


- Pediatric Endocrinology Unit, Department of Women’s and Children’s Health, Karolinska Institutet, Stockholm, Sweden
The epiphyseal growth plate consists of a layer of cartilage present only during the growth period and vanishes soon after puberty in long bones. It is divided to three well-defined zones, from epiphyses; resting, proliferative, and hypertrophic zones. Chondrocyte proliferation and differentiation and subsequent bone formation in this cartilage are controlled by various endocrine, autocrine, and paracrine factors which finally results into elimination of the cartilaginous tissue and promotion of the epiphyseal fusion. As chondrocytes differentiate from round, quiescent, and single structure to flatten and proliferative and then large and terminally differentiated, they experience changes in their gene expression pattern which allow them to transform from cartilaginous tissue to bone. This review summarizes the literature in this area and shortly describes different factors that affect growth plate cartilage both at the local and systemic levels. This may eventually help us to develop new treatment strategies of different growth disorders.
Introduction
The bone and cartilage tissues of our skeletal system provide both mechanical support and protection of the vital organs of the body, including the heart, lungs, and brain. Consequently, normal growth and development of skeletal bone is essential. These processes involve two distinct pathways: membranous ossification, which gives rise to calvarial bones directly from osteoblastic cells; and endochondral ossification. The latter, is an important process in the replacement of the fetal skeleton during organogenesis and bone elongation until adult height is achieved.
Bone elongation occurs through proliferation and differentiation of the cells located in the cartilage of the epiphyseal growth plates at the ends of the long bones. These plates are present only during the growth period and vanish when sexual maturation is complete.
The growth plate is divided into three well-defined zones. Closest to the epiphysis, the resting zone (also known as the germinal zone) contains single or pairs of small, uniformly round, and relatively quiescent cells embedded in a large volume of extracellular matrix (ECM; Ballock and O’Keefe, 2003; Melrose et al., 2008). Immediately beneath this zone lies the proliferative zone, where the chondrocytes flatten, begin to divide and form ladders parallel to the bone alignment, and synthesize collagen of types II and XI (Ballock and O’Keefe, 2003). In the underlying zone of maturation, referred to as the hypertrophic zone, the chondrocytes which are larger and more swollen than in the other zones, begin their terminal differentiation. The characteristic features of these hypertrophic chondrocytes include a lack of cell division, a pronounced increase in alkaline phosphatase activity and synthesis of large amounts of various elements of the ECM, including type-X collagen, a unique short-chain collagen found only in this zone (O’Keefe et al., 1994; Karimian et al., 2008). This morphological transformation is regulated by various hormones and growth factors, both at systemic and local levels (Figure 1).
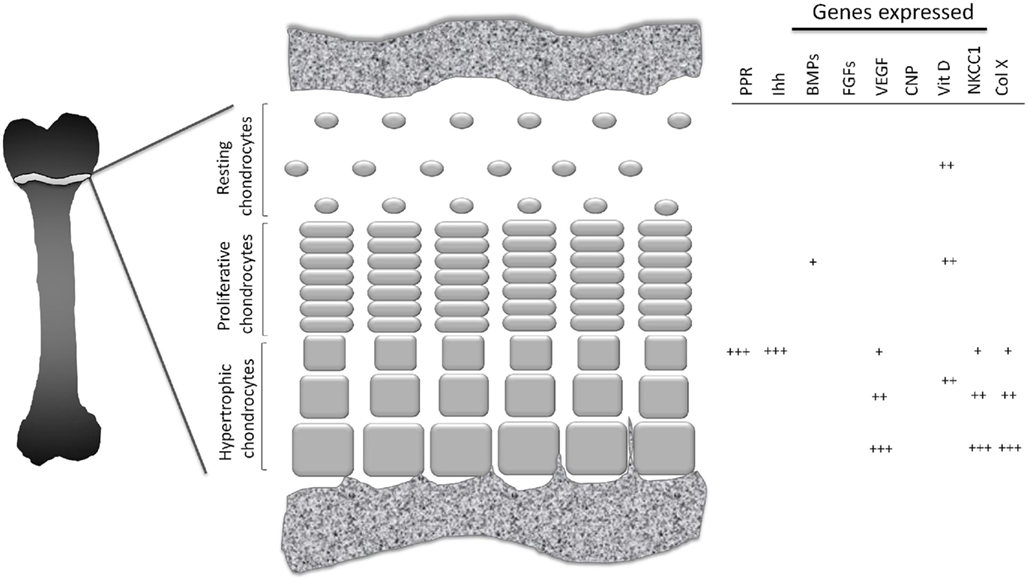
Figure 1. Schematic picture of growth plate cartilage. At different stages in their development growth plate chondrocytes exhibit different pattern of gene expression.
Local Regulation of the Growth Plate
The key para/autocrine regulators of bone formation are bone morphogenic proteins (BMPs), fibroblast growth factors (FGFs), hedgehog proteins and parathyroid hormone-related peptide (PTHrP), C-type natriuretic peptide (CNP), vascularization factors and vitamin D, transforming growth factor beta (TGF-β), CCAART/enhancing binding protein beta (C/EBP beta), pannexin 3, cilia and glycosylphosphatidylinositol (GPI).
Bone Morphogenic Proteins
Bone morphogenic proteins are growth and differentiation factors which have pivotal roles at every stage of endochondral bone formation and angiogenesis (Moser and Patterson, 2005; Zhang et al., 2009). Lack of BMPs and/or their receptors results into failure in mesenchymal condensation or digit formation in mice (Storm and Kingsley, 1999; Baur et al., 2000; Pizette and Niswander, 2000). At later stages, these proteins are expressed in the perichondrium as well as hypertrophic and proliferative chondrocytes. Indian hedgehog (Ihh) expression in prehypertrophic chondrocytes increases through BMP signaling (Minina et al., 2001, 2002), thereby increasing both the rate of chondrocyte proliferation and the length of proliferative columns. Furthermore, in vitro these factors directly trigger chondrogenesis by human mesenchymal stem cells to induce the formation of hypertrophic chondrocytes (Steinert et al., 2009).
Fibroblast Growth Factors
Genetic studies have shown that FGF-signaling play a key role in regulating chondrogenesis and 22 different FGFs and four target receptor (FGFR) genes are expressed at every stage of endochondral ossification (Ornitz and Marie, 2002; Ornitz, 2005). FGFs acting via FGF receptor-3 (FGFR3) are the key negative regulators in chondrocyte proliferation. An activation mutation in this receptor inhibits linear bone growth and may cause achondroplasia, hypochondroplasia, or type I or II thanatophoric dysplasia (Horton et al., 2007). In human growth plate cartilage predominant FGF ligands are FGF 1, 2, 17, and 19 (Krejci et al., 2007). All FGF ligands have the potential to bind FGFR3 as demonstrated in experimental studies (Ornitz et al., 1996; Zhang et al., 2006; Krejci et al., 2007). Inhibition of proliferation by FGF-signaling through FGFR3 involves, at least partially, activation of Janus kinase signaling and activator of transcription-1 (JAK–STAT1; Colvin et al., 1996). The expression of FGFs and their receptors in postnatal growth plate cartilage suggests that these proteins contribute to growth plate senescence and thus help to determine the size of the adult skeleton (Lazarus et al., 2007).
Signaling by Indian Hedgehog/Parathyroid Hormone-Related Protein
Indian hedgehog, the key paracrine regulator of bone development, orchestrates chondrocyte proliferation, and differentiation, as well as osteoblast differentiation. During bone formation, Ihh is expressed and secreted by post-mitotic hypertrophic chondrocytes simultaneously with expression of the parathyroid hormone-related protein receptor (PPR; Figure 1). Ihh diffuses throughout the growth plate and binds to its receptor, Patched-1 (Ptc-1) expressed by chondrocytes in the resting zone. This, in turn, activates downstream signaling via Smoothened (Smo) and transcription factors of the Gli family and leads to elevation of PTHrP expression (St-Jacques et al., 1999). PTHrP is expressed by periarticular chondrocytes, diffuses throughout the growth plate and delays differentiation of columnar chondrocytes into hypertrophic, thereby increasing the distance between the Ihh-expressing domain and the Ihh-targeted cell population. Thereby the Ihh/PTHrP feedback loop is formed which controls fetal growth plate development (Kronenberg, 2003).
The importance of the Ihh/PTHrP feedback loop for postnatal growth was recently revealed. Inactivation of either Ihh or PPR in postnatal chondrocytes leads to abrupt fusion of the epiphyseal growth plate in mice (Maeda et al., 2007; Hirai et al., 2011), thus suggesting that the loop is crucial for maintaining the growth plate in the open phase.
