
 David J. Rossi2
David J. Rossi2
- 1 Division of Neuroscience, Oregon National Primate Research Center, Beaverton, OR, USA
- 2 Department of Behavioral Neuroscience, Oregon Health and Science University, Portland, OR, USA
This review will highlight a variety of mechanisms by which neurosteroids affect sensitivity to ethanol, including physiological states associated with activity of the hypothalamic–pituitary–adrenal (HPA) and hypothalamic–pituitary–gonadal (HPG) axes, and the effects of chronic exposure to ethanol, in addition to behavioral implications. To date, γ-aminobutyric acid (GABAA) receptor mechanisms are a major focus of the modulation of ethanol effects by neuroactive steroids. While NMDA receptor mechanisms are gaining prominence in the literature, these complex data would be best discussed separately. Accordingly, GABAA receptor mechanisms are emphasized in this review with brief mention of some NMDA receptor mechanisms to point out contrasting neuroactive steroid pharmacology. Overall, the data suggest that neurosteroids are virtually ubiquitous modulators of inhibitory neurotransmission. Neurosteroids appear to affect sensitivity to ethanol in specific brain regions and, consequently, specific behavioral tests, possibly related to the efficacy and potency of ethanol to potentiate the release of GABA and increase neurosteroid concentrations. Although direct interaction of ethanol and neuroactive steroids at common receptor binding sites has been suggested in some studies, this proposition is still controversial. It is currently difficult to assign a specific mechanism by which neuroactive steroids could modulate the effects of ethanol in particular behavioral tasks.
Background
In the brain, fast inhibitory electrical signaling is mediated primarily by the family of γ-aminobutyric acid (GABA)A receptors, making them important modulators of all neural processing. Harrison and Simmonds (1984) provided the first clear evidence that neuroactive steroids act at specific neurotransmitter receptors, investigating the ability of alphaxalone (3α-hydroxy-5α-pregnane l l,20-dione) to potentiate responses at GABAA receptors induced by muscimol in cortical slices. Within 2 years, a progesterone metabolite (3α-hydroxy-5α-pregnan-20-one, 3α,5α-THP, allopregnanolone) and a deoxycorticosterone metabolite (3α,21-dihydroxy-5α-pregnan-20-one, 3α,5α-THDOC, tetrahydrodeoxycorticosterone) were shown to be positive modulators of GABAA receptors (Majewska et al., 1986). That same year, publications showed that ethanol has direct action at GABAA receptors (Allan and Harris, 1986; Suzdak et al., 1986). Thus, the GABAA receptor complex was identified as a membrane-bound target providing a pharmacological basis for shared sensitivity between neurosteroids and ethanol. The common behavioral effects of ethanol and neuroactive steroids were compared directly using drug discrimination procedures (Ator et al., 1993). The N-methyl-D-aspartate (NMDA) receptor complex, a membrane-bound ionophore important for excitatory glutamate neurotransmission, was shown to be antagonized by low concentrations of ethanol (Lovinger et al., 1989). Soon, data was showing that neurosteroids act at NMDA receptors (Wu et al., 1991). Thus, by the mid-1990s, the stage was set for the suggestion that neurosteroids, and physiological states that alter circulating neuroactive steroids, could affect sensitivity to ethanol (Grant et al., 1997).
