 Nicola J. Smith1,2*
Nicola J. Smith1,2*- 1 Molecular Cardiology, Victor Chang Cardiac Research Institute, Darlinghurst, NSW, Australia
- 2 St. Vincent’s Clinical School, University of New South Wales, Sydney, NSW, Australia
The discovery that a number of metabolites and metabolic intermediates can act through G protein-coupled receptors has attracted great interest in the field and has led to new therapeutic targets for diseases such as hypertension, type 2 diabetes, inflammation, and metabolic syndrome. However, the low apparent affinity of these ligands for their cognate receptors poses a number of challenges for pharmacologists interested in investigating receptor structure, function or physiology. Furthermore, the endogenous ligands matched to their receptors have other, well established metabolic roles and thus selectivity is difficult to achieve. This review discusses some of the issues researchers face when working with these receptors and highlights the ways in which a number of these obstacles have been overcome.
G protein-coupled receptors (GPCRs) respond to a diverse array of stimuli, making them obvious targets for therapeutic exploitation. Indeed, ligands for GPCRs currently account for ∼30% of FDA-approved pharmaceuticals (Overington et al., 2006). Despite great progress, few GPCRs are targeted therapeutically and more than 80 non-olfactory GPCRs remain to be deorphanized (matched to their cognate endogenous ligand; Allen and Roth, 2011). Initial deorphanization approaches involved screening a small group of known transmitters against a larger group of cloned receptors (Civelli, 2005). However, as the number of un-partnered transmitters dwindled, the concept of “reverse pharmacology” was born, where a known GPCR of therapeutic interest (most likely due to similarity to an existing therapeutically relevant GPCR or due to a distinct tissue expression pattern) is screened against larger libraries of potential endogenous mediators (Civelli, 2005). The reverse pharmacology approach yielded a number of additional ligand:receptor pairs, although some were found to come from unusual sources, such as metabolic intermediates (Table 1). In this review, “metabolic intermediate” refers to both true metabolic intermediates which have been assigned no biological effect other than comprising part of an essential metabolic pathway (e.g., succinate and β-hydroxybutyrate) and also to metabolites that are produced from digestion and until recently were thought to mediate their effects exclusively within the cell (e.g., free fatty acids, FFAs). The GPCRs discussed herein are also notable in that their apparent endogenous ligands interact at the receptors with low affinity. Whilst broadening the therapeutic options for a variety of diseases, such as diabetes and metabolic syndrome, these low affinity receptors have generated a number of additional challenges, discussed below.
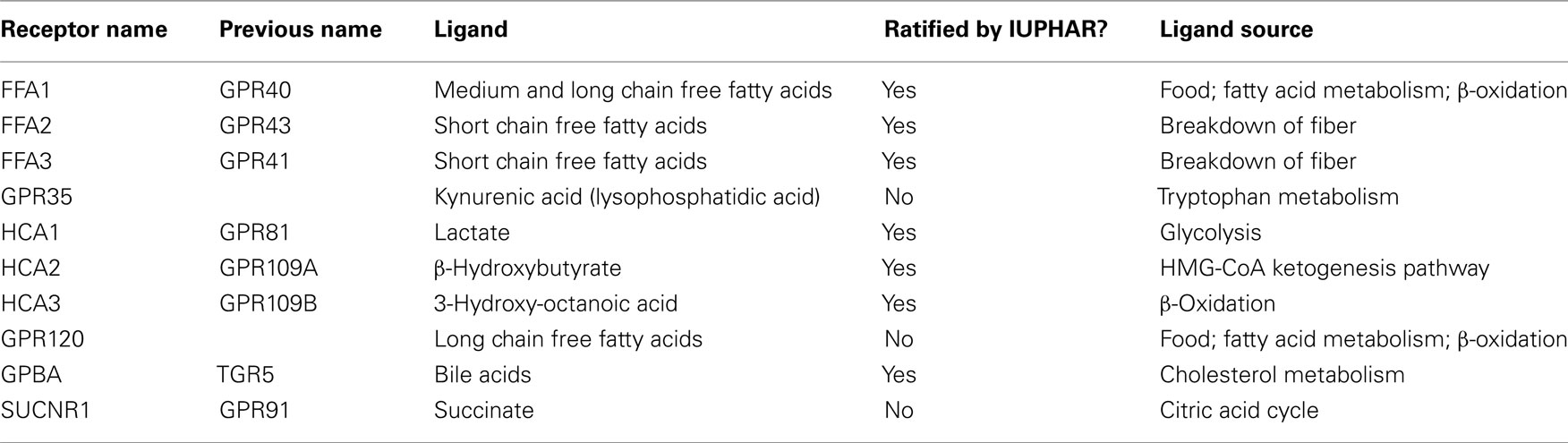
Table 1. Low affinity GPCRs for metabolites and metabolic intermediates.
Target Specificity/Selectivity
Target specificity or selectivity, the narrowness of the range of targets in an organism, is by definition a challenge at metabolic intermediate receptors. For example, succinate is a well established intermediate in the tricarboxylic acid/citric acid cycle (Krebs, 1970), while long chain fatty acids have for years been considered as nutrients (Yaney and Corkey, 2003) rather than cell surface ligands. Thus, distinguishing between GPCR-specific and non-GPCR functions for these endogenous ligands can be particularly challenging.