The importance of the Ihh/PTHrP loop for human physiology is exemplified by the following observations. Both Ihh and PTHrP are expressed in the human growth plate and expression levels correlate with pubertal progression (Kindblom et al., 2002). Inactivating mutation in Ihh results in acrocapitofemoral dysplasia, which is associated with premature closure of the growth plates (Hellemans et al., 2003). Inactivating mutation in the PTH/PTHrP receptor results in Blomstrand dysplasia (Jobert et al., 1998; Karaplis et al., 1998; Zhang et al., 1998). Patients with Blomstrand dysplasia die in uterus, but their skeletal abnormalities closely resembling those of the PTH/PTHrP receptor deficient mice (Lanske et al., 1996; Karaplis et al., 1998; Zhang et al., 1998). Furthermore, humans with Jansen chondro-osteodystrophy have point mutations in the gene encoding the PTH/PTHrP receptor that render the receptor active even in the absence of ligand (Schipani et al., 1995, 1996). Such people have growth abnormalities due to delay in hypertrophic chondrocyte differentiation and resultant short stature. Recently, two reports of families with inactivating, heterozygous mutations of the PTHrP gene show that such heterozygosity for PTHrP causes brachydactyly type E associated with premature cessation of growth in many cases (Klopocki et al., 2010; Maass et al., 2010). Finally, loss-of-function mutation in the GNAS1 gene, which encodes the α-subunit of the stimulatory G-protein (Gsα), downstream mediator of PPR in chondrocytes (Bastepe et al., 2004), leads to Albright hereditary osteodystrophy (AHO), which often display brachydactyly and premature closure of the epiphyseal growth plate (Steinbach and Young, 1966; Poznanski et al., 1977; Graudal et al., 1986). Altogether, existing data suggest that both in mice and humans the Ihh/PTHrP loop is crucial for maintaining the growth plate in an open phase.
C-Type Natriuretic Peptide
C-type natriuretic peptide is a member of the natriuretic peptide family, together with ANP and BNP proteins (Rosenzweig and Seidman, 1991), and it is widely expressed in many tissues including growth plate cartilages (Hagiwara et al., 1994). It has been identified as a crucial regulator of endochondral bone growth (Chusho et al., 2001; Olney, 2006). CNP promotes chondrogenesis by stimulating expression of molecules involved in cell adhesion and glycosaminoglycan synthesis (Woods et al., 2007). It acts through natriuretic peptide receptor 2 (NPr2). Disruption of the genes encoding CNP or NPr2 results into dwarfism and impaired endochondral ossification (Komatsu et al., 2002; Tamura et al., 2004) in mice.
Vascularization Factors
During limb bud development, chondrocytes in the middle of the bud undergo hypertrophic differentiation. Hypertrophic chondrocytes express several chemoattractant proteins, including vascular endothelial growth factor (VEGF; Zelzer et al., 2001), high mobility group box 1 protein (HMG-1; Taniguchi et al., 2007), and receptor activator of NFκB ligand (RANK-L; Kishimoto et al., 2006). This, in turn, attracts blood vessel invasion and brings in endothelial cells, osteoclasts, and osteoblasts leading to formation of the primary ossification center and bone development.
During bone elongation, vascularization is also of extreme importance. Despite the growth plate is an avascular structure, vascularization is important for substitution of cartilage tissue by bone tissue during endochondral ossification. Indeed, inactivating VEGF in mice utilizing soluble VEGF receptor impairs cartilage resorption leading to expansion of the hypertrophic zone and impaired formation of trabecular bone (Gerber et al., 1999). In addition, conditional inactivation of the Vegfa gene in the mouse growth plate leads to delayed invasion of blood vessels into the primary ossification center and delayed removal of terminal hypertrophic chondrocytes together with massive cell death in chondrocytes throughout the growth plate (Zelzer et al., 2004). Interestingly, estrogens can modulate the amount of VEGF expressed by growth plate chondrocytes, thus providing a new plausible mechanism for estrogen-dependent bone formation (Emons et al., 2010). Supporting such a mechanism is the fact that mice overexpressing VEGF in the growth plate dramatically increase their trabecular bone mass whereas the growth plate itself is not affected (Maes et al., 2010). Thus, vascularization is unequivocally important for bone elongation and new bone formation. However, if this process is utilized by the endocrine system to modulate fusion of the growth plate remains to be elucidated.
Vitamin D
Vitamin D needs double hydroxylation, 1α, 25-(OH)2D3, to exert its biological activity (DeLuca, 2004). Vitamin D deficiency results in rickets with disorganized growth plate, impaired mineralization, and widening of the growth plate. 1α, 25-(OH)2D3, locally regulates the growth plate cartilage, partially through a mechanism which involves the nuclear vitamin D receptor (Boyan et al., 2003). Less differentiated chondrocytes, including those in the resting zone, respond to 24,25-(OH)2D3 (Boyan et al., 2002, 2003; Denison et al., 2009) whereas more differentiated cells, including prehypertrophic and hypertrophic chondrocytes, respond primarily to 1α, 25-(OH)2D3 (Boyan et al., 2003). It has been reported that vitamin D receptor knockout mice (VDR−/−) arrests longitudinal bone growth 5 weeks after birth and peripheral quantitative computed tomography analysis of the femur midshaft show a significant decrease in all bone parameters (Xue and Fleet, 2009) when compared to the control mice. Disorganized, widened, and expanded growth plates in VDR−/− mice indicate the important role of vitamin D on longitudinal bone growth (Xue and Fleet, 2009).
Transforming Growth Factor Beta
The significance of TGF-β during skeletal development has been well established in different species (Gatherer et al., 1990; Pelton et al., 1990). It has been shown that a mutation in a TGF-β superfamily member causes skeletal dysplasia (Thomas et al., 1996). Recently it has been shown that E-selectin ligand-1 regulates growth plate homeostasis in mice by inhibiting the intracellular processing and secretion of mature TGF-β and therefore prevents skeletal dysplasia (Yang et al., 2010). In addition TGF-β is involved in biomineralization through ankyrin, an important inorganic pyrophosphate transporter (Skubutyte et al., 2010).
CCAART/Enhancing Binding Protein Beta and Pannexin 3
Transition of the chondrocytes from resting to proliferative and from proliferative to hypertrophic phase is a crucial step for endochondral ossification. The transcription factor (C/EBP beta) promotes the transition of proliferative chondrocytes to hypertrophic chondrocytes through transactivation of p57 (Hirata et al., 2009). Pannexin 3, a member of the gap junction protein family, promotes chondrocytes differentiation from proliferative to hypertrophic by regulating the intracellular ATP/cAMP levels (Iwamoto et al., 2010).
Cilia and Glycosylphosphatidylinositol (GPI)
The primary cilia may be partially responsible for polarization gradients in the growth plate allowing ladder alignment of chondrocytes in the proliferative zone. In addition, primary cilia might be important for transducing Ihh signaling (Koyama et al., 2007). Glycosylphosphatidylinositol (GPI) presumably contributes to this process via cell signaling and adhesion (Ahrens et al., 2009).
Systemic Regulation of the Growth Plate
During childhood longitudinal bone growth is regulated by a number of key hormones including growth hormone (GH), insulin-like growth factor-I (IGF-I), glucocorticoids, and thyroid hormone. During sexual maturation sex steroids (androgens and estrogens) contribute significantly to this process.
The GH/IGF-I System
From the second trimester of gestation GH is synthesized by the anterior pituitary gland (Kaplan et al., 1972). The physiological role of GH during fetal life is not well understood. During fetal life, IGF-I and IGF-II are generally believed to be the key determinants of embryonic growth (Honnebier and Swaab, 1973; Woods et al., 1996) although a later report indicated that GH may influence fetal development (Waters and Kaye, 2002). On the other hand, following birth and throughout puberty and adulthood GH is well known to play a critical role for longitudinal bone growth (Isaksson et al., 1987; Guler et al., 1988; van der Eerden et al., 2003), exerting anabolic effects on both trabecular and cortical bone (Giustina et al., 2008).
Growth hormone exerts its effects directly by binding to a single-chain trans-membrane glycoprotein receptor (GHR) expressed at high levels in almost all organs including growth plate cartilage (Gevers et al., 2002). At the same time, this hormone stimulates the production of IGF-I (formerly known as somatomedin C) in the liver (Melmed, 1999), which is the major target organ for GH and the principal site of IGF-I production. Also, several extrahepatic tissues synthesize this hormone under the local control of various hormones including GH (Ohlsson et al., 2009).