Neuroactive Steroids Modulate Neurotransmission in an Isomeric-Dependent Manner
Neuroactive steroids are produced in peripheral organs, including testes, ovaries, adrenal glands, lung, and liver, but can cross the blood–brain barrier and be metabolized in the brain. Further, neurosteroids are produced de novo in the brain. Whether of peripheral or central origin, all neuroactive steroids are derived from cholesterol. The transport of cholesterol to the inner mitochondrial membrane, mediated by steroidogenic acute regulatory protein (StAR), is the rate-limiting step in steroidogenesis (Stocco and Clark, 1996). At the inner mitochondrial membrane, the enzyme P450 side-chain cleavage transforms cholesterol to pregnenolone. Neuroactive steroids with activity at GABAA receptors are metabolites of the precursor steroids deoxycorticosterone, progesterone, testosterone, and androstenedione (Figure 1). Transformation of these precursors, respectively, first by 5α-reductase and then by 3α-hydroxysteroid dehydrogenase (HSD) produces 3α,5α-THDOC, 3α,5α-P, and 5α-androstan-3α-ol-17-one (3α,5α-A; androsterone). Each of these steroids, in addition to the 5β-reduced pregnane steroid 3α-hydroxy-5β-pregnan-20-one (3α,5β-P, pregnanolone), positively modulates responses to GABA at GABAA receptors (Puia et al., 1990; Park-Chung et al., 1999). Specifically, this means that the efficacy of GABA is increased by concentrations of steroid that do not, by themselves, activate the receptor. In contrast, muscimol and gaboxadol [4,5,6,7-tetrahydroisoxazolo (5,4-c)pyridin-3(-ol); THIP] are direct agonists of GABAA receptors because they open the Cl− channel in the absence of GABA. The pharmacological activity of neuroactive steroids depends on their isomeric configuration, which is determined by the steroidogenic enzymes that are present. Transformation by 5β-reductase and then by 3β-HSD results in neuroactive steroids that have distinct pharmacological activity resembling neuroactive steroids to which a sulfate group has been added (Wang et al., 2002). Sulfation by sulfotransferase enzymes is a low-energy strategy by which the effects of neuroactive steroids on neurotransmission may be adjusted (Gibbs et al., 2006). Sulfated or 3β-reduced steroids negatively modulate GABAA receptors (e.g., pregnanolone sulfate, epipregnanolone sulfate, androsterone sulfate, epiandrosterone sulfate). Other activity includes negative modulation (e.g., allopregnanolone sulfate, epipregnanolone sulfate) or positive modulation of NMDA receptor responses (e.g., epiallopregnanolone sulfate). Thus, sulfation may rapidly and transiently enhance excitatory neurotransmission, as suggested by Wu et al. (1991). The effects of sulfated steroids on glutamate transmission are hypothesized to regulate sensory processing and memory (Valenzuela et al., 2007). Overall, steroidogenesis allows for modulation of excitatory and inhibitory neurotransmission related to endocrine activity that may be an important regulator of sensitivity to environmental stimuli and behavior.
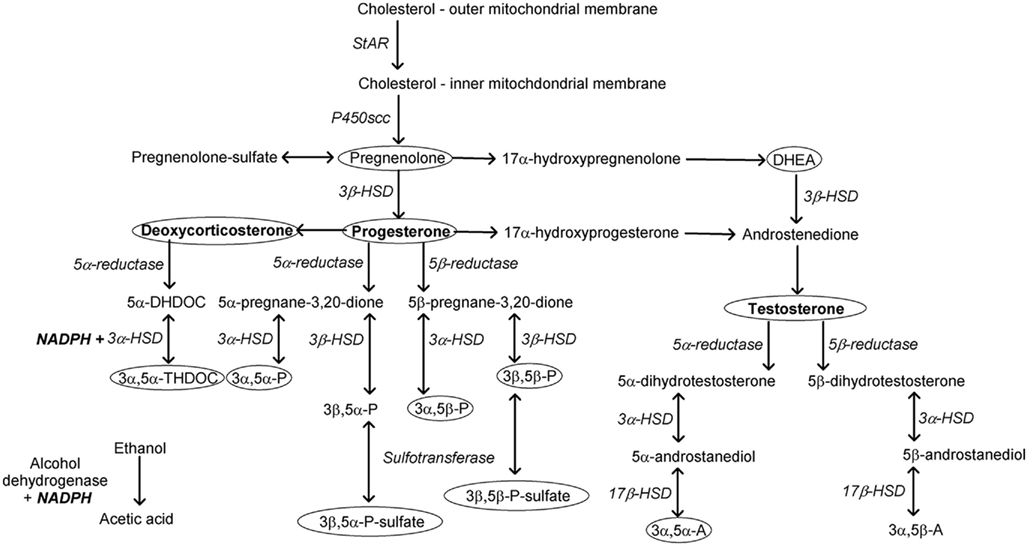
Figure 1. Neurosteroids modulating GABAergic neurotransmission are produced by the precursor steroids deoxycorticosterone (3α,5α-THDOC, allotetrahydrodeoxycorticosterone), progesterone (3α,5α-P, allopregna-nolone; 3α,5β-P, pregnenolone; 3β,5α-P, epiallopregnanolone; 3β,5β-P, epipregnanolone), and testosterone (3α,5α-A, androsterone; 3α,5β-A, etiocholanolone) via the sequential actions of reductase and hydroxy-steroid dehydrogenase enzymes. Ellipses highlight steroids known to influence sensitivity to the behavioral or endocrine effects of ethanol. Alcohol dehydrogenase and 3α-hydroxysteroid dehydrogenase require common co-factors. Sulfotransferase enzymes adding a sulfate group rapidly alter the pharmacological activity of neurosteroids. DHDOC, dihydrodeoxycortico-sterone; HSD, hydroxysteroid dehydrogenase; NADPH, nicotinamide adenine dinucleotide phosphate-oxidase; NADH, nicotinamide adenine dinucleotide; scc, side-chain cleavage; StAR, steroidogenic acute regulatory protein.