Coupled to the issue of lack of specificity is the apparent low affinity of the endogenous ligand:GPCR pairings. This has led many to question whether certain ligands are correctly assigned as the endogenous partner to the previously orphan receptor (Milligan et al., 2009; Milligan, 2011). Indeed, how can we determine whether ligand:receptor pairings are correct? A number of criteria must be met before a receptor is officially deorphanized by the International Union of Basic and Clinical Pharmacology Nomenclature Committee (NC-IUPHAR), including reproducibility of the original observation, consistent ligand and receptor pharmacology (often confirmed by transgenic mouse models) and, particularly pertinent to low affinity GPCRs, that the tissue concentrations achieved by each ligand are compatible with activating the cognate receptor (Sharman et al., 2011). This last point is often the most contentious. For example, all three FFA receptors are expressed in tissues where the local concentration of ligands are accepted to reach concentrations required for receptor activation, such as after a meal (Stoddart et al., 2008b; Hudson et al., 2011). In contrast, based upon reported potency values for succinate in vitro (He et al., 2004), the SUCNR1 receptor is unlikely to be activated except in cases where energy homeostasis has become dysregulated, such as in diabetes or metabolic syndrome (Deen and Robben, 2011), leading to it being viewed as a local sensor of metabolic and oxidative stress (see Ariza et al., 2012). Similarly, the lactate hydroxyl–carboxylic acid (HCA) receptor HCA1 (previously known as GPR81) is most likely activated after exercise or hyperglycemia as this is when lactate plasma concentrations are commensurate with activation (Ahmed et al., 2010). In contrast, kynurenic acid (KYNA) has been questioned as GPR35’s legitimate ligand as a result of failing the above criteria. Although KYNA agonism has been repeated by others, receptor activation occurs with micromolar potency (Jenkins et al., 2010, 2011; Zhao et al., 2010) while endogenous plasma concentrations of KYNA remain in the nanomolar range (see MacKenzie et al., 2011). The vast discrepancy in agonism at rodent versus human GPR35 (see below) is another concern, as is the discovery that lysophosphatidic acid can equally activate the receptor (Oka et al., 2010). NC-IUPHAR has recently released a statement indicating their reasons for not yet ratifying KYNA as the ligand for GPR351. For researchers new to the field, the NC-IUPHAR is a useful starting point for understanding legitimate versus spurious findings at GPCRs as their database is curated by a panel of esteemed pharmacologists (Foord et al., 2005).
Discrimination between specific effects of the metabolic intermediates at GPCRs versus their other physiological roles (here considered to be off-target effects) is hampered by a number of confounding issues. First, a number of the targets have overlapping tissue distributions. For example, the LCFA FFA1 receptor is found in the pancreas and is a promising target for the treatment of type 2 diabetes (Swaminath, 2008; Alquier and Poitout, 2009; Kebede et al., 2009; Hara et al., 2011). Although its predominant site of action as an insulin sensitizer is at adipose tissue, the widely expressed peroxisome proliferator-activated receptor, PPARγ, which also responds to LCFAs, is similarly expressed in the pancreas (Michalik et al., 2006). Furthermore, a selection of the insulin-sensitizing thiazolidinedione PPARγ agonists also activate FFA1 (Kotarsky et al., 2003; Smith et al., 2009), indicating similarity of both expression patterns and ligand pharmacophores. FFA1 expression patterns also overlap with that of another LCFA GPCR, GPR120, in taste buds (Matsumura et al., 2007; Cartoni et al., 2010), and enteroendocrine cells (Edfalk et al., 2008; Liou et al., 2011). In another example, the overlap of expression patterns is a source of debate. FFA3 has alternately been reported as having adipose tissue expression (Brown et al., 2003; Xiong et al., 2004) or lacking it (Hong et al., 2005; Zaibi et al., 2010), with mounting evidence pointing toward an FFA2-, not FFA3-, mediated role for adipocyte short chain FFAs (SCFAs; Hong et al., 2005; Ge et al., 2008; Zaibi et al., 2010; Dewulf et al., 2011). Second, some low affinity GPCRs are co-expressed and mediate the same physiological pathways as their non-GPCR counterparts. Bile acids, for example, activate both the GPCR, GPBA (also known as TGR5), and the nuclear hormone receptor, farnesoid X receptor (FXR), both of which are highly expressed in the liver and intestine where they play complementary roles in bile acid homeostasis and signaling (Chen et al., 2011). Third, both FFA2 and FFA3 receptors display overlapping tissue expression, respond to the same SCFAs and couple to Gαi/o signaling pathways (Brown et al., 2003; Stoddart et al., 2008b; Milligan et al., 2009), indicating that true physiological characterization requires knockout mice or the development of selective ligands.
