 Colin W. Ward1
Colin W. Ward1 Michael C. Lawrence1,2*
Michael C. Lawrence1,2*- 1 Walter and Eliza Hall Institute of Medical Research, Parkville, VIC, Australia
- 2 Department of Medical Biology, University of Melbourne, Parkville, VIC, Australia
Ever since the discovery of insulin and its role in the regulation of glucose uptake and utilization, there has been great interest in insulin, its structure and the way in which it interacts with its receptor and effects signal transduction. As the 90th anniversary of the discovery of insulin approaches, it is timely to provide an overview of the landmark discoveries relating to the structure and function of this remarkable molecule and its receptor.
Insulin
Discovery of Insulin: 1922
Frederick Banting made the first public presentation of the discovery of insulin to the Association of American Physicians in 1922 (Banting et al., 1922). The remarkable story of the Toronto group of Banting, Charles Best, James Collip, and John Macleod and their monumental finding is now well documented (Bliss, 1982; Rosenfeld, 2002; King, 2003). The discovery was followed shortly after by the successful large-scale production of insulin in 1923 by the USA company Eli Lilly, resulting from a collaboration between the Toronto researchers and the company’s director of biochemical research George Clowes. This was followed rapidly by the treatment of patients with insulin produced in Copenhagen. August Krogh, a doctor and researcher in metabolic diseases, and his wife Marie, a type II diabetic, had heard about Banting and Best’s research while touring the USA in late 1922 and were granted permission to produce insulin in Denmark. On his return to Denmark, Krogh, together with Hans Christian Hagedorn, founded the Nordisk Insulinlaboratorium with the financial support of pharmacist August Kongsted. In December 1922, the Nordisk laboratory successfully extracted a small quantity of insulin from a bovine pancreas and the first patients were treated in 1923 (Novo Nordisk, 2009). The commercial availability of insulin then rapidly revolutionized the treatment of diabetes. The various forms of insulin developed over the next 80 years included mono-component forms, human mono-component forms (derived from porcine insulin and converted chemically to human insulin), biosynthetic human insulins (produced by genetic engineering and microbial expression), and long-acting insulins (Brange et al., 1990; King, 2003).
Physicochemical Characterization: 1926–1953
Early insulin research centered on methods for its extraction in the purest possible form from animal pancreas tissue and on methods for its large-scale production. The physicochemical characterization of insulin then followed over the subsequent two-and-a-half decades. Milestones included the crystallization of insulin (Abel, 1926), the determination of its molecular weight (Sjögren and Svedberg, 1931), and the demonstration that it consisted of a pair of disulfide-linked polypeptide chains, namely the acidic chain A and the basic chain B (Sanger, 1949). In 1953, Dorothy Crowfoot demonstrated that insulin crystals diffracted X rays (Crowfoot, 1935), deducing at the same time the likely dimensions of the insulin molecule.
Primary Structure Determination: 1953
The chemical structure of the two-chains of the mature human insulin molecule was determined by Frederick Sanger and colleagues and described in a series of four papers in the early 1950s (Sanger and Tuppy, 1951a,b; Sanger and Thompson, 1953a,b). Insulin was the first protein to have its sequence determined and in 1958 Sanger was awarded his first Nobel Prize in Chemistry for this achievement. The pioneering study showed that human insulin B-chain consisted of 30 amino acids and the A-chain of 21 amino acids, with the B- and A-chains being held together by the two disulfide bonds CysB7 to CysA7 and CysB19 to CysA20, with a third intra-chain disulfide bond linking CysA6 to CysA11 (Figure 1).
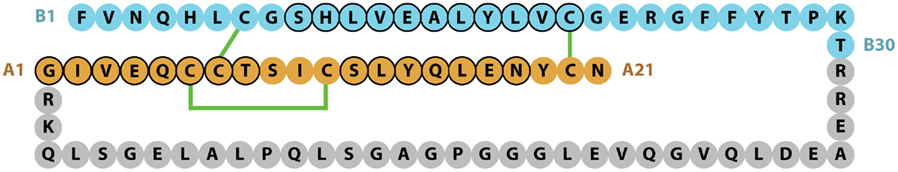
Figure 1. Sequence of human insulin precursor. The B- and A-chain segments of the processed protein are shown in cyan and light brown color, respectively, with the proteolytically removed C-peptide in gray. Disulfide bonds are indicated in green, while residues adopting a helical conformation in the mature protein are circled in black.
Demonstration of Biosynthesis from a Single-Chain Precursor: 1967
The processes involved in insulin biosynthesis were elucidated by Donald Steiner and colleagues in the late 1960s and have been recently reviewed by him in a “reflections” article (Steiner, 2011). Steiner showed, by incubating tissue slices from a rare insulin-producing adenoma of the pancreas in a medium containing tritium-labeled leucine and phenylalanine, that extracts from these incubated samples contained insulin as well as a higher molecular weight component of 9–10 kDa. This higher molecular weight component was then shown to consist of a single-chain polypeptide that he called “proinsulin.” Proinsulin contained the B-chain at its N-terminus and the A-chain at its C-terminus, with a connecting segment, the C-peptide, in the middle (Figure 1). The first reports of these findings were in 1967 (Steiner and Oyer, 1967; Steiner et al., 1967), with the sequence of porcine proinsulin being reported in 1968 by Chance et al. (1968) from Eli Lilly.
The identification and characterization of the subtilisin-like convertases that carry out the proteolytic removal of the C-peptide to yield the two-chain insulin molecule (Smeekens and Steiner, 1990; Smeekens et al., 1991) was a major challenge and took a further 20 years (Steiner, 2011).
It is now recognized that human insulin is part of a larger family of sequence-related hormones that comprises insulin, the insulin-like growth factors IGF-I and IGF-II, the relaxin peptides relaxin-1, -2, and -3 and the insulin-like peptides INSL3, INSL4, INSL5, and INSL6 (Shabanpoor et al., 2009). The two IGFs each contain a single polypeptide chain, while each of the remaining members of the family contains a pair of chains resulting from the proteolytic processing of a single-chain precursor. All members of the family exhibit the same disulfide-bonding pattern that was determined originally for human insulin.
Determination of the Three-Dimensional Structure: 1969
The crystal structure of porcine insulin was first determined by Dorothy Hodgkin (née Crowfoot) and colleagues in 1969 as the 2Zn2+-stabilized hexamer at 2.8 Å resolution (Adams et al., 1969). The structure revealed that the insulin monomer consists of a two-layered sandwich structure with the B-chain overlaying the A-chain. The B-chain consists of an N-terminal segment (residues B1–B6), a type II β-turn (B7–B10) a central α-helix (B9–B19), a type I β-turn (B20–B23), and a C-terminal β-strand (B24–B28), followed by residues B29 and B30, which are less well ordered (Figure 2A). The A-chain consists of an N-terminal α-helix (A1–A8), a non-canonical turn (A9–A11), a second α-helix (A12–A18), and a C-terminal segment (A19–A21).
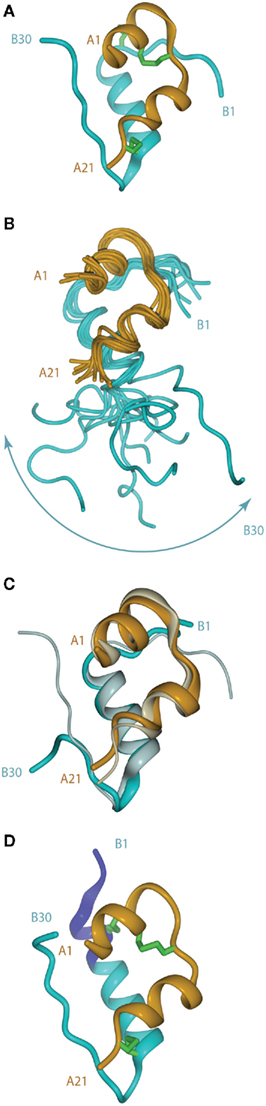
Figure 2. (A) Tertiary structure of the porcine insulin monomer in its 2 Zn rhombohedral form (PDB entry 4INS). (B) Structure of the human insulin mutant GlyB24 (PDB entry 1HIT), showing the flexibility of the B-chain C-terminus. (C) Structure of a high affinity N-MeAlaB26 human insulin mutant (PDB entry 2WRX, solid colors) overlaid on that of wild-type hormone (gray). (D) Structure of the so-called R state form of porcine insulin (PDB entry 1ZNI), with the N-terminal extension of the B-chain helix shown in blue. For clarity, disulfide bonds are omitted in (B,C).
