 Brian P. Kenealy1
Brian P. Kenealy1 E. Terasawa1,2*
E. Terasawa1,2*- 1 Wisconsin National Primate Research Center, University of Wisconsin, Madison, WI, USA
- 2 Department of Pediatrics, University of Wisconsin, Madison, WI, USA
Estradiol plays a pivotal role in the control of gonadotropin-releasing hormone (GnRH) neuronal function and female reproduction. While positive and negative feedback actions of estradiol that enhance and suppress release of GnRH and LH are primarily mediated through estrogen receptor alpha located in interneurons, a series of recent studies in our laboratory indicate that rapid excitatory actions of estradiol also directly modify GnRH neuronal activity. We observed this phenomenon in cultured primate GnRH neurons, but similar rapid direct actions of estradiol are also described in cultured GnRH neurons and green fluorescent protein-labeled GnRH neurons of mice. Importantly, rapid direct action of estradiol in GnRH neurons is mediated through membrane or membrane associated receptors, such as GPR30, STX-sensitive receptors, and ERβ. In this review, possible implications of this rapid estradiol action in GnRH neurons are discussed.
Introduction
Estradiol (E2) is a vital regulator of female reproduction. In addition to trophic effects of E2 on breast, ovarian, and uterine tissue, E2 plays a key role in regulating the function of gonadotropin-releasing hormone (GnRH) neurons in the hypothalamus and gonadotrophs in the pituitary throughout the female reproductive cycle. E2 released from the ovary induces positive and negative feedback effects on GnRH neurons in the hypothalamus. Historically, it has been viewed that E2 controls the GnRH neuronal system through interneurons or glial cells (Herbison, 2006), because earlier studies with autoradiography combined with immunocytochemistry showed that GnRH neurons do not contain nuclear estrogen receptors (ER; Shivers et al., 1983), whereas interneurons, such as those synthesizing neuropeptide Y (NPY), catecholamines, glutamate, GABA, and kisspeptin express nuclear ER (Stumpf and Jennes, 1984; Leranth et al., 1992; Smith et al., 2005; Franceschini et al., 2006).
After the discovery of ERβ (Kuiper et al., 1996), several studies found that ERβ colocalizes in GnRH neurons in several species including mice, rats, sheep, and humans (Hrabovszky et al., 2000, 2001, 2007; Herbison and Pape, 2001; Sharifi et al., 2002; Skinner and Dufourny, 2005). More recently, direct action of E2 on GnRH neurons has been shown by several groups including our own (Abrahám et al., 2003; Temple et al., 2004; Abe and Terasawa, 2005; Abe et al., 2008; Chu et al., 2009; Noel et al., 2009; Sun et al., 2010). Direct action of E2 is rapid, mediated through receptors associated with the plasma membrane, and seen in many types of neurons (Terasawa et al., 2009). In this article, we will review the mechanism of rapid, direct action of E2 on GnRH neuronal activity and possible implications of direct E2 action, focusing on our studies in non-human primates.
Models for Studying Direct E2 Action in GnRH Neurons
The number of GnRH neurons in the brain is small and they are diffusely distributed in the preoptic area and basal hypothalamus. Thus, to study cellular and molecular mechanisms of E2 action in GnRH neurons we need to utilize a relatively simple population of GnRH neurons that can be directly visualized. To date three in vitro models have been described (Terasawa, 2001): (1) GT-1 and GN11 cell lines (Mellon et al., 1990; Radovick et al., 1991), (2) cultured GnRH neurons derived from the nasal placode regions in monkeys, sheep, rats, and mice (Terasawa et al., 1993; Daikoku and Koide, 1994; Fueshko and Wray, 1994; Duittoz et al., 1997), and (3) sliced preoptic-hypothalamic green fluorescent protein (GFP) labeled mouse GnRH neurons (Spergel et al., 1999; Suter et al., 2000) or mouse and guinea pig GnRH neurons identified with single cell RT-PCR or immunocytochemistry (Lagrange et al., 1995; Zhang et al., 2007). In these models, demonstration of pulsatile GnRH release or periodic activity recorded from GnRH neurons is essential, because GnRH neurons release the decapeptide hormone into the pituitary portal circulation in a pulsatile manner (Knobil, 1980; Gearing and Terasawa, 1988; Kokoris et al., 1988) and this pulsatility is crucial for the maintenance of normal reproductive function (Knobil, 1980). Importantly, the pulse frequency of GnRH release or periodic activity in a model should reflect the species origin of these GnRH neurons. In the GT-1 cells and cultured GnRH neurons, pulsatile release of GnRH peptide with species specific frequency (i.e., 20–30 min in mouse and rats, 40–60 min in sheep and monkeys) has been shown (Krsmanovic et al., 1992; Martinez de la Escalera et al., 1992; Wetsel et al., 1992; Terasawa et al., 1999a; Duittoz and Batailler, 2000; Funabashi et al., 2000; Constantin et al., 2009). In GFP-labeled mouse GnRH neurons and GT-1 cells, periodic burst firing activity similar to GnRH pulses has also been reported (Costantin and Charles, 1999; Nunemaker et al., 2003). Among these three models, we have been using cultured GnRH neurons derived from the embryonic nasal placode of rhesus monkey fetuses, which are obtained from time-mated pregnancies (Terasawa et al., 1993). These GnRH neurons exhibit a spontaneous oscillatory pattern of [Ca2+]i levels with variable peak amplitude and duration of each oscillation unique to each cell (Terasawa et al., 1999b). On average, the interpulse interval between [Ca2+]i oscillations is 8 min with synchronization of [Ca2+]i oscillations among GnRH neurons occurring at ∼60 min intervals (Terasawa et al., 1999b) and release GnRH peptide also at ∼60 min intervals (Terasawa et al., 1999a). Using our cultured primate GnRH neuron model we have reported several important discoveries (see Terasawa et al., 2009; Terasawa et al., 2010). Based on comprehensive comparisons between the embryonic GnRH neuron model (in both monkeys and mice) and GFP-labeled mice GnRH neuron model, Jasoni et al. (2010) and Constantin (2011) conclude that physiological characteristics of the two models are quite similar.
E2 Rapidly Stimulates Firing Activity, Intracellular Calcium Oscillations, and GnRH Release
To determine if E2 causes direct action in primate GnRH neurons, we first examined the effects of E2 on firing activity. Application of E2 (1 nM) to cultured primate GnRH neurons for 10 min, induces a 250% increase in action potential firing frequency, an increase in the number of action potentials per burst, and an increase in burst duration (Abe and Terasawa, 2005). E2, however, did not change the timing of bursts (interburst interval) nor did it alter the cluster pattern, suggesting that E2 modulates overall firing intensity, but not the firing pattern.