As pharmacologists, how can we discriminate between GPCR and off-target effects of these low affinity metabolic intermediates? For SCFA receptors, there are subtle differences in the rank order of potency between FFA2 and FFA3 (Brown et al., 2003; Le Poul et al., 2003; Schmidt et al., 2011), allowing for subtype distinction based upon pharmacological parameters. Likewise, primary and secondary bile acids display different rank orders of potency for GPBA and FXR (Maruyama et al., 2002; Kawamata et al., 2003). Signaling outcomes are another way of discriminating between specific and non-specific effects. Although FFA2 and FFA3 both couple to Gαi/o, FFA2 additionally couples to Gαq/11, so inositol phosphates generation by SCFAs should be specific to FFA2 (Brown et al., 2003; Le Poul et al., 2003). Similarly, although FFA1 and GPR120 are both activated by LCFAs and signal through Gαq/11, only GPR120 robustly recruits β-arrestins in in vitro translocation assays (see Holliday et al., 2012). The time-course of signaling can also assist with discriminating between GPCR and non-GPCR functions, as nuclear receptors such as PPARγ and FXR result in transcriptional activation/repression (a process which takes hours) whilst FFA1 and GPBA activate second messengers within seconds/minutes.
Although an effective way to discriminate between targets, the abovementioned distinctions are cumbersome at the experimental level. More practical approaches rely upon the removal or absence of the off-target effector, either pharmacologically or genetically. A commonly used method is heterologous expression of the GPCR of interest in a cell line lacking each target or the identification of an endogenous cell line expressing only one of the targets. Arguably the most successfully applied expression system is the inducible FlpIN TRex method from Invitrogen2 where the gene of interest is stably integrated into the same genomic site of a polyclonal pool of cells and receptor expression is induced by the addition of tetracycline (Ward et al., 2011). Because of the likelihood of off-target effects being present at the high concentrations required to activate the metabolic intermediate receptors, each experiment can be performed in parallel in cells lacking the induced receptor. This method has been used to investigate the pharmacology of FFA1 (Stoddart et al., 2007; Smith et al., 2009), FFA2 and FFA3 (Schmidt et al., 2011; Smith et al., 2011b), GPR35 (Jenkins et al., 2010, 2011), and SUCNR1 (Robben et al., 2009). An alternative to parallel experimentation is the use of antagonists to the off-target protein. For example, the rapid and transient effects of LCFAs and thiazolidinediones on ERK1/2 activation were demonstrated to occur through FFA1 and not PPARγ by the use of various PPARγ and FFA1 antagonists (Smith et al., 2009). Unfortunately such selective ligands are not always available.
Despite the precautions being taken by pharmacologists to establish selectivity/specificity of each of the endogenous ligand:receptor interactions, new challenges continue to arise. For example, a very recent study demonstrated that while FFA3 responded to SCFAs to induce sympathetic outflow after a meal, this receptor was antagonized by the ketone β-hydroxybutyrate, thought to be the HCA2 endogenous agonist (Taggart et al., 2005), in times of starvation or in diabetes (Kimura et al., 2011). In another case, the apparent constitutive activity of FFA1 in [35S]GTPγS activation assays was demonstrated to actually result from ligand-mediated receptor activation due to endogenous agonists released during membrane preparation (Stoddart et al., 2007; Stoddart and Milligan, 2010). It seems likely that further unexpected interactions at these receptors will be discovered in the future.
Determination of Ligand Affinity and Receptor Binding Sites
Traditionally, receptor affinity has been determined by radiolabeling high affinity ligands (preferably antagonists) and subsequent saturation and competition binding assays (Bylund and Toews, 1993; Keen, 1995; Bylund et al., 2004). These experiments yield a plethora of information, including receptor expression levels, affinity of both the radiolabeled ligand and competing non-labeled ligands and association and dissociation kinetics. However, metabolic intermediates are largely unsuitable for development as radioligands, not least because of their low affinity, but also because of their numerous targets within the cell and, in some cases, their capacity to integrate into the plasma membrane. The exception to this is [3H]-nicotinic acid which has been used as a radiolabel at the HCA2 receptor since its discovery (Wise et al., 2003; Offermanns et al., 2011). Thus, in the absence of high affinity small molecules amenable to radiolabeling, novel approaches are required to demonstrate ligand binding and affinity.
While some radioligands have recently been reported in the patent literature for metabolic intermediate GPCRs, including FFA1 (Amgen, US patent 2010/0298367A1) and FFA2 (Euroscreen, WO2011092284A1), several groups have turned to alternative methods for determining ligand affinity. At FFA1, for example, Hara et al. (2009) employed a flow cytometry-based binding assay to measure BODIPY-C12 binding to immunoprecipitated FFA1 receptors. A more radical approach came from Bartoschek et al. (2010) who measured ligand binding of unlabeled FFA1 agonists using saturation transfer difference 1H NMR spectroscopy. Although informative these studies both had limitations, such as the potential loss of affinity by incorporation of the BOPIDY tag into the ligand and the limited chemical diversity permitted for the spectroscopy approach.