Ninety-nine percent of all circulating IGF-I is part of a 150-kD ternary complex also containing its dominant circulatory binding proteins, IGFBP-3 or IGFBP-5, and the acid labile subunit (ALS; Boisclair et al., 2001). This ternary complex stabilizes IGF-I, prolonging its half-life and availability to target organs (Boisclair et al., 2001). Genetic ablation of individual components of this complex (i.e., liver-specific IGF-I, ALS, and IGFBP-3) only partially reduces serum levels of IGF-I and yields only minor skeletal abnormalities, indicating that the remaining serum IGF-I still exerts a substantial impact on the skeleton (Yakar et al., 2009), alternatively, that an elevation in the level of GH is sufficient to compensate for the defect. However, triple-knockout mice lacking liver-specific IGF-1, ALS, and IGFBP-3 exhibit a 97% reduction in their serum level of IGF-I and obvious destruction of bone (Yakar et al., 2009). At the same time, this destruction was markedly less severe than in IGF-I-null mice (Liu et al., 1993), strongly suggesting that local IGF-I and its complexes in tissues, rather than circulating IGF-I, play a major role in skeletal development. Interestingly, a more recent study by Stratikopoulos et al. (2008) showed that mice expressing IGF-I in their livers only attain no more than approximately 30% of the normal adult body size during postnatal development, i.e., that endocrine IGF-I plays a highly significant role in murine growth.
Glucocorticoids
Glucocorticoid (GC) therapies often impair growth in both humans and animals (Altman et al., 1992; Allen, 1996; Chrysis et al., 2003), while hereditary deficiencies in these steroids are associated with tall stature (Elias et al., 2000). In rats and humans, epiphyseal cartilage and bone cells express the GC receptor (Silvestrini et al., 1999; Abu et al., 2000) suggesting that GCs exert direct effects on these tissues.
Even low doses of GCs suppress bone growth in both humans and animals, an effect believed to be mediated through disturbance of the GH/IGF-I axis at different levels (Allen, 1996; Smink et al., 2003). Smink et al. (2003) showed that such short-term treatment of mice inhibits growth significantly, decreases the width of the growth plate and the rate of chondrocyte proliferation, promotes apoptosis in hypertrophic chondrocytes, and reduces the local level of IGF-I in the growth plate. Baron et al. (1992) found that local infusion of dexamethasone into the epiphyseal growth plate of one leg of rabbits inhibits the growth of this leg and following dexamethasone withdrawal, catch-up growth in comparison to the contralateral leg occurs. Furthermore, we have recently demonstrated that both dexamethasone-dependent growth inhibition as well as followed catch-up growth occurs in cultured rat metatarsal bones (Chagin et al., 2010). These observations suggest that both the inhibitory effects of GCs on growth, as well as catch-up growth following GC withdrawal are characteristic of the growth plate, even though alterations in the GH/IGF system also play a role in this connection.
Thyroid Hormones
Thyroid hormones are important positive regulators of chondrocyte proliferation and hypertrophy (Shao et al., 2006; Mackie et al., 2011). In humans, hypothyroidism leads to retardation of longitudinal bone growth (Rivkees et al., 1988). In vitro studies have demonstrated that triiodothyronine (T3) stimulates chondrocytes hypertrophy together with molecular markers of differentiated chondrocytes (Burch and Lebovitz, 1982; Bohme et al., 1992; Wang et al., 2007). Mice lacking thyroid hormone receptor alfa-1 manifest typical features of skeletal hypothyroidism, postnatal growth retardation, and delayed endochondral ossification (O’Shea et al., 2005).
Androgens
Androgens are important for male gonadal differentiation during fetal life, for sexual maturation and maintenance during and after puberty as well as genital function and spermatogenesis during adulthood. However, their impact on bone growth and development in men remains not fully understood.
The androgen receptor (AR) is widely expressed in growth plate cartilage of several species, including rat, rabbit, and human (Abu et al., 1997; Ben-Hur et al., 1997; Nilsson et al., 2003a). Unilateral injection of testosterone into the epiphyseal growth plate of the rat tibia expands the width of this growth plate in comparison to the contralateral one (Ren et al., 1989). Moreover, non-aromatizable androgens such as dihydrotestosterone (DHT) and oxandrolone accelerate the rate of bone growth in patients without affecting systemic levels of GH (Stanhope et al., 1988; Veldhuis et al., 1997). In girls with Turner syndrome, oxandrolone has been used in combination with GH therapy and has been shown to increase both growth rate and adult height when given alone or in combination with GH (Batch, 2002). All mentioned evidences suggest that androgens have the capacity to stimulate bone growth, an effect which could be systemically and/or locally mediated. Oxandrolone has been found not to influence the growth of cultured fetal rat metatarsal bones suggesting that androgens in vivo might stimulate longitudinal bone growth by acting indirectly, rather than directly on growth plate chondrocytes (Chagin et al., 2009). Interestingly, Sun et al. (2011) recently reported that dehydroepiandrosterone (DHEA) suppresses the growth of cultured metatarsal bones by acting directly at the growth plate level through estrogen receptor (ER) interaction. This effect is however not surprising as DHEA is easily aromatized to estrogens, a process which may occur locally in the growth plate where the P450 aromatase (CYP19) is known to be expressed (Oz et al., 2001).
The evidence accumulated so far suggests that testosterone affects bone growth only after being, locally in the growth plate, aromatized to estrogens. Furthermore, despite demonstrating normal levels of testosterone both aromatase-deficient males and females exhibit the characteristic features of estrogen deficiency, i.e., an unfused epiphysis, markedly delayed bone development and severe osteopenia in adulthood (Morishima et al., 1995; Carani et al., 1997). Together, these findings indicate that estrogens should not be considered to be solely female hormones, but more generally as sex steroids required for normal bone growth and development in both females and males.
Estrogens and the Estrogen Receptors
Estrogens influence target organs through two known nuclear receptors, estrogen receptor-α (ERα; Green et al., 1986; Greene et al., 1986) and estrogen receptor-β (ERβ; Kuiper et al., 1996; Figure 2). These two receptors considered as classical ligand-activated transcription factors, located in the cytosol until binding their ligand. Then estrogen–ER complexes are translocated into the nucleus and interact with estrogen response elements (EREs) in the promoter regions of target genes. Additionally, estrogens bind to subpopulations of ERα and ERβ associated with the plasma membrane, thereby rapidly activating a variety of intracellular signaling cascades.
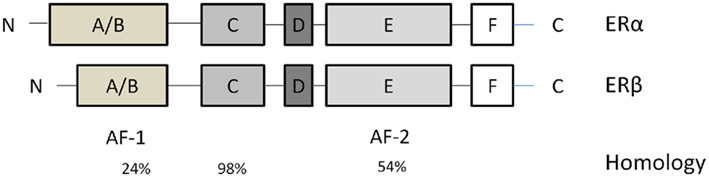
Figure 2. Estrogen receptors (ERs) structures. They contain six domains (A–F). From the N- to the C-terminus: the A/B domain mediates transactivation in the absence of ligand (AF-1). The C domain contains the DNA-binding domain that binds to EREs in target genes. The D domain is a hinge region that provides flexibility between the C- and N- termini. The E and F domains contain the site that binds estrogen and estrogenic compounds. The percentage homology between different domains in these two isoforms is indicated below these domains.
More recently, a membranous G-protein-coupled estrogen receptor (GPER; formerly named GPR30) that rapidly mediates estrogen signaling was discovered (Revankar et al., 2005). This receptor is widely expressed in the hypertrophic zone of human growth plate cartilage and the expression level decreases during pubertal progression, suggesting that GPER is involved in modulating longitudinal bone growth (Chagin and Sävendahl, 2007). Indeed, in vivo studies using genetically modified mice with targeted deletion of this receptor showed that GPER is required for normal estrogenic responses in the growth plate (Windahl et al., 2009).
Endogenous Production of Estrogen
Based on numerous in vivo and in vitro studies, it is now well established that chondrocytes can produce estrogens (Oz et al., 2001; Sylvia et al., 2002; van der Eerden et al., 2002). Expression of P450 aromatase in growth plate cartilage of both humans (Oz et al., 2001) and rats (van der Eerden et al., 2002; Chagin et al., 2006) further supports that chondrocytes have the potential to synthesize this hormone. Furthermore, local production of estrogens have been reported to be of importance for chondrocyte proliferation, protecting these cells from spontaneous cell death, and thereby regulating longitudinal bone growth (Chagin et al., 2006).