Release of GnRH is also modulated by E2. In primate GnRH neurons, exposure to 1 nM E2 for 20 min results in a rapid increase of GnRH peptide release, which is initiated within 10 min of E2 application and lasts for 40 min (Noel et al., 2009). In GT-1 cells, it has been reported that exposure to a picomolar dose of E2 for 4 h suppresses the frequency of GnRH release (Navarro et al., 2003). However, the mechanism of E2 action between these two studies may differ, as the E2 exposure time in the study of GT-1 cells is much longer and may lead to nuclear receptor mediated genomic action.
E2 causes potent stimulatory effects on [Ca2+]i oscillations. A 10 min exposure to E2 at 1 nM induces a 180–200% increase in the frequency of [Ca2+]i oscillations, returning to baseline levels 40–60 min after initiation of E2 treatment (Abe et al., 2008). E2 also increases the number of activated cells from 30 to 70%. Additionally, E2 stimulates the average number of synchronized [Ca2+]i oscillations from 1 synchronization event/hour in control samples to ∼2.7 events/hour in E2 treated samples (Abe et al., 2008). Similar E2 effects on [Ca2+]i oscillations have also been reported in cultured mouse GnRH neurons (Temple et al., 2004) and we also confirmed similar E2 effects in additional studies (Noel et al., 2009; Kenealy et al., 2011a,b). Importantly, tetrodotoxin does not change the pattern of the E2-induced [Ca2+]i oscillations (Abe et al., 2008), consistent with this E2 action causing direct effects on GnRH neurons and not through interneurons.
Rapid Stimulatory E2 Action in GnRH Neurons is a Membrane-Initiated Mechanism
In order to assess the mechanism of rapid E2 action, we have examined the effects of a plasma membrane impermeable form of E2, E2-17 hemisuccinate BSA (E2-BSA). E2-BSA (1 nM) increases the frequencies of firing activity (Abe and Terasawa, 2005) and [Ca2+]i oscillations (Noel et al., 2009), and stimulates GnRH release (Noel et al., 2009), similar to the increase observed with E2, suggesting that rapid action of E2 occurs at the cell membrane. Moreover, exposure of GnRH neurons to the nuclear impermeable E2 dendrimer conjugate (EDC, 1 nM), described by Harrington et al. (2006), also causes an increased frequency of [Ca2+]i oscillations and elevated GnRH release (Noel et al., 2009), indicating that E2 causes rapid effects without entering the nucleus. These observations indicate that rapid excitatory E2 action is a membrane-initiated mechanism, and does not require genomic action of E2. However, the amplitude and duration of the EDC- and E2-BSA-induced GnRH release are smaller and shorter (Noel et al., 2009), indicating the presence of multiple mechanisms of E2 action (see below).
E2 Action is Not Mediated by ERα or ERβ
In general, E2 action through genomic changes occurs in the order of an hour to several hours or even days. In contrast, the effects elicited by E2 in GnRH neurons described above, i.e., the increase in firing activity, [Ca2+]i oscillations, and GnRH release, occurs at a time scale of seconds to minutes. Therefore, it is speculated that the mechanism of rapid E2 action differs from nuclear ER mediated genomic action.
To identify the type of ER mediating rapid E2 action in GnRH neurons, we first examined the role of ERα or ERβ using the ER antagonist, ICI182,780. To our surprise, ICI182,780 blocks neither the E2-induced [Ca2+]i oscillations nor synchronization (Abe et al., 2008). Moreover, ICI182,780 did not influence the E2-induced release of GnRH peptide (Noel et al., 2009). Considering the results of rapid direct E2 action through ERβ in mouse GnRH neurons (Abrahám et al., 2003; Temple et al., 2004; Chu et al., 2009), we further examined E2’s effects in GnRH neurons, in which ERα and ERβ were respectively deleted by an siRNA knockdown approach. The results show that knockdown of ERα and ERβ do not interfere with the E2-induced changes in [Ca2+]i oscillations nor synchronization (Kenealy et al., 2011a), confirming our findings with ICI182,780 on [Ca2+]i oscillations. Collectively, these findings suggest that ERα and ERβ are neither involved in E2-induced [Ca2+]i oscillations nor rapid release of GnRH peptide in primate GnRH neurons.
Rapid E2 Action is Mediated by Multiple Receptors
In addition to ERα and ERβ, several types of ERs, such as ER-X (Toran-Allerand et al., 2002), G protein coupled receptor 30 (GPR30, Thomas et al., 2010), and membrane ER sensitive to the diphenylacrylamide compound STX (STX-R, Qiu et al., 2003) participate in membrane-initiated E2 action. Interestingly, these membrane ERs require G-protein coupled receptor (GPCR) signaling mechanisms for estrogen action (Terasawa et al., 2009). Rapid action through membrane ERα and ERβ also requires a companion GPCR, metabotropic glutamate receptors (Boulware et al., 2005, 2007; Grove-Strawser et al., 2010). In primate GnRH neurons, a GPCR is also involved in rapid E2 action, as pertussis toxin, a broad inhibitor of GPCR signaling, blocks the E2-induced changes in [Ca2+]i oscillations, their synchronization, and GnRH release (Noel et al., 2009).
GPR30, an orphan receptor coupled to Gαs proteins, binds E2 (Revankar et al., 2005; Thomas et al., 2005). However, E2 action through GPR30 does not require ERα or ERβ, as knockdown of GPR30 in ERα and ERβ negative cancer cells blocks E2 action (Thomas et al., 2005). In cancer cell lines, E2 action mediated by GPR30 mobilizes Gαs to stimulate adenylyl cyclase resulting in cyclic AMP synthesis and activates Gβγ signaling resulting in transactivation of epidermal growth factor receptor (Filardo et al., 2000, 2002). Importantly, unlike ER-X and STX-R, the sequence of GPR30 is known. Consequently, G1, a specific agonist for GPR30 (Bologa et al., 2006) and G15, a specific antagonist for GPR30 (Dennis et al., 2009) have been synthesized. Using these tools, we have studied the role of GPR30 in membrane-initiated E2 action in primate GnRH neurons and found that GPR30 is, at least in part, responsible for rapid excitatory E2 action. First, exposure of GnRH neurons to the GPR30 receptor agonist, G1, at 10 nM, but not 1 nM, induces an increase in [Ca2+]i oscillations (Noel et al., 2009) and G1 at 100 nM stimulates GnRH release (Kenealy and Terasawa, unpublished observations). Second, siRNA knockdown of GPR30 completely blocks the E2- and EDC-induced changes in [Ca2+]i oscillations and their synchronization (Noel et al., 2009). Third, a high dose of ICI182,780 (1 μM) alone elicits changes in [Ca2+]i oscillations similar to cancer cells, in which a high dose (1 μM) of ICI182,780 is an agonist for GPR30 (Filardo et al., 2000). Fourth, treatment with the GPR30 antagonist G15 blocks both the E2-induced increase in [Ca2+]i oscillations, their synchronization, and E2-induced GnRH release, while G15 alone showed no significant effects (Kenealy and Terasawa, unpublished observations). Finally, GPR30 colocalizes in one-third of GnRH neurons in the monkey hypothalamus (Noel et al., 2009).