To circumvent the lack of radioligand binding assays, indirect measures of receptor function have been employed to obtain estimates of affinity. Based upon the assumption that signals most proximal to ligand binding and receptor activation are subjected to the least amplification, and therefore represent a 1:1:1 ratio of ligand:receptor:readout, potency values are increasingly used as proxy measures of affinity. One common assay employed for this purpose is the [35S]GTPγS activation assay, either using endogenous or co-transfected G proteins and rapid filtration or immunocapture (Labrecque et al., 2009; Strange, 2010), or a receptor-G protein fusion construct, which forces a strict 1:1 ratio (Milligan, 2000; Milligan et al., 2004). These assays have been used extensively for the metabolic intermediate GPCRs (for example, Stoddart et al., 2007, 2008a; Smith et al., 2009, 2011b) but do generally rely upon knowledge of the preferred G protein partner (this can be overcome in some cases using the receptor-G protein fusions). Another approach that avoids the problem of deciding which G protein subunit to assay is that of β-arrestin recruitment assays (Kocan and Pfleger, 2011). In assays where β-arrestin is recruited to an occupied receptor and the signal readout is unamplified [e.g., bioluminescence resonance energy transfer (BRET) and high content imaging of β-arrestin–GFP translocation] the measured potency should be equivalent to the affinity of the ligand for the receptor. Of course, these approaches are not guaranteed to work as not all GPCRs recruit β-arrestins (Liu et al., 2009, see Holliday et al., 2012). Finally, to measure antagonist affinity, any assay that allows generation of consecutive agonist curves in the presence of various concentrations of antagonist, such that a Schild plot or global analysis can be performed (Hall and Langmead, 2010), should yield affinity estimates of the antagonist. However, the low affinity of the metabolic intermediate GPCRs can prohibit the resolution of complete concentration–response curves at sufficient intervals.
In the absence of radioligands, how have the binding pockets of these GPCRs been investigated? For a number of receptors, sequence conservation and knowledge of the chemical structure of the ligand enabled mapping of the binding pocket. All three HCA receptors, for example, require a conserved arginine residue in transmembrane helix 3 (TM3) for receptor activation (Tunaru et al., 2005; Liu et al., 2009), presumably because the positive charge of the amino acid interacts with the negative charge of the carboxylic acid. This same residue (Arg3.36 according to the numbering of Ballesteros and Weinstein, 1995) was later shown to be critical for both KYNA and zaprinast activity at GPR35 (Jenkins et al., 2011). Similar observations were made for the FFA family, where Arg5.39 and Arg7.35 were absolutely required for FFA1 (Sum et al., 2007; Tikhonova et al., 2007; Smith et al., 2009), FFA2 (Stoddart et al., 2008a; Schmidt et al., 2011; Smith et al., 2011b), and FFA3 (Stoddart et al., 2008a; Schmidt et al., 2011) activation. Although none of these studies were able to directly demonstrate loss of binding rather than a global perturbation of receptor activation, molecular modeling (Sum et al., 2007; Tikhonova et al., 2007; Stoddart et al., 2008a; Schmidt et al., 2011) placed the ligands and residues within the generally conserved GPCR binding pocket. Additional evidence was also provided by the fact that an allosteric agonist for FFA2, 4-CMTB (Lee et al., 2008), retained the ability to activate the receptor at both single and double Arg5.39 and Arg7.35 mutant constructs (Smith et al., 2011b; Swaminath et al., 2011), demonstrating that the receptors themselves were correctly folded and still responsive to activation. Using their model, Tikhonova et al. (2008) developed and refined a pharmacophore model such that novel ligands could be screened in silico, leading to the identification of 15 new compounds active at FFA1. For GPR120, in silico library screening was possible in the absence of existing mutational data and although the model wasn’t confirmed by mutagenesis, the discovery of novel GPR120 agonists suggests the pharmacophore was correct (Sun et al., 2010). Thus, despite the obvious difficulties faced when working with low affinity GPCRs, significant inroads can still be achieved.
Species Differences
A final cautionary note is provided by studies using species orthologs of the metabolic intermediate receptors. Traditional drug discovery pipelines involve original hit discovery, often at human receptors, followed by in vitro characterization and then rodent models both to confirm efficacy in various disease states and to determine ligand pharmacokinetics/pharmacodynamics. Such a pipeline relies upon the assumption that each species ortholog displays similar pharmacology to the human receptor. This is not necessarily the case. A prime example is GPR35, which displays markedly lower potency for zaprinast and KYNA at human when compared to rat GPR35. This presents the very real possibility that lead compounds with efficacy in mice or rats could fail once introduced into humans (for a nice review of this issue, refer to Milligan, 2011). Fortunately, subsequent screening campaigns have identified ligands with either human selectivity (Jenkins et al., 2010; Zhao et al., 2010) or equivalent selectivity at both human and rat GPR35 (Jenkins et al., 2010). Such a phenomenon at species orthologs is not limited to GPR35 – mouse FFA1 has been shown to respond to both SCFAs and medium chain fatty acids when expressed heterologously in Xenopus oocytes, in addition to the LCFAs that are characteristic agonists for human FFA1 (Stewart et al., 2006).
Concluding Remarks
There is a growing appreciation that orphan and poorly characterized deorphanized GPCRs provide a largely untapped source of new therapeutic targets. However, the metabolic intermediate GPCRs provide a number of additional challenges to pharmacologists beyond those faced for high affinity GPCRs. However, after issues of low affinity, lack of specificity/selectivity and the need to use indirect measures of ligand binding have been considered, a great deal of progress has been made in our understanding of the physiological significance of these receptors. Subtype selectivity will remain a challenge to the field and the use of bitopic ligands or allosteric agonists/inhibitors (Smith et al., 2011a) may provide an alternative to pharmacological targeting of the orthosteric binding site in the future.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Nicola J. Smith is an NHMRC/NHF C. J. Martin Fellow. She thanks Professor Graeme Milligan (University of Glasgow, UK) for his guidance with low affinity GPCRs.