Role of Estrogens on Bone Maturation and Growth Plate Fusion
Estrogens were first considered as female hormones primarily responsible for pubertal growth and sexual organ development in girls, acting similarly as androgens do in boys. This theory was challenged by Smith and his colleagues in the 1990s when they described a unique case of an extremely tall 28-year-old man who still had unfused epiphyses, a bone age of 15 years, and a lumbar spine BMD that was 3 SD below the appropriate control mean. He was 204 cm tall, had an arm span of 213 cm and eunuchoidal appearance structure, and, due to his open epiphyses, was still growing slowly during the third decade of his life. This patient did not respond to high doses of transdermal ethinyl estradiol (Smith et al., 1994). Laboratory tests revealed an inactivation mutation in the ERα gene, which led to a substitution of thymine for cytosine. He was described to have a normal serum androgen level supporting that the genetic estrogen resistance indeed was responsible for this skeletal disorder (Smith et al., 1994). Histomorphometrical analysis of his bones revealed markedly decreased mineral content and impaired bone structure although the periosteal circumference was unaffected (Smith et al., 2008).
A year later, in 1995, a similar phenotype appearance was described by Morishima and his colleagues. DNA tests revealed a homozygous mutation of the aromatase P450 (CYP19) gene that led to high levels of androgens and undetectable levels of estradiol in the serum (Morishima et al., 1995; Zirilli et al., 2009). In contrast to the ERα-mutated man, this patient responded to high-dose estrogens.
These case reports indicate that androgen alone is not enough to advance skeletal maturation and maintain bone mass. Moreover, they demonstrate that estrogens play an important role in bone mineralization in both sexes. The expression of ERs in growth plate chondrocytes of different species (Nilsson et al., 1999, 2002) suggests that estrogens directly affect growth plate chondrocytes.
Estrogen Treatment for Modulation of Bone Growth
High-dose estrogen therapy has been documented to effectively reduce adult height in extremely tall girls (Goldzieher, 1956; Venn et al., 2008). However, the long-term effects of such treatment has to be considered. It has been reported that high-dose estrogen therapy may reduce fertility later in life (Venn et al., 2004; Hendriks et al., 2011) and even induce premature ovarian failure (Hendriks et al., 2011), increase the risk for deep vein thrombosis (Weimann and Brack, 1996), as well as, possibly, increase the risk for breast and gynecological cancers (Genazzani et al., 2001). Conversely, inhibition of estrogen biosynthesis by administering an aromatase inhibitor in patients with idiopathic short stature successfully delays skeletal maturation and enhances adult height (Hero et al., 2005). Later, it was reported that such treatment with an aromatase inhibitor may increase bone resorption (Hero et al., 2009). Furthermore, 2 years treatment of pre- or early- pubertal idiopathic short stature males with an aromatase inhibitor led to vertebral deformities (Hero et al., 2010), again emphasizing the importance of estrogens for bone health, not only in females but also in males.
Selective Estrogen Receptor Modulators
Unlike estrogens, which are all agonists, and antiestrogens, which are all antagonists, selective estrogen receptor modulators (SERMs) include a diversity of compounds that can act as either ER agonists or antagonists in a tissue-specific manner (Cho and Nuttall, 2001). They dissociate desirable estrogen effects on bone from undesirable stimulatory effects on the breast and endometrium. Despite lacking the steroid structure of estrogens, their tertiary structure allows them to bind to the ERs. Most, of these distinct activities on target tissues for estrogens can be explained by three major interacting factors: differences in expression levels of ERα and ERβ, differences in the conformational changes upon binding of ligand to ERs, and differences in the expression of coregulator proteins (coactivators or corepressors; Riggs and Hartmann, 2003).
Tamoxifen, a first generation SERM, has been used to treat pubertal gynecomastia in adolescent boys (Derman et al., 2003; Lawrence et al., 2004) and precocious puberty in patients with McCune–Albright Syndrome (Eugster et al., 2003). In these studies, longitudinal growth was either unaffected or diminished following months of treatment (Derman et al., 2003; Eugster et al., 2003; Lawrence et al., 2004). In an ex vivo model of cultured fetal rat metatarsal bones we earlier reported that tamoxifen may induce permanent growth arrest (Chagin et al., 2007). Indeed, in vivo data in male rats verified that tamoxifen impairs longitudinal bone growth, an effect which is accompanied by cortical bone growth destruction (Karimian et al., 2008).
Raloxifen, another SERM, acts as an estrogen agonist in bone and as an estrogen antagonist in breast and uterine tissues (Barrett-Connor, 2001). In rats, this compound inhibits longitudinal bone growth (Evans et al., 1994) while in ovariectomized rabbits it acts as an estrogen agonist on the growth plate accelerating chondrocyte senescence and thereby hastening epiphyseal fusion (Nilsson et al., 2003b). In contrast, Zirilli et al. (2009) recently reported that, unlike in rats and rabbits, raloxifene do not accelerate epiphyseal fusion, but does enhance BMD and circulating levels of gonadotropins in aromatase-deficient humans.
Conclusion
A better understanding of the genetic regulation of growth plate cartilage could open for novel therapeutic strategies in children with different growth disorders. As illustrated in this review, it is difficult to extrapolate data obtained in experimental animals to the human growth plate. This emphasizes the urgent need for new experimental models that will allow studies in human growth plate cartilage.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abu, E. O., Horner, A., Kusec, V., Triffitt, J. T., and Compston, J. E. (1997). The localization of androgen receptors in human bone. J. Clin. Endocrinol. Metab. 82, 3493–3497.
Abu, E. O., Horner, A., Kusec, V., Triffitt, J. T., and Compston, J. E. (2000). The localization of the functional glucocorticoid receptor alpha in human bone. J. Clin. Endocrinol. Metab. 85, 883–889.
Ahrens, M. J., Li, Y., Jiang, H., and Dudley, A. T. (2009). Convergent extension movements in growth plate chondrocytes require gpi-anchored cell surface proteins. Development 136, 3463–3474.
Allen, D. B. (1996). Growth suppression by glucocorticoid therapy. Endocrinol. Metab. Clin. North Am. 25, 699–717.
Altman, A., Hochberg, Z., and Silbermann, M. (1992). Interactions between growth hormone and dexamethasone in skeletal growth and bone structure of the young mouse. Calcif. Tissue Int. 51, 298–304.
Ballock, R. T., and O’Keefe, R. J. (2003). The biology of the growth plate. J. Bone Joint Surg. Am. 85-A, 715–726.
Baron, J., Huang, Z., Oerter, K. E., Bacher, J. D., and Cutler, G. B. Jr. (1992). Dexamethasone acts locally to inhibit longitudinal bone growth in rabbits. Am. J. Physiol. 263, E489–E492.
Bastepe, M., Weinstein, L. S., Ogata, N., Kawaguchi, H., Juppner, H., Kronenberg, H. M., and Chung, U. I. (2004). Stimulatory G protein directly regulates hypertrophic differentiation of growth plate cartilage in vivo. Proc. Natl. Acad. Sci. U.S.A. 101, 14794–14799.
Batch, J. (2002). Turner syndrome in childhood and adolescence. Best Pract. Res. Clin. Endocrinol. Metab. 16, 465–482.
Baur, S. T., Mai, J. J., and Dymecki, S. M. (2000). Combinatorial signaling through BMP receptor IB and GDF5: shaping of the distal mouse limb and the genetics of distal limb diversity. Development 127, 605–619.
Ben-Hur, H., Thole, H. H., Mashiah, A., Insler, V., Berman, V., Shezen, E., Elias, D., Zuckerman, A., and Ornoy, A. (1997). Estrogen, progesterone and testosterone receptors in human fetal cartilaginous tissue: immunohistochemical studies. Calcif. Tissue Int. 60, 520–526.
Bohme, K., Conscience-Egli, M., Tschan, T., Winterhalter, K. H., and Bruckner, P. (1992). Induction of proliferation or hypertrophy of chondrocytes in serum-free culture: the role of insulin-like growth factor-I, insulin, or thyroxine. J. Cell Biol. 116, 1035–1042.
Boisclair, Y. R., Rhoads, R. P., Ueki, I., Wang, J., and Ooi, G. T. (2001). The acid-labile subunit (ALS) of the 150 kDa IGF-binding protein complex: an important but forgotten component of the circulating IGF system. J. Endocrinol. 170, 63–70.