Remarkably, at the same time and in isolation during the Cultural Revolution in China, the so-called “Beijing Insulin Structure Group” was also working to determine the structure of insulin. The details of the structure were finally published in Scientia Sinica in 1972 (Beijing Insulin Structure Group, 1972).
Since then, the three-dimensional structures of the insulin-like growth factors, the relaxin peptides and the insulin-like peptides have also been determined and, as expected from their close sequence relationship, all have the same three-helix tertiary structure as insulin (Shabanpoor et al., 2009).
Structural Changes in Insulin Upon Receptor Binding: 1991–2010
Following the landmark determination of the structure of porcine insulin, numerous structures of mutant insulins, insulin analogs, and insulins from other species have also been determined by either X-ray crystallography or NMR spectroscopy (Weiss, 2009). This extensive body of knowledge provides overwhelming evidence that insulin undergoes a conformational change upon binding the insulin receptor (IR). The first such evidence came from studies of a single-chain B29–A1 peptide-linked insulin molecule (Derewenda et al., 1991). This molecule was completely devoid of biological activity yet retained the same crystal structure as wild-type insulin. The authors concluded that receptor binding required a separation of the B-chain C-terminus away from the core body of insulin. Shortly afterward, an NMR study of the active GlyB24 insulin mutant revealed that the C-terminal region of its B-chain (residues B20–B30) were disordered in solution (Figure 2B), supporting the contention that displacement of the B-chain C-terminus away from the core structure may occur upon receptor binding (Hua et al., 1991).
The detachment model for insulin binding (Hua et al., 1991; De Meyts, 1994, 2004; Ludvigsen et al., 1998; Glendorf et al., 2008; Xu et al., 2009; Jiráček et al., 2010) proposes that the B-chain C-terminal region (residues B21–B30) reorganizes to expose the side chains of residues IleA2 and ValA3 at the N-terminus of the A-chain and in so doing exposes a mostly hydrophobic surface comprised of residues GlyA1, IleA2, ValA3, GlnA5, ThrA8, TyrA19, AsnA21, ValB12, TyrB16, GlyB23, PheB24, PheB25, and TyrB26. This so-called “classical” binding surface consists of residues predominantly from the dimer interface of insulin and represents binding surface 1. With regards to insulin’s transition from an inactive to active form and the unmasking of previously buried amino acids required for IR binding, Jiráček et al. (2010) have presented indirect evidence that suggests that a key element in this process may be the formation of a β-turn at residues B25–B26 (Figure 2C) which is associated with a trans to cis switch at the B25–B26 peptide bond.
The B-chain N-terminus of insulin is also thought to change on binding from the extended, more stable, but less active, T state to the less stable, but more active, R state. The T state is that observed in the original insulin crystal structure mentioned above (Adams et al., 1969) and is similar to that adopted by the insulin monomer in solution (Hua et al., 1991). In the R state (Figure 2D), the N-terminal portion of the B-chain alters conformation to become part of an extended α-helix B1–B19, as first observed in a crystal structure of porcine insulin produced under high-salt conditions (Bentley et al., 1976). Structural studies of insulin analogs suggest that the classical insulin T state represents an inactive conformation of the hormone that favors storage and stability, while the R state is less stable, but is the state that facilitates receptor binding and signal transduction (Nakagawa et al., 2005).
Mutagenesis data indicate that there is a second receptor binding surface (surface 2) on insulin that involves residues SerA12, LeuA13, GluA17, HisB10, GluB13, and LeuB17 from the hexamer-forming face of insulin (Schäffer, 1994; Jensen, 2000; De Meyts, 2004; Glendorf et al., 2008).
Similar, though less detailed, information now exists for other molecules in the insulin family. IGF-I binds the Type 1 insulin-like growth factor receptor (IGF-1R) via surfaces related to those employed by insulin in binding the IR, but the relative contribution of the equivalent individual residues varies between the two ligands. Binding surface 1 in IGF-I includes residues Phe23, Tyr24, Val44, Tyr60, and Ala62 (corresponding to insulin surface 1 residues PheB24, PheB25, ValA3, TyrA19, and AsnA21, respectively); additional residues employed by IGF-I in binding to surface 1 of IGF-1R are the B-domain residue Ala8, the C-domain residues Tyr31, Arg36 and Arg37, and the A-domain residue Met59 (Gauguin et al., 2008). Residues involved in the second binding surface of IGF-I have been identified using alanine-scanning mutagenesis (Gauguin et al., 2008) – these are Glu9, Asp12, Leu54, and Glu58 (corresponding to the insulin surface 2 residues HisB10, GluB13, LeuA13, and GluA17, respectively). IGF-II has been shown to bind the A, but not the B, isoform of IR with similar high affinity to insulin (Frasca et al., 1999). In contrast, IGF-I is shown to bind both the IR isoforms relatively weakly. Comparison of a series of chimeric IGFs shows that the C and D domains of the IGFs are primarily responsible for their differential binding properties to the IR isoforms and IGF-1R (Denley et al., 2004). An analysis of selected candidates for the IGF-II binding site residues confirmed that Val14, Phe28, and Val43 (equivalent to insulin surface 1 residues) are critical to both IGF-1R and IR binding, whereas mutation to alanine of Gln18 affects only IGF-1R and not IR binding (Alvino et al., 2009). Alanine substitutions at Glu12, Asp15, Phe19, Leu53, and Glu57 result in significant (>two-fold) decreases in affinity for both IGF-1R and IR. Residues equivalent to insulin surface 2 are found to be Glu12, Phe19, Leu53, and Glu57 (Alvino et al., 2009).
Insulin Receptor
Discovery of Insulin Binding to Cell Surfaces: 1970
The early literature on the discovery of the IR has been summarized by Pierre de Meyts in a review describing his more than three decades of research in the field (De Meyts, 2004). Levine in 1949 postulated that insulin interacted with the cell membrane to facilitate the uptake of hexoses into cells, rather than playing an enzymatic role in carbohydrate metabolism (Levine et al., 1949). The first description of radio-labeled insulin binding to liver cell membranes was by the Australians House and Weidemann in 1970, followed by more detailed reports from two USA laboratories in 1971 (House and Weidemann, 1970; Cuatrecasas et al., 1971; Freychet et al., 1971). Independently, and around the same time, Gammeltoft and Gliemann (1973) in Copenhagen developed an assay to detect insulin binding to isolated fat cells.
The Receptor is a Disulfide-Linked Homodimer: 1980–1981
The next major discovery was the demonstration in 1980–1981 that the IR is a dimer of apparent molecular weight ∼350 kDa and composed of two α-subunits (∼120–130 kDa) and two β-subunits (∼90 kDa) that are linked by disulfide bonds (Massague et al., 1980). Sixteen years later, Lindsay Sparrow et al. (1997) showed there was a single disulfide bond (Cys685–Cys872) connecting the α- and β-chains of each monomer and two sites of inter-monomer α–α disulfide bonds in the IR dimer, namely, at Cys524–Cys524 and at a site comprising at least one of the potential disulfide bonds Cys682–Cys682, Cys683–Cys683, and Cys685–Cys685. Partial reduction studies (Finn et al., 1990; Chiacchia, 1991) had shown earlier that there were only two α–α disulfide bonds in the IR dimer, suggesting that, within the (Cys682, Cys683, Cys685) triplet, the remaining two residues form an intra-chain disulfide.
