
 Latrice D. Faulkner2
Latrice D. Faulkner2
- 1 Department of Pediatrics, Children’s Nutrition Research Center, Baylor College of Medicine, Houston, TX, USA
- 2 Department of Physiology and Pharmacology, Center for Diabetes and Endocrine Research, College of Medicine, The University of Toledo, Toledo, OH, USA
- 3 Department of Obstetrics and Gynecology, College of Medicine, The University of Toledo, Toledo, OH, USA
Energy homeostasis and reproduction require tight coordination, but the mechanisms underlying their interaction are not fully understood. Two sets of hypothalamic neurons, namely pro-opiomelanocortin (POMC) neurons in the arcuate nucleus and steroidogenic factor-1 (SF1) neurons in the ventromedial hypothalamic nucleus, are emerging as critical nodes where metabolic and reproductive signals communicate. This view is supported by recent genetic studies showing that disruption of metabolic signals (e.g., leptin and insulin) or reproductive signals (e.g., estradiol) in these neurons leads to impaired regulation of both energy homeostasis and fertility. In this review, we will examine the potential mechanisms of neuronal communication between POMC, SF1, and gonadotropin-releasing hormone neurons in the regulation of metabolism and reproduction.
Introduction
Since animals under metabolic stress must invest their energy in survival rather than reproduction, the reproductive axis has the capacity to respond to changes in caloric status. The hypothalamic signals driving the reproductive axis are suppressed when a mammal is in negative energy balance, whether that state is caused by inadequate food intake, excessive locomotor activity, or heavy thermoregulatory costs. Likewise, the energy demands of maintaining fertility and successful reproduction require increased food consumption and appropriate regulation of energy expenditure. Therefore, hypothalamic control of metabolism must be responsive to the reproductive state of the animal. However, despite the passage of 40 years since the discovery of gonadotropin-releasing hormone (GnRH; Schally et al., 1971), the afferent neuronal groups and pathways through which gonadal steroids and nutrient signals regulate GnRH release remain unresolved.
Much attention has focused on the role of hypothalamic neurons expressing kisspeptin in coordinating GnRH neuronal function and the physiological state of the animal. However, while the central role of kisspeptin in steroid feedback to the hypothalamus is clear, evidence of it conveying metabolic signals to the reproductive axis is equivocal. In addition, data suggest that other hypothalamic neurons also convey gonadal steroid input to GnRH circuitry, either directly or via the kisspeptin network.
Here we will discuss new findings resulting from genetically modifying pro-opiomelanocortin (POMC) and steroidogenic factor-1 (SF1) neurons of the hypothalamus. While primarily understood to function as metabolic regulators, these neurons are emerging as critical nodes of communication that respond to both metabolic and reproductive cues and directly interact with reproductive circuitry. In particular, we will focus on their ability to transmit information gleaned from circulating factors, specifically leptin, insulin, and estradiol (E2).
Canonical Reproductive Circuits
Hypothalamic GnRH neurons produce the final output of a complex neuronal system regulating fertility. Adult mammals possess a loose field of GnRH neurons stretching from the olfactory bulbs to the medial basal hypothalamus with a highly dense population of GnRH neurons within the preoptic area (POA) and adjacent to the organum vasculosum of the lamina terminalis (OVLT). The axons of GnRH neurons project to the median eminence (ME) where GnRH is secreted in pulses into the pituitary portal bloodstream. GnRH neurons can possess dendrites extending millimeters away from their cell bodies (Campbell et al., 2005; Cottrell et al., 2006), with the average extending over 550 μm (Roberts et al., 2006). Interestingly, many GnRH dendrites follow routes similar to those of their axons toward the ME. Dendrites of GnRH neurons frequently initiate action potentials due to their expression of voltage-gated sodium channels (Rhodes and Llinas, 2005). As a result of the morphology of these dendrites, highly proficient action potential initiation in the distal dendrites is possible even when synaptic potentials are quite small (Witkin and Silverman, 1985; Witkin et al., 1995). Distal portions of the GnRH dendrite, for instance segments located in the arcuate nucleus of the hypothalamus (ARC), thus provide synaptic input and can potently affect GnRH hormone secretion (Campbell and Suter, 2010; Zoli et al., 1998; Herbison, 2006).
A great deal of the recent focus on afferent control of GnRH secretion has been on kisspeptin neurons in the hypothalamus, based on the ability of mutations of the kisspeptin receptor to cause hypogonadotropic hypogonadism in humans and animal models (de Roux et al., 2003; Seminara et al., 2003). Kisspeptin exerts an extremely powerful and long-lasting depolarizing stimulus upon GnRH neurons (Gottsch et al., 2004; Han et al., 2005; Messager et al., 2005; Pielecka-Fortuna et al., 2008) and plays a central role in the physiological regulation of GnRH release (Oakley et al., 2009; Ohkura et al., 2009). Confocal images have shown kisspeptin terminals in direct apposition to GnRH cell bodies (Clarkson and Herbison, 2006; Ramaswamy et al., 2008). In addition, in the rat and monkey, kisspeptin terminals form close contacts with GnRH terminals in the ME (Krajewski et al., 2005, 2010; Ciofi et al., 2006; Decourt et al., 2008; Lehman et al., 2010). Recent studies on the stimulatory effects of kisspeptin are consistent with actions at both the ME (d’Anglemont de Tassigny et al., 2008) and GnRH cell bodies in the POA (Pielecka-Fortuna et al., 2008; Moenter and Pielecka-Fortuna, 2010). Two populations of kisspeptin neurons exist in the hypothalamus; one is located in the anteroventral periventricular nucleus (AVPV; Smith et al., 2005), and the other is located in the ARC. Most ARC kisspeptin neurons co-express neurokinin B (NKB) and dynorphin (Dyn) in rat (Burke et al., 2006), mouse (Navarro et al., 2009), sheep (Goodman et al., 2007), goat (Wakabayashi et al., 2010), and possibly human (Rance, 2009) leading to these neurons acquiring the moniker of KNDy neurons (Cheng et al., 2010). Each of these three neuropeptides has been strongly implicated in the feedback regulation of GnRH neurons (Rance and Young, 1991; Sahu and Kalra, 1992; Rance and Bruce, 1994; Goodman et al., 2004; Foradori et al., 2005). Since the kisspeptin network is not the focus of this review, readers interested in this topic are referred to some excellent recent publications (Oakley et al., 2009; Navarro and Tena-Sempere, 2011).
POMC Neurons Link Metabolic and Reproductive Circuits
POMC Neurons Innervate Reproductive Circuits
Besides kisspeptin neurons, other hypothalamic populations may also provide afferent signals on the hypothalamo-pituitary gonadal (HPG) axis to regulate reproduction. Particularly, recent evidence suggests that hypothalamic sites that regulate energy balance have important inputs to GnRH neurons. One example is POMC neurons in the ARC. POMC neurons have long been believed to be a primary central regulator of energy homeostasis (Huszar et al., 1997; Cone, 1999). The anorexigenic property of POMC neurons has been well established, as the deletion of POMC gene causes hyperphagia and obesity (Yaswen et al., 1999).
Evidence also suggests that POMC neurons innervate the reproductive circuits in the central nervous system (CNS). For example, POMC neurons make direct synaptic contact with GnRH neurons (Leranth et al., 1988; Thind and Goldsmith, 1988; Chen et al., 1989a). The major neurotransmitters released from POMC neurons include two POMC gene products: the anorectic peptide α-melanocyte-stimulating hormone (α-MSH) and the endogenous opioid β-endorphin (Cheung et al., 1997; Broberger et al., 1998; Hahn et al., 1998; Vrang et al., 1999). Fibers specifically immunoreactive for β-endorphin have been identified within the immediate vicinity of GnRH neurons, and based upon electron microscopic evidence, the β-endorphin-immunoreactive terminals synapse on the GnRH neuron soma in the rat (Leranth et al., 1988; Chen et al., 1989b), sheep (Goodman et al., 2004), and monkey (Thind and Goldsmith, 1988). About 20–30% of POMC neurons in the ARC co-express estrogen receptor-α (ERα; Lehman et al., 1993; de Souza et al., 2011; Xu et al., 2011), and an ERα-positive subpopulation of ARC POMC neurons has been shown to project to the POA, where GnRH neurons are concentrated (Simonian et al., 1999). Furthermore, GnRH neurons express receptors for β-endorphin. For example, μ-opioid receptors, have been indentified in GnRH neurons of the guinea pig (Lagrange et al., 1995). In addition, δ-opioid receptors have been identified in mouse GnRH neuron-derived GT-1 cells and in a fraction of rat GnRH nerve terminals, including in the ME (Pimpinelli et al., 2006). Collectively, these findings indicate that POMC neurons are well positioned to provide synaptic inputs to GnRH neurons.
At the functional level, neurotransmitters released from POMC neurons have been shown to regulate the HPG axis. In both monkeys and rats, β-endorphin and other opioids exert inhibitory effects on GnRH and LH secretion (Bruni et al., 1977; Kinoshita et al., 1982; Gilbeau et al., 1985; Leadem and Kalra, 1985b,a; Wiesner et al., 1985; Leadem and Yagenova, 1987; Wardlaw and Ferin, 1990; Kalra and Kalra, 1996). This inhibitory effects appear to be tonic, as administration of naloxone, an opiate receptor antagonist, to rats increases LH production (Babu et al., 1987). Reducing the opioid inhibition also facilitates the production of the GnRH surge on proestrus (Lustig et al., 1988; Masotto et al., 1990; Hashimoto and Kimura, 1991; Zhang and Gallo, 2002). This inhibition by opioids can be interpreted as increased overall suppression of GnRH and LH release or as augmentation of steroid negative feedback. Indeed, opioid antagonism can also stimulate GnRH release independent of E2 levels (Babu et al., 1987; Karahalios and Levine, 1988; Goodman et al., 1995). However, during the rat estrous cycle, β-endorphin levels fluctuate in the ARC, POA, and ME. The maximum levels are seen during diestrus while the lowest occurs on proestrus leading up to the LH surge (Gallo and Drouva, 1979), suggesting a role in negative feedback. While these studies strongly support an inhibitory effect of opioids on GnRH release, genetic evidence is less convincing. Mutant mice lacking the classical endogenous opioids (dynorphin, enkephalin, and endorphin; Konig et al., 1996; Rubinstein et al., 1996; Sharifi et al., 2001) as well as mutants of the three opioid receptors (Matthes et al., 1996; Sora et al., 1997; Roy et al., 1998; Simonin et al., 1998; Schuller et al., 1999; Zhu et al., 1999; Filliol et al., 2000) are fertile, although μ-opioid receptor-deficient mice display reduced spermatogenesis and impaired sexual function (Tian et al., 1997). Thus, at least when absent during the development of hypothalamic circuitry, opioid inhibition of GnRH release is not required for fertility.
In contrast, it has been demonstrated that α-MSH exerts an excitatory effect on the GnRH system, likely by acting on central melanocortin receptors (MCs; Celis, 1985; Backholer et al., 2009). A robust connection has recently been demonstrated between MCH neurons, which express MC4 receptors and receive direct input from POMC neurons, and GnRH neurons (Wu et al., 2009). Agouti-related peptide (AgRP), an endogenous receptor antagonist of MCs, decreases the magnitude of the LH surges in normally fed rats (Watanobe et al., 1999; Schioth et al., 2001; Schioth and Watanobe, 2002), while anti-AgRP serum partially but significantly enlarges the LH surge (Schioth et al., 2001). Indeed, melanocortin 4 receptor (MC4R) deficient mice exhibit erectile dysfunction and changed sexual behavior in males secondary to obesity (Van der Ploeg et al., 2002) and reduced ovulation rates and fertility accompanied by increased follicular atresia by 6 months of age in females (Sandrock et al., 2009). Furthermore, agouti mice, which congenitally overproduce AgRP, have adult-onset infertility (Granholm et al., 1986). Collectively, these findings point to α-MSH as a potential afferent signal to the HPG axis.