In addition to modulating receptor responses, neurosteroids have been shown to regulate neurotransmitter release. For example, pregnenolone sulfate increased glutamate release in neonatal cerebellar synapses (Zamudio-Bulcock and Valenzuela, 2011) and cultured hippocampal neurons (Meyer et al., 2002). Likewise, an increased probability of GABA release after application of pregnanolone was observed in studies using spinal motor neurons in toad embryos (Reith and Sillar, 1997). Haage et al. (2002) reported increased GABA release in isolated neurons from the medial preoptic area of rats after application of allopregnanolone. Thus, neurosteroids can modulate excitatory and inhibitory synaptic transmission by modulating receptor activity and neurotransmitter release. However, the efficacious actions of neuroactive steroids at GABAA receptors are particularly powerful (Park-Chung et al., 1999).
Ethanol Affects Neuroactive Steroid and Neurosteroid Concentrations
The ability of neuroactive steroids to modulate sensitivity to ethanol via GABAA receptors has been reviewed recently (Morrow et al., 2006; Biggio et al., 2007) and was the topic of a recent symposium on the interactions between stress and ethanol (Morrow et al., 2009). In addition to potentially sharing common receptor mechanisms, a major idea explored in these publications was that acute ethanol increases the release of neuroactive steroids. One mechanism involves stimulation of the hypothalamic–pituitary–adrenal (HPA) axis. Stimulation of the HPA axis results in the release of adrenocorticotropic hormone (ACTH) from the pituitary into circulation. ACTH can then stimulate the expression of StAR protein and the mitochondrial enzyme CYP11A1 (P450scc) through a second messenger pathway that involves cyclic adenosine monophosphate. As a result, ACTH promotes the production of pregnenolone from cholesterol in the adrenal cortex (Lavoie and King, 2009). As described, pregnenolone can be metabolized to all other neuroactive steroids depending on which steroidal enzymes are present (Figure 1). Also shown in Figure 1, hydroxysteroid dehydrogenase is a key enzyme involved in the production of neuroactive steroids, and this enzyme requires nicotinamide adenine dinucleotide phosphate (NADPH) or nicotinamide adenine dinucleotide (NADH) as a co-factor (Krause and Karavolas, 1980). Because the metabolism of ethanol also requires this co-factor, one possibility that requires additional investigation is that steroidogenesis could be altered in the presence of high doses of ethanol as the necessary co-factors are depleted (Crabb et al., 2004).
Several studies have shown that acute ethanol increases circulating neuroactive steroids (e.g., VanDoren et al., 2000; Porcu et al., 2010). As mentioned, ethanol appears to have this effect by increasing pituitary release of ACTH in conjunction with vasopressin and corticotropin releasing hormone (CRH; Lee et al., 2004). Adrenalectomy largely eliminates neuroactive steroids that are induced by ethanol (Porcu et al., 2004), indicating that ACTH stimulates adrenal production of neuroactive steroids. In rats, HPA axis activation and de novo synthesis of StAR protein are necessary for a threshold dose of 1.5 g/kg ethanol to increase GABAergic neuroactive steroids in plasma and cerebral cortex (Boyd et al., 2010b). Ethanol also stimulates de novo steroidogenesis within the brain in principal excitatory neurons and glial cells (Chisari et al., 2010). In hippocampal slices from 3-week-old rats, ethanol (50 mM) stimulated GABA release and increased the amplitude of postsynaptic currents upon application, with a second amplitude increase 20–30 min after application of ethanol. Because the secondary increase was blocked by an inhibitor of the enzyme 5α-reductase (finasteride), enzymatic conversion of neuroactive steroids appears to have mediated this effect of ethanol (Sanna et al., 2004). These data highlight the possibility of indirect effects of ethanol on GABAergic neurotransmission via steroidogenesis, for which variability in these processes could account for individual differences in sensitivity to ethanol.