Footnotes
References
Ahmed, K., Tunaru, S., Tang, C., Muller, M., Gille, A., Sassmann, A., Hanson, J., and Offermanns, S. (2010). An autocrine lactate loop mediates insulin-dependent inhibition of lipolysis through GPR81. Cell Metab. 11, 311–319.
Allen, J. A., and Roth, B. L. (2011). Strategies to discover unexpected targets for drugs active at G protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol. 51, 117–144.
Ariza, A. C., Deen, P. M. T., and Robben, J. H. (2012). The succinate receptor as a novel therapeutic target for oxidative and metabolic stress-related conditions. Front. Endocrin. (in press).
Ballesteros, J. A., and Weinstein, H. (1995). Integrated methods for the constructions of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 25, 366–428.
Bartoschek, S., Klabunde, T., Defossa, E., Dietrich, V., Stengelin, S., Griesinger, C., Carlomagno, T., Focken, I., and Wendt, K. U. (2010). Drug design for G-protein-coupled receptors by a ligand-based NMR method. Angew. Chem. Int. Ed. Engl. 49, 1426–1429.
Brown, A. J., Goldsworthy, S. M., Barnes, A. A., Eilert, M. M., Tcheang, L., Daniels, D., Muir, A. I., Wigglesworth, M. J., Kinghorn, I., Fraser, N. J., Pike, N. B., Strum, J. C., Steplewski, K. M., Murdock, P. R., Holder, J. C., Marshall, F. H., Szekeres, P. G., Wilson, S., Ignar, D. M., Foord, S. M., Wise, A., and Dowell, S. J. (2003). The orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J. Biol. Chem. 278, 11312–11319.
Bylund, D. B., Deupree, J. D., and Toews, M. L. (2004). Radioligand-binding methods for membrane preparations and intact cells. Methods Mol. Biol. 259, 1–28.
Bylund, D. B., and Toews, M. L. (1993). Radioligand binding methods: practical guide and tips. Am. J. Physiol. 265(Pt 1), L421–L429.
Cartoni, C., Yasumatsu, K., Ohkuri, T., Shigemura, N., Yoshida, R., Godinot, N., Le Coutre, J., Ninomiya, Y., and Damak, S. (2010). Taste preference for fatty acids is mediated by GPR40 and GPR120. J. Neurosci. 30, 8376–8382.
Chen, X., Lou, G., Meng, Z., and Huang, W. (2011). TGR5: a novel target for weight maintenance and glucose metabolism. Exp. Diabetes Res. 2011, 853501.
Civelli, O. (2005). GPCR deorphanizations: the novel, the known and the unexpected transmitters. Trends Pharmacol. Sci. 26, 15–19.
Deen, P. M., and Robben, J. H. (2011). Succinate receptors in the kidney. J. Am. Soc. Nephrol. 22, 1416–1422.
Dewulf, E. M., Cani, P. D., Neyrinck, A. M., Possemiers, S., Van Holle, A., Muccioli, G. G., Deldicque, L., Bindels, L. B., Pachikian, B. D., Sohet, F. M., Mignolet, E., Francaux, M., Larondelle, Y., and Delzenne, N. M. (2011). Inulin-type fructans with prebiotic properties counteract GPR43 overexpression and PPARgamma-related adipogenesis in the white adipose tissue of high-fat diet-fed mice. J. Nutr. Biochem. 22, 712–722.
Edfalk, S., Steneberg, P., and Edlund, H. (2008). Gpr40 is expressed in enteroendocrine cells and mediates free fatty acid stimulation of incretin secretion. Diabetes 57, 2280–2287.
Foord, S. M., Bonner, T. I., Neubig, R. R., Rosser, E. M., Pin, J. P., Davenport, A. P., Spedding, M., and Harmar, A. J. (2005). International Union of Pharmacology. XLVI. G protein-coupled receptor list. Pharmacol. Rev. 57, 279–288.
Ge, H., Li, X., Weiszmann, J., Wang, P., Baribault, H., Chen, J. L., Tian, H., and Li, Y. (2008). Activation of G protein-coupled receptor 43 in adipocytes leads to inhibition of lipolysis and suppression of plasma free fatty acids. Endocrinology 149, 4519–4526.
Hall, D. A., and Langmead, C. J. (2010). Matching models to data: a receptor pharmacologist’s guide. Br. J. Pharmacol. 161, 1276–1290.
Hara, T., Hirasawa, A., Ichimura, A., Kimura, I., and Tsujimoto, G. (2011). Free fatty acid receptors FFAR1 and GPR120 as novel therapeutic targets for metabolic disorders. J. Pharm. Sci. 100, 3594–3601.
Hara, T., Hirasawa, A., Sun, Q., Koshimizu, T. A., Itsubo, C., Sadakane, K., Awaji, T., and Tsujimoto, G. (2009). Flow cytometry-based binding assay for GPR40 (FFAR1; free fatty acid receptor 1). Mol. Pharmacol. 75, 85–91.
He, W., Miao, F. J., Lin, D. C., Schwandner, R. T., Wang, Z., Gao, J., Chen, J. L., Tian, H., and Ling, L. (2004). Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature 429, 188–193.