In addition to neuropeptides, POMC neurons release classical neurotransmitters GABA and glutamate (Hentges et al., 2004, 2009), both of which have been shown to regulate GnRH neurons (Kusano et al., 1995; Shepherd, 1996; Spergel et al., 1999; Sim et al., 2000; Sorra and Harris, 2000; Simonian and Herbison, 2001; DeFazio et al., 2002; Fiala et al., 2002; Han et al., 2002; Kuehl-Kovarik et al., 2002; Ottem et al., 2002; Moenter and DeFazio, 2005). In addition to direct synaptic inputs, POMC neurons may also influence GnRH neurons via kisspeptin intermediary neurons. Supporting this possibility, kisspeptin fibers have been shown in close apposition with ARC POMC neurons in ewes (Backholer et al., 2009, 2010), and double-label fluorescent immunohistochemistry showed that reciprocal connections exist between kisspeptin neurons and POMC neurons (Backholer et al., 2010).
The studies reviewed above emphasize the critical “choice” of POMC neurons to express β-endorphin or α-MSH. The former, possibly acting in concert with other opioids such as dynorphin (via its own kappa-opioid receptor; Navarro et al., 2009), is intimately involved in the negative feedback regulation of GnRH release. The latter, through activation of second-order metabolic circuitry, is involved in the gating of fertility during times of energy deprivation. The control of β-endorphin vs. α-MSH production by POMC neurons is an area of ongoing study (Wardlaw, 2011).
Leptin and Insulin Act on POMC Neurons to Regulate both Reproduction and Energy Homeostasis
Emerging evidence indicates that POMC neurons respond to metabolic cues to provide coordinated control of metabolism and reproduction. One example of these metabolic cues is leptin. Leptin is a circulating adiposity-related factor that informs the CNS regarding energy stores. Released by adipocytes when stored fat is plentiful, leptin acts in the hypothalamus to suppress body weight gain and to improve insulin sensitivity (Morton et al., 2003, 2005; Balthasar et al., 2004; Coppari et al., 2005; Dhillon et al., 2006; van de Wall et al., 2008). Mice lacking leptin or leptin receptors (LepRs) develop hyperphagic morbid obesity, insulin resistant diabetes, and hypothermia (Coleman, 1978). Leptin reduces food intake and body weight when administered to leptin-null mice (Campfield et al., 1995; Halaas et al., 1995; Pelleymounter et al., 1995), and brain-specific deletion of LepRs leads to obesity (Cohen et al., 2001).
Leptin is also a prerequisite for pubertal development and successful reproduction. Humans and mice carrying leptin gene mutations fail to go through puberty, have low LH levels, and are infertile (Montague et al., 1997), and leptin administration, but not weight loss alone, allows pubertal progression and restores their fertility (Barash et al., 1996; Chehab et al., 1996; Mounzih et al., 1997; Ziotopoulou et al., 2000). Leptin also overrides the fasting-induced suppression of LH secretion and fertility (Nagatani et al., 1998, 2000; Gonzalez et al., 1999; Kohsaka et al., 1999). In anorectic females and in athletes with extreme decreases in body adiposity, leptin can increase levels of luteinizing hormone (Licinio et al., 1998) and restore the menstrual cycle (Welt et al., 2004). Re-expression of LepRs in the brain of LepR-null mice restores fertility completely in males and partially in females (Kowalski et al., 2001; de Luca et al., 2005). In addition, AAV-induced expression of the LepR gene in the POA or ARC of LepR-null rats normalizes their estrous cycle length and increases GnRH concentrations in the hypothalamus (Keen-Rhinehart et al., 2005). Collectively, this evidence indicates that leptin, while primarily acting as a metabolic signal to maintain normal energy and glucose homeostasis, also plays essential roles in reproduction.
Insulin, another circulating factor related to adiposity, is also implicated in the coordinated control of metabolism and reproduction. Insulin levels in the circulation are proportional to adipose tissue in most mammals (Woods et al., 1979). Intracerebroventricular (icv) insulin administration results in a dose-dependent reduction in food intake and body weight (Woods et al., 1979), and neuron-specific deletion of insulin receptors (IRs) leads to increased body fat deposition (Bruning et al., 2000).
A variety of mouse models have demonstrated insulin’s essential role in the central control of reproduction. For example, increased circulating levels of insulin during a hyperinsulinemic clamp stimulate LH secretion (Burcelin et al., 2003a). In addition, mice lacking IRs in the brain exhibit decreased spermatogenesis and follicular maturation, resulting in only 42–46% of matings successfully producing offspring (Bruning et al., 2000). The primary deficit in these mice was found to be a reduction in GnRH release and consequent reductions in pituitary gonadotropin secretion and gonadal function. In another study, expression of IRs in liver and pancreas alone was sufficient to maintain fertility in males, but females also required IR expression in the brain (Okamoto et al., 2004). Finally, a study of IR substrate 2 (IRS-2) knockout mice found that only 9% of IRS-2−/− females and 89% of IRS-2−/− males were fertile. IRS-2−/− females showed a supranormal response to GnRH, consistent with hypothalamic hypogonadism (Burks et al., 2000). These studies show that, particularly in the female, IRs in the brain are required for fertility.
While it is clear that both leptin and insulin could signal the brain to coordinate energy status and reproductive demands, the exact brain sites where these signals are integrated remained unclear. GnRH neurons do not appear to be the direct target of these hormones. For example, double-labeling experiments using in situ hybridization and immunohistochemistry have shown few GnRH neurons, if any, to express LepRs in rats (Burcelin et al., 2003a) and monkeys (Finn et al., 1998). Insulin has a direct stimulatory effect on the output of GnRH in hypothalamic cells in vitro and in vivo (Kovacs et al., 2002; Burcelin et al., 2003b). However, mice lacking IRs specifically in GnRH neurons displayed normal pubertal timing and fertility (Divall et al., 2010), suggesting that the insulin responsiveness of GnRH neurons is low. While early reports suggested that 40% of kisspeptin mRNA-expressing cells in the ARC of mice express LepRs (Smith et al., 2006), other laboratories have found fewer than 5% of kisspeptin neurons exhibit LepRs (Donato et al., 2011; Louis et al., 2011). The latter results appear to be borne out by the lack of a reproductive phenotype in mice with a targeted deletion of LepRs from kisspeptin neurons (Donato et al., 2011). Similarly, our preliminary data suggest that insulin sensing directly by kisspeptin neurons plays a minor role in mouse fertility (Qiu et al., 2011). Thus, leptin/insulin sensing outside of the dedicated reproductive circuitry is likely to play a role in their effects on reproduction.
POMC neurons are well positioned to be a direct target of leptin and insulin signals. POMC neurons express LepRs (Cheung et al., 1997; Elmquist et al., 1998; Baskin et al., 1999) and IRs (Benoit et al., 2002). LepRs in POMC neurons mediate a portion of leptin actions on energy homeostasis, as mice lacking LepRs specifically in POMC neurons are mildly obese and hyperleptinemic (Balthasar et al., 2004). Although deletion of IRs from POMC neurons does not affect body weight (Konner et al., 2007), simultaneous deletion of IRs and LepRs from POMC neurons produces more severe insulin resistance and diabetes than deletion of each individual receptor alone (Hill et al., 2010). Therefore, POMC neurons appear to be one important site where insulin and leptin signals interact to regulate energy and glucose homeostasis.
Our recent studies also pinpointed POMC neurons as a target of leptin/insulin actions important for fertility. We have reported that female mice lacking both leptin and IRs in POMC neurons (IR/LepRPOMC) exhibit lengthened reproductive cycles, follicular arrest, hyperandrogenemia, and infertility. These mice lack IRs and LepRs in POMC-expressing cells in the hypothalamus and pituitary corticotrophs and melanotrophs, but retain them in other cell types and tissues, such as liver and ovary (Hill et al., 2010). These results were confirmed by an absence of altered IR and LepR expression in these tissues using qPCR. Despite the expression of POMC in corticotrophs, we found no alteration in corticosterone release. Pup numbers born to experimental females older than 4 months were significantly reduced (Hill et al., 2010). These females also showed a lengthened estrous cycle. In addition, the percentage of matings not producing a litter was higher for experimental females across all maternal ages. These reproductive deficits were not caused by abnormal prolactin or E2 levels, and no pups born to IR/LepRPOMC dams died after birth. While hypothalamic GnRH gene expression was comparable among the groups, LH levels were significantly increased in IR/LepRPOMC females. Histological examination of their ovaries showed that double knockout females exhibited more degenerating follicles. Serum testosterone levels were significantly elevated in experimental females, accompanied by a significant elevation in the expression of ovarian 3β-HSD I gene, which produces androstenedione. CYP17 gene expression was also slightly increased (p = 0.0530; Hill et al., 2010). Interestingly, males also exhibit reduced numbers of successful pairings with wild-type females despite an enthusiastic mounting response and increased testes weights (Figures 1A–C). In addition, a subset of males exhibited dramatically increased LH levels (Figure 1D) with normal FSH concentrations (data not shown). The heterogeneity in these mice may be due to their mixed strain background. Collectively, these results suggest that the absence of leptin and insulin signaling in POMC neurons may reduce the inhibitory opioid tone on GnRH neurons and cause basal LH levels to increase, disrupting reproductive function. Indeed, the absence of leptin and insulin signaling would both be expected to reduce β-endorphin production from its POMC precursor (Wardlaw, 2011). Further studies are required to determine whether β-endorphin production in these mice is reduced, and the signaling mechanisms involved.
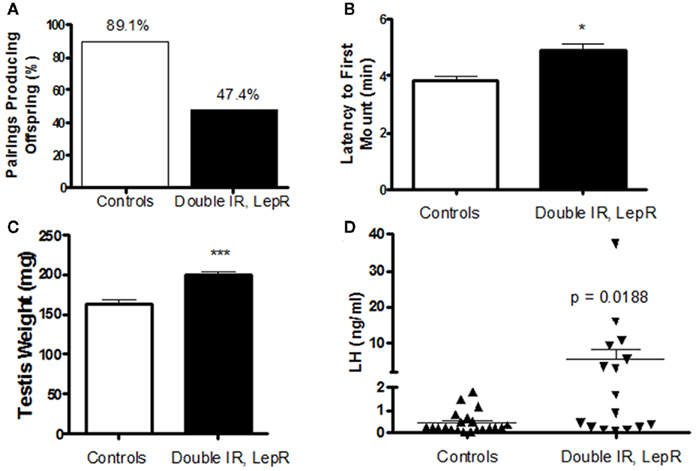
Figure 1. Reproductive phenotype of 4-month-old IR, LepRPOMC mice. (A) Male mice lacking insulin and leptin receptors in POMC neurons were paired with genetically normal control females and the number of pairings producing pups were quantified. (B) The amount of time before experimental and control males attempted to mount a novel control female was quantified. Latency was scored during a 10-min period after male was introduced to the cage of a singly housed female in Proestrus under red lighting during the first half hour after lights out (1800 h). (C). Testes weights were measured at time of sacrifice. (D). Serum levels of luteinizing hormone were measured by RIA by the University of Virginia Ligand Assay and Analysis Core.
Estrogens Act on POMC Neurons to Regulate both Reproduction and Energy Homeostasis
Steroid hormones such as E2 exert potent feedback at both the neural and pituitary levels to regulate the HPG axis. During much of the female reproductive cycle, E2 reduces the GnRH pulse amplitude (Sarkar and Fink, 1980; Caraty et al., 1989; Chongthammakun and Terasawa, 1993; Evans et al., 1994) and inhibits LH release via negative feedback actions on the hypothalamus as well as the pituitary gonadotropes (Shupnik et al., 1988; Shupnik, 1996). A similar mechanism appears to be at work in males via aromatization of testosterone to E2 (Veldhuis and Dufau, 1987; Finkelstein et al., 1991a,b; Bagatell et al., 1994; Hayes et al., 2000; Schnorr et al., 2001). In the course of the later stage of the follicular phase, circulating E2 levels rise resulting in a biphasic effect on GnRH secretion, causing a noticeable suppression of pulsatile GnRH and LH secretion, followed by an induction of a high amplitude GnRH surge supplementary to the LH surge, a process termed positive feedback (Crowder and Nett, 1984; Moenter et al., 1990). Lesion studies performed in the rat and hamster (Shander and Barraclough, 1980; Wiegand et al., 1980) have implicated anatomically distinct brain regions for the conveyance of the positive and negative feedback. These studies localized negative feedback to the ARC and ME and positive feedback to the POA and the suprachiasmatic nucleus.