Species differences in the efficacy of ethanol to increase neuroactive steroid concentrations have been reported, but procedural differences make comparison difficult. For example, acute ethanol increased plasma neuroactive steroids when given by injection to rodents [mean blood-ethanol concentration (BEC), 212 mg/dl] but did not change, or decreased neuroactive steroid concentrations following intragastric administration in macaque monkeys (mean BEC, 125 mg/dl; Porcu et al., 2010). Another confound is interactions with innate biological rhythms, as some neuroactive steroids have a diurnal rhythm, similar to cortisol (Weitzman et al., 1971). Porcu et al. (2008) demonstrated that, in humans, deoxycorticosterone has a diurnal rhythm in which concentrations are greater in the morning compared to the evening. In contrast, concentrations of pregnenolone sulfate were similar in the morning and evening, suggesting that diurnal activity of specific steroidogenic enzymes could have contributed to deoxycorticosterone concentrations. Whether ethanol affects circulating neuroactive steroid concentrations could depend on the time of day. In one study, young adult men that drank 0.80 g/kg ethanol at 6:00 pm had similar serum concentrations of allopregnanolone (0.145 ng/ml) and pregnenolone (0.297 ng/ml) compared to baseline (0.125 and 0.262 ng/ml, respectively; Porcu et al., 2010). In contrast, Pierucci-Lagha et al. (2006) reported that when young adult men drank the same dose of ethanol at 11:00 am, plasma allopregnanolone (0.07 ng/ml) decreased and pregnenolone (4.56 ng/ml) increased compared to baseline (allopregnanolone, 0.13 ng/ml; pregnenolone, 4.05 ng/ml). Further studies of ethanol effects on circulating neuroactive steroids are needed that include a range of BECs and times of day.
Possible species differences in ethanol sensitivity could relate to basal levels of neuroactive steroids. For example, basal pregnenolone was much greater in male cynomolgus monkeys (945 pg/ml) compared to humans (297 pg/ml), male rats (165 pg/ml), or male mice (49 pg/ml), but 3α,5α-THP, 3α,5β-THP, and 3α,5α-THDOC were much lower in cynomolgus monkeys compared to rats, mice, and adult men (Porcu et al., 2010). The relatively high basal pregnenolone concentration observed in cynomolgus macaques suggests the potential for high concentrations of pregnenolone sulfate following sulfation that could negatively modulate GABAA receptors (Park-Chung et al., 1999) and potentiate NMDA responses to glutamate (Wu et al., 1991; Park-Chung et al., 1997). High basal pregnenolone may predict a lower potency of ethanol in behavioral assays. For example, 30 and 56 mg/kg pregnenolone decreased responding for ethanol and ethanol consumption in rats (Besheer et al., 2010). Although the equivalent primate studies have not been conducted, and the levels of neuroactive metabolites can make interpretation challenging, it is possible that circulating levels of pregnenolone contribute to individual and species differences in the effects of ethanol.
GABAA Receptor-Mediated Inhibitory Signaling
GABAA receptor-mediated inhibition can be roughly grouped into two forms, “phasic” and “tonic” inhibition (Cavelier et al., 2005; Farrant and Nusser, 2005; Glykys and Mody, 2007a). Phasic inhibition refers to the long known, traditional inhibitory postsynaptic currents (IPSCs), generated by GABAA receptors in the postsynaptic membrane that are transiently activated (hence the term phasic) by vesicular release of GABA. The phasic nature of IPSCs is due to the rapid removal of GABA from the synaptic cleft by diffusion and uptake on plasma-membrane GABA transporters, as well as desensitization of postsynaptic GABAA receptors. Conversely, tonic inhibition is a more recently discovered form of steady state inhibitory current, generated primarily by extrasynaptically located GABAA receptors that are persistently activated (hence the term tonic) by the ambient extracellular concentration of GABA (Hamann et al., 2002; Cavelier et al., 2005; Farrant and Nusser, 2005; Glykys and Mody, 2007a,b; Choi et al., 2008; Lee et al., 2011). The extracellular concentration of GABA that drives tonic GABAA inhibition is determined by the balance between GABA release (via vesicle exocytosis and various forms of non-vesicular release) and GABA uptake by GABA transporters (Rossi et al., 2003; Glykys and Mody, 2007b; Lee et al., 2010). Accordingly, the magnitude of tonic GABAA inhibition is increased by blocking GABA uptake, and decreased by blocking any of the various forms of GABA release. Conversely, increasing GABA release increases the magnitude of tonic GABAA inhibition. Importantly, since a main source of GABA release is the vesicular release that also generates phasic IPSCs, the magnitude of tonic GABAA inhibition often varies in parallel with changes in IPSC frequency (Glykys and Mody, 2007b).