Holliday, N. D., Watson, S.-J., and Brown, A. J. H. (2012). Drug discovery opportunities and challenges at G protein coupled receptors for long chain free fatty acids. Front. Endocrin. 2:112. doi: 10.3389/fendo.2011.00112
Hong, Y. H., Nishimura, Y., Hishikawa, D., Tsuzuki, H., Miyahara, H., Gotoh, C., Choi, K. C., Feng, D. D., Chen, C., Lee, H. G., Katoh, K., Roh, S. G., and Sasaki, S. (2005). Acetate and propionate short chain fatty acids stimulate adipogenesis via GPCR43. Endocrinology 146, 5092–5099.
Hudson, B. D., Smith, N., and Milligan, G. (2011). Experimental challenges to targeting poorly characterized GPCRs: uncovering the therapeutic potential for free fatty acid receptors. Adv. Pharmacol. 62, 175–218.
Jenkins, L., Alvarez-Curto, E., Campbell, K., de Munnik, S., Canals, M., Schlyer, S., and Milligan, G. (2011). Agonist activation of the G protein-coupled receptor GPR35 involves transmembrane domain III and is transduced via Gα and β-arrestin-2. Br. J. Pharmacol. 162, 733–748.
Jenkins, L., Brea, J., Smith, N. J., Hudson, B. D., Reilly, G., Bryant, N. J., Castro, M., Loza, M. I., and Milligan, G. (2010). Identification of novel species-selective agonists of the G-protein-coupled receptor GPR35 that promote recruitment of β-arrestin-2 and activate Galpha13. Biochem. J. 432, 451–459.
Kawamata, Y., Fujii, R., Hosoya, M., Harada, M., Yoshida, H., Miwa, M., Fukusumi, S., Habata, Y., Itoh, T., Shintani, Y., Hinuma, S., Fujisawa, Y., and Fujino, M. (2003). A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 278, 9435–9440.
Kebede, M. A., Alquier, T., Latour, M. G., and Poitout, V. (2009). Lipid receptors and islet function: therapeutic implications? Diabetes Obes. Metab. 11(Suppl. 4), 10–20.
Kimura, I., Inoue, D., Maeda, T., Hara, T., Ichimura, A., Miyauchi, S., Kobayashi, M., Hirasawa, A., and Tsujimoto, G. (2011). Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41). Proc. Natl. Acad. Sci. U.S.A. 108, 8030–8035.
Kocan, M., and Pfleger, K. D. (2011). Study of GPCR-protein interactions by BRET. Methods Mol. Biol. 746, 357–371.
Kotarsky, K., Nilsson, N. E., Flodgren, E., Owman, C., and Olde, B. (2003). A human cell surface receptor activated by free fatty acids and thiazolidinedione drugs. Biochem. Biophys. Res. Commun. 301, 406–410.
Labrecque, J., Wong, R. S., and Fricker, S. P. (2009). A time-resolved fluorescent lanthanide (Eu)-GTP binding assay for chemokine receptors as targets in drug discovery. Methods Mol. Biol. 552, 153–169.
Le Poul, E., Loison, C., Struyf, S., Springael, J. Y., Lannoy, V., Decobecq, M. E., Brezillon, S., Dupriez, V., Vassart, G., Van Damme, J., Parmentier, M., and Detheux, M. (2003). Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J. Biol. Chem. 278, 25481–25489.
Lee, T., Schwandner, R., Swaminath, G., Weiszmann, J., Cardozo, M., Greenberg, J., Jaeckel, P., Ge, H., Wang, Y., Jiao, X., Liu, J., Kayser, F., Tian, H., and Li, Y. (2008). Identification and functional characterization of allosteric agonists for the G protein-coupled receptor FFA2. Mol. Pharmacol. 74, 1599–1609.
Liou, A. P., Lu, X., Sei, Y., Zhao, X., Pechhold, S., Carrero, R. J., Raybould, H. E., and Wank, S. (2011). The G-protein-coupled receptor GPR40 directly mediates long-chain fatty acid-induced secretion of cholecystokinin. Gastroenterology 140, 903–912.
Liu, C., Wu, J., Zhu, J., Kuei, C., Yu, J., Shelton, J., Sutton, S. W., Li, X., Yun, S. J., Mirzadegan, T., Mazur, C., Kamme, F., and Lovenberg, T. W. (2009). Lactate inhibits lipolysis in fat cells through activation of an orphan G-protein-coupled receptor, GPR81. J. Biol. Chem. 284, 2811–2822.
MacKenzie, A. E., Lappin, J. E., Taylor, D. L., Nicklin, S. A., and Milligan, G. (2011). GPR35 as a novel therapeutic target. Front. Endocrin. 2:68. doi: 10.3389/fendo.2011.00068
Maruyama, T., Miyamoto, Y., Nakamura, T., Tamai, Y., Okada, H., Sugiyama, E., Itadani, H., and Tanaka, K. (2002). Identification of membrane-type receptor for bile acids (M-BAR). Biochem. Biophys. Res. Commun. 298, 714–719.
Matsumura, S., Mizushige, T., Yoneda, T., Iwanaga, T., Tsuzuki, S., Inoue, K., and Fushiki, T. (2007). GPR expression in the rat taste bud relating to fatty acid sensing. Biomed. Res. 28, 49–55.
Michalik, L., Auwerx, J., Berger, J. P., Chatterjee, V. K., Glass, C. K., Gonzalez, F. J., Grimaldi, P. A., Kadowaki, T., Lazar, M. A., O’Rahilly, S., Palmer, C. N., Plutzky, J., Reddy, J. K., Spiegelman, B. M., Staels, B., and Wahli, W. (2006). International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol. Rev. 58, 726–741.