Both positive and negative feedback regulation are primarily mediated by ERα, but not by estrogen receptor-β (ERβ). This is demonstrated by observations that global deletion of ERα in mice abolishes the positive and negative feedback responses, while mice with global ERβ deficiency appear to retain normal feedback responses (Couse et al., 1995, 2003; Wintermantel et al., 2006). However, the sites where estrogens act to regulate the HPG axis have not been fully revealed. Wintermantel et al. (2006) demonstrated that female mice lacking ERα only in the brain show blunted positive feedback and impaired fertility. While these findings identified brain ERα as the key mediator for the positive feedback, the exact ERα populations in the brain responsible for this regulation remain unknown. Singh et al. (2009) showed that deletion of ERα in the pituitary LH cells blunts E2-induced inhibition on LH secretion and results in infertility in female mice. Therefore, pituitary ERα is at least partly responsible for the negative feedback, but redundant ERα populations likely exist in the brain. Given that GnRH neurons do not appear to express ERα (Shivers et al., 1983), interneurons must exist to relay E2/ERα signals on GnRH neurons.
Recent evidence suggests that POMC neurons may serve as interneurons mediating E2 actions. First, POMC neurons co-express ERα (Miller et al., 1995; de Souza et al., 2011), suggesting that POMC neurons could be the direct target of estrogenic actions. E2 treatment increases POMC expression (Hammer et al., 1994; Cheung and Hammer, 1997). β-endorphin positive fibers and opioid receptors in the POA also increase upon E2 treatment (Hammer et al., 1994). In addition, E2 regulates the excitability of POMC neurons. Using electron microscopy, Gao et al. (2007) reported that the number of excitatory synaptic inputs to ARC POMC neurons rises as mice enter proestrus when E2 levels are high. Further, central E2 administration rapidly increases the excitatory synapses on POMC neurons, an effect that is also reflected by increased miniature excitatory postsynaptic current recorded from POMC neurons (Gao et al., 2007). Similarly, Malyala et al. (2008) reported that E2 administration in hypothalamic slices increases excitability of POMC neurons by rapidly uncoupling GABAB receptors from their G-protein-gated inwardly rectifying K+ channels. These studies demonstrated that E2 directly acts on POMC neurons and regulates their cellular activity.
The physiological relevance of E2 sensing by POMC neurons was further established by our recent findings in mice lacking ERα specifically in POMC neurons. First, we found that mutant females have a modest increase in plasma E2, raising the possibility of impaired negative feedback on the HGP axis (Xu et al., 2011). Thus, while E2 replacement suppresses FSH and LH expression in the pituitary of ovariectomized (OVX) wild-type mice, this E2-induced suppression is blunted in OVX mutant mice (Xu et al., 2011). Interestingly, only 30% of female mice lacking ERα in POMC neurons successfully delivered pups (Xu et al., 2011). The averaged size of litters from these 30% mutant dams was significantly reduced compared to those from control dams (Xu et al., 2011). In addition, the mating time required for these mutant females to conceive was significantly increased (Xu et al., 2011). These findings indicate that E2/ERα signals in POMC neurons are at least partially required to mediate the negative feedback regulation of the HPG axis and to maintain normal fertility.
Besides being a reproductive cue, estrogens also exert important anti-obesity effects in women (Flegal et al., 2002; Freedman et al., 2002; Carr, 2003) and female mammals (Drewett, 1973; Blaustein and Wade, 1976; Wallen et al., 2001; Rogers et al., 2009). E2 reduces food intake and body adiposity and increase energy expenditure in animals and humans of both sexes through a hypothalamic mechanism (Dubuc, 1985; Wade et al., 1985; Hess et al., 1997; Heine et al., 2000). Effects of E2 on energy balance are primarily mediated by ERα, as women or female mice with mutations in the ERα gene display hyperadiposity (Heine et al., 2000; Okura et al., 2003), whereas ERβ-null mice have no body weight phenotype (Ohlsson et al., 2000). Interestingly, we recently found that a portion of anti-obesity effects of estrogens are mediated by ERα expressed by POMC neurons. For example, we found that female mice lacking ERα only in POMC neurons develop chronic hyperphagia and increased body weight (Xu et al., 2011). In addition, the leptin-induced suppression in food intake was blunted in female mice lacking ERα in POMC neurons (Xu et al., 2011). Collectively, these findings indicate that E2/ERα signals within POMC neurons are not only important to mediate the negative feedback and maintain normal reproduction, but also are physiologically relevant in the regulation of feeding behavior. Therefore, E2 signals within POMC neurons may coordinate the regulation of energy balance and reproductive demands.
SF1 Neurons Link Metabolic and Reproductive Circuits
SF1 Neurons Innervate Reproductive Circuits
In addition to POMC neurons in the ARC, steroidogenic factor-1 (SF1) neurons, located in the ventromedial hypothalamic nucleus (VMH) may serve as an important connection point relaying metabolic and reproductive cues.
The physiological relevance of VMH neurons to the regulation of body weight homeostasis is well recognized. Ikeda et al. (1995) discovered that a transcription factor, SF1, is expressed exclusively in the VMH neurons within the brain. SF1 neurons constitute the majority of VMH neurons (Stallings et al., 2002). During early development, SF1 is essential for the formation of the VMH architecture, as mice with embryonic deletion of SF1 gene do not form a VMH (Dellovade et al., 2000). These SF1 knockout mice develop massive obesity (Majdic et al., 2002). It is important to note that SF1 is also abundant in a number of endocrine organs, such as the pituitary gland, the adrenal gland and gonads (Zhao et al., 2001). Therefore, the obesity phenotype in global SF1 knockout mice may have been confounded by the dysfunctions of these tissues (Majdic et al., 2002). To circumvent this issue, a brain-specific SF1 knockout mouse line was generated to determine the role of VMH SF1 neurons in the context of body weight control (Kim et al., 2011). This brain-specific SF1 deletion also leads to disruption of the VMH structure and obesity in mice (Kim et al., 2011). Thus, these findings demonstrate that VMH neurons are physiologically important for the regulation of energy homeostasis.
VMH neurons may also regulate the central reproductive circuits. For example, the VMH has well defined projections to the medial central gray and periaqueductal gray regions that have been implicated in lordosis (Canteras et al., 1994). In addition, VMH neurons are also found to project to GnRH neurons (Boehm et al., 2005; Yoon et al., 2005). Further, numerous studies have suggested that VMH neurons regulate sexual behaviors (Blaustein and Feder, 1980; Rubin and Barfield, 1983a,b; Canteras et al., 1994; Mani et al., 1997; Sinchak et al., 2007). Mice with genetic ablation of VMH SF1 neurons show a significantly blunted lordosis quotient and receptivity (Kim et al., 2010). The role of the VMH in reproduction may be more extensive than just behavior. For example, the mutant female mice with genetic ablation of VMH SF1 neurons show severely irregular estrus cycles, and are infertile or subfertile (Kim et al., 2010). The impaired female fertility is likely due to defective ovulation, as demonstrated by decreased or absent corpora lutea in the ovary (Kim et al., 2010). Interestingly, exogenous administration of gonadotropins induced normal ovulation in these mice, demonstrating that the ovaries are functionally intact (Kim et al., 2010). Further, when the mutant females were stimulated with a synthetic GnRH agonist after priming, they exhibited markedly reduced LH secretion compared with wild-type littermates, arguing that disorganization in and around the VMH caused by SF1 ablation interferes with the GnRH priming process or gonadotrope LH capacity (Kim et al., 2010).
Collectively, these findings indicate that functional VMH SF1 neurons are required to maintain not only normal energy balance but also reproduction. Therefore, these SF1 neurons could serve as an import point of intersection for these two systems.
Estrogens Act on SF1 Neurons to Regulate both Reproduction and Energy Homeostasis
E2 acts in the VMH to regulate energy balance. Abundant ERα is found in the ventrolateral subdivision of the VMH (Osterlund et al., 1998; Merchenthaler et al., 2004; Schlenker and Hansen, 2006). To determine whether ERα in the VMH mediate the anti-obesity effects of estrogens, Musatov and colleagues used shRNA-mediated gene silencing approach to knock-down ERα in the VMH in female rodents. They found that VMH-specific ERα knock-down leads to obesity and metabolic syndrome primarily due to decreased energy expenditure (Musatov et al., 2007). We recently crossed ERα floxed mice (Feng et al., 2007) to SF1-Cre transgenic mice (Dhillon et al., 2006), which resulted in mice lacking ERα specifically in VMH SF1 neurons (Xu et al., 2011). This SF1-specific deletion of ERα results in modest body weight gain and hyperadiposity solely due to decreased energy expenditure in female mice (Xu et al., 2011). We further demonstrate that both basal metabolic rate and diet-induced thermogenesis in these mice are reduced, while the energy expenditure associated with physical activity is not altered (Xu et al., 2011). Interestingly, female mice lacking ERα in SF1 neurons also show increased visceral fat distribution, while the subcutaneous fat distribution is reduced (Xu et al., 2011). Consistent with this abdominal obesity, these mice predictably develop glucose intolerance (Xu et al., 2011). Collectively, our findings support a model in which E2 acts on ERα in VMH SF1 neurons to stimulate energy expenditure and inhibit visceral fat expansion. As we also found that norepinephrine is decreased in mice lacking ERα in SF1 neurons (Xu et al., 2011), the effects of E2/ERα in SF1 neurons on energy expenditure and fat distribution are presumably mediated by elevated sympathetic outflow.
The actions of E2 in VMH neurons are also important for reproduction. For example, administration of E2 in the VMH has been shown to modulate female sexual behaviors (Rubin and Barfield, 1983a). In addition, E2 actions in the VMH may mediate induction of progesterone receptors in the VMH (McGinnis et al., 1981; Olster and Blaustein, 1989; Kalra, 1993; Moffatt et al., 1998), thereby permitting progesterone to reduce basal GnRH and LH release (Chappell et al., 1997). The role of VMH E2 sensing in reproduction was further supported by our findings from mice lacking ERα only in SF1 neurons. These mutant female mice have irregular estrus cycles (Figure 2A). Further, most of mutant females (9 out of 10 mice tested) are infertile (Figure 2B). The only mutant dam that successfully conceived and delivered had smaller litter size than controls (Figure 2C). The impaired fertility in these females is likely due to anovulation, demonstrated by the lack of corpora lutea in the ovaries (Figure 2D). One caveat of this model is that ERα may also be deleted from SF1 cells in the pituitary, adrenal gland, and gonads, in addition to SF1 neurons in the VMH, which makes it difficult to attribute the fertility and ovarian phenotypes solely to the loss of ERα in SF1 neurons in the VMH. However, we did not found any significant reduction of ERα mRNA in the pituitary, adrenal gland, and ovaries from the mutant mice (Xu et al., 2011), which argues that the infertility/subfertility and anovulation phenotypes are likely due to the loss of ERα in VMH neurons. Interestingly, the brain-specific ERα knockout model shows the same fertility and ovarian phenotypes as in our mice lacking ERα in SF1 cells (Wintermantel et al., 2006), suggesting that ERα in the brain, such as in VMH SF1 neurons, is required to trigger ovulation in the ovaries. Collectively, these findings support a model in which adequate E2 signaling in VMH SF1 neurons is required both for fertility and normal energy balance.
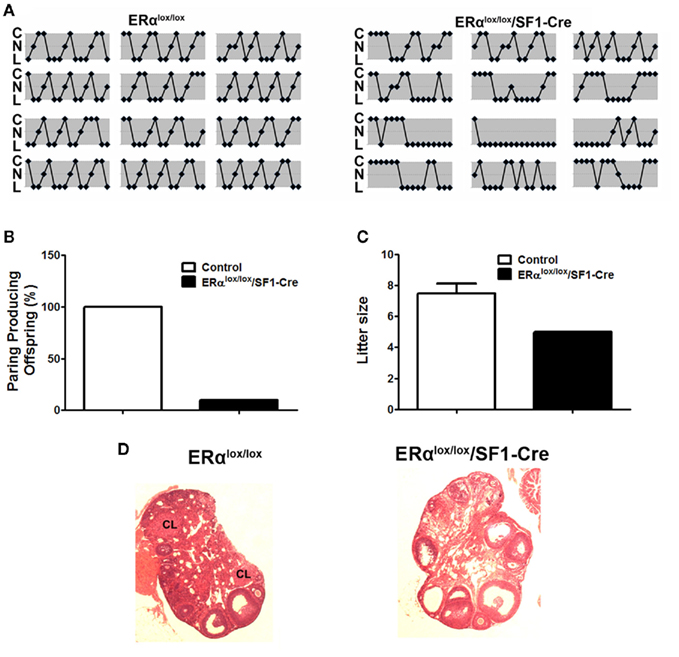
Figure 2. Reproductive phenotype of female mice lacking ERα in SF1 neurons (ERαlox/lox/SF1–Cre). (A). Estrus cycles from 12-week-old female mice lacking ERα in SF1 neurons and their wild-type controls were evaluated by vaginal cytology. Note: C, cornified cells (estrus); N, nucleated epithelial cells (proestrus); L, leukocytes (diestrus). (B). Female mice lacking ERα in SF1 neurons and their wild-type controls were paired with genetically normal control males and the numbers of pairings producing pups were quantified. (C). Litter size from dams that successfully delivered pups in (B). (D) H&E staining of ovaries from adult female mice. There was no corpus luteum in the ovary from mice lacking ERα in SF1 neurons.