Boyan, B. D., Sylvia, V. L., Dean, D. D., Del Toro, F., and Schwartz, Z. (2002). Differential regulation of growth plate chondrocytes by 1alpha,25-(OH)2D3 and 24R,25-(OH)2D3 involves cell-maturation-specific membrane-receptor-activated phospholipid metabolism. Crit. Rev. Oral Biol. Med. 13, 143–154.
Boyan, B. D., Sylvia, V. L., Mckinney, N., and Schwartz, Z. (2003). Membrane actions of vitamin D metabolites 1alpha,25(OH)2D3 and 24R,25(OH)2D3 are retained in growth plate cartilage cells from vitamin D receptor knockout mice. J. Cell. Biochem. 90, 1207–1223.
Burch, W. M., and Lebovitz, H. E. (1982). Triiodothyronine stimulates maturation of porcine growth-plate cartilage in vitro. J. Clin. Invest. 70, 496–504.
Carani, C., Qin, K., Simoni, M., Faustini-Fustini, M., Serpente, S., Boyd, J., Korach, K. S., and Simpson, E. R. (1997). Effect of testosterone and estradiol in a man with aromatase deficiency. N. Engl. J. Med. 337, 91–95.
Chagin, A. S., Chrysis, D., Takigawa, M., Ritzen, E. M., and Savendahl, L. (2006). Locally produced estrogen promotes fetal rat metatarsal bone growth; an effect mediated through increased chondrocyte proliferation and decreased apoptosis. J. Endocrinol. 188, 193–203.
Chagin, A. S., Karimian, E., Sundstrom, K., Eriksson, E., and Savendahl, L. (2010). Catch-up growth after dexamethasone withdrawal occurs in cultured postnatal rat metatarsal bones. J. Endocrinol. 204, 21–29.
Chagin, A. S., Karimian, E., Zaman, F., Takigawa, M., Chrysis, D., and Savendahl, L. (2007). Tamoxifen induces permanent growth arrest through selective induction of apoptosis in growth plate chondrocytes in cultured rat metatarsal bones. Bone 40, 1415–1424.
Chagin, A. S., and Sävendahl, L. (2007). GPR30 estrogen receptor expression in the growth plate declines as puberty progresses. J. Clin. Endocrinol. Metab. 92, 4873–4877.
Chagin, A. S., Vannesjo, J., and Sävendahl, L. (2009). Androgen receptor modulation does not affect longitudinal growth of cultured fetal rat metatarsal bones. Horm. Res. 71, 219–227.
Cho, C. H., and Nuttall, M. E. (2001). Therapeutic potential of oestrogen receptor ligands in development for osteoporosis. Expert Opin. Emerg. Drugs 6, 137–154.
Chrysis, D., Ritzen, E. M., and Savendahl, L. (2003). Growth retardation induced by dexamethasone is associated with increased apoptosis of the growth plate chondrocytes. J. Endocrinol. 176, 331–337.
Chusho, H., Tamura, N., Ogawa, Y., Yasoda, A., Suda, M., Miyazawa, T., Nakamura, K., Nakao, K., Kurihara, T., Komatsu, Y., Itoh, H., Tanaka, K., Saito, Y., and Katsuki, M. (2001). Dwarfism and early death in mice lacking C-type natriuretic peptide. Proc. Natl. Acad. Sci. U.S.A. 98, 4016–4021.
Colvin, J. S., Bohne, B. A., Harding, G. W., Mcewen, D. G., and Ornitz, D. M. (1996). Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat. Genet. 12, 390–397.
DeLuca, H. F. (2004). Overview of general physiologic features and functions of vitamin D. Am. J. Clin. Nutr. 80, 1689S–1696S.
Denison, T. A., Koch, C. F., Shapiro, I. M., Schwartz, Z., and Boyan, B. D. (2009). Inorganic phosphate modulates responsiveness to 24,25(OH)2D3 in chondrogenic ATDC5 cells. J. Cell. Biochem. 107, 155–162.
Derman, O., Kanbur, N. O., and Kutluk, T. (2003). Tamoxifen treatment for pubertal gynecomastia. Int. J. Adolesc. Med. Health 15, 359–363.
Elias, L. L., Huebner, A., Metherell, L. A., Canas, A., Warne, G. L., Bitti, M. L., Cianfarani, S., Clayton, P. E., Savage, M. O., and Clark, A. J. (2000). Tall stature in familial glucocorticoid deficiency. Clin. Endocrinol. (Oxf.) 53, 423–430.
Emons, J., Chagin, A. S., Malmlof, T., Lekman, M., Tivesten, A., Ohlsson, C., Wit, J. M., Karperien, M., and Savendahl, L. (2010). Expression of vascular endothelial growth factor in the growth plate is stimulated by estradiol and increases during pubertal development. J. Endocrinol. 205, 61–68.
Eugster, E. A., Rubin, S. D., Reiter, E. O., Plourde, P., Jou, H. C., and Pescovitz, O. H. (2003). Tamoxifen treatment for precocious puberty in McCune-Albright syndrome: a multicenter trial. J. Pediatr. 143, 60–66.
Evans, G., Bryant, H. U., Magee, D., Sato, M., and Turner, R. T. (1994). The effects of raloxifene on tibia histomorphometry in ovariectomized rats. Endocrinology 134, 2283–2288.
Gatherer, D., Ten Dijke, P., Baird, D. T., and Akhurst, R. J. (1990). Expression of TGF-beta isoforms during first trimester human embryogenesis. Development 110, 445–460.
Genazzani, A. R., Gadducci, A., and Gambacciani, M. (2001). Controversial issues in climacteric medicine II. Hormone replacement therapy and cancer. Maturitas 40, 117–130.
Gerber, H. P., Vu, T. H., Ryan, A. M., Kowalski, J., Werb, Z., and Ferrara, N. (1999). VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat. Med. 5, 623–628.
Gevers, E. F., Van Der Eerden, B. C., Karperien, M., Raap, A. K., Robinson, I. C., and Wit, J. M. (2002). Localization and regulation of the growth hormone receptor and growth hormone-binding protein in the rat growth plate. J. Bone Miner. Res. 17, 1408–1419.
Giustina, A., Mazziotti, G., and Canalis, E. (2008). Growth hormone, insulin-like growth factors, and the skeleton. Endocr. Rev. 29, 535–559.
Goldzieher, M. A. (1956). Treatment of excessive growth in the adolescent female. J. Clin. Endocrinol. Metab. 16, 249–252.
Graudal, N., Milman, N., Nielsen, L. S., Niebuhr, E., and Bonde, J. (1986). Coexistent pseudohypoparathyroidism and D brachydactyly in a family. Clin. Genet. 30, 449–455.
Green, S., Walter, P., Greene, G., Krust, A., Goffin, C., Jensen, E., Scrace, G., Waterfield, M., and Chambon, P. (1986). Cloning of the human oestrogen receptor cDNA. J. Steroid Biochem. 24, 77–83.
Greene, G. L., Gilna, P., Waterfield, M., Baker, A., Hort, Y., and Shine, J. (1986). Sequence and expression of human estrogen receptor complementary DNA. Science 231, 1150–1154.
Guler, H. P., Zapf, J., Scheiwiller, E., and Froesch, E. R. (1988). Recombinant human insulin-like growth factor I stimulates growth and has distinct effects on organ size in hypophysectomized rats. Proc. Natl. Acad. Sci. U.S.A. 85, 4889–4893.
Hagiwara, H., Sakaguchi, H., Lodhi, K. M., Suda, K., and Hirose, S. (1994). Subtype switching of natriuretic peptide receptors in rat chondrocytes during in vitro culture. J. Biochem. 116, 606–609.
Hellemans, J., Coucke, P. J., Giedion, A., De Paepe, A., Kramer, P., Beemer, F., and Mortier, G. R. (2003). Homozygous mutations in IHH cause acrocapitofemoral dysplasia, an autosomal recessive disorder with cone-shaped epiphyses in hands and hips. Am. J. Hum. Genet. 72, 1040–1046.
Hendriks, A. E., Laven, J. S., Valkenburg, O., Fong, S. L., Fauser, B. C., De Ridder, M. A., De Jong, F. H., Visser, J. A., Van Ginneken, A. M., Boot, A. M., and Drop, S. L. (2011). Fertility and ovarian function in high-dose estrogen-treated tall women. J. Clin. Endocrinol. Metab. 96, 1098–1105.
Hero, M., Makitie, O., Kroger, H., Nousiainen, E., Toiviainen-Salo, S., and Dunkel, L. (2009). Impact of aromatase inhibitor therapy on bone turnover, cortical bone growth and vertebral morphology in pre- and peripubertal boys with idiopathic short stature. Horm. Res. 71, 290–297.