The GABAA receptor is composed of five trans-membrane subunits arranged to form an anion-conducting pore, and the makeup of subunits, from a family of 19 unique subunits (α1–6, β1–3, γ1–3, δ, ε, þ, π, and ρ1–3), determines the sub-cellular location of the receptor (synaptic versus extrasynaptic). The subunit composition of GABAA receptors also determines their kinetic and pharmacological properties, including sensitivity to neurosteroids, ethanol, and other GABAA modulatory compounds. Thus, interpreting the behavioral impact and interactions of ethanol and neurosteroids requires an understanding of the subunit properties, and their distribution pattern in various brain regions.
One consistent and important GABAA receptor theme is that δ subunits are located exclusively extrasynaptically, and it is such δ subunit-containing, extrasynaptic GABAA receptors that mediate tonic inhibition (Rossi and Hamann, 1998; Hamann et al., 2002; Stell et al., 2003; Farrant and Nusser, 2005; Kelm et al., 2008). The δ subunit is primarily expressed by a limited number of cell types in a few brain regions, including cerebellar and hippocampal granule cells, thalamic relay neurons, and possibly more restrictedly in subsets of cells elsewhere, including non-granule hippocampal cells and some cortical cells (Wisden et al., 1992). Thus, tonic inhibition is generally restricted to specific cell types in particular brain regions. In these brain regions the δ subunit generally pairs with α4 or α6 subunits, which together endows the parent receptor with properties idealized for sensing ambient GABA to generate tonic inhibition: a higher affinity for GABA and less receptor desensitization than other subtypes of GABAA receptors (Saxena and Macdonald, 1994, 1996). However, although the δ subunit is solely expressed extrasynaptically and typically co-assembles with α4 or α6 subunits, the latter subunits can also be part of functional, synaptic receptors that do not contain δ subunits. Thus, while pharmacological or genetic manipulations of the δ subunit can be unambiguously attributed to effects on tonic GABAA inhibition, α4- or α6-specific manipulations may affect synaptic transmission (i.e., IPSCs), extrasynaptic transmission (i.e., tonic inhibition), or both. On the other hand, the α6 subunit is expressed almost exclusively by cerebellar granule cells, making actions of α6-specific manipulations attributable to actions at cerebellar granule cells. Other than the α5 subunit, which is largely restricted to the hippocampus and cortex, the rest of the GABAA subunits are more ubiquitously expressed making attribution of subunit-specific manipulations to specific brain region processing more complicated. Finally, although expression of the δ subunit is restricted to only a few brain regions, because of the constant nature of the tonic inhibition they generate, tonic inhibition is significantly more powerful than phasic inhibition, mediating ~75% of total inhibition in cells that exhibit tonic inhibition (Hamann et al., 2002). Thus, the δ subunit is potentially a powerful target for neural modulation.
Neurosteroid Actions at GABAA Receptors
As described in the introduction, at physiologically relevant concentrations, neurosteroids can either potently enhance GABAA receptor function, or, in the case of sulfated neurosteroids, powerfully suppress them. The diversity of neurosteroids combined with their potency and respective abilities to enhance or suppress GABAA receptor function enables a powerful fine-tuning of the GABAA system across behavioral states and across brain regions. Early behavioral and electrophysiological studies suggested that the δ subunit played a crucial role in mediating the actions of neurosteroids, with many studies indicating that the δ subunit imparted an increased sensitivity to neurosteroids (Mihalek et al., 1999; Belelli et al., 2002; Wohlfarth et al., 2002; Stell et al., 2003), although other studies suggested that the δ subunit decreased sensitivity to neurosteroids (Zhu et al., 1996; Hamann et al., 2002). However, more recent work on recombinant GABAA receptors has determined that there is a conserved neurosteroid binding site on α subunits (α1–5; Hosie et al., 2009). Thus, because all GABAA receptors contain α subunits, neurosteroids are potent modulators of most, if not all, known GABAA receptors, regardless of subunit makeup. Such studies suggested that earlier data implicating the δ subunit in neurosteroid sensitivity resulted from the δ subunit increasing neurosteroid efficacy rather than receptor affinity for neurosteroids. Thus, in principle, systemic neurosteroids could impact both phasic and tonic GABAA inhibition throughout the brain, and selective actions on particular cell types could be dependent on local synthesis. Nonetheless, in situ studies showed that very low concentrations of exogenous neurosteroid (<20 nM) powerfully enhanced tonic GABAA currents but not phasic GABAA currents in the same preparation, and that knocking out the δ subunit eliminated tonic GABAA current sensitivity to neurosteroids (Stell et al., 2003). The contrast with data from recombinant receptors suggests that in situ receptors may behave differently, and that the δ subunit does affect receptor affinity in situ. In fact, studies examining GABAA currents in different brain regions indicate that the sensitivity of both phasic and tonic GABAA currents to neurosteroids varies across cell types, even when apparently identical GABAA receptor subunits are involved (Belelli et al., 2006). Such variability may be due to the phosphorylation state of the GABAA receptor and/or local neurosteroid metabolism, two factors that add further control and complexity to neurosteroid actions at GABAA receptors. Thus, although the presence of the δ subunit or specific α, β, or γ subunit may impart subtle differences to the parent receptor that are physiologically relevant, it is unlikely that exogenous neurosteroids can be used as subunit-selective modulators to identify crucial subunits or brain regions involved in a particular behavioral response. Similarly, if selective modulation of a specific GABAA subunit (such as genetic deletion) has a significant impact on a behavioral response to neurosteroids, it should be interpreted with respect to the importance of the brain region(s) in which the subunit is expressed to the behavior, rather than to the particular sensitivity of the subunit to neurosteroids.