Conclusion
While POMC and SF1 neurons have previously been thought to serve primarily in the control of food intake and energy balance, new genetic rodent models have highlighted their crucial roles in the maintenance of fertility. While additional work is needed to clarify the mechanisms by which these neuronal circuits modulate the GnRH system, these studies highlight the profound integration of reproductive and metabolic control. In particular, POMC and SF1 neurons are emerging as critical sites that both respond to circulating factors and directly interact with reproductive circuitry. Future studies of POMC and SF1 neurons may shed light on the nature of positive and negative steroid feedback as well as integration of signals of adiposity in the control of the reproductive axis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the NIH (R00DK085330, 1R01DK093587, and P30 DK079638-03 to Yong Xu, and R00HD056491 to Jennifer Wootton Hill), from American Diabetes Association (Yong Xu), from the Naman Family Fund for Basic Research and the Curtis Hankamer Basic Research Fund (Yong Xu).
References
Babu, G., NMarco, J., Bona-Gallo, A., and Gallo, R. V. (1987). Steroid-independent endogenous opioid peptide suppression of pulsatile luteinizing hormone release between estrus and diestrus in the rat estrous cycle. Brain Res. 416, 235–242.
Backholer, K., Smith, J., and Clarke, I. J. (2009). Melanocortins may stimulate reproduction by activating orexin neurons in the dorsomedial hypothalamus and kisspeptin neurons in the preoptic area of the ewe. Endocrinology 150, 5488–5497.
Backholer, K., Smith, J. T., Rao, A., Pereira, A., Iqbal, J., Ogawa, S., Li, Q., and Clarke, I. J. (2010). Kisspeptin cells in the ewe brain respond to leptin and communicate with neuropeptide Y and proopiomelanocortin cells. Endocrinology 151, 2233–2243.
Backholer, K., Smith, J. T., Rao, A., Pereira, A., Iqbal, J., Ogawa, S., Li, Q., and Clarke, I. J. (2010). Kisspeptin cells in the ewe brain respond to leptin and communicate with neuropeptide Y and proopiomelanocortin cells. Endocrinology 151, 2233–2243.
Bagatell, C. J., Dahl, K. D., and Bremner, W. J. (1994). The direct pituitary effect of testosterone to inhibit gonadotropin secretion in men is partially mediated by aromatization to estradiol. J. Androl. 15, 15–21.
Balthasar, N., Coppari, R., McMinn, J., Liu, S. M., Lee, C. E., Tang, V., Kenny, C. D., McGovern, R. A., Chua, S. C. Jr., Elmquist, J. K., and Lowell, B. B. (2004). Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron 42, 983–991.
Barash, I. A., Cheung, C. C., Cheung, C. C., Weigle, D. S., Ren, H., Kabigting, E. B., Kuijper, J. L., Clifton, D. K., and Steiner, R. A. (1996). Leptin is a metabolic signal to the reproductive system. Endocrinology 137, 3144–3147.
Baskin, D. G., Breininger, J. F., and Schwartz, M. W. (1999). Leptin receptor mRNA identifies a subpopulation of neuropeptide Y neurons activated by fasting in rat hypothalamus. Diabetes 48, 828–833.
Benoit, S. C., Air, E. L., Coolen, L. M., Strauss, R., Jackman, A., Clegg, D. J., Seeley, R. J., and Woods, S. C. (2002). The catabolic action of insulin in the brain is mediated by melanocortins. J. Neurosci. 22, 9048–9052.
Blaustein, J. D., and Feder, H. H. (1980). Nuclear progestin receptors in guinea pig brain measured by an in vitro exchange assay after hormonal treatments that affect lordosis. Endocrinology 106, 1061–1069.
Blaustein, J. D., and Wade, G. N. (1976). Ovarian influences on the meal patterns of female rats. Physiol. Behav. 17, 201–208.
Boehm, U., Zou, Z., and Buck, L. B. (2005). Feedback loops link odor and pheromone signaling with reproduction. Cell 123, 683–695.
Broberger, C., Johansen, J., Johansson, C., Schalling, M., and Hökfelt, T. (1998). The neuropeptide Y/agouti gene-related protein (AGRP) brain circuitry in normal, anorectic, and monosodium glutamate-treated mice. Proc. Natl. Acad. Sci. U.S.A. 95, 15043–15048.
Bruni, J. F., Van Vugt, D., Marshall, S., and Meites, J. (1977). Effects of naloxone, morphine and methionine enkephalin on serum prolactin, luteinizing hormone, follicle stimulating hormone, thyroid stimulating hormone and growth hormone. Life Sci. 21, 461–466.
Bruning, J. C., Gautam, D., Burks, D. J., Gillette, J., Schubert, M., Orban, P. C., Klein, R., Krone, W., Müller-Wieland, D., and Kahn, C. R. (2000). Role of brain insulin receptor in control of body weight and reproduction. Science 289, 2122–2125.
Burcelin, R., Thorens, B., Thorens, B., Glauser, M., Gaillard, R. C., and Pralong, F. P. (2003a). Gonadotropin-releasing hormone secretion from hypothalamic neurons: stimulation by insulin and potentiation by leptin. Endocrinology 144, 4484–4491.
Burcelin, R., Thorens, B., Glauser, M., Gaillard, R. C., and Pralong, F. P. (2003b). Gonadotropin-releasing hormone secretion from hypothalamic neurons: stimulation by insulin and potentiation by leptin. Endocrinology 144, 4484–4491.
Burke, M. C., Letts, P. A., Krajewski, S. J., and Rance, N. E. (2006). Coexpression of dynorphin and neurokinin B immunoreactivity in the rat hypothalamus: morphologic evidence of interrelated function within the arcuate nucleus. J. Comp. Neurol. 498, 712–726.
Burks, D. J., Font de Mora, J., Schubert, M., Withers, D. J., Myers, M. G., Towery, H. H., Altamuro, S. L., Flint, C. L., and White, M. F. (2000). IRS-2 pathways integrate female reproduction and energy homeostasis. Nature 407, 377–382.
Campbell, R. E., Han, S. K., and Herbison, A. E. (2005). Biocytin filling of adult gonadotropin-releasing hormone neurons in situ reveals extensive, spiny, dendritic processes. Endocrinology 146, 1163–1169.
Campbell, R. E., and Suter, K. J. (2010). Redefining the gonadotropin-releasing hormone neuron dendrite. J. Neuroendocrinol. 22, 650–658.
Campfield, L. A., Smith, F. J., Guisez, Y., Devos, R., and Burn, P. (1995). Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks. Science 269, 546–549.
Canteras, N. S., Simerly, R. B., and Swanson, L. W. (1994). Organization of projections from the ventromedial nucleus of the hypothalamus: a Phaseolus vulgaris-leucoagglutinin study in the rat. J. Comp. Neurol. 348, 41–79.
Caraty, A., Locatelli, A., and Martin, G. B. (1989). Biphasic response in the secretion of gonadotropin-releasing hormone in ovariectomized ewes injected with oestradiol. J. Endocrinol. 123, 375–382.
Carr, M. C. (2003). The emergence of the metabolic syndrome with menopause. J. Clin. Endocrinol. Metab. 88, 2404–2411.
Celis, M. E. (1985). Release of LH in response to alpha-MSH administration. Acta Physiol. Pharmacol. Latinoam. 35, 281–290.
Chappell, P. E., Lydon, J. P., Conneely, O. M., O’Malley, B. W., and Levine, J. E. (1997). Endocrine defects in mice carrying a null mutation for the progesterone receptor gene. Endocrinology 138, 4147–4152.
Chehab, F. F., Lim, M. E., and Lu, R. (1996). Correction of the sterility defect in homozygous obese female mice by treatment with the human recombinant leptin. Nat. Genet. 12, 318–320.
Chen, W. P., Witkin, J. W., and Silverman, A. J. (1989a). Gonadotropin-releasing hormone (Gnrh) neurons are directly innervated by catecholamine terminals. Synapse 3, 288–290.
Chen, W. P., Witkin, J. W., and Silverman, A. J. (1989b). beta-Endorphin and gonadotropin-releasing hormone synaptic input to gonadotropin-releasing hormone neurosecretory cells in the male rat. J. Comp. Neurol. 286, 85–95.
Cheng, G., Coolen, L. M., Padmanabhan, V., Goodman, R. L., and Lehman, M. N. (2010). The kisspeptin/neurokinin B/dynorphin (KNDy) cell population of the arcuate nucleus: sex differences and effects of prenatal testosterone in sheep. Endocrinology 151, 301–311.
Cheung, C. C., Clifton, D. K., and Steiner, R. A. (1997). Proopiomelanocortin neurons are direct targets for leptin in the hypothalamus. Endocrinology 138, 4489–4492.
Cheung, S., and Hammer, R. P. Jr. (1997). Gonadal steroid hormone regulation of proopiomelanocortin gene expression in neurons that innervate the median eminence of the rat. Neurosci. Lett. 224, 181–184.
Chongthammakun, S., and Terasawa, E. (1993). Negative feedback effects of E2 on luteinizing hormone-releasing hormone release occur in pubertal, but not prepubertal, ovariectomized female rhesus monkeys. Endocrinology 132, 735–743.
Ciofi, P., Leroy, D., and Tramu, G. (2006). Sexual dimorphism in the organization of the rat hypothalamic infundibular area. Neuroscience 141, 1731–1745.
Clarkson, J., and Herbison, A. E. (2006). Postnatal development of kisspeptin neurons in mouse hypothalamus; sexual dimorphism and projections to gonadotropin-releasing hormone neurons. Endocrinology 147, 5817–5825.
Cohen, P., Zhao, C., Cai, X., Montez, J. M., Rohani, S. C., Feinstein, P., Mombaerts, P., and Friedman, J. M. (2001). Selective deletion of leptin receptor in neurons leads to obesity. J. Clin. Invest. 108, 1113–1121.
Coleman, D. L. (1978). Obese and diabetes: two mutant genes causing diabetes-obesity syndromes in mice. Diabetologia 14, 141–148.
Cone, R. D. (1999). The central melanocortin system and energy homeostasis. Trends Endocrinol. Metab. 10, 211–216.
Coppari, R., Ichinose, M., Lee, C. E., Pullen, A. E., Kenny, C. D., McGovern, R. A., Tang, V., Liu, S. M., Ludwig, T., Chua, S. C. Jr., Lowell, B. B., and Elmquist, J. K. (2005). The hypothalamic arcuate nucleus: a key site for mediating leptin’s effects on glucose homeostasis and locomotor activity. Cell Metab. 1, 63–72.
Cottrell, E. C., Campbell, R. E., Han, S. K., and Herbison, A. E. (2006). Postnatal remodeling of dendritic structure and spine density in gonadotropin-releasing hormone neurons. Endocrinology 147, 3652–3661.
Couse, J. F., Curtis, S. W., Washburn, T. F., Eddy, E. M., Schomberg, D. W., and Korach, K. S. (1995). Disruption of the mouse oestrogen receptor gene: resulting phenotypes and experimental findings. Biochem. Soc. Trans. 23, 929–935.
Couse, J. F., Yates, M. M., Walker, V. R., and Korach, K. S. (2003). Characterization of the hypothalamic-pituitary-gonadal axis in estrogen receptor (ER) Null mice reveals hypergonadism and endocrine sex reversal in females lacking ERalpha but not ERbeta. Mol. Endocrinol. 17, 1039–1053.