The Receptor is a Tyrosine Kinase: 1982
The next key discovery was the demonstration by Kasuga et al. (1982a,b) that the IR is a tyrosine kinase (TK) that activates its β-subunit upon insulin binding. The first discovery of an intra-cellular substrate was by White et al. (1985) - this protein was subsequently characterized and called IR substrate 1, or IRS1 (Sun et al., 1991).
Sequence Determination: 1985
The cDNA sequence for the human IR was determined independently by two laboratories in 1985 (Ebina et al., 1985; Ullrich et al., 1985) and immediately revealed that there were two isoforms of the receptor that differed by the absence (in the IR-A isoform) or presence (in the IR-B isoform) of an additional 12 residues between residues 716 and 717. These, and subsequent amino acid analyses (reviewed in Adams et al., 2000), revealed that each receptor monomer consists, from its N-terminus to C-terminus (Figure 3), of a leucine-rich repeat domain (L1), a cysteine-rich region (CR), a second leucine-rich repeat domain (L2), and three fibronectin type III domains (FnIII-1, FnIII-2, and FnIII-3), with FnIII-2 containing a large (∼120 residues) insert domain (ID). The ID contains the furin cleavage site that yields the α-chain and β-chain of the mature receptor monomer. The intra-cellular C-terminal region of the IR monomer contains the TK catalytic domain, which is flanked by two regulatory regions (the juxtamembrane region and the C-tail).
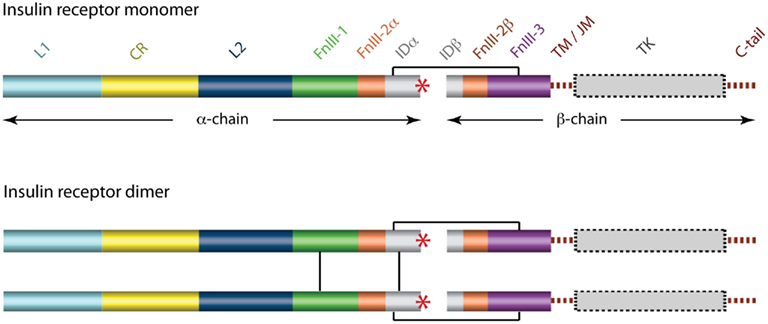
Figure 3. Domain structure of the human insulin receptor. L1, first leucine-rich repeat domain; CR, cysteine-rich region; L2, second leucine-rich repeat domain; FnIII-1, -2, -3, first, second, and third fibronectin type III domains; ID, insert domain; TM/JM, trans- and juxtamembrane regions; TK, tyrosine kinase domain. The second fibronectin Type III domain and the insert domain span both the α- and the β-chains of the mature protein. The critical C-terminal segment of the α-chain (αCT) is indicated by a red asterisk. Inter-chain and inter-monomer disulfide bonds are indicated by black line segments and membrane and cytoplasmic regions are shown in dashed outline.
The human IR is heavily glycosylated and estimated to contain 58–64 kDa of carbohydrate (Cosgrove et al., 1995). The sequence revealed 18 potential N-linked glycosylation sites, 16 of which were subsequently shown to have carbohydrate attached (Sparrow et al., 2007b). Further analysis showed the existence of six O-linked glycosylation sites, all of which lay near the N-terminus of the β-chain (Sparrow et al., 2007a).
There are 13 potential tyrosine phosphorylation sites in the intra-cellular IR β-subunit that provide potential docking sites for SH2-containing (Pawson et al., 2001; Machida et al., 2007) and PTB-containing signaling proteins (Wolf et al., 1995; Borg and Margolis, 1998). Some of these are located in the catalytic domain, rather than the juxtamembrane and C-tail regions, and some do not conform to the conventionally accepted recognition sequence motifs (Ward et al., 1996).
The cDNA sequence for IGF-1R was determined in 1985 and revealed extensive similarity in size and structural topology to that of IR (Ullrich et al., 1986). Nevertheless, regions of difference both within the respective receptor ectodomains and at the C-terminal tail of the β-subunit provided the first hints as to how specificity of ligand binding and specificity of signaling might occur.
The major signaling pathways by which insulin and the IGFs regulate metabolism and gene expression have been reviewed elsewhere (Johnston et al., 2003; Siddle, 2011) and include the serine/threonine kinases Akt/PKB and MEK whose activation is dependent on phosphorylation of IRS1 and Shc and the subsequent activation of PI3K and the small G-protein Ras. The roles of various accessory pathways involving PTPs, PTEN, CAP/cbl, other PI3Ks, PIKfyve, GABs, DOKs, and other signaling proteins are also reviewed (Siddle, 2011).
Determination of the Three-Dimensional Structure of the Kinase Domain: 1994
The crystal structure of the human IR TK domain in its unphosphorylated (basal) state was determined by Hubbard et al. (1994). This was the first structure of a TK to be reported, though the structure of a serine kinase (cAMP protein kinase) had been reported earlier (Knighton et al., 1991). Like the serine kinases, the IR TK is composed of two lobes with a single connection between them (Figure 4A). The N-terminal lobe comprises a twisted β-sheet of five anti-parallel β-strands (β1–β5) and one α-helix (αC). The larger C-terminal lobe comprises eight α-helices (αD, αE, αEF, αF, αG, αH, αI, αJ) and four β-strands (β7, β8, β10, β11). The human IR TK lacks β-strands β6 and β9 present in cAMP protein kinase.
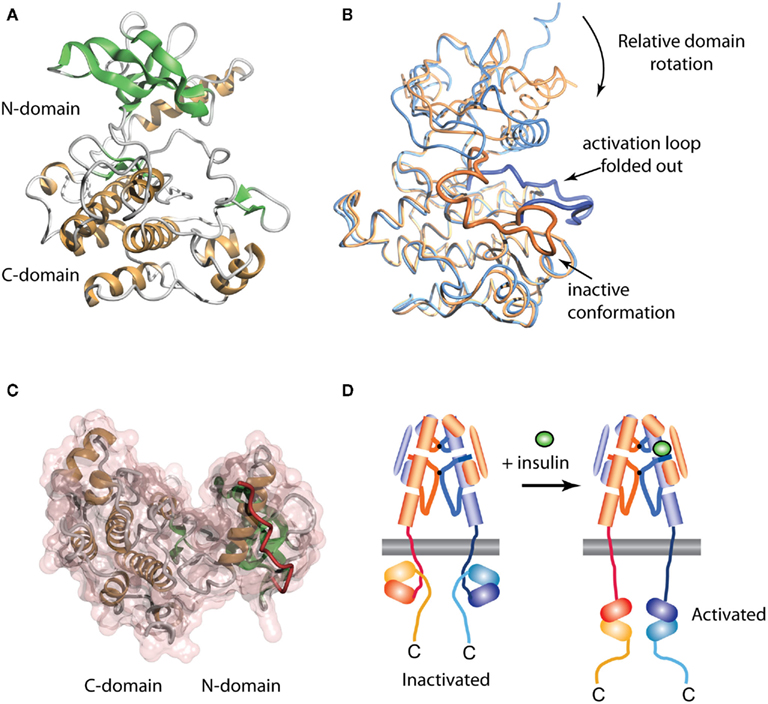
Figure 4. Three-dimensional crystal structure of the insulin receptor tyrosine kinase domain. (A) Inactive form, showing the secondary structure of the N- and C-terminal domains. (B) Overlay of the inactive (blue, PDB entry 1IRK) and activated (orange, PDB entry 1IR3) forms of the domain, showing the displacement from the catalytic site of the activation loop (residues 1149–1170, highlighted as a thicker ribbon within each form) and the concomitant domain rotation which are observed upon activation. (C) Interaction between the juxtamembrane residues 978–988 (red tube) and the N-terminal domain of the IR tyrosine kinase in its basal state (transparent pink surface, PDB entry 1P14). (D) Stylized cartoon showing the hypothesized sequestered disposition of the kinase domain as a consequence of its interaction with the proximal region juxtamembrane segment [see (C)], followed by its release upon activation.