Milligan, G. (2000). Insights into ligand pharmacology using receptor-G-protein fusion proteins. Trends Pharmacol. Sci. 21, 24–28.
Milligan, G. (2011). Orthologue selectivity and ligand bias: translating the pharmacology of GPR35. Trends Pharmacol. Sci. 32, 317–325.
Milligan, G., Feng, G. J., Ward, R. J., Sartania, N., Ramsay, D., McLean, A. J., and Carrillo, J. J. (2004). G protein-coupled receptor fusion proteins in drug discovery. Curr. Pharm. Des. 10, 1989–2001.
Milligan, G., Stoddart, L. A., and Smith, N. J. (2009). Agonism and allosterism: the pharmacology of the free fatty acid receptors FFA2 and FFA3. Br. J. Pharmacol. 158, 146–153.
Offermanns, S., Colletti, S. L., Lovenberg, T. W., Semple, G., Wise, A., and IJzerman, A. P. (2011). International Union of Basic and Clinical Pharmacology. LXXXII: nomenclature and classification of hydroxy-carboxylic acid receptors (GPR81, GPR109A, and GPR109B). Pharmacol. Rev. 63, 269–290.
Oka, S., Ota, R., Shima, M., Yamashita, A., and Sugiura, T. (2010). GPR35 is a novel lysophosphatidic acid receptor. Biochem. Biophys. Res. Commun. 395, 232–237.
Overington, J. P., Al-Lazikani, B., and Hopkins, A. L. (2006). How many drug targets are there? Nat. Rev. Drug Discov. 5, 993–996.
Robben, J. H., Fenton, R. A., Vargas, S. L., Schweer, H., Peti-Peterdi, J., Deen, P. M., and Milligan, G. (2009). Localization of the succinate receptor in the distal nephron and its signaling in polarized MDCK cells. Kidney Int. 76, 1258–1267.
Schmidt, J., Smith, N. J., Christiansen, E., Tikhonova, I. G., Grundmann, M., Hudson, B. D., Ward, R. J., Drewke, C., Milligan, G., Kostenis, E., and Ulven, T. (2011). Selective orthosteric free fatty acid receptor 2 (FFA2) agonists: identification of the structural and chemical requirements for selective activation of FFA2 versus FFA3. J. Biol. Chem. 286, 10628–10640.
Sharman, J. L., Mpamhanga, C. P., Spedding, M., Germain, P., Staels, B., Dacquet, C., Laudet, V., and Harmar, A. J. (2011). IUPHAR-DB: new receptors and tools for easy searching and visualization of pharmacological data. Nucleic Acids Res. 39, D534–D538.
Smith, N. J., Bennett, K. A., and Milligan, G. (2011a). When simple agonism is not enough: emerging modalities of GPCR ligands. Mol. Cell. Endocrinol. 331, 241–247.
Smith, N. J., Ward, R. J., Stoddart, L. A., Hudson, B. D., Kostenis, E., Ulven, T., Morris, J. C., Trankle, C., Tikhonova, I. G., Adams, D. R., and Milligan, G. (2011b). Extracellular loop 2 of the free fatty acid receptor 2 mediates allosterism of a phenylacetamide ago-allosteric modulator. Mol. Pharmacol. 80, 163–173.
Smith, N. J., Stoddart, L. A., Devine, N. M., Jenkins, L., and Milligan, G. (2009). The action and mode of binding of thiazolidinedione ligands at free fatty acid receptor 1. J. Biol. Chem. 284, 17527–17539.
Stewart, G., Hira, T., Higgins, A., Smith, C. P., and McLaughlin, J. T. (2006). Mouse GPR40 heterologously expressed in Xenopus oocytes is activated by short-, medium-, and long-chain fatty acids. Am. J. Physiol. Cell Physiol. 290, C785–C792.
Stoddart, L. A., Brown, A. J., and Milligan, G. (2007). Uncovering the pharmacology of the G protein-coupled receptor GPR40: high apparent constitutive activity in guanosine 5′-O-(3-[35S]thio)triphosphate binding studies reflects binding of an endogenous agonist. Mol. Pharmacol. 71, 994–1005.
Stoddart, L. A., and Milligan, G. (2010). Constitutive activity of GPR40/FFA1 intrinsic or assay dependent? Methods Enzymol. 484, 569–590.
Stoddart, L. A., Smith, N. J., Jenkins, L., Brown, A. J., and Milligan, G. (2008a). Conserved polar residues in transmembrane domains V, VI, and VII of free fatty acid receptor 2 and free fatty acid receptor 3 are required for the binding and function of short chain fatty acids. J. Biol. Chem. 283, 32913–32924.
Stoddart, L. A., Smith, N. J., and Milligan, G. (2008b). International Union of Pharmacology. LXXI. Free fatty acid receptors FFA1, -2, and -3: pharmacology and pathophysiological functions. Pharmacol. Rev. 60, 405–417.
Strange, P. G. (2010). Use of the GTPgammaS ([35S]GTPgammaS and Eu-GTPgammaS) binding assay for analysis of ligand potency and efficacy at G protein-coupled receptors. Br. J. Pharmacol. 161, 1238–1249.