Hero, M., Norjavaara, E., and Dunkel, L. (2005). Inhibition of estrogen biosynthesis with a potent aromatase inhibitor increases predicted adult height in boys with idiopathic short stature: a randomized controlled trial. J. Clin. Endocrinol. Metab. 90, 6396–6402.
Hero, M., Toiviainen-Salo, S., Wickman, S., Makitie, O., and Dunkel, L. (2010). Vertebral morphology in aromatase inhibitor-treated males with idiopathic short stature or constitutional delay of puberty. J. Bone Miner. Res. 25, 1536–1543.
Hirai, T., Chagin, A. S., Kobayashi, T., Mackem, S., and Kronenberg, H. M. (2011). Parathyroid hormone/parathyroid hormone-related protein receptor signaling is required for maintenance of the growth plate in postnatal life. Proc. Natl. Acad. Sci. U.S.A. 108, 191–196.
Hirata, M., Kugimiya, F., Fukai, A., Ohba, S., Kawamura, N., Ogasawara, T., Kawasaki, Y., Saito, T., Yano, F., Ikeda, T., Nakamura, K., Chung, U. I., and Kawaguchi, H. (2009). C/EBP beta Promotes transition from proliferation to hypertrophic differentiation of chondrocytes through transactivation of p57. PLoS ONE 4, e4543. doi:10.1371/journal.pone.0004543
Honnebier, W. J., and Swaab, D. F. (1973). The influence of anencephaly upon intrauterine growth of fetus and placenta and upon gestation length. J. Obstet. Gynaecol. Br. Commonw. 80, 577–588.
Isaksson, O. G., Lindahl, A., Nilsson, A., and Isgaard, J. (1987). Mechanism of the stimulatory effect of growth hormone on longitudinal bone growth. Endocr. Rev. 8, 426–438.
Iwamoto, T., Nakamura, T., Doyle, A., Ishikawa, M., De Vega, S., Fukumoto, S., and Yamada, Y. (2010). Pannexin 3 regulates intracellular ATP/cAMP levels and promotes chondrocyte differentiation. J. Biol. Chem. 285, 18948–18958.
Jobert, A. S., Zhang, P., Couvineau, A., Bonaventure, J., Roume, J., Le Merrer, M., and Silve, C. (1998). Absence of functional receptors for parathyroid hormone and parathyroid hormone-related peptide in Blomstrand chondrodysplasia. J. Clin. Invest. 102, 34–40.
Kaplan, S. L., Grumbach, M. M., and Shepard, T. H. (1972). The ontogenesis of human fetal hormones. I. Growth hormone and insulin. J. Clin. Invest. 51, 3080–3093.
Karaplis, A. C., He, B., Nguyen, M. T., Young, I. D., Semeraro, D., Ozawa, H., and Amizuka, N. (1998). Inactivating mutation in the human parathyroid hormone receptor type 1 gene in Blomstrand chondrodysplasia. Endocrinology 139, 5255–5258.
Karimian, E., Chagin, A. S., Gjerde, J., Heino, T., Lien, E. A., Ohlsson, C., and Savendahl, L. (2008). Tamoxifen impairs both longitudinal and cortical bone growth in young male rats. J. Bone Miner. Res. 23, 1267–1277.
Kindblom, J. M., Nilsson, O., Hurme, T., Ohlsson, C., and Savendahl, L. (2002). Expression and localization of Indian hedgehog (Ihh) and parathyroid hormone related protein (PTHrP) in the human growth plate during pubertal development. J. Endocrinol. 174, R1–R6.
Kishimoto, K., Kitazawa, R., Kurosaka, M., Maeda, S., and Kitazawa, S. (2006). Expression profile of genes related to osteoclastogenesis in mouse growth plate and articular cartilage. Histochem. Cell Biol. 125, 593–602.
Klopocki, E., Hennig, B. P., Dathe, K., Koll, R., De Ravel, T., Baten, E., Blom, E., Gillerot, Y., Weigel, J. F., Kruger, G., Hiort, O., Seemann, P., and Mundlos, S. (2010). Deletion and point mutations of PTHLH cause brachydactyly type E. Am. J. Hum. Genet. 86, 434–439.
Komatsu, Y., Chusho, H., Tamura, N., Yasoda, A., Miyazawa, T., Suda, M., Miura, M., Ogawa, Y., and Nakao, K. (2002). Significance of C-type natriuretic peptide (CNP) in endochondral ossification: analysis of CNP knockout mice. J. Bone Miner. Metab. 20, 331–336.
Koyama, E., Young, B., Nagayama, M., Shibukawa, Y., Enomoto-Iwamoto, M., Iwamoto, M., Maeda, Y., Lanske, B., Song, B., Serra, R., and Pacifici, M. (2007). Conditional Kif3a ablation causes abnormal hedgehog signaling topography, growth plate dysfunction, and excessive bone and cartilage formation during mouse skeletogenesis. Development 134, 2159–2169.
Krejci, P., Krakow, D., Mekikian, P. B., and Wilcox, W. R. (2007). Fibroblast growth factors 1, 2, 17, and 19 are the predominant FGF ligands expressed in human fetal growth plate cartilage. Pediatr. Res. 61, 267–272.
Kuiper, G. G., Enmark, E., Pelto-Huikko, M., Nilsson, S., and Gustafsson, J. A. (1996). Cloning of a novel receptor expressed in rat prostate and ovary. Proc. Natl. Acad. Sci. U.S.A. 93, 5925–5930.
Lanske, B., Karaplis, A. C., Lee, K., Luz, A., Vortkamp, A., Pirro, A., Karperien, M., Defize, L. H., Ho, C., Mulligan, R. C., Abou-Samra, A. B., Juppner, H., Segre, G. V., and Kronenberg, H. M. (1996). PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science 273, 663–666.
Lawrence, S. E., Faught, K. A., Vethamuthu, J., and Lawson, M. L. (2004). Beneficial effects of raloxifene and tamoxifen in the treatment of pubertal gynecomastia. J. Pediatr. 145, 71–76.
Lazarus, J. E., Hegde, A., Andrade, A. C., Nilsson, O., and Baron, J. (2007). Fibroblast growth factor expression in the postnatal growth plate. Bone 40, 577–586.
Liu, J. P., Baker, J., Perkins, A. S., Robertson, E. J., and Efstratiadis, A. (1993). Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell 75, 59–72.
Maass, P. G., Wirth, J., Aydin, A., Rump, A., Stricker, S., Tinschert, S., Otero, M., Tsuchimochi, K., Goldring, M. B., Luft, F. C., and Bahring, S. (2010). A cis-regulatory site downregulates PTHLH in translocation t(8;12)(q13;p11.2) and leads to Brachydactyly Type E. Hum. Mol. Genet. 19, 848–860.
Mackie, E. J., Tatarczuch, L., and Mirams, M. (2011). The growth plate chondrocyte and endochondral ossification. J. Endocrinol. 211, 109–121.
Maeda, Y., Nakamura, E., Nguyen, M. T., Suva, L. J., Swain, F. L., Razzaque, M. S., Mackem, S., and Lanske, B. (2007). Indian Hedgehog produced by postnatal chondrocytes is essential for maintaining a growth plate and trabecular bone. Proc. Natl. Acad. Sci. U.S.A. 104, 6382–6387.
Maes, C., Goossens, S., Bartunkova, S., Drogat, B., Coenegrachts, L., Stockmans, I., Moermans, K., Nyabi, O., Haigh, K., Naessens, M., Haenebalcke, L., Tuckermann, J. P., Tjwa, M., Carmeliet, P., Mandic, V., David, J. P., Behrens, A., Nagy, A., Carmeliet, G., and Haigh, J. J. (2010). Increased skeletal VEGF enhances beta-catenin activity and results in excessively ossified bones. EMBO J. 29, 424–441.
Melmed, S. (1999). Insulin-like growth factor I – a prototypic peripheral-paracrine hormone? Endocrinology 140, 3879–3880.
Melrose, J., Hayes, A. J., Whitelock, J. M., and Little, C. B. (2008). Perlecan, the “jack of all trades” proteoglycan of cartilaginous weight-bearing connective tissues. Bioessays 30, 457–469.
Minina, E., Kreschel, C., Naski, M. C., Ornitz, D. M., and Vortkamp, A. (2002). Interaction of FGF, Ihh/Pthlh, and BMP signaling integrates chondrocyte proliferation and hypertrophic differentiation. Dev. Cell 3, 439–449.