In the inactivated IR TK, one of the three tyrosines in the activation loop, Tyr1162, is bound in the active site but cannot be phosphorylated (in cis) because part of the A-loop interferes with the ATP binding site and the catalytic residue Asp1150 is improperly positioned to co-ordinate MgATP (Hubbard et al., 1994). Upon activation, auto-phosphorylation of residues Tyr1162, Tyr1158, and Tyr1163 occurs in trans by the kinase domain of the second monomer. Thus, in the basal state, Tyr1162 competes with the neighboring β-chain and other protein substrates for binding to the active site, but is not cis-phosphorylated because of steric constraints that prevent simultaneous binding of Tyr1162 and MgATP.
The structure of the activated phosphorylated IR kinase reveals that auto-phosphorylation of the three tyrosines in the A-loop leads to a dramatic change in its configuration (Hubbard, 1997). In the phosphorylated state, the A-loop is displaced by approximately 30 Å, resulting in unrestricted access to the binding site for ATP and protein substrates. This movement facilitates a functional spatial arrangement of Lys1030 and Glu1047, the residues involved in MgATP coordination, and of Asp1150, which is part of the highly conserved Asp–Phe–Gly triad. The A-loop rearrangement also leads to closure of the N and C-terminal lobes, which is necessary for productive ATP binding (Hubbard, 1997). This closure involves significant rotation of the N-terminal lobe (Figure 4B).
In 2003 Stevan Hubbard’s group (Li et al., 2003) described the structure of an extended IR kinase construct showing the molecular details of the interaction between the catalytic domain and the juxtamembrane region (Figure 4C). In IR, the juxtamembrane interactions dominate and are considerably stronger than the activation loop inhibition described earlier (Li et al., 2003; Craddock et al., 2007). In IR and IGF-1R, the proximal juxtamembrane regions show TK inhibition through the highly conserved residue Tyr984 (IR-B numbering, equivalent to Tyr972 in IR-A and Tyr957 in IGF-1R). This tyrosine residue interacts with several conserved residues in the N-terminal lobe of the IR kinase domain, stabilizing a non-productive position of the αC helix (Li et al., 2003). Mutation of this residue in the full-length IR or IGF-1R increases basal autophosphorylation substantially (Li et al., 2003; Craddock et al., 2007). This juxtamembrane inhibition in IR is more significant than that contributed by the activation loop since in the full-length IR found on cells; mutation of Tyr984 to Ala increases the basal phosphorylation state 30-fold, 10 times greater than the three-fold increase seen following mutation of the activation loop residue Tyr1162 to Asp (Li et al., 2003).
The direction of the juxtamembrane segment relative to the lobes of the catalytic domain as revealed in the three-dimensional structure of the extended IR kinase construct (Figure 4C) implies that, in the basal state, the catalytic domain is partially wrapped up and held inverted with respect to the cell membrane, as indicated schematically in Figure 4D. We thus proposed (Ward and Lawrence, 2009) that, in the basal state, the catalytic domain is unlikely to be exposed and/or suspended from the end of the 41-residue (∼140 Å) strand of juxtamembrane polypeptide. We suggest that ligand binding results in an as yet unknown domain re-arrangement within the ectodomain that in turn affects the juxtamembrane/catalytic domain interaction, resulting in the release of the sequestered kinase domains and their subsequent transphosphorylation, as summarized in Figure 4D.
The Three-Dimensional Structure of the First Three Domains of IGF-1R: 1998
The first information on the structure of the IR ligand binding region came in 1998 from the crystal structure of the closely related Type 1 IGF-1R (Garrett et al., 1998). This revealed that the IGF-1R L1 and L2 domains were, in fact, leucine-rich repeats with the characteristic solenoid shape comprised of three β-sheet surfaces with an irregular end (Figure 5A). The high degree of sequence relationship between IR and IGF-1R implied immediately that the corresponding three domains of IR had a closely similar structure to their respective IGF-1R counterparts. The structure confirmed the earlier predictions (Ward et al., 1995) that the CR domain was comprised of seven smaller disulfide-linked modules, each containing one or two disulfide bonds similar to those found in the epidermal growth factor, laminin, and the tumor necrosis factor receptor. The structure also revealed that the residues that are mutated in IR in many patients that have defective insulin binding, as well as all of the residues in the L1 domain that are compromised in insulin binding when mutated to alanine, lie on the central β-sheet of the L1 domain (Figure 5B).
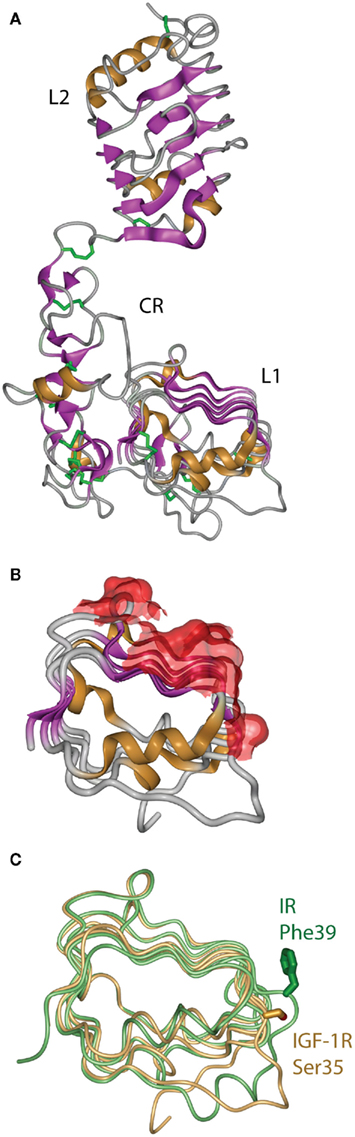
Figure 5. (A) Three-dimensional crystal structure of the L1–CR–L2 module of the Type 1 insulin-like growth factor receptor (IGF-1R). (B) View of the molecular surface of the central and third β-sheet of the L1 domain of IGF-1R, highlighting in red those portions of the surface that correspond to equivalent residues within the insulin receptor that have been implicated in hormone binding. (C) Overlay of the L1 domains of insulin receptor (light green) and IGF-1R (light orange), highlighting the difference in side chain disposition of the equivalent residues IR Phe39 and IGF-1R Ser35. Panels are based on PDB entries 1IGR and 2HR7.
The structure of the equivalent L1–CR–L2 fragment of IR was determined in 1999 and published in 2006 (Lou et al., 2006). As expected, it closely resembled the IGF-1R fragment structure, but with two important differences found at sites known to be involved in determining ligand specificity. The first site is at the corner of the L1 domain central β-sheet, where the side chain of IR Phe39 extends down to form part of the ligand binding surface, in contrast to the disposition of its counterpart IGF-1R Ser35 (Figure 5C). The second major difference is in the sixth module of the CR domain, where IR contains a longer loop that protrudes further into the binding pocket than its IGF-1R counterpart. This module, which contributes to IGF binding specificity, shows negligible sequence identity, significantly more α-helical secondary structure, an additional disulfide bond and an opposite electrostatic potential to that of the IGF-1R (Lou et al., 2006).
It is interesting to note that the receptors that bind relaxin and INSL3 also employ leucine-rich-repeat domains for ligand binding, despite belonging to the otherwise structurally and functionally distinct class of GPCRs (Ward and Lawrence, 2009).