Because (1) a higher dose of G1 and EDC is required for the induction of changes in [Ca2+]i oscillations and GnRH release, as compared to E2 changes (1 nM) and (2) the effects of a higher dose of E2 (10 nM) on changes in [Ca2+]i oscillations is only partly blocked by GPR30 siRNA treatment (Kenealy et al., 2011b), we have speculated that additional membrane ERs may be involved in rapid E2 action. STX-R was an enticing target to pursue, as STX (1) has been shown to mediate effects in hypothalamic cells in mutant mice lacking ERα, ERβ, and GPR30 receptors (Qiu et al., 2006, 2008), (2) modulates ion channels involved in membrane potential changes (Zhang et al., 2010), and (3) is potently inhibited by ICI182,780 (Qiu et al., 2003). Indeed, treatment of primate GnRH neurons with 10 nM STX induces an increase in [Ca2+]i oscillations, their synchronization, and increases the percent of cells stimulated, similar to both E2 (1 nM) and G1 (10 nM; Kenealy et al., 2011b). Interestingly, ICI182,780 blocks the STX-induced [Ca2+]i oscillations and their synchronization. Furthermore, GPR30 knockdown does not influence the STX-induced changes in [Ca2+]i oscillations and their synchronization. Finally, STX (10 and 100 nM) induces a dose dependent increase in GnRH release, although the effectiveness is smaller than that induced by E2 at 1 nM (Kenealy et al., 2011b). Therefore, E2 action in primate GnRH neurons is mediated through multiple receptor mechanisms (see Figure 1). This is not limited to primate GnRH neurons: in mouse GnRH neurons, in addition to E2 action through ERβ (Temple et al., 2004) and STX–R (Zhang et al., 2010), E2 through GPR30 modulates voltage gated calcium channels (Sun et al., 2010). Any interaction between STX-R and other ERs including GPR30 will become clear when the molecular nature of STX-R is identified.
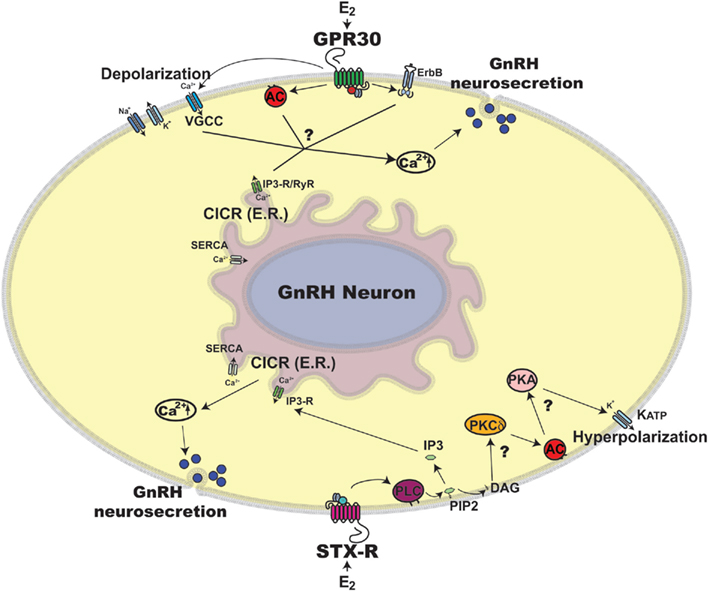
Figure 1. Schematic illustration of rapid estradiol (E2) action in primate GnRH neurons. Exposure of primate GnRH neurons to E2 rapidly induces [Ca2+]i oscillations and GnRH peptide release within 10 min (Noel et al., 2009; Kenealy et al., 2011b). Two possible mechanisms for the rapid E2 action through GPR30 and STX-R are discussed in this article, although many details are yet to be clarified, which are noted by question marks in the scheme. First, E2 binding to GPR30 may induce activation of two intracellular pathways: 1) E2 activation through GPR30 depolarizes GnRH neuronal membrane via VGCCs (Sun et al., 2010), which allows [Ca2+]e entry, resulting in CICR (Kenealy et al., 2011c) and 2) E2 transactivates AC and/or ErbB pathways (Filardo et al., 2002), which also results in CICR. Second, E2 binding to STX-R appears to cause 1) activation of CICR through a PLC and IP3-R mechanism leading to [Ca2+]i increase (Kenealy et al., 2011b) and 2) activation of a PKCδ-AC-PKA mechanism resulting in hyperpolarization of the GnRH neuronal membrane through KATP channels (Zhang et al., 2010), which are essential for burst firing of GnRH neurons, hence neurosecretion. Abbreviations: AC, adenylyl cyclase; Ca2+, calcium; [Ca2+]e, extracellular Ca2+; [Ca2+]i, intracellular Ca2+; CICR, calcium induced calcium release; DAG, diacylglycerol; E2, estradiol; ER, endoplasmic reticulum; ErbB, epidermal growth factor receptor; GnRH, gonadotropin-releasing hormone neuron; GPR30, G protein coupled estrogen receptor; IP3, inositol triphosphate; IP3-R, inositol triphosphate receptor; KATP, ATP sensitive potassium channel; PIP2, phosphatidylinositol biphosphate; PKA, protein kinase A; PKCδ, Protein kinase C delta; PLC, phospholipase C; RyR, ryanodine receptor; SERCA, sarco/endoplasmic reticulum Ca2+ ATPase; STX-R, membrane estrogen receptor sensitive to STX; VGCC, voltage gated calcium channel.
Physiological Significance of Rapid Action in GnRH Neurons
Rapid E2 action in hypothalamic neurons was first described nearly 40 years ago (Yagi, 1973; Kelly et al., 1976). During this period, a large number of reports on rapid steroid action in various types of neurons and in glia have been reported (Orchinik et al., 1991; Lagrange et al., 1995; Boulware et al., 2005, 2007; Fehrenbacher et al., 2009; Grove-Strawser et al., 2010; Kuo et al., 2010; Labombarda et al., 2010; Lebesgue et al., 2010). As discussed above, “rapid” timing of E2 action is a membrane-initiated phenomenon, rather than “long term” E2 action, which requires transcription after E2 binds to cytoplasmic/nuclear ER. However, the physiological significance of rapid E2 action in the hypothalamus, and specifically within the GnRH system, remains unclear.