Ethanol Actions at GABAA Receptors
Ethanol has also long been known to be a potent enhancer of GABAA receptor-mediated inhibition, but identifying the mechanism(s) of action has been controversial, and is clearly even more complex than for neurosteroid modulation. Several groups have reported that concentrations of ethanol that are likely to be achieved during moderate to abusive drinking (5–50 mM) can enhance tonic GABAA inhibition via directly increasing the affinity of certain GABAA receptors for GABA, including α6βδ subunits in cerebellar granule cells, α4βδ subunits in hippocampal granule cells and thalamic relay neurons, and α1βδ subunits in hippocampal interneurons (Wei et al., 2004; Hanchar et al., 2005; Glykys et al., 2007). Most of the studies showing direct actions of ethanol on GABAA receptors have observed this effect in receptors containing the δ subunit, leading to the notion that the δ subunit containing GABAA receptors are low-dose ethanol receptors (Hanchar et al., 2004; Olsen et al., 2007; Mody, 2008). Unfortunately, demonstration of direct actions of ethanol on GABAA receptors has been difficult to replicate, and most researchers, using similar, nearly identical techniques and preparations have been unable to show any direct actions of ethanol on GABAA receptors of any composition (Borghese et al., 2006; Borghese and Harris, 2007; Botta et al., 2007a,b). Despite intensive efforts, the reasons for this discrepancy have not yet been elucidated. One factor may relate, at least in part, to the phosphorylation status of the GABAA receptor, similar to neurosteroids. In particular, Choi et al. (2008) determined that enhancement of tonic GABAA currents mediated by GABAA receptors containing α4δ subunits required that the receptor be phosphorylated by protein kinase (PK) Cδ, and accordingly, knocking out PKCδ prevented ethanol from enhancing tonic GABAA currents in hippocampus and thalamus, and reduced behavioral sensitivity to ethanol. While it is not clear why the phosphorylation status of the GABAA receptor would be so starkly different across laboratories, it does provide a potential mechanism explaining in part the discrepant observations across laboratories regarding direct actions of ethanol on GABAA receptor currents. Intriguingly, although PKCδ expression correlates with the GABAA δ subunit expression in hippocampus and thalamus, it is not expressed in the cerebellar granule cell layer, where α6δ subunits are expressed. This raises the possibility that if phosphorylation status explains the discrepant observations of ethanol-induced potentiation of tonic GABAA currents in cerebellar granule cells, there must be kinases other than PKCδ to serve that role in the cerebellar granule cell layer.
In addition to the potential direct action of ethanol on some GABAA receptors, it is widely accepted that ethanol strongly enhances vesicular release of GABA. As a result, the frequency of IPSCs increases and, in cells that express extrasynaptic GABAA receptors, the magnitude of tonic GABAA currents is strongly enhanced due to the accumulation of vesicular release of GABA in the extrasynaptic space. This mechanism is observed in many brain regions, including the cerebellum, ventral tegmental area, substantia nigra, and amygdala, but lacking in others, including cortex, lateral septum, and thalamus (Kelm et al., 2011). The mechanisms underlying ethanol-induced vesicular GABA release are not fully understood, but often appears to involve G-protein coupled receptor systems (Kelm et al., 2011). Thus, regardless of whether ethanol acts directly on GABAA receptors, ethanol can increase both the frequency of IPSCs as well as the magnitude of the tonic GABAA current. Therefore, even if ethanol does not directly affect δ subunits in a particular brain region or under particular conditions, knocking out (or selectively modifying) the δ subunit could still strongly influence the response to ethanol, because the lack of extrasynaptic GABAA receptors will eliminate tonic GABAA currents that would normally be generated by increased vesicular release of GABA.