The Three-Dimensional Structure of the IR Ectodomain Dimer: 2006
The structure of the IR ectodomain dimer (approximately 1800 amino acids) was reported in a landmark paper in Nature in 2006 (McKern et al., 2006) and represented a major milestone in insulin research. The difficulty confronting researchers was the generation of crystals that diffracted sufficiently to allow the three-dimensional structure of the large, heavily glycosylated ectodomain to be solved. The Australian group made numerous receptor constructs using different cell lines for the protein production. They also explored subtle methods for high-resolution protein purification and a variety of protein treatment protocols before obtaining crystals of adequate quality. Final success came as a result of a series of improvements, including in particular antibody-mediated crystallization wherein the IR dimer was decorated with two monoclonal antibody fragments (Fabs) from the anti-IR antibody 83-7 and two from the anti-IR antibody 83-14 (Soos et al., 1986). The attachment of Fabs resulted in a crystal packing arrangement that involved almost solely Fab-to-Fab interfaces, thus overcoming the hindrance to crystallization posed by the surface glycans of the IR ectodomain. These crystals were obtained by Meizhen Lou (McKern et al., 2006), who, remarkably, had also been a technician within the Beijing Insulin Structure Group working on the crystallization of insulin in the early 1970s!
The three-dimensional structure revealed that the IR adopted a folded-over (inverted “V”) conformation that placed putative ligand binding regions in close juxtaposition. One leg of each monomer consists of the L1–CR–L2 module described above and the other of the FnIII-1, FnIII-2, and FnIII-3 domains in an extended, linear arrangement (Figure 6A). FnIII-2 contains the ID. The IR homodimer has a two-fold rotation axis that places the L2 domains of each monomer in contact with the FnIII-1 domain of the alternate monomer at the apex of the inverted “V” and places the L1 domains of the each monomer in contact with the FnIII-2 domains of the alternate monomer around the midpoint of the legs of the inverted “V” (McKern et al., 2006). At the base of the structure, the C-termini of the two FnIII-3 domains are poised to extend through the cell membrane to the intra-cellular juxtamembrane, kinase, and C-tail domains of the intact receptor. The FnIII-1 and FnIII-2 domains contain unusually large CC′ loops. In the FnIII-1 domain, the large CC′ loop enables formation of the Cys525–Cys524 dimer disulfide bond, while in the FnIII-2 domains the entire ID segment is contained within their respective CC′ loops. The α−β disulfide bond between Cys647 (near the start of the ID) and Cys860 (at the beginning of the C′E loop of FnIII-3) is visible in the structure. The Cys524 α–α disulfide bond lies in a very weakly ordered region of the polypeptide, whereas the α–α disulfide bond(s) involving Cys862, Cys863, and Cys865 lies in a region of the ID (residues 655–755) that is entirely disordered in the crystal (McKern et al., 2006).
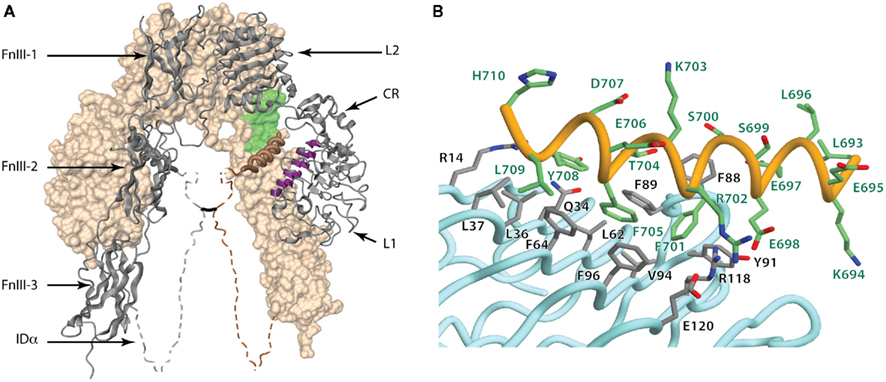
Figure 6. (A) Three-dimensional crystal structure of the IR ectodomain, showing the inverted “V” conformation with respect to the membrane. The background monomer is shown in molecular surface representation (beige) and the foreground monomer in secondary structure schematic representation (gray) with the constituent domains labeled. The disordered portions of the α-chain components of the insert domain are shown as dashed and their conformation shown here is speculative, with the approximate location of the inter-chain disulfide being indicated by a black line. The relative location of the components that make up the insulin binding site within one leg of the inverted “V” are highlighted and include the surface of the central β-sheet of the L1 domain of one monomer (purple), residues at the junction of FnIII-1 and FnIII-2 domains of the adjacent monomer (light green) and the helix formed by the C-terminal segment of α-chain (brown coil). (B) Detail of the conformation of the αCT segment on the surface the central β-sheet of the L1 domain in the crystal structure of the apo-form of the insulin receptor ectodomain. The Figure is based on PDB entry 3LOH; (B) is from Smith et al. (2010), used by permission.
An intriguing, though uninterpretable, segment of electron density was seen to be lying across the central β-sheet of each L1 domain in the original crystallographic maps of IR ectodomain (McKern et al., 2006). It was suggested that this segment most likely corresponded to the C-terminal region of the IR α-chain, the so-called “αCT” segment, that is known to be critical for insulin binding (Kurose et al., 1994; Mynarcik et al., 1996, 1997; Kristensen et al., 1998). Indeed, prior evidence existed for the proximity of these two elements: the adjacent insulin B-chain residues Phe24 and Phe25 can be cross-linked respectively to the L1 domain and CT region (Xu et al., 2004). The electron density segment was finally resolved by means of thermal-factor sharpening, revealing it to be α-helical in conformation and interpretable as IR residues 693–710 (Smith et al., 2010). The final nine residues of the IR α-chain (711–719; IR-A isoform numbering) remained disordered.
The key features of the interaction of the 693–710 segment with the surface of the central β-sheet of the L1 domain are shown in Figure 6B. In particular, (i) the side chains of residues Phe701 and Phe705 are packed adjacent to each other in a hydrophobic pocket formed by the side chains of the L1 domain residues Leu62, Phe64, Phe88, Phe89, Tyr91, Val94, Phe96, and Arg118 - these two aromatic residues are conserved in type in IGF-1R (as Tyr688 and Phe692, respectively); (ii) the side chain of Tyr708 is packed approximately parallel to the strands of the L1 central β-sheet, in proximity to the side chains of Arg14, Gln34, Leu36, and Phe88 – again this residue is conserved in type in IGF-1R (as Phe695); (iii) the side chains of the αCT residue pair Glu698/Arg702 lie close to each other and interact with the side chains of the L1 domain residue pair Arg118/Glu120 respectively, with the four side chains forming a charge-compensating cluster; and (iv) the side chain of Leu709 is in hydrophobic interaction with the side chains of Leu37 and Phe64 (Smith et al., 2010).
A remaining feature of interest is the fact that the αCT segment associated with each L1 domain in the IR dimer is contributed by the alternate monomer to that which contributes the L1 domain. This was first predicted by (Ward et al., 2007), based on the expectation that C-terminus of the α-chain most likely lies in proximity to the N-terminus of the β-chain, given that during IR biosynthesis, proteolytic cleavage of the pro-receptor into α and β chains occurs after dimer assembly (Bass et al., 1998). The relative proximity of the last observed residue in the αCT segment (His710) to the first observed residue of the β-chain (Glu755) within the CC′ loop of FnIII-2 thus defines these as being part of the same monomer. This trans arrangement of L1 and αCT has since been confirmed experimentally by complementation analysis (Chan et al., 2007) using co-expression of pairs of differentially tagged insulin midi-receptors carrying single mutations either in L1 or αCT, respectively, and by direct chemical crosslinking of two IR α-chains using doubly derivatized [PapB16, PapB25]-insulin or [PapB16, PapA3]-single-chain insulin (Smith et al., 2010).
Future Landmark: The Three-Dimensional Structure of the Insulin/IR Complex
This still remains an unachieved goal. However the 3D structures of the intact ectodomain and the L1–CR–L2 fragments from IR and IGF-1R along with the wealth of studies on mutant insulins and IGFs provide some insight into the nature of the ligand binding sites and the insulin/IR complexes. This has been reviewed in detail elsewhere (Lawrence et al., 2007; Ward et al., 2007, 2008; Ward and Lawrence, 2009).