It is unlikely that rapid, direct, excitatory action of E2 plays a significant role in the classical positive and negative feedback mechanisms during the reproductive cycle. Rather, the enhancing and suppressing effects of E2 are mediated by ERα. First, E2 fails to induce LH surges in ERα knockout mice as well as in mutant mice lacking estrogen response element (ERE)-dependent ERα signaling (Glidewell-Kenney et al., 2007). By contrast, in ERβ knockout mice E2 induces LH surges (Wintermantel et al., 2006), although these mice had ERβ splice variants (Krege et al., 1998), therefore, reexamination of the role of ERβ in positive feedback in mice with complete elimination of ERβ splice variants (Antal et al., 2008) is still needed. Similarly, cytochrome P450 aromatase knockout (ArKO) female mice, in which estradiol is absent, have acyclic estrus cycles characterized by elevated levels of LH, FSH, and testosterone (Fisher et al., 1998). Second, whereas convincing evidence for the presence of ERα in GnRH neurons has not been shown, the presence of ERα in interneurons that innervate GnRH neurons, such as neurons that synthesize kisspeptin, NPY, catecholamines, glutamate, and GABA have been consistently reported (Stumpf and Jennes, 1984; Leranth et al., 1992; Smith et al., 2005; Franceschini et al., 2006). Third, positive feedback effects of E2 on LH surges require 16–48 h (depending on species) after its administration (Yamaji et al., 1971; Karsch and Foster, 1975) and negative feedback effects of E2 require a minimum of 1–2 h lasting more than 12 h in rhesus monkeys (Chongthammakun and Terasawa, 1993; Mizuno and Terasawa, 2005). While it is possible that membrane ERα signaling may be in part responsible for negative feedback effects of E2 on LH release, as E2 can suppress LH levels in mice lacking ERE-dependent ERα signaling (Glidewell-Kenney et al., 2007), the genomic action of E2 through ERα is indispensable for LH/GnRH surges. Taken together, ERα within interneurons, such as kisspeptin neurons that directly innervate GnRH neurons, play a role in E2’s feedback action.
Based on the classical concept of positive and negative ovarian steroid feedback, doubt has been cast on the role of rapid excitatory E2 action in the reproductive cycle (Herbison, 2009). It is still unknown whether E2 directly alters secretory activity of GnRH neurons in vivo, as to date, in vivo effects of E2 on GnRH release are tested only after systemic administration of E2. Accumulating evidence suggests local effects of E2 in various brain functions (Woolley, 2007). For example, a recent report in male song birds showing that forebrain E2 concentrations rapidly (within 30 min) increase during social interaction with females (Remage-Healey et al., 2008) indicating the occurrence of rapid synthesis and release of E2 in the brain (Saldanha et al., 2011). Moreover, another recent study by Konkle and McCarthy (2011) reports that E2 levels of 20–30 pg/mg tissue are observed during the first 10 days after birth in the rat hypothalamus, regardless of the presence or absence of gonads. These observations raise the possibility that a relatively high amount of E2 can be synthesized and released locally and rapidly in the hypothalamus under certain physiological conditions, although local concentrations of estradiol in the monkey brain are presently unknown. Therefore, we propose the hypothesis that local E2 release in the hypothalamus may contribute to the activity of GnRH neurons.
As described above, our observations in vitro consistently show that E2 is a potent frequency modulator of GnRH neurons (Terasawa et al., 2009). Thus, we can extend our hypothesis to suggest that local E2 may modulate pulsatility of GnRH release in a subtle manner. Although the mechanism of pulsatile GnRH release is presently unclear, the concept that coordinated release of GnRH is due to synchronized activity among GnRH neurons is generally accepted. In fact, as a means to coordinate activity, GnRH neurons may communicate through dendro-dendritic interactions (Campbell et al., 2009). Because GnRH is released from the cell body and dendrites of GnRH neurons besides their neuroterminals (Fuenzalida et al., 2011), it is possible that rapid excitatory action of E2 on GnRH release may contribute to the communication within dendro-dendritic bundles and consequently, modulate pulsatile GnRH release in vivo.
In rhesus monkeys and sheep, prenatal treatments with testosterone, which is aromatizable to E2 in the brain, result in conditions similar to those of polycystic ovary syndrome (PCOS), one of the most common causes of infertility in women (Abbott et al., 1998; Padmanabhan et al., 2010). Importantly, this testosterone treatment in monkeys at a fetal age of E40-60 (early treatment), but not E100-115 (late treatment), results in LH hypersecretion, reduced hypothalamic sensitivity to negative steroid feedback, and follicular arrest with premature follicle differentiation (Dumesic et al., 2005), as seen in PCOS patients. Moreover, studies in humans show that PCOS women exhibit an accelerated frequency of LH pulses and presumably GnRH pulses (Marshall and Eagleson, 1999), indicating impairment of the GnRH pulse-generating mechanism. Importantly, the effective timing of early testosterone treatment (fetal age at E40-60) in rhesus monkeys (Dumesic et al., 2005) exactly corresponds to the period of GnRH neuronal maturation. After differentiation from progenitor cells at E32-34, GnRH neurons start migrating into the hypothalamus at E38 and settle down in the hypothalamus by E50 (Rønnekleiv and Resko, 1990; Quanbeck et al., 1997). In fact, this period appears critical for the maturation of GnRH neuronal function. Recently, we have shown that GnRH neurons gradually mature in vitro during the 3-week period, after GnRH neurons are dissected out from the nasal placode and started for culture at E36 (Kurian et al., 2010). It should be noted that although prenatal testosterone effects in monkeys may not solely be attributable to aromatized estrogens, it is also possible that rapid excitatory E2 action described in this review may reflect developmental programming influenced by maternal steroidal environments during fetal development and elevated concentrations of cortisol and androgens (Schneider et al., 2002; Padmanabhan et al., 2010; Dunn et al., 2011).
Conclusion
E2 action in the brain is not limited to the classical feedback control of the GnRH neuronal system. E2 could be involved in excitatory and inhibitory neuronal function in the hypothalamus, as shown by Mermelstein and his colleagues in hippocampal neurons (Boulware et al., 2005, 2007). Moreover, E2 action in GnRH neurons may be the consequence of events occurring in utero, such as developmental programming and sexual differentiation. Advances in technology and further detailed studies will ensure the importance of rapid action of estradiol in the hypothalamus.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by NIH grants: R01HD15433 and R01HD11355 for E. Terasawa and T32HD041921 for Brian P. Kenealy, and was possible to perform by NIH support (P51RR000167, RR15459, and RR020141) to the Wisconsin National Primate Research Center.
Abbreviations
[Ca2+]i, intracellular calcium; E2, estradiol; E2-BSA, E2-17 hemisuccinate bovine serum albumin; EDC, E2 dendrimer conjugate; ER, estrogen receptor; GFP, green fluorescent protein; GnRH, gonadotropin-releasing hormone; GPCR, G protein coupled receptor; GPR30, G protein coupled receptor 30; NPY, neuropeptide Y; PCOS, polycystic ovary syndrome; STX-R, membrane ER sensitive to STX.