Crowder, M. E., and Nett, T. M. (1984). Pituitary content of gonadotropins and receptors for gonadotropin-releasing hormone (GnRH) and hypothalamic content of GnRH during the periovulatory period of the ewe. Endocrinology 114, 234–239.
d’Anglemont de Tassigny, X., Fagg, L. A., Carlton, M. B., and Colledge, W. H. (2008). Kisspeptin can stimulate gonadotropin-releasing hormone (GnRH) release by a direct action at GnRH nerve terminals. Endocrinology 149, 3926–3932.
de Luca, C., Kowalski, T. J., Zhang, Y., Elmquist, J. K., Lee, C., Kilimann, M. W., Ludwig, T., Liu, S. M., and Chua, S. C. Jr. (2005). Complete rescue of obesity, diabetes, and infertility in db/db mice by neuron-specific LEPR-B transgenes. J. Clin. Invest. 115, 3484–3493.
de Roux, N., Genin, E., Carel, J. C., Matsuda, F., Chaussain, J. L., and Milgrom, E. (2003). Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc. Natl. Acad. Sci. U.S.A. 100, 10972–10976.
de Souza, F. S., Nasif, S., López-Leal, R., Levi, D. H., Low, M. J., and Rubinsten, M. (2011). The estrogen receptor alpha colocalizes with proopiomelanocortin in hypothalamic neurons and binds to a conserved motif present in the neuron-specific enhancer nPE2. Eur. J. Pharmacol. 660, 181–187.
Decourt, C., Tillet, Y., Caraty, A., Franceschini, I., and Briant, C. (2008). Kisspeptin immunoreactive neurons in the equine hypothalamus Interactions with GnRH neuronal system. J. Chem. Neuroanat. 36, 131–137.
DeFazio, R. A., Heger, S., Ojeda, S. R., and Moenter, S. M. (2002). Activation of A-type gamma-aminobutyric acid receptors excites gonadotropin-releasing hormone neurons. Mol. Endocrinol. 16, 2872–2891.
Dellovade, T. L., Young, M., Ross, E. P., Henderson, R., Caron, K., Parker, K., and Tobet, S. A. (2000). Disruption of the gene encoding SF-1 alters the distribution of hypothalamic neuronal phenotypes. J. Comp. Neurol. 423, 579–589.
Dhillon, H., Zigman, J. M., Ye, C., Lee, C. E., McGovern, R. A., Tang, V., Kenny, C. D., Christiansen, L. M., White, R. D., Edelstein, E. A., Coppari, R., Balthasar, N., Cowley, M. A., Chua, S. Jr., Elmquist, J. K., and Lowell, B. B. (2006). Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron 49, 191–203.
Divall, S. A., Williams, T. R., Carver, S. E., Koch, L., Brüning, J. C., Kahn, C. R., Wondisford, F., Radovick, S., and Wolfe, A. (2010). Divergent roles of growth factors in the GnRH regulation of puberty in mice. J. Clin. Invest. 120, 2900–2909.
Donato, J. Jr., Cravo, R. M., Frazão, R., Gautron, L., Scott, M. M., Lachey, J., Castro, I. A., Margatho, L. O., Lee, S., Lee, C., Richardson, J. A., Friedman, J., Chua, S. Jr., Coppari, R., Zigman, J. M., Elmquist, J. K., and Elias, C. F. (2011). Leptin’s effect on puberty in mice is relayed by the ventral premammillary nucleus and does not require signaling in Kiss1 neurons. J. Clin. Invest. 121, 355–368.
Drewett, R. F. (1973). Sexual behaviour and sexual motivation in the female rat. Nature 242, 476–477.
Dubuc, P. U. (1985). Effects of E2 on food intake, body weight, and temperature of male and female obese mice. Proc. Soc. Exp. Biol. Med. 180, 468–473.
Elmquist, J. K., Bjørbaek, C., Ahima, R. S., Flier, J. S., and Saper, C. B. (1998). Distributions of leptin receptor mRNA isoforms in the rat brain. J. Comp. Neurol. 395, 535–547.
Evans, N. P., Dahl, G. E., Glover, B. H., and Karsch, F. J. (1994). Central regulation of pulsatile gonadotropin-releasing hormone (GnRH) secretion by estradiol during the period leading up to the preovulatory GnRH surge in the ewe. Endocrinology 134, 1806–1811.
Feng, Y., Manka, D., Wagner, K. U., and Khan, S. A. (2007). Estrogen receptor-alpha expression in the mammary epithelium is required for ductal and alveolar morphogenesis in mice. Proc. Natl. Acad. Sci. U.S.A. 104, 14718–14723.
Fiala, J. C., Spacek, J., and Harris, K. M. (2002). Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res. Brain Res. Rev. 39, 29–54.
Filliol, D., Ghozland, S., Chluba, J., Martin, M., Matthes, H. W., Simonin, F., Befort, K., Gavériaux-Ruff, C., Dierich, A., LeMeur, M., Valverde, O., Maldonado, R., and Kieffer, B. L. (2000). Mice deficient for delta- and mu-opioid receptors exhibit opposing alterations of emotional responses. Nat. Genet. 25, 195–200.
Finkelstein, J. S., Whitcomb, R. W., O’Dea, L. S., Longcope, C., Schoenfeld, D. A., and Crowley, W. F. Jr. (1991a). Sex steroid control of gonadotropin secretion in the human male. I. Effects of testosterone administration in normal and gonadotropin-releasing hormone-deficient men. J. Clin. Endocrinol. Metab. 73, 609–620.
Finkelstein, J. S., O’Dea, L. S., Whitcomb, R. W., and Crowley, W. F. Jr. (1991b). Sex steroid control of gonadotropin secretion in the human male. II. Effects of estradiol administration in normal and gonadotropin-releasing hormone-deficient men. J. Clin. Endocrinol. Metab. 73, 621–628.
Finn, P. D., Cunningham, M. J., Pau, K. Y., Spies, H. G., Clifton, D. K., and Steiner, R. A. (1998). The stimulatory effect of leptin on the neuroendocrine reproductive axis of the monkey. Endocrinology 139, 4652–4662.
Flegal, K. M., Carroll, M. D., Ogden, C. L., and Johnson, C. L. (2002). Prevalence and trends in obesity among US adults, 1999–2000. JAMA 288, 1723–1727.
Foradori, C. D., Goodman, R. L., Adams, V. L., Valent, M., and Lehman, M. N. (2005). Progesterone increases dynorphin a concentrations in cerebrospinal fluid and preprodynorphin messenger ribonucleic acid levels in a subset of dynorphin neurons in the sheep. Endocrinology 146, 1835–1842.
Freedman, D. S., Khan, L. K., Serdula, M. K., Galuska, D. A., and Dietz, W. H. (2002). Trends and correlates of class 3 obesity in the United States from 1990 through 2000. JAMA 288, 1758–1761.
Gallo, R. V., and Drouva, S. V. (1979). Effect of intraventricular infusion of catecholamines on luteinizing hormone release in ovariectomized and ovariectomized, steroid-primed rats. Neuroendocrinology 29, 149–162.
Gao, Q., Mezei, G., Nie, Y., Rao, Y., Choi, C. S., Bechmann, I., Leranth, C., Toran-Allerand, D., Priest, C. A., Roberts, J. L., Gao, X. B., Mobbs, C., Shulman, G. I., Diano, S., and Horvath, T. L. (2007). Anorectic E2 mimics leptin’s effect on the rewiring of melanocortin cells and Stat3 signaling in obese animals. Nat. Med. 13, 89–94.
Gilbeau, P. M., Almirez, R. G., Holaday, J. W., and Smith, C. G. (1985). Opioid effects on plasma concentrations of luteinizing hormone and prolactin in the adult male rhesus monkey. J. Clin. Endocrinol. Metab. 60, 299–305.
Gonzalez, L. C., Pinilla, L., Tena-Sempere, M., and Aguilar, E. (1999). Leptin(116–130) stimulates prolactin and luteinizing hormone secretion in fasted adult male rats. Neuroendocrinology 70, 213–220.
Goodman, R. L., Coolen, L. M., Anderson, G. M., Hardy, S. L., Valent, M., Connors, J. M., Fitzgerald, M. E., and Lehman, M. N. (2004). Evidence that dynorphin plays a major role in mediating progesterone negative feedback on gonadotropin-releasing hormone neurons in sheep. Endocrinology 145, 2959–2967.
Goodman, R. L., Lehman, M. N., Smith, J. T., Coolen, L. M., de Oliveira, C. V., Jafarzadehshirazi, M. R., Pereira, A., Iqbal, J., Caraty, A., Ciofi, P., and Clarke, I. J. (2007). Kisspeptin neurons in the arcuate nucleus of the ewe express both dynorphin a and neurokinin B. Endocrinology 148, 5752–5760.
Goodman, R. L., Parfitt, D. B., Evans, N. P., Dahl, G. E., and Karsch, F. J. (1995). Endogenous opioid peptides control the amplitude and shape of gonadotropin-releasing hormone pulses in the ewe. Endocrinology 136, 2412–2420.
Gottsch, M. L., Cunningham, M. J., Smith, J. T., Popa, S. M., Acohido, B. V., Crowley, W. F., Seminara, S., Clifton, D. K., and Steiner, R. A. (2004). A role for kisspeptins in the regulation of gonadotropin secretion in the mouse. Endocrinology 145, 4073–4077.
Granholm, N. H., Jeppesen, K. W., and Japs, R. A. (1986). Progressive infertility in female lethal yellow mice (Ay/a strain C57bl/6j). J. Reprod. Fertil. 76, 279–287.
Hahn, T. M., Breininger, J. F., Baskin, D. G., and Schwartz, M. W. (1998). Coexpression of Agrp and NPY in fasting-activated hypothalamic neurons. Nat. Neurosci. 1, 271–272.
Halaas, J. L., Gajiwala, K. S., Maffei, M., Cohen, S. L., Chait, B. T., Rabinowitz, D., Lallone, R. L., Burley, S. K., and Friedman, J. M. (1995). Weight-reducing effects of the plasma protein encoded by the obese gene. Science 269, 543–546.
Hammer, R. P. Jr., Zhou, L., and Cheung, S. (1994). Gonadal steroid hormones and hypothalamic opioid circuitry. Horm. Behav. 28, 431–437.
Han, S. K., Abraham, I. M., and Herbison, A. E. (2002). Effect of GABA on GnRH neurons switches from depolarization to hyperpolarization at puberty in the female mouse. Endocrinology 143, 1459–1466.
Han, S. K., Gottsch, M. L., Lee, K. J., Popa, S. M., Smith, J. T., Jakawich, S. K., Clifton, D. K., Steiner, R. A., and Herbison, A. E. (2005). Activation of gonadotropin-releasing hormone neurons by kisspeptin as a neuroendocrine switch for the onset of puberty. J. Neurosci. 25, 11349–11356.
Hashimoto, R., and Kimura, F. (1991). Effects of intraventricular injection of delta receptor antagonist ICI 154, 129 on the secretion of luteinizing hormone and prolactin in the proestrous rat. Endocrinol. Jpn. 38, 213–218.
Hayes, F. J., Seminara, S. B., Decruz, S., Boepple, P. A., and Crowley, W. F. Jr. (2000). Aromatase inhibition in the human male reveals a hypothalamic site of E2 feedback. J. Clin. Endocrinol. Metab. 85, 3027–3035.
Heine, P. A., Taylor, J. A., Iwamoto, G. A., Lubahn, D. B., and Cooke, P. S. (2000). Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proc. Natl. Acad. Sci. U.S.A. 97, 12729–12734.
Hentges, S. T., Nishiyama, M., Overstreet, L. S., Stenzel-Poore, M., Williams, J. T., and Low, M. J. (2004). GABA release from proopiomelanocortin neurons. J. Neurosci. 24, 1578–1583.
Hentges, S. T., Otero-Corchon, V., Pennock, R. L., King, C. M., and Low, M. J. (2009). Proopiomelanocortin expression in both GABA and glutamate neurons. J. Neurosci. 29, 13684–13690.
Herbison, A. (2006). “Physiology of the gonadotropin-releasing hormone neuronal network,” in Knobil and Neil’s Physiology of Reproduction, 3rd Edn, ed. J. D. Neill (St. Louis, MO: Raven Press), 1415–1482.