To date, no successful high-resolution crystallizations of ligand-receptor complexes have been reported, despite the availability of a number of domain-minimized versions of the IR that are known to retain insulin binding capacity (Kristensen et al., 1998, 2002; Surinya et al., 2002; Menting et al., 2009) and which might assist in the production of crystals for X-ray diffraction studies. The reasons for this lack of success are not obvious. From our own experience of trying to produce such crystals now for over two decades, we speculate that this might relate to the limited range of pH over which insulin/IR complexes are stable (and hence over which they might be amenable to crystallization), or to further intrinsic structural instabilities in the receptor/ligand complex that are not yet understood. Nevertheless, given the progress in understanding the structural biology of the apo-receptor and in automated crystallization technology, we are confident that in the near future these problems will be overcome.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported in part by the Australian National Health and Medical Research Council (NHMRC) Project Grant No. 1005896, NHMRC Independent Research Institutes Infrastructure Support Scheme Grant 361646 and a Victorian State Government Operational Infrastructure Support Grant. Figures were produced using DINO (http://www.dino3d.org).
References
Adams, M. J., Blundell, T. L., Dodson, E. J., Dodson, G. G., Vijayan, M., Baker, E. N., Harding, M. M., Hodgkin, D. C., Rimmer, B., and Sheat, S. (1969). Structure of rhombohedral 2 zinc insulin crystals. Nature 224, 491–495.
Adams, T. E., Epa, V. C., Garrett, T. P., and Ward, C. W. (2000). Structure and function of the type 1 insulin-like growth factor receptor. Cell. Mol. Life Sci. 57, 1050–1093.
Alvino, C. L., McNeil, K. A., Ong, S. C., Delaine, C., Booker, G. W., Wallace, J. C., Whittaker, J., and Forbes, B. E. (2009). A novel approach to identify two distinct receptor binding surfaces of insulin-like growth factor II. J. Biol. Chem. 284, 7656–7664.
Banting, F. G., Best, C. H., Collip, J. B., Campbell, W. R., Fletcher, A. A., MacLeod, J. J. R., and Noble, E. C. (1922). The effect produced on diabetes by extracts of pancreas. Trans. Assoc. Am. Physicians 37, 337–347.
Bass, J., Chiu, G., Argon, Y., and Steiner, D. F. (1998). Folding of insulin receptor monomers is facilitated by the molecular chaperones calnexin and calreticulin and impaired by rapid dimerization. J. Cell Biol. 141, 637–646.
Beijing Insulin Structure Group. (1972). Studies on crystal structure of insulin at 2.5Å. Sci. Sin. 1, 3–30.
Bentley, G., Dodson, E., Dodson, G., Hodgkin, D., and Mercola, D. (1976). Structure of insulin in 4-zinc insulin. Nature 261, 166–168.
Borg, J. P., and Margolis, B. (1998). Function of PTB domains. Curr. Top. Microbiol. Immunol. 228, 23–38.
Brange, J., Owens, D. R., Kang, S., and Volund, A. (1990). Monomeric insulins and their experimental and clinical implications. Diabetes Care 13, 923–954.
Chan, S. J., Nakagawa, S., and Steiner, D. F. (2007). Complementation analysis demonstrates that insulin cross-links both a subunits in a truncated insulin receptor dimer. J. Biol. Chem. 282, 13754–13758.
Chance, R. E., Ellis, R. M., and Bromer, W. W. (1968). Porcine proinsulin: characterization and amino acid sequence. Science 161, 165–167.
Chiacchia, K. B. (1991). Quantitation of the class I disulfides of the insulin receptor. Biochem. Biophys. Res. Commun. 176, 1178–1182.
Cosgrove, L., Lovrecz, G. O., Verkuylen, A., Cavaleri, L., Black, L. A., Bentley, J. D., Howlett, G. J., Gray, P. P., Ward, C. W., and McKern, N. M. (1995). Purification and properties of insulin receptor ectodomain from large-scale mammalian cell culture. Protein Expr. Purif. 6, 789–798.
Craddock, B. P., Cotter, C., and Miller, W. T. (2007). Autoinhibition of the insulin-like growth factor I receptor by the juxtamembrane region. FEBS Lett. 581, 3235–3240.
Cuatrecasas, P., Desbuquois, B., and Krug, F. (1971). Insulin-receptor interactions in liver cell membranes. Biochem. Biophys. Res. Commun. 44, 333–339.
De Meyts, P. (1994). The structural basis of insulin and insulin-like growth factor-I receptor binding and negative co-operativity, and its relevance to mitogenic versus metabolic signalling. Diabetologia 37(Suppl. 2), S135–S148.
De Meyts, P. (2004). Insulin and its receptor: structure, function and evolution. Bioessays 26, 1351–1362.
Denley, A., Bonython, E. R., Booker, G. W., Cosgrove, L. J., Forbes, B. E., Ward, C. W., and Wallace, J. C. (2004). Structural determinants for high-affinity binding of insulin-like growth factor II to insulin receptor (IR)-A, the exon 11 minus isoform of the IR. Mol. Endocrinol. 18, 2502–2512.
Derewenda, U., Derewenda, Z., Dodson, E. J., Dodson, G. G., Bing, X., and Markussen, J. (1991). X-ray analysis of the single chain B29-A1 peptide-linked insulin molecule. A completely inactive analogue. J. Mol. Biol. 220, 425–433.
Ebina, Y., Ellis, L., Jarnagin, K., Edery, M., Graf, L., Clauser, E., Ou, J. H., Masiarz, F., Kan, Y. W., Goldfine, I. D., Roth, R. A., and Rutter, W. J. (1985). The human insulin receptor cDNA: the structural basis for hormone-activated transmembrane signalling. Cell 40, 747–758.
Finn, F. M., Ridge, K. D., and Hofmann, K. (1990). Labile disulfide bonds in human placental insulin receptor. Proc. Natl. Acad. Sci. U.S.A. 87, 419–423.
Frasca, F., Pandini, G., Scalia, P., Sciacca, L., Mineo, R., Costantino, A., Goldfine, I. D., Belfiore, A., and Vigneri, R. (1999). Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol. Cell. Biol. 19, 3278–3288.
Freychet, P., Roth, J., and Neville, D. M. Jr. (1971). Insulin receptors in the liver: specific binding of [125I]insulin to the plasma membrane and its relation to insulin bioactivity. Proc. Natl. Acad. Sci. U.S.A. 68, 1833–1837.
Gammeltoft, S., and Gliemann, J. (1973). Binding and degradation of 125I-labelled insulin by isolated rat fat cells. Biochim. Biophys. Acta 320, 16–32.
Garrett, T. P., McKern, N. M., Lou, M., Frenkel, M. J., Bentley, J. D., Lovrecz, G. O., Elleman, T. C., Cosgrove, L. J., and Ward, C. W. (1998). Crystal structure of the first three domains of the type-1 insulin-like growth factor receptor. Nature 394, 395–399.
Gauguin, L., Delaine, C., Alvino, C. L., McNeil, K. A., Wallace, J. C., Forbes, B. E., and De Meyts, P. (2008). Alanine scanning of a putative receptor binding surface of insulin-like growth factor-I. J. Biol. Chem. 283, 20821–20829.
Glendorf, T., Sorensen, A. R., Nishimura, E., Pettersson, I., and Kjeldsen, T. (2008). Importance of the solvent-exposed residues of the insulin B chain alpha-helix for receptor binding. Biochemistry 47, 4743–4751.
House, P. D., and Weidemann, M. J. (1970). Characterization of an [125 I]-insulin binding plasma membrane fraction from rat liver. Biochem. Biophys. Res. Commun. 41, 541–548.
Hua, Q. X., Shoelson, S. E., Kochoyan, M., and Weiss, M. A. (1991). Receptor binding redefined by a structural switch in a mutant human insulin. Nature 354, 238–241.
Hubbard, S. R. (1997). Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J. 16, 5572–5581.