Interactions between Neurosteroids and Ethanol at GABAA Receptors
Given that neurosteroids and ethanol converge on GABAA receptors, it is worth considering how they may interact with each other at the GABAA receptor. Given the more consistent and ubiquitous direct actions of neurosteroids on GABAA receptors compared to ethanol, it seems unlikely that the two compounds share a binding site, and thus will not compete or interact directly. It is, however, possible that for those GABAA receptors that are directly modulated by both neurosteroids and ethanol (see above), combined exposure will have a synergistic or other interactive impact. However, to our knowledge there are no reports of such interactions, and given current controversies about direct actions for both ethanol and neuroactive steroids, it may be difficult to ascertain such interactions at the receptor level. Perhaps the most obvious way in which neurosteroids and ethanol are likely to interact at the GABAA receptor is via ethanol-induced increase in vesicular GABA release as described above. It is clear that neurosteroid effects on GABAA receptors are relatively ubiquitous across receptors with different subunit compositions. Therefore, it is likely that neurosteroids will be active at GABAA receptors in brain regions in which ethanol increases GABA release. Indeed, there is overlap between the documented actions of neurosteroids at GABAA receptors and documented ethanol-induced vesicular GABA release in the cerebellum, hippocampus, thalamus, and amygdala (Hamann et al., 2002; Stell et al., 2003; Carta et al., 2004; Belelli et al., 2006; Criswell et al., 2008; Kelm et al., 2008, 2011). Accordingly, in those brain regions, since neurosteroids enhance the actions of GABA at GABAA receptors and ethanol enhances GABA release, there will likely be a synergistic interaction if cells in those brain regions are exposed to both simultaneously. Importantly, since ethanol increases neurosteroid release both systemically and locally (see earlier sections), such a synergistic enhancement of GABAA currents is likely. Irrespective of the cellular/molecular mechanisms at play, since both neurosteroids and ethanol enhance GABAA currents, it is likely that they may substitute for each other behaviorally. Whereas both tonic and phasic inhibition may decrease the magnitude and duration of excitatory input, because tonic inhibition is persistent and distributed, it may increase the threshold of depolarization required for a cell to fire (Farrant and Kaila, 2007, p. 70). This leads to the hypothesis that neurosteroids and ethanol could modulate the threshold for stimulation by sensory input by enhancing tonic inhibition in brain areas containing δ subunits, whereas the activity of neurosteroids and ethanol in other brain areas could affect the magnitude of physiological or behavioral response to a stimulus.
Common Receptor Activity of Ethanol and Neuroactive Steroids: Evidence from Behavioral Pharmacology
Changes in sensory thresholds are a major aspect of sleep, which involves modulation of thalamocortical excitability (Huguenard and McCormick, 2007). Ethanol (50 mM) has been shown to suppress the activity of thalamocortical relay neurons (Jia et al., 2008), and as described above, these contain δ subunits. However, few studies have examined the interactive effects of ethanol and neuroactive steroids on psychophysical parameters and these have mainly been restricted to response magnitude or latency, rather than response threshold. Nonetheless, the idea that the effects of ethanol on sensorimotor processing are influenced by neuroactive steroids is not well-supported by the limited data available. For example, a very high dose of ethanol (5.25 g/kg, i.g.) administered to rats on post-natal days 4–9 did not alter the magnitude of startle to a 110-db noise, or the attenuation of this startle by a preceding 60-db tone (i.e., prepulse inhibition) compared to vehicle controls when the animals were tested as adolescents or adults (70 days of age; Woolfrey et al., 2005). A single study of neuroactive steroids in healthy women found that neither startle magnitude nor prepulse inhibition were affected by allopregnanolone (0.05 mg/kg; Kask et al., 2009). In contrast, the magnitude of prepulse inhibition was increased in adult rats by administration of allopregnanolone (but not pregnenolone sulfate) to hippocampal CA1 (Darbra et al., 2011), but decreased by systemic administration of allopregnanolone (10 mg/kg) during post-natal days 5–9 (Darbra and Pallarès, 2010). Thus, administration of allopregnanolone (Darbra and Pallarès, 2010) but not ethanol (Woolfrey et al., 2005) to neonates decreased prepulse inhibition in adulthood, at least at the doses tested. Because none of these studies examined the interaction between ethanol and neuroactive steroids, it is unknown whether neuroactive steroids could have a modulatory role under conditions in which ethanol decreased arousal and stimulus processing.