References
Abbott, D. H., Dumesic, D. A., Eisner, J. R., Colman, R. J., and Kemnitz, J. W. (1998). Insights into the development of polycystic ovary syndrome (PCOS) from studies of prenatally androgenized female rhesus monkeys. Trends Endocrinol. Metab. 9, 62–67.
Abe, H., Keen, K. L., and Terasawa, E. (2008). Rapid action of estrogens on intracellular calcium oscillations in primate luteinizing hormone-releasing hormone-1 neurons. Endocrinology 149, 1155–1162.
Abe, H., and Terasawa, E. (2005). Firing pattern and rapid modulation of activity by estrogen in primate luteinizing hormone releasing hormone-1 neurons. Endocrinology 146, 4312–4320.
Abrahám, I. M., Han, S. K., Todman, M. G., Korach, K. S., and Herbison, A. E. (2003). Estrogen receptor beta mediates rapid estrogen actions on gonadotropin-releasing hormone neurons in vivo. J. Neurosci. 23, 5771–5777.
Antal, M. C., Krust, A., Chambon, P., and Mark, M. (2008). Sterility and absence of histopathological defects in nonreproductive organs of a mouse ERbeta-null mutant. Proc. Natl. Acad. Sci. U.S.A. 105, 2433–2438.
Bologa, C. G., Revankar, C. M., Young, S. M., Edwards, B. S., Arterburn, J. B., Kiselyov, A. S., Parker, M. A., Tkachenko, S. E., Savchuck, N. P., Sklar, L. A., Oprea, T. I., and Prossnitz, E. R. (2006). Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat. Chem. Biol. 2, 207–212.
Boulware, M. I., Kordasiewicz, H., and Mermelstein, P. G. (2007). Caveolin proteins are essential for distinct effects of membrane estrogen receptors in neurons. J. Neurosci. 27, 9941–9950.
Boulware, M. I., Weick, J. P., Becklund, B. R., Kuo, S. P., Groth, R. D., and Mermelstein, P. G. (2005). Estradiol activates group I and II metabotropic glutamate receptor signaling, leading to opposing influences on cAMP response element-binding protein. J. Neurosci. 25, 5066–5078.
Campbell, R. E., Gaidamaka, G., Han, S. K., and Herbison, A. E. (2009). Dendro-dendritic bundling and shared synapses between gonadotropin-releasing hormone neurons. Proc. Natl. Acad. Sci. U.S.A. 106, 10835–10840.
Chongthammakun, S., and Terasawa, E. (1993). Negative feedback effects of estrogen on luteinizing hormone-releasing hormone release occur in pubertal, but not prepubertal, ovariectomized female rhesus monkeys. Endocrinology 132, 735–743.
Chu, Z., Andrade, J., Shupnik, M. A., and Moenter, S. M. (2009). Differential regulation of gonadotropin-releasing hormone neuron activity and membrane properties by acutely applied estradiol: dependence on dose and estrogen receptor subtype. J. Neurosci. 29, 5616–5627.
Constantin, S. (2011). Physiology of the gonadotrophin-releasing hormone (GnRH) neurone: studies from embryonic GnRH neurons. J. Neuroendocrinol. 23, 542–553.
Constantin, S., Caraty, A., Wray, S., and Duittoz, A. H. (2009). Development of gonadotropin-releasing hormone-1 secretion in mouse nasal explants. Endocrinology 150, 3221–3227.
Costantin, J. L., and Charles, A. C. (1999). Spontaneous action potentials initiate rhythmic intercellular calcium waves in immortalized hypothalamic (Gt1-1) neurons. J. Neurophysiol. 82, 429–435.
Daikoku, S., and Koide, I. (1994). In vitro development of placode-derived LHRH neurons: possible involvement of alpha-fetoprotein. Horm. Behav. 28, 328–335.
Dennis, M. K., Burai, R., Ramesh, C., Petrie, W. K., Alcon, S. N., Nayak, T. K., Bologa, C. G., Leitao, A., Brailoiu, E., Deliu, E., Dun, N. J., Sklar, L. A., Hathaway, H. J., Arterburn, J. B., Oprea, T. I., and Prossnitz, E. R. (2009). In vivo effects of a GPR30 antagonist. Nat. Chem. Biol. 5, 421–427.
Duittoz, A. H., and Batailler, M. (2000). Pulsatile GnRH secretion from primary cultures of sheep olfactory placode explants. J. Reprod. Fertil. 120, 391–396.
Duittoz, A. H., Batailler, M., and Caldani, M. (1997). Primary cell culture of LHRH neurons from embryonic olfactory placode in the sheep (Ovis aries). J. Neuroendocrinol. 9, 669–675.
Dumesic, D. A., Schramm, R. D., and Abbott, D. H. (2005). Early origins of polycystic ovary syndrome. Reprod. Fertil. Dev. 17, 349–360.
Dunn, G. A., Morgan, C. P., and Bale, T. L. (2011). Sex-specificity in transgenerational epigenetic programming. Horm. Behav. 59, 290–295.
Fehrenbacher, J. C., LoVerme, J., Clarke, W., Hargreaves, K. M., Piomelli, D., and Taylor, B. K. (2009). Rapid pain modulation with nuclear receptor ligands. Brain Res. Rev. 60, 114–124.
Filardo, E. J., Quinn, J. A., Bland, K. I., and Frackelton, A. R. Jr. (2000). Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol. Endocrinol. 14, 1649–1660.
Filardo, E. J., Quinn, J. A., Frackelton, A. R. Jr., and Bland, K. I. (2002). Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol. Endocrinol. 16, 70–84.
Fisher, C. R., Graves, K. H., Parlow, A. F., and Simpson, E. R. (1998). Characterization of mice deficient in aromatase (ArKO) because of targeted disruption of the cyp19 gene. Proc. Natl. Acad. Sci. U.S.A. 95, 6965–6970.
Franceschini, I., Lomet, D., Cateau, M., Delsol, G., Tillet, Y., and Caraty, A. (2006). Kisspeptin immunoreactive cells of the ovine preoptic area and arcuate nucleus co-express estrogen receptor alpha. Neurosci. Lett. 401, 225–230.
Fuenzalida, L. C., Keen, K. L., and Terasawa, E. (2011). Colocalization of FM1-43, Bassoon and GnRH-1: GnRH-1 release from cell bodies and their neuroprocesses. Endocrinology 152, 4310–4321.
Fueshko, S., and Wray, S. (1994). LHRH cells migrate on peripherin fibers in embryonic olfactory explant cultures: an in vitro model for neurophilic neuronal migration. Dev. Biol. 166, 331–348.