Hubbard, S. R., Wei, L., Ellis, L., and Hendrickson, W. A. (1994). Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature 372, 746–754.
Jensen, A.-M. (2000). Analysis of Structure-Activity Relationships of the Insulin Molecule by Alanine Scanning Mutagenesis. Master’s Thesis, The University of Copenhagen, Copenhagen.
Jiráček, J., Zakova, L., Antolikova, E., Watson, C. J., Turkenburg, J. P., Dodson, G. G., and Brzozowski, A. M. (2010). Implications for the active form of human insulin based on the structural convergence of highly active hormone analogues. Proc. Natl. Acad. Sci. U.S.A. 107, 1966–1970.
Johnston, A. M., Pirola, L., and Van Obberghen, E. (2003). Molecular mechanisms of insulin receptor substrate protein-mediated modulation of insulin signalling. FEBS Lett. 546, 32–36.
Kasuga, M., Zick, Y., Blith, D. L., Karlsson, F. A., Haring, H. U., and Kahn, C. R. (1982a). Insulin stimulation of phosphorylation of the beta subunit of the insulin receptor. Formation of both phosphoserine and phosphotyrosine. J. Biol. Chem. 257, 9891–9894.
Kasuga, M., Zick, Y., Blithe, D. L., Crettaz, M., and Kahn, C. R. (1982b). Insulin stimulates tyrosine phosphorylation of the insulin receptor in a cell-free system. Nature 298, 667–669.
King, K. M. (2003). A history of insulin: from discovery to modern alternatives. Br. J. Nurs. 12, 1137–1141.
Knighton, D. R., Zheng, J. H., Ten Eyck, L. F., Xuong, N. H., Taylor, S. S., and Sowadski, J. M. (1991). Structure of a peptide inhibitor bound to the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 253, 414–420.
Kristensen, C., Andersen, A. S., Østergaard, S., Hansen, P. H., and Brandt, J. (2002). Functional reconstitution of insulin receptor binding site from non-binding receptor fragments. J. Biol. Chem. 277, 18340–18345.
Kristensen, C., Wiberg, F. C., Schäffer, L., and Andersen, A. S. (1998). Expression and characterization of a 70-kDa fragment of the insulin receptor that binds insulin. Minimizing ligand binding domain of the insulin receptor. J. Biol. Chem. 273, 17780–17786.
Kurose, T., Pashmforoush, M., Yoshimasa, Y., Carroll, R., Schwartz, G. P., Burke, G. T., Katsoyannis, P. G., and Steiner, D. F. (1994). Cross-linking of a B25 azidophenylalanine insulin derivative to the carboxyl-terminal region of the α-subunit of the insulin receptor. Identification of a new insulin-binding domain in the insulin receptor. J. Biol. Chem. 269, 29190–29197.
Lawrence, M. C., McKern, N. M., and Ward, C. W. (2007). Insulin receptor structure and its implications for the IGF-1 receptor. Curr. Opin. Struct. Biol. 17, 699–705.
Levine, R., Goldstein, M., Klein, S., and Huddlestun, B. (1949). The action of insulin on the distribution of galactose in eviscerated nephrectomized dogs. J. Biol. Chem. 179, 985.
Li, S., Covino, N. D., Stein, E. G., Till, J. H., and Hubbard, S. R. (2003). Structural and biochemical evidence for an autoinhibitory role for tyrosine 984 in the juxtamembrane region of the insulin receptor. J. Biol. Chem. 278, 26007–26014.
Lou, M., Garrett, T. P., McKern, N. M., Hoyne, P. A., Epa, V. C., Bentley, J. D., Lovrecz, G. O., Cosgrove, L. J., Frenkel, M. J., and Ward, C. W. (2006). The first three domains of the insulin receptor differ structurally from the insulin-like growth factor 1 receptor in the regions governing ligand specificity. Proc. Natl. Acad. Sci. U.S.A. 103, 12429–12434.
Ludvigsen, S., Olsen, H. B., and Kaarsholm, N. C. (1998). A structural switch in a mutant insulin exposes key residues for receptor binding. J. Mol. Biol. 279, 1–7.
Machida, K., Thompson, C. M., Dierck, K., Jablonowski, K., Karkkainen, S., Liu, B., Zhang, H., Nash, P. D., Newman, D. K., Nollau, P., Pawson, T., Renkema, G. H., Saksela, K., Schiller, M. R., Shin, D. G., and Mayer, B. J. (2007). High-throughput phosphotyrosine profiling using SH2 domains. Mol. Cell 26, 899–915.
Massague, J., Pilch, P. F., and Czech, M. P. (1980). Electrophoretic resolution of three major insulin receptor structures with unique subunit stoichiometries. Proc. Natl. Acad. Sci. U.S.A. 77, 7137–7141.
McKern, N. M., Lawrence, M. C., Streltsov, V. A., Lou, M. Z., Adams, T. E., Lovrecz, G. O., Elleman, T. C., Richards, K. M., Bentley, J. D., Pilling, P. A., Hoyne, P. A., Cartledge, K. A., Pham, T. M., Lewis, J. L., Sankovich, S. E., Stoichevska, V., Da Silva, E., Robinson, C. P., Frenkel, M. J., Sparrow, L. G., Fernley, R. T., Epa, V. C., and Ward, C. W. (2006). Structure of the insulin receptor ectodomain reveals a folded-over conformation. Nature 443, 218–221.
Menting, J. G., Ward, C. W., Margetts, M. B., and Lawrence, M. C. (2009). A thermodynamic study of ligand binding to the first three domains of the human insulin receptor: relationship between the receptor α-chain C-terminal peptide and the site 1 insulin mimetic peptides. Biochemistry 48, 5492–5500.
Mynarcik, D. C., Williams, P. F., Schäffer, L., Yu, G. Q., and Whittaker, J. (1997). Identification of common ligand binding determinants of the insulin and insulin-like growth factor 1 receptors. Insights into mechanisms of ligand binding. J. Biol. Chem. 272, 18650–18655.
Mynarcik, D. C., Yu, G. Q., and Whittaker, J. (1996). Alanine-scanning mutagenesis of a C-terminal ligand binding domain of the insulin receptor α subunit. J. Biol. Chem. 271, 2439–2442.
Nakagawa, S. H., Zhao, M., Hua, Q. X., Hu, S. Q., Wan, Z. L., Jia, W., and Weiss, M. A. (2005). Chiral mutagenesis of insulin. Foldability and function are inversely regulated by a stereospecific switch in the B chain. Biochemistry 44, 4984–4999.
Novo Nordisk. (2009). History of Novo Nordisk. Available: http://www.novonordisk.com.au/documents/article_page/document/History.asp
Pawson, T., Gish, G. D., and Nash, P. (2001). SH2 domains, interaction modules and cellular wiring. Trends Cell Biol. 11, 504–511.
Sanger, F., and Thompson, E. O. (1953a). The amino-acid sequence in the glycyl chain of insulin. I. The identification of lower peptides from partial hydrolysates. Biochem. J. 53, 353–366.
Sanger, F., and Thompson, E. O. (1953b). The amino-acid sequence in the glycyl chain of insulin. II. The investigation of peptides from enzymic hydrolysates. Biochem. J. 53, 366–374.
Sanger, F., and Tuppy, H. (1951a). The amino-acid sequence in the phenylalanyl chain of insulin. 1. The identification of lower peptides from partial hydrolysates. Biochem. J. 49, 463–481.
Sanger, F., and Tuppy, H. (1951b). The amino-acid sequence in the phenylalanyl chain of insulin. 2. The investigation of peptides from enzymic hydrolysates. Biochem. J. 49, 481–490.
Schäffer, L. (1994). A model for insulin binding to the insulin receptor. Eur. J. Biochem. 221, 1127–1132.
Shabanpoor, F., Separovic, F., and Wade, J. D. (2009). The human insulin superfamily of polypeptide hormones. Vitam. Horm. 80, 1–31.
