density of 3,000 cells/well. The cells were stimulated with different Tpo concentrations, or with 5 ng IL3/ml or left unstimulated during 72 h. After 72 h, cell proliferation was determined by the DNA synthesis assay reflected by the measurements of the thymidine incorporation as described in Experimental procedures. Results are the mean ± variation of triplicate samples. One of two independent experiments is depicted.
Keywords: cytokine receptor, thrombopoietin, JAK2, N-glycosylation, signal transduction, endoglycosidase H, ER maturation, cell surface traffic
Citation: Albu RI and Constantinescu SN (2011) Extracellular domain N-glycosylation controls human thrombopoietin receptor cell surface levels. Front. Endocrin. 2:71. doi: 10.3389/fendo.2011.00071
Received: 13 September 2011; Paper pending published: 10 October 2011;
Accepted: 21 October 2011; Published online: 11 November 2011.
Edited by:
Shoshana J. Wodak, Hospital for Sick Children, CanadaReviewed by:
Milka Vrecl, University in Ljubljana, SloveniaChengcheng Alec “Zhang”, UT Southwestern Medical Center, USA
Copyright: © 2011 Albu and Constantinescu. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Stefan N. Constantinescu, Ludwig Institute for Cancer Research, de Duve Institute, Université catholique de Louvain, Avenue Hippocrate 74, UCL 75-4, Brussels B-1200, Belgium. e-mail:c3RlZmFuLmNvbnN0YW50aW5lc2N1QGJydS5saWNyLm9yZw==