Funabashi, T., Daikoku, S., Shinohara, K., and Kimura, F. (2000). Pulsatile gonadotropin-releasing hormone (GnRH) secretion is an inherent function of GnRH neurons, as revealed by the culture of medial olfactory placode obtained from embryonic rats. Neuroendocrinology 71, 138–144.
Gearing, M., and Terasawa, E. (1988). Luteinizing hormone releasing hormone (LHRH) neuroterminals mapped using the push-pull perfusion method in the rhesus monkey. Brain Res. Bull. 21, 117–121.
Glidewell-Kenney, C., Hurley, L. A., Pfaff, L., Weiss, J., Levine, J. E., and Jameson, J. L. (2007). Nonclassical estrogen receptor alpha signaling mediates negative feedback in the female mouse reproductive axis. Proc. Natl. Acad. Sci. U.S.A. 104, 8173–8177.
Grove-Strawser, D., Boulware, M. I., and Mermelstein, P. G. (2010). Membrane estrogen receptors activate the metabotropic glutamate receptors mGluR5 and mGluR3 to bidirectionally regulate CREB phosphorylation in female rat striatal neurons. Neuroscience 170, 1045–1055.
Harrington, W. R., Kim, S. H., Funk, C. C., Madak-Erdogan, Z., Schiff, R., Katzenellenbogen, J. A., and Katzenellenbogen, B. S. (2006). Estrogen dendrimer conjugates that preferentially activate extranuclear, nongenomic versus genomic pathways of estrogen action. Mol. Endocrinol. 20, 491–502.
Herbison, A. E. (2006). “Physiology of the GnRH neuronal network,” in Knobil and Neill’s Physiology of Reproduction, ed. J. D. Neill (San Diego, CA: Academic Press/Elsevier), 1415–1482.
Herbison, A. E. (2009). Rapid actions of oestrogen on gonadotropin-releasing hormone neurons; from fantasy to physiology? J. Physiol. 587, 5025–5030.
Herbison, A. E., and Pape, J. R. (2001). New evidence for estrogen receptors in gonadotropin-releasing hormone neurons. Front. Neuroendocrinol. 22, 292–308.
Hrabovszky, E., Kalló, I., Szlávik, N., Keller, E., Merchenthaler, I., and Liposits, Z. (2007). Gonadotropin-releasing hormone neurons express estrogen receptor-beta. J. Clin. Endocrinol. Metab. 92, 2827–2830.
Hrabovszky, E., Shughrue, P. J., Merchenthaler, I., Hajszán, T., Carpenter, C. D., Liposits, Z., and Petersen, S. L. (2000). Detection of estrogen receptor-beta messenger ribonucleic acid and 125I-estrogen binding sites in luteinizing hormone-releasing hormone neurons of the rat brain. Endocrinology 141, 3506–3509.
Hrabovszky, E., Steinhauser, A., Barabás, K., Shughrue, P. J., Petersen, S. L., Merchenthaler, I., and Liposits, Z. (2001). Estrogen receptor-beta immunoreactivity in luteinizing hormone-releasing hormone neurons of the rat brain. Endocrinology 142, 3261–3264.
Jasoni, C. L., Romanó, N., Constantin, S., Lee, K., and Herbison, A. E. (2010). Calcium dynamics in gonadotropin-releasing hormone neurons. Front. Neuroendocrinol. 31, 259–269.
Karsch, F. J., and Foster, D. L. (1975). Sexual differentiation of the mechanism controlling the preovulatory discharge of luteinizing hormone in sheep. Endocrinology 97, 373–379.
Kelly, M. J., Moss, R. L., and Dudley, C. A. (1976). Differential sensitivity of the preoptic-septal neurons to microelectrophoresed estrogen during the estrous cycle. Brain Res. 114, 152–157.
Kenealy, B. P., Keen, K. L., and Terasawa, E. (2011a). Rapid action of estradiol in primate GnRH neurons: the role of estrogen receptor alpha and estrogen receptor beta. Steroids 76, 861–866.
Kenealy, B. P., Keen, K. L., Rønnekleiv, O. K., and Terasawa, E. (2011b). STX a novel non-steroidal estrogenic compound, induces rapid action in primate GnRH neuronal calcium dynamics and peptide release. Endocrinology 152, 3182–3191.
Kenealy, B. P., Keen, K. L., and Terasawa, E. (2011c). “Role of intracellular and extracellular sources of calcium in the rapid estrogen action in primate GnRH neurons,” in Abstracts of the 41th Annual Meeting of the Society for Neuroscience (No. 500.08), Held November 12–16, 2011, Washington DC.
Knobil, E. (1980). The neuroendocrine control of the menstrual cycle. Recent Prog. Horm. Res. 36, 53–88.
Kokoris, G. J., Lam, N. Y., Ferin, M., Silverman, A. J., and Gibson, M. J. (1988). Transplanted gonadotropin-releasing hormone neurons promote pulsatile luteinizing hormone secretion in congenitally hypogonadal (hpg) male mice. Neuroendocrinology 48, 45–52.
Konkle, A. T., and McCarthy, M. M. (2011). Developmental time course of estradiol, testosterone, and dihydrotestosterone levels in discrete regions of male and female rat brain. Endocrinology 152, 223–235.
Krege, J. H., Hodgin, J. B., Couse, J. F., Enmark, E., Warner, M., Mahler, J. F., Sar, M., Korach, K. S., Gustafsson, J. A., and Smithies, O. (1998). Generation and reproductive phenotypes of mice lacking estrogen receptor beta. Proc. Natl. Acad. Sci. U.S.A. 95, 15677–15682.
Krsmanovic, L. Z., Stojilkovic, S. S., Merelli, F., Dufour, S. M., Virmani, M. A., and Catt, K. J. (1992). Calcium signaling and episodic secretion of gonadotropin-releasing hormone in hypothalamic neurons. Proc. Natl. Acad. Sci. U.S.A. 89, 8462–8466.
Kuiper, G. G., Enmark, E., Pelto-Huikko, M., and Gustafsson, J. A. (1996). Cloning of a novel receptor expressed in rat prostate and ovary. Proc. Natl. Acad. Sci. U.S.A. 93, 5925–5930.
Kuo, J., Hamid, N., Bondar, G., Prossnitz, E. R., and Micevych, P. (2010). Membrane estrogen receptors stimulate intracellular calcium release and progesterone synthesis in hypothalamic astrocytes. J. Neurosci. 30, 12950–12957.
Kurian, J. R., Keen, K. L., and Terasawa, E. (2010). Epigenetic changes during rhesus monkey (Macaca mulatta) GnRH neuronal development. Endocrinology 51, 5359–5368.