Siddle, K. (2011). Signalling by insulin and IGF receptors: supporting acts and new players. J. Mol. Endocrinol. 47, R1–R10.
Sjögren, B., and Svedberg, T. (1931). The molecular weight of insulin. J. Am. Chem. Soc. 53, 2657–2661.
Smeekens, S. P., Avruch, A. S., LaMendola, J., Chan, S. J., and Steiner, D. F. (1991). Identification of a cDNA encoding a second putative prohormone convertase related to PC2 in AtT20 cells and islets of Langerhans. Proc. Natl. Acad. Sci. U.S.A. 88, 340–344.
Smeekens, S. P., and Steiner, D. F. (1990). Identification of a human insulinoma cDNA encoding a novel mammalian protein structurally related to the yeast dibasic processing protease Kex2. J. Biol. Chem. 265, 2997–3000.
Smith, B. J., Huang, K., Kong, G., Chan, S. J., Nakagawa, S., Menting, J. G., Hu, S.-Q., Whittaker, J., Steiner, D. F., Katsoyannis, P. G., Ward, C. W., Weiss, M. A., and Lawrence, M. C. (2010). Structural resolution of a tandem hormone-binding element in the insulin receptor and its implications for design of peptide agonists. Proc. Natl. Acad. Sci. U.S.A. 107, 6771–6776.
Soos, M. A., Siddle, K., Baron, M. D., Heward, J. M., Luzio, J. P., Bellatin, J., and Lennox, E. S. (1986). Monoclonal antibodies reacting with multiple epitopes on the human insulin receptor. Biochem. J. 235, 199–208.
Sparrow, L. G., Gorman, J. J., Strike, P. M., Robinson, C. P., McKern, N. M., Epa, V. C., and Ward, C. W. (2007a). The location and characterisation of the O-linked glycans of the human insulin receptor. Proteins Struct. Funct. Bioinform. 66, 261–265.
Sparrow, L. G., Lawrence, M. C., Gorman, J. J., Strike, P. M., Robinson, C. P., McKern, N. M., and Ward, C. W. (2007b). N-linked glycans of the human insulin receptor and their distribution over the crystal structure. Proteins Struct. Funct. Bioinform. 71, 426–439.
Sparrow, L. G., McKern, N. M., Gorman, J. J., Strike, P. M., Robinson, C. P., Bentley, J. D., and Ward, C. W. (1997). The disulfide bonds in the C-terminal domains of the human insulin receptor ectodomain. J. Biol. Chem. 272, 29460–29467.
Steiner, D. F. (2011). Adventures with Insulin in the Islets of Langerhans. J. Biol. Chem. 286, 17399–17241.
Steiner, D. F., Cunningham, D., Spigelman, L., and Aten, B. (1967). Insulin biosynthesis: evidence for a precursor. Science 157, 697–700.
Steiner, D. F., and Oyer, P. E. (1967). The biosynthesis of insulin and a probable precursor of insulin by a human islet cell adenoma. Proc. Natl. Acad. Sci. U.S.A. 57, 473–480.
Sun, X. J., Rothenberg, P., Kahn, C. R., Backer, J. M., Araki, E., Wilden, P. A., Cahill, D. A., Goldstein, B. J., and White, M. F. (1991). Structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature 352, 73–77.
Surinya, K. H., Molina, L., Soos, M. A., Brandt, J., Kristensen, C., and Siddle, K. (2002). Role of insulin receptor dimerization domains in ligand binding, cooperativity, and modulation by anti-receptor antibodies. J. Biol. Chem. 277, 16718–16725.
Ullrich, A., Bell, J. R., Chen, E. Y., Herrera, R., Petruzzelli, L. M., Dull, T. J., Gray, A., Coussens, L., Liao, Y. C., Tsubokawa, M., Mason, A., Seeburg, P. H., Grunfeld, C., Rosen, O. M., and Ramachandran, J. (1985). Human insulin receptor and its relationship to the tyrosine kinase family of oncogenes. Nature 313, 756–761.
Ullrich, A., Gray, A., Tam, A. W., Yang-Feng, T., Tsubokawa, M., Collins, C., Henzel, W., Le Bon, T., Kathuria, S., Chen, E., Jaobs, S., Francke, U., Ramachandran, J., and Fujita-Yamaguchi, Y. (1986). Insulin-like growth factor I receptor primary structure: comparison with insulin receptor suggests structural determinants that define functional specificity. EMBO J. 5, 2503–2512.
Ward, C., Lawrence, M., Streltsov, V., Garrett, T., McKern, N., Lou, M. Z., Lovrecz, G., and Adams, T. (2008). Structural insights into ligand-induced activation of the insulin receptor. Acta Physiol. (Oxf.) 192, 3–9.
Ward, C. W., Gough, K. H., Rashke, M., Wan, S. S., Tribbick, G., and Wang, J. (1996). Systematic mapping of potential binding sites for Shc and Grb2 SH2 domains on insulin receptor substrate-1 and the receptors for insulin, epidermal growth factor, platelet-derived growth factor, and fibroblast growth factor. J. Biol. Chem. 271, 5603–5609.
Ward, C. W., Hoyne, P. A., and Flegg, R. H. (1995). Insulin and epidermal growth factor receptors contain the cysteine repeat motif found in the tumor necrosis factor receptor. Proteins 22, 141–153.
Ward, C. W., and Lawrence, M. C. (2009). Ligand-induced activation of the insulin receptor: a multi-step process involving structural changes in both the ligand and the receptor. Bioessays 31, 422–434.
Ward, C. W., Lawrence, M. C., Streltsov, V. A., Adams, T. E., and McKern, N. M. (2007). The insulin and EGF receptor structures: new insights into ligand-induced receptor activation. Trends Biochem. Sci. 32, 129–137.
Weiss, M. A. (2009). The structure and function of insulin decoding the TR transition. Vitam. Horm. 80, 33–49.
White, M. F., Maron, R., and Kahn, C. R. (1985). Insulin rapidly stimulates tyrosine phosphorylation of a Mr-185,000 protein in intact cells. Nature 318, 183–186.
Wolf, G., Trub, T., Ottinger, E., Groninga, L., Lynch, A., White, M. F., Miyazaki, M., Lee, J., and Shoelson, S. E. (1995). PTB domains of IRS-1 and Shc have distinct but overlapping binding specificities. J. Biol. Chem. 270, 27407–27410.
Xu, B., Hu, S. Q., Chu, Y. C., Huang, K., Nakagawa, S. H., Whittaker, J., Katsoyannis, P. G., and Weiss, M. A. (2004). Diabetes-associated mutations in insulin: consecutive residues in the B chain contact distinct domains of the insulin receptor. Biochemistry 43, 8356–8372.
Xu, B., Huang, K., Chu, Y. C., Hu, S. Q., Nakagawa, S., Wang, S., Wang, R. Y., Whittaker, J., Katsoyannis, P. G., and Weiss, M. A. (2009). Decoding the cryptic active conformation of a protein by synthetic photo-scanning. Insulin inserts a detachable arm between receptor domains. J. Biol. Chem. 284, 14597–14608.
Keywords: insulin, insulin receptor, tyrosine kinase receptor
Citation: Ward CW and Lawrence MC (2011) Landmarks in insulin research. Front. Endocrin. 2:76. doi: 10.3389/fendo.2011.00076
Received: 26 September 2011;
Paper pending published: 11 October 2011;
Accepted: 31 October 2011;
Published online: 22 November 2011.
Edited by:
Briony Forbes, The University of Adelaide, AustraliaReviewed by:
Jeff S. Davies, Swansea University, UKBriony Forbes, The University of Adelaide, Australia
Andrzej Marek Brzozowski, University of York, UK
Copyright: © 2011 Ward and Lawrence. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Michael C. Lawrence, Walter and Eliza Hall Institute, 1G Royal Parade, Parkville, VIC 3052, Australia. e-mail:bGF3cmVuY2VAd2VoaS5lZHUuYXU=