Hess, R. A., Bunick, D., Lee, K. H., Bahr, J., Taylor, J. A., Korach, K. S., and Lubahn, D. B. (1997). A role for oE2s in the male reproductive system. Nature 390, 509–512.
Hill, J. W., Elias, C. F., Fukuda, M., Williams, K. W., Berglund, E. D., Holland, W. L., Cho, Y. R., Chuang, J. C., Xu, Y., Choi, M., Lauzon, D., Lee, C. E., Coppari, R., Richardson, J. A., Zigman, J. M., Chua, S., Scherer, P. E., Lowell, B. B., Brüning, J. C., and Elmquist, J. K. (2010). Direct insulin and leptin action on pro-opiomelanocortin neurons is required for normal glucose homeostasis and fertility. Cell Metab. 11, 286–297.
Huszar, D., Lynch, C. A., Fairchild-Huntress, V., Dunmore, J. H., Fang, Q., Berkemeier, L. R., Gu, W., Kesterson, R. A., Boston, B. A., Cone, R. D., Smith, F. J., Campfield, L. A., Burn, P., and Lee, F. (1997). Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell 88, 131–141.
Ikeda, Y., Luo, X., Abbud, R., Nilson, J. H., and Parker, K. L. (1995). The nuclear receptor steroidogenic factor 1 is essential for the formation of the ventromedial hypothalamic nucleus. Mol. Endocrinol. 9, 478–486.
Kalra, S. P. (1993). Mandatory neuropeptide-steroid signaling for the preovulatory luteinizing hormone-releasing hormone discharge. Endocr. Rev. 14, 507–538.
Kalra, S. P., and Kalra, P. S. (1996). Nutritional infertility: the role of the interconnected hypothalamic neuropeptide Y-galanin-opioid network. Front. Neuroendocrinol. 17, 371–401.
Karahalios, D. G., and Levine, J. E. (1988). Naloxone stimulation of in vivo LHRH release is not diminished following ovariectomy. Neuroendocrinology 47, 504–510.
Keen-Rhinehart, E., Kalra, S. P., and Kalra, P. S. (2005). AAV-mediated leptin receptor installation improves energy balance and the reproductive status of obese female Koletsky rats. Peptides 26, 2567–2578.
Kim, K. W., Li, S., Zhao, H., Peng, B., Tobet, S. A., Elmquist, J. K., Parker, K. L., and Zhao, L. (2010). CNS-specific ablation of steroidogenic factor 1 results in impaired female reproductive function. Mol. Endocrinol. 24, 1240–1250.
Kim, K. W., Zhao, L., Donato, J. Jr., Kohno, D., Xu, Y., Elias, C. F., Lee, C., Parker, K. L., and Elmquist, J. K. (2011). Steroidogenic factor 1 directs programs regulating diet-induced thermogenesis and leptin action in the ventral medial hypothalamic nucleus. Proc. Natl. Acad. Sci. U.S.A. 108, 10673–10678.
Kinoshita, F., Nakai, Y., Katakami, H., and Imura, H. (1982). Suppressive effect of dynorphin-(1-13) on luteinizing hormone release in conscious castrated rats. Life Sci. 30, 1915–1919.
Kohsaka, A., Watanobe, H., Kakizaki, Y., Habu, S., and Suda, T. (1999). A significant role of leptin in the generation of steroid-induced luteinizing hormone and prolactin surges in female rats. Biochem. Biophys. Res. Commun. 254, 578–581.
Konig, M., Zimmer, A. M., Steiner, H., Holmes, P. V., Crawley, J. N., Brownstein, M. J., and Zimmer, A. (1996). Pain responses, anxiety and aggression in mice deficient in pre-proenkephalin. Nature 383, 535–538.
Konner, A. C., Janoschek, R., Plum, L., Jordan, S. D., Rother, E., Ma, X., Xu, C., Enriori, P., Hampel, B., Barsh, G. S., Kahn, C. R., Cowley, M. A., Ashcroft, F. M., and Brüning, J. C. (2007). Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab. 5, 438–449.
Kovacs, P., Parlow, A. F., and Karkanias, G. B. (2002). Effect of centrally administered insulin on gonadotropin-releasing hormone neuron activity and luteinizing hormone surge in the diabetic female rat. Neuroendocrinology 76, 357–365.
Kowalski, T. J., Liu, S. M., Leibel, R. L., and Chua, S. C. Jr. (2001). Transgenic complementation of leptin-receptor deficiency. I. Rescue of the obesity/diabetes phenotype of LEPR-null mice expressing a LEPR-B transgene. Diabetes 50, 425–435.
Krajewski, S. J., Anderson, M. J., Iles-Shih, L., Chen, K. J., Urbanski, H. F., and Rance, N. E. (2005). Morphologic evidence that neurokinin B modulates gonadotropin-releasing hormone secretion via neurokinin 3 receptors in the rat median eminence. J. Comp. Neurol. 489, 372–386.
Krajewski, S. J., Burke, M. C., Anderson, M. J., McMullen, N. T., and Rance, N. E. (2010). Forebrain projections of arcuate neurokinin B neurons demonstrated by anterograde tract-tracing and monosodium glutamate lesions in the rat. Neuroscience 166, 680–697.
Kuehl-Kovarik, M. C., Pouliot, W. A., Halterman, G. L., Handa, R. J., Dudek, F. E., and Partin, K. M. (2002). Episodic bursting activity and response to excitatory amino acids in acutely dissociated gonadotropin-releasing hormone neurons genetically targeted with green fluorescent protein. J. Neurosci. 22, 2313–2322.
Kusano, K., Fueshko, S., Gainer, H., and Wray, S. (1995). Electrical and synaptic properties of embryonic luteinizing hormone-releasing hormone neurons in explant cultures. Proc. Natl. Acad. Sci. U.S.A. 92, 3918–3922.
Lagrange, A. H., Rønnekleiv, O. K., and Kelly, M. J. (1995). Estradiol-17 beta and mu-opioid peptides rapidly hyperpolarize GnRH neurons: a cellular mechanism of negative feedback? Endocrinology 136, 2341–2344.
Leadem, C. A., and Kalra, S. P. (1985a). Effects of endogenous opioid peptides and opiates on luteinizing hormone and prolactin secretion in ovariectomized rats. Neuroendocrinology 41, 342–352.
Leadem, C. A., and Kalra, S. P. (1985b). Reversal of beta-endorphin-induced blockade of ovulation and luteinizing hormone surge with prostaglandin E2. Endocrinology 117, 684–689.
Leadem, C. A., and Yagenova, S. V. (1987). Effects of specific activation of mu-, delta- and kappa-opioid receptors on the secretion of luteinizing hormone and prolactin in the ovariectomized rat. Neuroendocrinology 45, 109–117.
Lehman, M. N., Coolen, L. M., and Goodman, R. L. (2010). Minireview: kisspeptin/neurokinin B/dynorphin (KNDy) cells of the arcuate nucleus: a central node in the control of gonadotropin-releasing hormone secretion. Endocrinology 151, 3479–3489.
Lehman, M. N., Ebling, F. J., Moenter, S. M., and Karsch, F. J. (1993). Distribution of estrogen receptor-immunoreactive cells in the sheep brain. Endocrinology 133, 876–886.
Leranth, C., MacLusky, N. J., Shanabrough, M., and Naftolin, F. (1988). Immunohistochemical evidence for synaptic connections between pro-opiomelanocortin-immunoreactive axons and Lh-Rh neurons in the preoptic area of the rat. Brain Res. 449, 167–176.
Licinio, J., Negrao, A. B., Mantzoros, C., Kaklamani, V., Wong, M. L., Bongiorno, P. B., Mulla, A., Cearnal, L., Veldhuis, J. D., Flier, J. S., McCann, S. M., and Gold, P. W. (1998). Synchronicity of frequently sampled, 24-h concentrations of circulating leptin, luteinizing hormone, and estradiol in healthy women. Proc. Natl. Acad. Sci. U.S.A. 95, 2541–2546.
Louis, G. W., Greenwald-Yarnell, M., Phillips, R., Coolen, L. M., Lehman, M. N., and Myers, M. G. (2011). Molecular mapping of the neural pathways linking leptin to the neuroendocrine reproductive axis. Endocrinology 152, 2302–2310.
Lustig, R. H., Pfaff, D. W., and Fishman, J. (1988). Opioidergic modulation of the oestradiol-induced LH surge in the rat: roles of ovarian steroids. J. Endocrinol. 116, 55–69.
Majdic, G., Young, M., Gomez-Sanchez, E., Anderson, P., Szczepaniak, L. S., Dobbins, R. L., McGarry, J. D., and Parker, K. L. (2002). Knockout mice lacking steroidogenic factor 1 are a novel genetic model of hypothalamic obesity. Endocrinology 143, 607–614.
Malyala, A., Zhang, C., Bryant, D. N., Kelly, M. J., and Rønnekleiv, O. K. (2008). PI3K signaling effects in hypothalamic neurons mediated by E2. J. Comp. Neurol. 506, 895–911.
Mani, S. K., Blaustein, J. D., and O’Malley, B. W. (1997). Progesterone receptor function from a behavioral perspective. Horm. Behav. 31, 244–255.
Masotto, C., Sahu, A., Dube, M. G., and Kalra, S. P. (1990). A decrease in opioid tone amplifies the luteinizing hormone surge in E2-treated ovariectomized rats: comparisons with progesterone effects. Endocrinology 126, 18–25.
Matthes, H. W., Maldonado, R., Simonin, F., Valverde, O., Slowe, S., Kitchen, I., Befort, K., Dierich, A., Le Meur, M., Dollé, P., Tzavara, E., Hanoune, J., Roques, B. P., and Kieffer, B. L. (1996). Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature 383, 819–823.
McGinnis, M. Y., Krey, L. C., MacLusky, N. J., and McEwen, B. S. (1981). Steroid receptor levels in intact and ovariectomized E2-treated rats: an examination of quantitative, temporal and endocrine factors influencing the efficacy of an estradiol stimulus. Neuroendocrinology 33, 158–165.
Merchenthaler, I., Lane, M. V., Numan, S., and Dellovade, T. L. (2004). Distribution of estrogen receptor alpha and beta in the mouse central nervous system: in vivo autoradiographic and immunocytochemical analyses. J. Comp. Neurol. 473, 270–291.
Messager, S., Chatzidaki, E. E., Ma, D., Hendrick, A. G., Zahn, D., Dixon, J., Thresher, R. R., Malinge, I., Lomet, D., Carlton, M. B., Colledge, W. H., Caraty, A., and Aparicio, S. A. (2005). Kisspeptin directly stimulates gonadotropin-releasing hormone release via G protein-coupled receptor 54. Proc. Natl. Acad. Sci. U.S.A. 102, 1761–1766.
Miller, M. M., Tousignant, P., Yang, U., Pedvis, S., and Billiar, R. B. (1995). Effects of age and long-term ovariectomy on the E2-receptor containing subpopulations of beta-endorphin-immunoreactive neurons in the arcuate nucleus of female C57BL/6J mice. Neuroendocrinology 61, 542–551.
Moenter, S. M., Caraty, A., and Karsch, F. J. (1990). The estradiol-induced surge of gonadotropin-releasing hormone in the ewe. Endocrinology 127, 1375–1384.
Moenter, S. M., and DeFazio, R. A. (2005). Endogenous gamma-aminobutyric acid can excite gonadotropin-releasing hormone neurons. Endocrinology 146, 5374–5379.
Moenter, S. M., and Pielecka-Fortuna, J. (2010). Kisspeptin increases gamma-aminobutyric acidergic and glutamatergic transmission directly to gonadotropin-releasing hormone neurons in an estradiol-dependent manner. Endocrinology 151, 291–300.
Moffatt, C. A., Rissman, E. F., Shupnik, M. A., and Blaustein, J. D. (1998). Induction of progestin receptors by estradiol in the forebrain of estrogen receptor-alpha gene-disrupted mice. J. Neurosci. 18, 9556–9563.
Montague, C. T., Farooqi, I. S., Whitehead, J. P., Soos, M. A., Rau, H., Wareham, N. J., Sewter, C. P., Digby, J. E., Mohammed, S. N., Hurst, J. A., Cheetham, C. H., Earley, A. R., Barnett, A. H., Prins, J. B., and O’Rahilly, S. (1997). Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 387, 903–908.