Minina, E., Wenzel, H. M., Kreschel, C., Karp, S., Gaffield, W., Mcmahon, A. P., and Vortkamp, A. (2001). BMP and Ihh/PTHrP signaling interact to coordinate chondrocyte proliferation and differentiation. Development 128, 4523–4534.
Morishima, A., Grumbach, M. M., Simpson, E. R., Fisher, C., and Qin, K. (1995). Aromatase deficiency in male and female siblings caused by a novel mutation and the physiological role of estrogens. J. Clin. Endocrinol. Metab. 80, 3689–3698.
Moser, M., and Patterson, C. (2005). Bone morphogenetic proteins and vascular differentiation: BMPing up vasculogenesis. Thromb. Haemost. 94, 713–718.
Nilsson, L. O., Boman, A., Savendahl, L., Grigelioniene, G., Ohlsson, C., Ritzen, E. M., and Wroblewski, J. (1999). Demonstration of estrogen receptor-beta immunoreactivity in human growth plate cartilage. J. Clin. Endocrinol. Metab. 84, 370–373.
Nilsson, O., Abad, V., Chrysis, D., Ritzen, E. M., Savendahl, L., and Baron, J. (2002). Estrogen receptor-alpha and -beta are expressed throughout postnatal development in the rat and rabbit growth plate. J. Endocrinol. 173, 407–414.
Nilsson, O., Chrysis, D., Pajulo, O., Boman, A., Holst, M., Rubinstein, J., Martin Ritzen, E., and Savendahl, L. (2003a). Localization of estrogen receptors-alpha and -beta and androgen receptor in the human growth plate at different pubertal stages. J. Endocrinol. 177, 319–326.
Nilsson, O., Falk, J., Ritzen, E. M., Baron, J., and Savendahl, L. (2003b). Raloxifene acts as an estrogen agonist on the rabbit growth plate. Endocrinology 144, 1481–1485.
Ohlsson, C., Mohan, S., Sjogren, K., Tivesten, A., Isgaard, J., Isaksson, O., Jansson, J. O., and Svensson, J. (2009). The role of liver-derived insulin-like growth factor-I. Endocr. Rev. 30, 494–535.
O’Keefe, R. J., Puzas, J. E., Loveys, L., Hicks, D. G., and Rosier, R. N. (1994). Analysis of type II and type X collagen synthesis in cultured growth plate chondrocytes by in situ hybridization: rapid induction of type X collagen in culture. J. Bone Miner. Res. 9, 1713–1722.
Olney, R. C. (2006). C-type natriuretic peptide in growth: a new paradigm. Growth Horm. IGF Res. 16(Suppl. A), S6–S14.
Ornitz, D. M. (2005). FGF signaling in the developing endochondral skeleton. Cytokine Growth Factor Rev. 16, 205–213.
Ornitz, D. M., and Marie, P. J. (2002). FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 16, 1446–1465.
Ornitz, D. M., Xu, J., Colvin, J. S., Mcewen, D. G., Macarthur, C. A., Coulier, F., Gao, G., and Goldfarb, M. (1996). Receptor specificity of the fibroblast growth factor family. J. Biol. Chem. 271, 15292–15297.
O’Shea, P. J., Bassett, J. H., Sriskantharajah, S., Ying, H., Cheng, S. Y., and Williams, G. R. (2005). Contrasting skeletal phenotypes in mice with an identical mutation targeted to thyroid hormone receptor alpha1 or beta. Mol. Endocrinol. 19, 3045–3059.
Oz, O. K., Millsaps, R., Welch, R., Birch, J., and Zerwekh, J. E. (2001). Expression of aromatase in the human growth plate. J. Mol. Endocrinol. 27, 249–253.
Pelton, R. W., Dickinson, M. E., Moses, H. L., and Hogan, B. L. (1990). In situ hybridization analysis of TGF beta 3 RNA expression during mouse development: comparative studies with TGF beta 1 and beta 2. Development 110, 609–620.
Pizette, S., and Niswander, L. (2000). BMPs are required at two steps of limb chondrogenesis: formation of prechondrogenic condensations and their differentiation into chondrocytes. Dev. Biol. 219, 237–249.
Poznanski, A. K., Werder, E. A., Giedion, A., Martin, A., and Shaw, H. (1977). The pattern of shortening of the bones of the hand in PHP and PPHP – a comparison with brachydactyly E, Turner Syndrome, and acrodysostosis. Radiology 123, 707–718.
Ren, S. G., Malozowski, S., Sanchez, P., Sweet, D. E., Loriaux, D. L., and Cassorla, F. (1989). Direct administration of testosterone increases rat tibial epiphyseal growth plate width. Acta Endocrinol. 121, 401–405.
Revankar, C. M., Cimino, D. F., Sklar, L. A., Arterburn, J. B., and Prossnitz, E. R. (2005). A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 307, 1625–1630.
Riggs, B. L., and Hartmann, L. C. (2003). Selective estrogen-receptor modulators – mechanisms of action and application to clinical practice. N. Engl. J. Med. 348, 618–629.
Rivkees, S. A., Bode, H. H., and Crawford, J. D. (1988). Long-term growth in juvenile acquired hypothyroidism: the failure to achieve normal adult stature. N. Engl. J. Med. 318, 599–602.
Rosenzweig, A., and Seidman, C. E. (1991). Atrial natriuretic factor and related peptide hormones. Annu. Rev. Biochem. 60, 229–255.
Schipani, E., Kruse, K., and Juppner, H. (1995). A constitutively active mutant PTH-PTHrP receptor in Jansen-type metaphyseal chondrodysplasia. Science 268, 98–100.
Schipani, E., Langman, C. B., Parfitt, A. M., Jensen, G. S., Kikuchi, S., Kooh, S. W., Cole, W. G., and Juppner, H. (1996). Constitutively activated receptors for parathyroid hormone and parathyroid hormone-related peptide in Jansen’s metaphyseal chondrodysplasia. N. Engl. J. Med. 335, 708–714.
Shao, Y. Y., Wang, L., and Ballock, R. T. (2006). Thyroid hormone and the growth plate. Rev. Endocr. Metab. Disord. 7, 265–271.
Silvestrini, G., Mocetti, P., Ballanti, P., Di Grezia, R., and Bonucci, E. (1999). Cytochemical demonstration of the glucocorticoid receptor in skeletal cells of the rat. Endocr. Res. 25, 117–128.
Skubutyte, R., Markova, D., Freeman, T. A., Anderson, D. G., Dion, A. S., Williams, C. J., Shapiro, I. M., and Risbud, M. V. (2010). Hypoxia-inducible factor regulation of ANK expression in nucleus pulposus cells: possible implications in controlling dystrophic mineralization in the intervertebral disc. Arthritis Rheum. 62, 2707–2715.
Smink, J. J., Gresnigt, M. G., Hamers, N., Koedam, J. A., Berger, R., and Van Buul-Offers, S. C. (2003). Short-term glucocorticoid treatment of prepubertal mice decreases growth and IGF-I expression in the growth plate. J. Endocrinol. 177, 381–388.
Smith, E. P., Boyd, J., Frank, G. R., Takahashi, H., Cohen, R. M., Specker, B., Williams, T. C., Lubahn, D. B., and Korach, K. S. (1994). Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N. Engl. J. Med. 331, 1056–1061.
Smith, E. P., Specker, B., Bachrach, B. E., Kimbro, K. S., Li, X. J., Young, M. F., Fedarko, N. S., Abuzzahab, M. J., Frank, G. R., Cohen, R. M., Lubahn, D. B., and Korach, K. S. (2008). Impact on bone of an estrogen receptor-alpha gene loss of function mutation. J. Clin. Endocrinol. Metab. 93, 3088–3096.
Stanhope, R., Buchanan, C. R., Fenn, G. C., and Preece, M. A. (1988). Double blind placebo controlled trial of low dose oxandrolone in the treatment of boys with constitutional delay of growth and puberty. Arch. Dis. Child. 63, 501–505.
Steinbach, H. L., and Young, D. A. (1966). The roentgen appearance of pseudohypoparathyroidism (PH) and pseudo-pseudohypoparathyroidism (PPH). Differentiation from other syndromes associated with short metacarpals, metatarsals, and phalanges. Am. J. Roentgenol. Radium Ther. Nucl. Med. 97, 49–66.
Steinert, A. F., Proffen, B., Kunz, M., Hendrich, C., Ghivizzani, S. C., Noth, U., Rethwilm, A., Eulert, J., and Evans, C. H. (2009). Hypertrophy is induced during the in vitro chondrogenic differentiation of human mesenchymal stem cells by bone morphogenetic protein-2 and bone morphogenetic protein-4 gene transfer. Arthritis Res. Ther. 11, R148.