Labombarda, F., Meffre, D., Delespierre, B., Krivokapic-Blondiaux, S., Chastre, A., Thomas, P., Pang, Y., Lydon, J. P., Gonzalez, S. L., De Nicola, A. F., Schumacher, M., and Guennoun, R. (2010). Membrane progesterone receptors localization in the mouse spinal cord. Neuroscience 166, 94–106.
Lagrange, A. H., Rønnekleiv, O. K., and Kelly, M. J. (1995). Estradiol-17 beta and mu-opioid peptides rapidly hyperpolarize GnRH neurons: a cellular mechanism of negative feedback? Endocrinology 136, 2341–2344.
Lebesgue, D., Traub, M., De Butte-Smith, M., Chen, C., Zukin, R. S., Kelly, M. J., and Etgen, A. M. (2010). Acute administration of non-classical estrogen receptor agonists attenuates ischemia-induced hippocampal neuron loss in middle aged female rats. PLoS ONE 5, e8642. doi:10.1371/journal.pone.0008642
Leranth, C., Maclusky, N. J., Brown, T. J., Chen, E. C., Redmond, D. E. Jr., and Naftolin, F. (1992). Transmitter content and afferent connections of estrogen-sensitive progestin receptor-containing neurons in the primate hypothalamus. Neuroendocrinology 55, 667–682.
Marshall, J. C., and Eagleson, C. A. (1999). Neuroendocrine aspects of polycystic ovary syndrome. Endocrinol. Metab. Clin. North Am. 28, 295–324.
Martinez de la Escalera, G., Choi, A. L. H., and Weiner, R. I. (1992). Generation and synchronization of gonadotropin-releasing hormone (GnRH) pulses: intrinsic properties of the GT1-1 GnRH neuronal cell line. Proc. Natl. Acad. Sci. U.S.A. 89, 1852–1855.
Mellon, P. L., Windle, J. J., Goldsmith, P. C., Padula, C. A., Roberts, J. L., and Weiner, R. I. (1990). Immortalization of hypothalamic GnRH neurons by genetically targeted tumorigenesis. Neuron 5, 1–10.
Mizuno, M., and Terasawa, E. (2005). Search for neural substrates mediating inhibitory effects of oestrogen on pulsatile luteinizing hormone-releasing hormone release in vivo in ovariectomized female rhesus monkeys (Macaca mulatta). J. Neuroendocrinol. 17, 238–245.
Navarro, C. E., Saeed, S. A., Murdock, C., Martinez-Fuentes, A. J., Arora, K. K., Krsmanovic, L. Z., and Catt, K. J. (2003). Regulation of cyclic adenosine 3′,5′- monophosphate signaling and pulsatile neurosecretion by Gi-coupled plasma membrane estrogen receptors in immortalized gonadotrophin-releasing hormone neurons. Mol. Endocrinol. 17, 1792–1804.
Noel, S. D., Keen, K. L., Baumann, D. I., Filardo, E. J., and Terasawa, E. (2009). Involvement of G-protein coupled receptor 30 (GPR30) in rapid action of estrogen in primate LHRH neurons. Mol. Endocrinol. 23, 349–359.
Nunemaker, C. S., Straume, M., DeFazio, R. A., and Moenter, S. M. (2003). Gonadotropin-releasing hormone neurons generate interacting rhythms in multiple time domains. Endocrinology 144, 823–831.
Orchinik, M., Murray, T. F., and Moore, F. L. (1991). A corticosteroid receptor in neuronal membranes. Science 252, 1848–1851.
Padmanabhan, V., Sarma, H. N., Savabieasfahani, M., Steckler, T. L., and Veiga-Lopez, A. (2010). Developmental reprogramming of reproductive and metabolic dysfunction in sheep: native steroids vs. environmental steroid receptor modulators. Int. J. Androl. 33, 394–404.
Qiu, J., Bosch, M. A., Tobias, S. C., Grandy, D. K., Scanlan, T. S., Rønnekleiv, O. K., and Kelly, M. J. (2003). Rapid signaling of estrogen in hypothalamic neurons involves a novel G-protein-coupled estrogen receptor that activates protein kinase C. J. Neurosci. 23, 9529–9540.
Qiu, J., Bosch, M. A., Tobias, S. C., Krust, A., Graham, S. M., Murphy, S. J., Korach, K. S., Chambon, P., Scanlan, T. S., Rønnekleiv, O. K., and Kelly, M. J. (2006). A G-protein-coupled estrogen receptor is involved in hypothalamic control of energy homeostasis. J. Neurosci. 26, 5649–5655.
Qiu, J., Rønnekleiv, O. K., and Kelly, M. J. (2008). Modulation of hypothalamic neuronal activity through a novel G-protein-coupled estrogen membrane receptor. Steroids 73, 985–991.
Quanbeck, C., Sherwood, N. M., Millar, R. P., and Terasawa, E. (1997). Two populations of luteinizing hormone-releasing hormone neurons in the forebrain of the rhesus macaque during embryonic development. J. Comp. Neurol. 380, 293–309.
Radovick, S., Wray, S., Lee, E., Nicols, D. K., Nakayama, Y., Weintraub, B. D., Westphal, H., Cutler, G. B. Jr., and Wondisford, F. E. (1991). Migratory arrest of gonadotropin-releasing hormone neurons in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 88, 3402–3406.
Remage-Healey, L., Maidment, N. T., and Schlinger, B. A. (2008). Forebrain steroid levels fluctuate rapidy during social interactions. Nat. Neurosci. 11, 1327–1334.
Revankar, C. M., Cimino, D. F., Sklar, L. A., Arterburn, J. B., and Prossnitz, E. R. (2005). A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 307, 1625–1630.
Rønnekleiv, O. K., and Resko, J. A. (1990). Ontogeny of gonadotropin releasing hormone-containing neurons in early fetal development of rhesus macaques. Endocrinology 126, 498–511.
Saldanha, C. J., Remage-Healey, L., and Schlinger, B. A. (2011). Synaptocrine signaling: steroid synthesis and action at the synapse. Endocr. Rev. 32, 532–549.
Schneider, M. L., Moore, C. F., Kraemer, G. W., Roberts, A. D., and DeJesus, O. T. (2002). The impact of prenatal stress, fetal alcohol exposure, or both on development: perspectives from a primate model. Psychoneuroendocrinology 27, 285–298.
Sharifi, N., Reuss, A. E., and Wray, S. (2002). Prenatal LHRH neurons in nasal explant cultures express estrogen receptor beta transcript. Endocrinology 143, 2503–2507.
Shivers, B. D., Harlan, R. E., Morrell, J. I., and Pfaff, D. W. (1983). Absence of oestradiol concentration in cell nuclei of LHRH-immunoreactive neurons. Nature 304, 345–347.
Skinner, D. C., and Dufourny, L. (2005). Oestrogen receptor beta-immunoreactive neurons in the ovine hypothalamus: distribution and colocalisation with gonadotropin-releasing hormone. J. Neuroendocrinol. 17, 29–39.