Morton, G., Niswender, K. D., Rhodes, C. J., Myers, M. G. Jr., Blevins, J. E., Baskin, D. G., and Schwartz, M. W. (2003). Arcuate nucleus-specific leptin receptor gene therapy attenuates the obesity phenotype of Koletsky (fa(k)/fa(k)) rats. Endocrinology 144, 2016–2024.
Morton, G. J., Gelling, R. W., Niswender, K. D., Morrison, C. D., Rhodes, C. J., and Schwartz, M. W. (2005). Leptin regulates insulin sensitivity via phosphatidylinositol-3-OH kinase signaling in mediobasal hypothalamic neurons. Cell Metab. 2, 411–420.
Mounzih, K., Lu, R., and Chehab, F. F. (1997). Leptin treatment rescues the sterility of genetically obese ob/ob males. Endocrinology 138, 1190–1193.
Musatov, S., Chen, W., Pfaff, D. W., Mobbs, C. V., Yang, X. J., Clegg, D. J., Kaplitt, M. G., and Ogawa, S. (2007). Silencing of estrogen receptor alpha in the ventromedial nucleus of hypothalamus leads to metabolic syndrome. Proc. Natl. Acad. Sci. U.S.A. 104, 2501–2506.
Nagatani, S., Guthikonda, P., Thompson, R. C., Tsukamura, H., Maeda, K. I., and Foster, D. L. (1998). Evidence for GnRH regulation by leptin: leptin administration prevents reduced pulsatile LH secretion during fasting. Neuroendocrinology 67, 370–376.
Nagatani, S., Zeng, Y., Keisler, D. H., Foster, D. L., and Jaffe, C. A. (2000). Leptin regulates pulsatile luteinizing hormone and growth hormone secretion in the sheep. Endocrinology 141, 3965–3975.
Navarro, V. M., Gottsch, M. L., Chavkin, C., Okamura, H., Clifton, D. K., and Steiner, R. A. (2009). Regulation of gonadotropin-releasing hormone secretion by kisspeptin/dynorphin/neurokinin B neurons in the arcuate nucleus of the mouse. J. Neurosci. 29, 11859–11866.
Navarro, V. M., and Tena-Sempere, M. (2011). Kisspeptins and the neuroendocrine control of reproduction. Front. Biosci. (Schol. Ed.) 3, 267–275.
Oakley, A. E., Clifton, D. K., and Steiner, R. A. (2009). Kisspeptin signaling in the brain. Endocr. Rev. 30, 713–743.
Ohkura, S., Uenoyama, Y., Yamada, S., Homma, T., Takase, K., Inoue, N., Maeda, K., and Tsukamura, H. (2009). Physiological role of metastin/kisspeptin in regulating gonadotropin-releasing hormone (GnRH) secretion in female rats. Peptides 30, 49–56.
Ohlsson, C., Hellberg, N., Parini, P., Vidal, O., Bohlooly, Y. M., Rudling, M., Lindberg, M. K., Warner, M., Angelin, B., and Gustafsson, J. A. (2000). Obesity and disturbed lipoprotein profile in estrogen receptor-alpha-deficient male mice. Biochem. Biophys. Res. Commun. 278, 640–645.
Okamoto, H., Nakae, J., Kitamura, T., Park, B. C., Dragatsis, I., and Accili, D. (2004). Transgenic rescue of insulin receptor-deficient mice. J. Clin. Invest. 114, 214–223.
Okura, T., Koda, M., Ando, F., Niino, N., Ohta, S., and Shimokata, H. (2003). Association of polymorphisms in the estrogen receptor alpha gene with body fat distribution. Int. J. Obes. Relat. Metab. Disord. 27, 1020–1027.
Olster, D. H., and Blaustein, J. D. (1989). Development of progesterone-facilitated lordosis in female guinea pigs: relationship to neural E2 and progestin receptors. Brain Res. 484, 168–176.
Osterlund, M., Kuiper, G. G., Gustafsson, J. A., and Hurd, Y. L. (1998). Differential distribution and regulation of estrogen receptor-alpha and -beta mRNA within the female rat brain. Brain Res. Mol. Brain Res. 54, 175–180.
Ottem, E. N., Godwin, J. G., and Petersen, S. L. (2002). Glutamatergic signaling through the N-methyl-D-aspartate receptor directly activates medial subpopulations of luteinizing hormone-releasing hormone (LHRH) neurons, but does not appear to mediate the effects of estradiol on LHRH gene expression. Endocrinology 143, 4837–4845.
Pelleymounter, M. A., Cullen, M. J., Baker, M. B., Hecht, R., Winters, D., Boone, T., and Collins, F. (1995). Effects of the obese gene product on body weight regulation in ob/ob mice. Science 269, 540–543.
Pielecka-Fortuna, J., Chu, Z., and Moenter, S. M. (2008). Kisspeptin acts directly and indirectly to increase gonadotropin-releasing hormone neuron activity and its effects are modulated by estradiol. Endocrinology 149, 1979–1986.
Pimpinelli, F., Parenti, M., Guzzi, F., Piva, F., Hokfelt, T., and Maggi, R. (2006). Presence of delta opioid receptors on a subset of hypothalamic gonadotropin releasing hormone (GnRH) neurons. Brain Res. 1070, 15–23.
Qiu, X., Dowling, A., Marino, J. S., Nedorezov, L., and Hill, J. W. (2011). “Insulin action in Kiss1 neurons and the regulation of puberty and reproduction,” in The Endocrine Society’s 93rd Annual Meeting, Boston, MA.
Ramaswamy, S., Guerriero, K. A., Gibbs, R. B., and Plant, T. M. (2008). Structural interactions between kisspeptin and GnRH neurons in the mediobasal hypothalamus of the male rhesus monkey (Macaca mulatta) as revealed by double immunofluorescence and confocal microscopy. Endocrinology 149, 4387–4395.
Rance, N. E. (2009). Menopause and the human hypothalamus: evidence for the role of kisspeptin/neurokinin B neurons in the regulation of E2 negative feedback. Peptides 30, 111–122.
Rance, N. E., and Bruce, T. R. (1994). Neurokinin-B gene-expression is increased in the arcuate nucleus of ovariectomized rats. Neuroendocrinology 60, 337–345.
Rance, N. E., and Young, W. S. (1991). Hypertrophy and increased gene-expression of neurons containing neurokinin-B and substance-P messenger ribonucleic-acids in the hypothalami of postmenopausal women. Endocrinology 128, 2239–2247.
Rhodes, P. A., and Llinas, R. (2005). A model of thalamocortical relay cells. J. Physiol. (Lond.) 565, 765–781.
Roberts, C. B., Best, J. A., and Suter, K. J. (2006). Dendritic processing of excitatory synaptic input in hypothalamic gonadotropin releasing-hormone neurons. Endocrinology 147, 1545–1555.
Rogers, N. H., Perfield, J. W. II, Strissel, K. J., Obin, M. S., and Greenberg, A. S. (2009). Reduced energy expenditure and increased inflammation are early events in the development of ovariectomy-induced obesity. Endocrinology 150, 2161–2168.
Roy, S., Barke, R. A., and Loh, H. H. (1998). MU-opioid receptor-knockout mice: role of mu-opioid receptor in morphine mediated immune functions. Brain Res. Mol. Brain Res. 61, 190–194.
Rubin, B. S., and Barfield, R. J. (1983a). Induction of estrous behavior in ovariectomized rats by sequential replacement of E2 and progesterone to the ventromedial hypothalamus. Neuroendocrinology 37, 218–224.
Rubin, B. S., and Barfield, R. J. (1983b). Progesterone in the ventromedial hypothalamus facilitates estrous behavior in ovariectomized, E2-primed rats. Endocrinology 113, 797–804.
Rubinstein, M., Mogil, J. S., Japón, M., Chan, E. C., Allen, R. G., and Low, M. J. (1996). Absence of opioid stress-induced analgesia in mice lacking beta-endorphin by site-directed mutagenesis. Proc. Natl. Acad. Sci. U.S.A. 93, 3995–4000.
Sahu, A., and Kalra, S. P. (1992). Effects of tachykinins on luteinizing-hormone release in female rats – potent inhibitory-action of neuropeptide-K. Endocrinology 130, 1571–1577.
Sandrock, M., Schulz, A., Merkwitz, C., Schöneberg, T., Spanel-Borowski, K., and Ricken, A. (2009). Reduction in corpora lutea number in obese melanocortin-4-receptor-deficient mice. Reprod. Biol. Endocrinol. 7, 24.
Sarkar, D. K., and Fink, G. (1980). Luteinizing hormone releasing factor in pituitary stalk plasma from long-term ovariectomized rats: effects of steroids. J. Endocrinol. 86, 511–524.
Schally, A. V., Arimura, A., Kastin, A. J., Matsuo, H., Baba, Y., Redding, T. W., Nair, R. M., Debeljuk, L., and White, W. F. (1971). Gonadotropin-releasing hormone – one polypeptide regulates secretion of luteinizing and follicle-stimulating hormones. Science 173, 1036–1038.
Schioth, H. B., Kakizaki, Y., Kohsaka, A., Suda, T., and Watanobe, H. (2001). Agouti-related peptide prevents steroid-induced luteinizing hormone and prolactin surges in female rats. Neuroreport 12, 687–690.
Schioth, H. B., and Watanobe, H. (2002). Melanocortins and reproduction. Brain Res. Brain Res. Rev. 38, 340–350.
Schlenker, E. H., and Hansen, S. N. (2006). Sex-specific densities of estrogen receptors alpha and beta in the subnuclei of the nucleus tractus solitarius, hypoglossal nucleus and dorsal vagal motor nucleus weanling rats. Brain Res. 1123, 89–100.
Schnorr, J. A., Bray, M. J., and Veldhuis, J. D. (2001). Aromatization mediates testosterone’s short-term feedback restraint of 24-hour endogenously driven and acute exogenous gonadotropin-releasing hormone-stimulated luteinizing hormone and follicle-stimulating hormone secretion in young men. J. Clin. Endocrinol. Metab. 86, 2600–2606.
Schuller, A. G., King, M. A., Zhang, J., Bolan, E., Pan, Y. X., Morgan, D. J., Chang, A., Czick, M. E., Unterwald, E. M., Pasternak, G. W., and Pintar, J. E. (1999). Retention of heroin and morphine-6 beta-glucuronide analgesia in a new line of mice lacking exon 1 of MOR-1. Nat. Neurosci. 2, 151–156.
Seminara, S. B., Messager, S., Chatzidaki, E. E., Thresher, R. R., Acierno, J. S. Jr., Shagoury, J. K., Bo-Abbas, Y., Kuohung, W., Schwinof, K. M., Hendrick, A. G., Zahn, D., Dixon, J., Kaiser, U. B., Slaugenhaupt, S. A., Gusella, J. F., O’Rahilly, S., Carlton, M. B., Crowley, W. F. Jr., Aparicio, S. A., and Colledge, W. H. (2003). The GPR54 gene as a regulator of puberty. N. Engl. J. Med. 349, U1614–U1618.
Shander, D., and Barraclough, C. A. (1980). Role of the preoptic brain in the regulation of preovulatory gonadotropin surges in the hamster. Exp. Brain Res. 40, 123–130.
Sharifi, N., Diehl, N., Yaswen, L., Brennan, M. B., and Hochgeschwender, U. (2001). Generation of dynorphin knockout mice. Brain Res. Mol. Brain Res. 86, 70–75.
Shepherd, G. M. (1996). The dendritic spine: a multifunctional integrative unit. J. Neurophysiol. 75, 2197–2210.
Shivers, B. D., Harlan, R. E., Morrell, J. I., and Pfaff, D. W. (1983). Absence of oestradiol concentration in cell nuclei of LHRH-immunoreactive neurones. Nature 304, 345–347.
Shupnik, M. A. (1996). Gonadotropin gene modulation by steroids and gonadotropin-releasing hormone. Biol. Reprod. 54, 279–286.
Shupnik, M. A., Gharib, S. D., and Chin, W. W. (1988). E2 suppresses rat gonadotropin gene transcription in vivo. Endocrinology 122, 1842–1846.
Sim, J. A., Skynner, M. J., Pape, J. R., and Herbison, A. E. (2000). Late postnatal reorganization of GABA(A) receptor signalling in native GnRH neurons. Eur. J. Neurosci. 12, 3497–3504.