St-Jacques, B., Hammerschmidt, M., and Mcmahon, A. P. (1999). Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 13, 2072–2086.
Storm, E. E., and Kingsley, D. M. (1999). GDF5 coordinates bone and joint formation during digit development. Dev. Biol. 209, 11–27.
Stratikopoulos, E., Szabolcs, M., Dragatsis, I., Klinakis, A., and Efstratiadis, A. (2008). The hormonal action of IGF1 in postnatal mouse growth. Proc. Natl. Acad. Sci. U.S.A. 105, 19378–19383.
Sun, H., Zang, W., Zhou, B., Xu, L., and Wu, S. (2011). DHEA suppresses longitudinal bone growth by acting directly at growth plate through estrogen receptors. Endocrinology 152, 1423–1433.
Sylvia, V. L., Gay, I., Hardin, R., Dean, D. D., Boyan, B. D., and Schwartz, Z. (2002). Rat costochondral chondrocytes produce 17beta-estradiol and regulate its production by 1alpha,25(OH)(2)D(3). Bone 30, 57–63.
Tamura, N., Doolittle, L. K., Hammer, R. E., Shelton, J. M., Richardson, J. A., and Garbers, D. L. (2004). Critical roles of the guanylyl cyclase B receptor in endochondral ossification and development of female reproductive organs. Proc. Natl. Acad. Sci. U.S.A. 101, 17300–17305.
Taniguchi, N., Yoshida, K., Ito, T., Tsuda, M., Mishima, Y., Furumatsu, T., Ronfani, L., Abeyama, K., Kawahara, K., Komiya, S., Maruyama, I., Lotz, M., Bianchi, M. E., and Asahara, H. (2007). Stage-specific secretion of HMGB1 in cartilage regulates endochondral ossification. Mol. Cell. Biol. 27, 5650–5663.
Thomas, J. T., Lin, K., Nandedkar, M., Camargo, M., Cervenka, J., and Luyten, F. P. (1996). A human chondrodysplasia due to a mutation in a TGF-beta superfamily member. Nat. Genet. 12, 315–317.
van der Eerden, B. C., Karperien, M., and Wit, J. M. (2003). Systemic and local regulation of the growth plate. Endocr. Rev. 24, 782–801.
van der Eerden, B. C., Van De Ven, J., Lowik, C. W., Wit, J. M., and Karperien, M. (2002). Sex steroid metabolism in the tibial growth plate of the rat. Endocrinology 143, 4048–4055.
Veldhuis, J. D., Metzger, D. L., Martha, P. M. Jr., Mauras, N., Kerrigan, J. R., Keenan, B., Rogol, A. D., and Pincus, S. M. (1997). Estrogen and testosterone, but not a nonaromatizable androgen, direct network integration of the hypothalamo-somatotrope (growth hormone)-insulin-like growth factor I axis in the human: evidence from pubertal pathophysiology and sex-steroid hormone replacement. J. Clin. Endocrinol. Metab. 82, 3414–3420.
Venn, A., Bruinsma, F., Werther, G., Pyett, P., Baird, D., Jones, P., Rayner, J., and Lumley, J. (2004). Oestrogen treatment to reduce the adult height of tall girls: long-term effects on fertility. Lancet 364, 1513–1518.
Venn, A., Hosmer, T., Hosmer, D., Bruinsma, F., Jones, P., Lumley, J., Pyett, P., Rayner, J. A., and Werther, G. (2008). Oestrogen treatment for tall stature in girls: estimating the effect on height and the error in height prediction. Clin. Endocrinol. (Oxf.) 68, 926–929.
Wang, L., Shao, Y. Y., and Ballock, R. T. (2007). Thyroid hormone interacts with the Wnt/beta-catenin signaling pathway in the terminal differentiation of growth plate chondrocytes. J. Bone Miner. Res. 22, 1988–1995.
Waters, M. J., and Kaye, P. L. (2002). The role of growth hormone in fetal development. Growth Horm. IGF Res. 12, 137–146.
Weimann, E., and Brack, C. (1996). Severe thrombosis during treatment with ethinylestradiol for tall stature. Horm. Res. 45, 261–263.
Windahl, S. H., Andersson, N., Chagin, A. S., Martensson, U. E., Carlsten, H., Olde, B., Swanson, C., Moverare-Skrtic, S., Savendahl, L., Lagerquist, M. K., Leeb-Lundberg, L. M., and Ohlsson, C. (2009). The role of the G protein-coupled receptor GPR30 in the effects of estrogen in ovariectomized mice. Am. J. Physiol. Endocrinol. Metab. 296, E490–E496.
Woods, A., Khan, S., and Beier, F. (2007). C-type natriuretic peptide regulates cellular condensation and glycosaminoglycan synthesis during chondrogenesis. Endocrinology 148, 5030–5041.
Woods, K. A., Camacho-Hubner, C., Savage, M. O., and Clark, A. J. (1996). Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N. Engl. J. Med. 335, 1363–1367.
Xue, Y., and Fleet, J. C. (2009). Intestinal vitamin D receptor is required for normal calcium and bone metabolism in mice. Gastroenterology 136, 1317–1327, e1311–1312.
Yakar, S., Rosen, C. J., Bouxsein, M. L., Sun, H., Mejia, W., Kawashima, Y., Wu, Y., Emerton, K., Williams, V., Jepsen, K., Schaffler, M. B., Majeska, R. J., Gavrilova, O., Gutierrez, M., Hwang, D., Pennisi, P., Frystyk, J., Boisclair, Y., Pintar, J., Jasper, H., Domene, H., Cohen, P., Clemmons, D., and Leroith, D. (2009). Serum complexes of insulin-like growth factor-1 modulate skeletal integrity and carbohydrate metabolism. FASEB J. 23, 709–719.
Yang, T., Mendoza-Londono, R., Lu, H., Tao, J., Li, K., Keller, B., Jiang, M. M., Shah, R., Chen, Y., Bertin, T. K., Engin, F., Dabovic, B., Rifkin, D. B., Hicks, J., Jamrich, M., Beaudet, A. L., and Lee, B. (2010). E-selectin ligand-1 regulates growth plate homeostasis in mice by inhibiting the intracellular processing and secretion of mature TGF-beta. J. Clin. Invest. 120, 2474–2485.
Zelzer, E., Glotzer, D. J., Hartmann, C., Thomas, D., Fukai, N., Soker, S., and Olsen, B. R. (2001). Tissue specific regulation of VEGF expression during bone development requires Cbfa1/Runx2. Mech. Dev. 106, 97–106.
Zelzer, E., Mamluk, R., Ferrara, N., Johnson, R. S., Schipani, E., and Olsen, B. R. (2004). VEGFA is necessary for chondrocyte survival during bone development. Development 131, 2161–2171.
Zhang, F., Qiu, T., Wu, X., Wan, C., Shi, W., Wang, Y., Chen, J. G., Wan, M., Clemens, T. L., and Cao, X. (2009). Sustained BMP signaling in osteoblasts stimulates bone formation by promoting angiogenesis and osteoblast differentiation. J. Bone Miner. Res. 24, 1224–1233.
Zhang, P., Jobert, A. S., Couvineau, A., and Silve, C. (1998). A homozygous inactivating mutation in the parathyroid hormone/parathyroid hormone-related peptide receptor causing Blomstrand chondrodysplasia. J. Clin. Endocrinol. Metab. 83, 3365–3368.
Zhang, X., Ibrahimi, O. A., Olsen, S. K., Umemori, H., Mohammadi, M., and Ornitz, D. M. (2006). Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J. Biol. Chem. 281, 15694–15700.
Keywords: growth plate, cartilage, epiphyseal fusion, chondrocyte
Citation: Karimian E, Chagin AS and Sävendahl L (2012) Genetic regulation of the growth plate. Front. Endocrin. 2:113. doi: 10.3389/fendo.2011.00113
Received: 29 September 2011;
Paper pending published: 31 October 2011;
Accepted: 20 December 2011;
Published online: 09 January 2012.
Edited by:
Silvano Bertelloni, S. Chiara University-Hospital, ItalyReviewed by:
Revi P. Mathew, Vanderbilt University Medical School, USAFrancesco Chiarelli, University of Chieti, Italy
Copyright: © 2012 Karimian, Chagin and Sävendahl. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Elham Karimian, Pediatric Endocrinology Unit Q2:08, Karolinska University Hospital, 171 76 Stockholm, Sweden. e-mail:ZWxoYW0ua2FyaW1pYW5Aa2kuc2U=