Sum, C. S., Tikhonova, I. G., Neumann, S., Engel, S., Raaka, B. M., Costanzi, S., and Gershengorn, M. C. (2007). Identification of residues important for agonist recognition and activation in GPR40. J. Biol. Chem. 282, 29248–29255.
Sun, Q., Hirasawa, A., Hara, T., Kimura, I., Adachi, T., Awaji, T., Ishiguro, M., Suzuki, T., Miyata, N., and Tsujimoto, G. (2010). Structure-activity relationships of GPR120 agonists based on a docking simulation. Mol. Pharmacol. 78, 804–810.
Swaminath, G. (2008). Fatty acid binding receptors and their physiological role in type 2 diabetes. Arch. Pharm. (Weinheim) 341, 753–761.
Swaminath, G., Jaeckel, P., Guo, Q., Cardozo, M., Weiszmann, J., Lindberg, R., Wang, Y., Schwandner, R., and Li, Y. (2011). Mutational analysis of G-protein coupled receptor – FFA2. Biochem. Biophys. Res. Commun. 405, 122–127.
Taggart, A. K., Kero, J., Gan, X., Cai, T. Q., Cheng, K., Ippolito, M., Ren, N., Kaplan, R., Wu, K., Wu, T. J., Jin, L., Liaw, C., Chen, R., Richman, J., Connolly, D., Offermanns, S., Wright, S. D., and Waters, M. G. (2005). (D)-beta-hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA-G. J. Biol. Chem. 280, 26649–26652.
Tikhonova, I. G., Sum, C. S., Neumann, S., Engel, S., Raaka, B. M., Costanzi, S., and Gershengorn, M. C. (2008). Discovery of novel agonists and antagonists of the free fatty acid receptor 1 (FFAR1) using virtual screening. J. Med. Chem. 51, 625–633.
Tikhonova, I. G., Sum, C. S., Neumann, S., Thomas, C. J., Raaka, B. M., Costanzi, S., and Gershengorn, M. C. (2007). Bidirectional, iterative approach to the structural delineation of the functional “chemoprint” in GPR40 for agonist recognition. J. Med. Chem. 50, 2981–2989.
Tunaru, S., Lattig, J., Kero, J., Krause, G., and Offermanns, S. (2005). Characterization of determinants of ligand binding to the nicotinic acid receptor GPR109A (HM74A/PUMA-G). Mol. Pharmacol. 68, 1271–1280.
Ward, R. J., Alvarez-Curto, E., and Milligan, G. (2011). Using the Flp-In T-Rex system to regulate GPCR expression. Methods Mol. Biol. 746, 21–37.
Wise, A., Foord, S. M., Fraser, N. J., Barnes, A. A., Elshourbagy, N., Eilert, M., Ignar, D. M., Murdock, P. R., Steplewski, K., Green, A., Brown, A. J., Dowell, S. J., Szekeres, P. G., Hassall, D. G., Marshall, F. H., Wilson, S., and Pike, N. B. (2003). Molecular identification of high and low affinity receptors for nicotinic acid. J. Biol. Chem. 278, 9869–9874.
Xiong, Y., Miyamoto, N., Shibata, K., Valasek, M. A., Motoike, T., Kedzierski, R. M., and Yanagisawa, M. (2004). Short-chain fatty acids stimulate leptin production in adipocytes through the G protein-coupled receptor GPR41. Proc. Natl. Acad. Sci. U.S.A. 101, 1045–1050.
Yaney, G. C., and Corkey, B. E. (2003). Fatty acid metabolism and insulin secretion in pancreatic beta cells. Diabetologia 46, 1297–1312.
Zaibi, M. S., Stocker, C. J., O’Dowd, J., Davies, A., Bellahcene, M., Cawthorne, M. A., Brown, A. J., Smith, D. M., and Arch, J. R. (2010). Roles of GPR41 and GPR43 in leptin secretory responses of murine adipocytes to short chain fatty acids. FEBS Lett. 584, 2381–2386.
Zhao, P., Sharir, H., Kapur, A., Cowan, A., Geller, E. B., Adler, M. W., Seltzman, H. H., Reggio, P. H., Heynen-Genel, S., Sauer, M., Chung, T. D., Bai, Y., Chen, W., Caron, M. G., Barak, L. S., and Abood, M. E. (2010). Targeting of the orphan receptor GPR35 by pamoic acid: a potent activator of extracellular signal-regulated kinase and β-arrestin2 with antinociceptive activity. Mol. Pharmacol. 78, 560–568.
Keywords: G protein-coupled receptor, molecular pharmacology, metabolism, low affinity
Citation: Smith NJ (2012) Low affinity GPCRs for metabolic intermediates: challenges for pharmacologists. Front. Endocrin. 3:1. doi: 10.3389/fendo.2012.00001
Received: 28 November 2011;
Accepted: 03 January 2012;
Published online: 13 January 2012.
Edited by:
Brian Hudson, University of Glasgow, UKCopyright: © 2012 Smith. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Nicola J. Smith, Molecular Cardiology, Victor Chang Cardiac Research Institute, Lowy Packer Building, 405 Liverpool Street, Darlinghurst, NSW 2010, Australia. e-mail:bi5zbWl0aEB2aWN0b3JjaGFuZy5lZHUuYXU=