Simonian, S. X., and Herbison, A. E. (2001). Differing, spatially restricted roles of ionotropic glutamate receptors in regulating the migration of gnrh neurons during embryogenesis. J. Neurosci. 21, 934–943.
Simonian, S. X., Spratt, D. P., and Herbison, A. E. (1999). Identification and characterization of estrogen receptor alpha-containing neurons projecting to the vicinity of the gonadotropin-releasing hormone perikarya in the rostral preoptic area of the rat. J. Comp. Neurol. 411, 346–358.
Simonin, F., Valverde, O., Smadja, C., Slowe, S., Kitchen, I., Dierich, A., Le Meur, M., Roques, B. P., Maldonado, R., and Kieffer, B. L. (1998). Disruption of the kappa-opioid receptor gene in mice enhances sensitivity to chemical visceral pain, impairs pharmacological actions of the selective kappa-agonist U-50,488H and attenuates morphine withdrawal. EMBO J. 17, 886–897.
Sinchak, K., Dewing, P., Cook, M., and Micevych, P. (2007). Release of orphanin FQ/nociceptin in the medial preoptic nucleus and ventromedial nucleus of the hypothalamus facilitates lordosis. Horm. Behav. 51, 406–412.
Singh, S. P., Wolfe, A., Ng, Y., DiVall, S. A., Buggs, C., Levine, J. E., Wondisford, F. E., and Radovick, S. (2009). Impaired E2 feedback and infertility in female mice with pituitary-specific deletion of estrogen receptor alpha (ESR1). Biol. Reprod. 81, 488–496.
Smith, J. T., Acohido, B. V., Clifton, D. K., and Steiner, R. A. (2006). KiSS-1 neurones are direct targets for leptin in the ob/ob mouse. J. Neuroendocrinol. 18, 298–303.
Smith, J. T., Cunningham, M. J., Rissman, E. F., Clifton, D. K., and Steiner, R. A. (2005). Regulation of Kiss1 gene expression in the brain of the female mouse. Endocrinology 146, 3686–3692.
Sora, I., Takahashi, N., Funada, M., Ujike, H., Revay, R. S., Donovan, D. M., Miner, L. L., and Uhl, G. R. (1997). Opiate receptor knockout mice define mu receptor roles in endogenous nociceptive responses and morphine-induced analgesia. Proc. Natl. Acad. Sci. U.S.A. 94, 1544–1549.
Sorra, K. E., and Harris, K. M. (2000). Overview on the structure, composition, function, development, and plasticity of hippocampal dendritic spines. Hippocampus 10, 501–511.
Spergel, D. J., Krüth, U., Hanley, D. F., Sprengel, R., and Seeburg, P. H. (1999). GABA- and glutamate-activated channels in green fluorescent protein-tagged gonadotropin-releasing hormone neurons in transgenic mice. J. Neurosci. 19, 2037–2050.
Stallings, N. R., Hanley, N. A., Majdic, G., Zhao, L., Bakke, M., and Parker, K. L. (2002). Development of a transgenic green fluorescent protein lineage marker for steroidogenic factor 1. Endocr. Res. 28, 497–504.
Thind, K. K., and Goldsmith, P. C. (1988). Infundibular gonadotropin-releasing hormone neurons are inhibited by direct opioid and autoregulatory synapses in juvenile monkeys. Neuroendocrinology 47, 203–216.
Tian, M., Broxmeyer, H. E., Fan, Y., Lai, Z., Zhang, S., Aronica, S., Cooper, S., Bigsby, R. M., Steinmetz, R., Engle, S. J., Mestek, A., Pollock, J. D., Lehman, M. N., Jansen, H. T., Ying, M., Stambrook, P. J., Tischfield, J. A., and Yu, L. (1997). Altered hematopoiesis, behavior, and sexual function in mu opioid receptor-deficient mice. J. Exp. Med. 185, 1517–1522.
van de Wall, E., Leshan, R., Xu, A. W., Balthasar, N., Coppari, R., Liu, S. M., Jo, Y. H., MacKenzie, R. G., Allison, D. B., Dun, N. J., Elmquist, J., Lowell, B. B., Barsh, G. S., de Luca, C., Myers, M. G. Jr., Schwartz, G. J., and Chua, S. C. Jr. (2008). Collective and individual functions of leptin receptor modulated neurons controlling metabolism and ingestion. Endocrinology 149, 1773–1785.
Van der Ploeg, L. H., Martin, W. J., Howard, A. D., Nargund, R. P., Austin, C. P., Guan, X., Drisko, J., Cashen, D., Sebhat, I., Patchett, A. A., Figueroa, D. J., DiLella, A. G., Connolly, B. M., Weinberg, D. H., Tan, C. P., Palyha, O. C., Pong, S. S., MacNeil, T., Rosenblum, C., Vongs, A., Tang, R., Yu, H., Sailer, A. W., Fong, T. M., Huang, C., Tota, M. R., Chang, R. S., Stearns, R., Tamvakopoulos, C., Christ, G., Drazen, D. L., Spar, B. D., Nelson, R. J., and MacIntyre, D. E. (2002). A role for the melanocortin 4 receptor in sexual function. Proc. Natl. Acad. Sci. U.S.A. 99, 11381–11386.
Veldhuis, J. D., and Dufau, M. L. (1987). Estradiol modulates the pulsatile secretion of biologically active luteinizing hormone in man. J. Clin. Invest. 80, 631–638.
Vrang, N., Larsen, P. J., Clausen, J. T., and Kristensen, P. (1999). Neurochemical characterization of hypothalamic cocaine- amphetamine-regulated transcript neurons. J. Neurosci. 19, RC5.
Wade, G. N., Gray, J. M., and Bartness, T. J. (1985). Gonadal influences on adiposity. Int. J. Obes. 9(Suppl. 1), 83–92.
Wakabayashi, Y., Nakada, T., Murata, K., Ohkura, S., Mogi, K., Navarro, V. M., Clifton, D. K., Mori, Y., Tsukamura, H., Maeda, K., Steiner, R. A., and Okamura, H. (2010). Neurokinin B and dynorphin A in kisspeptin neurons of the arcuate nucleus participate in generation of periodic oscillation of neural activity driving pulsatile gonadotropin-releasing hormone secretion in the goat. J. Neurosci. 30, 3124–3132.
Wallen, W. J., Belanger, M. P., and Wittnich, C. (2001). Sex hormones and the selective estrogen receptor modulator tamoxifen modulate weekly body weights and food intakes in adolescent and adult rats. J. Nutr. 131, 2351–2357.
Wardlaw, S. L. (2011). Hypothalamic proopiomelanocortin processing and the regulation of energy balance. Eur. J. Pharmacol. 660, 213–219.
Wardlaw, S. L., and Ferin, M. (1990). Interaction between beta-endorphin and alpha-melanocyte-stimulating hormone in the control of prolactin and luteinizing hormone secretion in the primate. Endocrinology 126, 2035–2040.
Watanobe, H., Schiöth, H. B., Wikberg, J. E., and Suda, T. (1999). The melanocortin 4 receptor mediates leptin stimulation of luteinizing hormone and prolactin surges in steroid-primed ovariectomized rats. Biochem. Biophys. Res. Commun. 257, 860–864.
Welt, C. K., Chan, J. L., Bullen, J., Murphy, R., Smith, P., DePaoli, A. M., Karalis, A., and Mantzoros, C. S. (2004). Recombinant human leptin in women with hypothalamic amenorrhea. N. Engl. J. Med. 351, 987–997.
Wiegand, S., Terasawa, E., Bridson, W. E., and Goy, R. W. (1980). Effects of discrete lesions of preoptic and suprachiasmatic structures in the female rat. Alterations in the feedback regulation of gonadotropin secretion. Neuroendocrinology 31, 147–157.
Wiesner, J. B., Koenig, J. I., Krulich, L., and Moss, R. L. (1985). Possible delta receptor mediation of the effect of beta-endorphin on luteinizing hormone (LH) release, but not on prolactin (PRL) release, in the ovariectomized rat. Endocrinology 116, 475–477.
Wintermantel, T. M., Campbell, R. E., Porteous, R., Bock, D., Gröne, H. J., Todman, M. G., Korach, K. S., Greiner, E., Pérez, C. A., Schütz, G., and Herbison, A. E. (2006). Definition of estrogen receptor pathway critical for E2 positive feedback to gonadotropin-releasing hormone neurons and fertility. Neuron 52, 271–280.
Witkin, J. W., O’Sullivan, H., and Silverman, A. J. (1995). Novel associations among gonadotropin-releasing hormone neurons. Endocrinology 136, 4323–4330.
Witkin, J. W., and Silverman, A. J. (1985). Synaptology of luteinizing hormone-releasing hormone neurons in rat preoptic area. Peptides 6, 263–271.
Woods, S. C., Lotter, E. C., McKay, L. D., and Porte, D. Jr. (1979). Chronic intracerebroventricular infusion of insulin reduces food intake and body weight of baboons. Nature 282, 503–505.
Wu, M., Dumalska, I., Morozova, E., van den Pol, A., and Alreja, M. (2009). Melanin-concentrating hormone directly inhibits GnRH neurons and blocks kisspeptin activation, linking energy balance to reproduction. Proc. Natl. Acad. Sci. U.S.A. 106, 17217–17222.
Xu, Y., Nedungadi, T. P., Zhu, L., Sobhani, N., Irani, B. G., Davis, K. E., Zhang, X., Zou, F., Gent, L. M., Hahner, L. D., Khan, S. A., Elias, C. F., Elmquist, J. K., and Clegg, D. J. (2011). Distinct hypothalamic neurons mediate E2ic effects on energy homeostasis and reproduction. Cell Metab. 14, 453–465.
Yaswen, L., Diehl, N., Brennan, M. B., and Hochgeschwender, U. (1999). Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat. Med. 5, 1066–1070.
Yoon, H., Enquist, L. W., and Dulac, C. (2005). Olfactory inputs to hypothalamic neurons controlling reproduction and fertility. Cell 123, 669–682.
Zhang, Q., and Gallo, R. V. (2002). Effect of prodynorphin-derived opioid peptides on the ovulatory luteinizing hormone surge in the proestrous rat. Endocrine 18, 27–32.
Zhao, L., Bakke, M., Krimkevich, Y., Cushman, L. J., Parlow, A. F., Camper, S. A., and Parker, K. L. (2001). Steroidogenic factor 1 (SF1) is essential for pituitary gonadotrope function. Development 128, 147–154.
Zhu, Y., King, M. A., Schuller, A. G., Nitsche, J. F., Reidl, M., Elde, R. P., Unterwald, E., Pasternak, G. W., and Pintar, J. E. (1999). Retention of supraspinal delta-like analgesia and loss of morphine tolerance in delta opioid receptor knockout mice. Neuron 24, 243–252.
Ziotopoulou, M., Erani, D. M., Hileman, S. M., Bjørbaek, C., and Mantzoros, C. S. (2000). Unlike leptin, ciliary neurotrophic factor does not reverse the starvation-induced changes of serum corticosterone and hypothalamic neuropeptide levels but induces expression of hypothalamic inhibitors of leptin signaling. Diabetes 49, 1890–1896.
Keywords: hypothalamus, energy homeostasis, reproduction
Citation: Xu Y, Faulkner LD and Hill JW (2012) Cross-talk between metabolism and reproduction: the role of POMC and SF1 neurons. Front. Endocrin. 2:98. doi: 10.3389/fendo.2011.00098
Received: 04 August 2011; Accepted: 22 November 2011;
Published online: 04 January 2012.
Edited by:
Carol F. Elias, University of Texas Southwestern Medical Center, USAReviewed by:
Manuel Tena-Sempere, University of Cordoba, SpainRaul Miguel Luque Huertas, University of Cordoba, Spain
Copyright: © 2012 Xu, Faulkner and Hill. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Yong Xu, Department of Pediatrics, Children’s Nutrition Research Center, Baylor College of Medicine, Houston, TX 77030, USA e-mail:eW9uZ3hAYmNtLmVkdQ==; Jennifer W. Hill, Department of Physiology and Pharmacology, Center for Diabetes and Endocrine Research, College of Medicine, The University of Toledo, Toledo, OH, USA e-mail:amVubmlmZXIuaGlsbDRAdXRvbGVkby5lZHU=