Smith, J. T., Cunningham, M. J., Rissman, E. F., Clifton, D. K., and Steiner, R. A. (2005). Regulation of Kiss1 gene expression in the brain of the female mouse. Endocrinology 146, 3686–3692.
Spergel, D. J., Krüth, U., Hanley, D. F., Sprengel, R., and Seeburg, P. H. (1999). GABA- and glutamate-activated channels in green fluorescent protein-tagged gonadotropin-releasing hormone neurons in transgenic mice. J. Neurosci. 19, 2037–2050.
Stumpf, W. E., and Jennes, L. (1984). The A-B-C (Allocortex-Brainstem-Core) circuitry of endocrine-autonomic integration and regulation: a proposed hypothesis on the anatomical-functional relationships between estradiol sites of action and peptidergic-aminergic neuronal systems. Peptides 5(Suppl. 1), 221–226.
Sun, J., Chu, Z., and Moenter, S. M. (2010). Diurnal in vivo and rapid in vitro effects of estradiol on voltage-gated calcium channels in gonadotropin-releasing hormone neurons. J. Neurosci. 30, 3912–3923.
Suter, K. J., Song, W. J., Sampson, T. L., Wuarin, J. P., Saunders, J. T., Dudek, F. E., and Moenter, S. M. (2000). Genetic targeting of green fluorescent protein to gonadotropin-releasing hormone neurons: characterization of whole-cell electrophysiological properties and morphology. Endocrinology 141, 412–419.
Temple, J. L., Laing, E., Sunder, A., and Wray, S. (2004). Direct action of estradiol on gonadotropin-releasing hormone-1 neuronal activity via a transcription-dependent mechanism. J. Neurosci. 24, 6326–6333.
Terasawa, E. (2001). Luteinizing hormone-releasing hormone (LHRH) neurons: mechanism of pulsatile LHRH release. Vitam. Horm. 63, 91–129.
Terasawa, E., Keen, K. L., Mogi, K., and Claude, P. (1999a). Pulsatile release of luteinizing hormone-releasing hormone (LHRH) in cultured LHRH neurons derived from the embryonic olfactory placode of the rhesus monkey. Endocrinology 140, 1432–1441.
Terasawa, E., Schanhofer, W. K., Keen, K. L., and Luchansky, L. (1999b). Intracellular Ca(2+) oscillations in luteinizing hormone-releasing hormone neurons derived from the embryonic olfactory placode of the rhesus monkey. J. Neurosci. 19, 5898–5909.
Terasawa, E., Kurian, J. R., Guerriero, K. A., Kenealy, B. P., Hutz, E. D., and Keen, K. L. (2010). Recent discoveries on the control of primate GnRH neurons. J. Neuroendocrinol. 22, 630–638.
Terasawa, E., Noel, S. D., and Keen, K. L. (2009). Rapid action of oestrogen in luteinising hormone-releasing hormone neurones: the role of GPR30. J. Neuroendocrinol. 21, 316–321.
Terasawa, E., Quanbeck, C. D., Schulz, C. A., Burich, A. J., Luchansky, L. L., and Claude, P. (1993). A primary cell culture system of luteinizing hormone releasing hormone neurons derived from embryonic olfactory placode in the rhesus monkey. Endocrinology 133, 2379–2390.
Thomas, P., Alyea, R., Pang, Y., Peyton, C., Dong, J., and Berg, A. H. (2010). Conserved estrogen binding and signaling functions of the G protein-coupled estrogen receptor 1 (GPER) in mammals and fish. Steroids 75, 595–602.
Thomas, P., Pang, Y., Filardo, E. J., and Dong, J. (2005). Identity of an estrogen membrane receptor to a G protein in human breast cancer cells. Endocrinology 146, 624–632.
Toran-Allerand, C. D., Guan, X., MacLusky, N. J., Horvath, T. L., Diano, S., Singh, M., Connolly, E. S. Jr., Nethrapalli, I. S., and Tinnikov, A. A. (2002). ER-X: a novel, plasma membrane-associated, putative estrogen receptor that is regulated during development and after ischemic brain injury. J. Neurosci. 22, 8391–8401.
Wetsel, W. C., Valença, M. M., Merchenthaler, I., Liposits, Z., López, F. J., Weiner, R. I., Mellon, P. L., and Negro-Vilar, A. (1992). Intrinsic pulsatile secretory activity of immortalized luteinizing hormone-releasing hormone-secreting neurons. Proc. Natl. Acad. Sci. U.S.A. 89, 4149–4153.
Wintermantel, T. M., Campbell, R. E., Porteous, R., Bock, D., Gröne, H. J., Todman, M. G., Korach, K. S., Greiner, E., Pérez, C. A., Schütz, G., and Herbison, A. E. (2006). Definition of estrogen receptor pathway critical for the estrogen positive feedback to gonadotropin-releasing hormone neurons and fertility. Neuron 52, 271–280.
Woolley, C. S. (2007). Acute effects of estrogen on neuronal physiology. Annu. Rev. Pharmacol. Toxicol. 47, 657–680.
Yagi, K. (1973). Changes in firing rates of single preoptic and hypothalamic units following an intravenous administration of estrogen in the castrated female rat. Brain Res. 53, 343–352.
Yamaji, T., Dierschke, D. J., Hotchkiss, J., Bhattacharya, A. N., Surve, A. H., and Knobil, E. (1971). Estrogen induction of LH release in the rhesus monkey. Endocrinology 89, 1034–1041.
Zhang, C., Bosch, M. A., Levine, J. E., Rønnekleiv, O. K., and Kelly, M. J. (2007). Gonadotropin-releasing hormone neurons express K(ATP) channels that are regulated by estrogen and responsive to glucose and metabolic inhibition. J. Neurosci. 27, 10153–10164.
Keywords: GnRH neurons, rapid action of estradiol, membrane estrogen receptors, GPR30, primates
Citation: Kenealy BP and Terasawa E (2012) Rapid direct action of estradiol in GnRH neurons: findings and implications. Front. Endocrin. 2:106. doi: 10.3389/fendo.2011.00106
Received: 29 September 2011;
Paper pending published: 22 November 2011;
Accepted: 06 December 2011;
Published online: 03 January 2012.
Edited by:
Henryk Urbanski, Oregon National Primate Research Center, USAReviewed by:
Jeff Schwartz, Griffith University, AustraliaRamesh Ramachandran, Pennsylvania State University, USA
Copyright: © 2012 Kenealy and Terasawa. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: E. Terasawa, Wisconsin National Primate Research Center, University of Wisconsin, 1223 Capitol Court, Madison, WI 53715-1299, USA. e-mail:dGVyYXNhd2FAcHJpbWF0ZS53aXNjLmVkdQ==