Another set of studies measured cortical electrical responses after presentation of auditory stimuli (event-related potentials, ERPs). Allopregnanolone and ethanol decreased the amplitude of ERPs in the cortex and amygdala, consistent with decreased stimulus efficacy (Slawecki et al., 2000). The same study found that the combination of sub-threshold doses of allopregnanolone (5.0 mg/kg) and ethanol (0.50 g/kg) decreased peak amplitudes in the amygdala, indicative of decreased arousal, but no additive effects were observed in the cortex. Ethanol vapor inhalation for 14 h/day for 5 weeks did not affect ERPs, but tolerance developed to the decrease in ERPs produced by allopregnanolone (Slawecki et al., 2005), suggesting that chronic ethanol exposure caused molecular adaptations of GABAA receptors (e.g., subunit composition) resulting in altered neurosteroid pharmacology/sensitivity (Liang et al., 2009). To our knowledge, there are no studies of ERPs in humans evaluating the interaction between neuroactive steroids and ethanol. Overall, these limited data suggest that the additive effects of ethanol and neuroactive steroids on neural activity depend on the brain region examined, which could be due to the localization of ethanol-induced GABA release, or to the presence or absence of specific GABAA receptor subunits, including those that mediate tonic inhibition.
A much more extensive body of behavioral data that addresses common receptor activity of ethanol and neuroactive steroids includes consummatory behaviors and discriminative stimulus effects. Neurons within the circuitry mediating contextual, reinforcing, or other stimulus properties of ethanol are likely to regulate ethanol consumption. The threshold at which ethanol-associated stimuli affect neurotransmission could be modulated by neurosteroids, thus providing a mechanism by which neuroactive steroids regulate ethanol intake. With regard to consummatory behavior, compared to vehicle administration, mice administered the direct GABAA agonist gaboxadol [4,5,6,7-tetrahydroisoxazolo (5,4-c)pyridin-3(-ol); THIP] at a dose (8 mg/kg) presumably selective for δ-containing GABAA receptors, showed a selective decrease in ethanol (not water) intake, and a lower latency to drink either solution (Ramaker et al., 2011). These data implicate extrasynaptic GABAA receptors in regulating consummatory behavior generally (Kelley et al., 2005), and further suggest that tonic GABAA inhibition can selectively regulate ethanol intake. Thus, the ability of neuroactive steroids to modulate tonic inhibition could make them therapeutically useful for alcohol abuse. Consistent with this possibility, neuroactive steroids are associated with the conditioned aspects of consummatory behavior. For example, anticipation and consumption of food increased plasma and cerebrocortical allopregnanolone and THDOC (Pisu et al., 2006). Because Ramaker et al. (2011) did not measure response thresholds with gaboxadol, nor was there a measure of conditioned stimulus effects that could regulate drinking, the behavioral processes affected by gaboxadol that underlie the decrease in ethanol intake are not clear. Gaboxadol-induced tonic currents in cerebellar granule cells (α6δ; Meera et al., 2011) and thalamic neurons (Herd et al., 2009) are greatly reduced in δ knockout mice, indicating that this subunit is important for sensitivity to gaboxadol. Another study suggested that δ subunits in the striatum are involved in ethanol drinking. Nie et al. (2011) used viral-mediated RNA interference to decrease GABAA receptor δ subunit mRNA and protein approximately two-fold in the medial nucleus accumbens shell. Eighteen days after infusion, ethanol but not sucrose intake under intermittent access decreased from an average of 4.5 g/kg/day to 2.5 g/kg/day. Suppression of δ subunits in the medial shell was suggested to decrease ethanol-induced potentiation of tonic inhibition in a brain area that mediates ethanol reinforcement. By extension, variation in δ subunit expression in the medial shell and perhaps potentiation of tonic inhibition by neurosteroids could be mechanisms for variation of ethanol effects within and across individuals.
By far, the greatest amount of data addressing the role of neuroactive steroids in sensitivity to ethanol has been obtained using drug discrimination procedures. In these procedures, a specific response is reinforced (e.g., pressing the one of two levers results in food presentation) following administration of a given dose of a drug such as ethanol, and another response (e.g., pressing the other lever) is reinforced following administration of vehicle. The animal’s perception of the effect of the drug is the only cue guiding its choice of which response to perform. Once the animals reliably respond on the drug-associated lever after training drug administration, test drugs can be administered. A test drug may be shown to substitute for the training drug, i.e., produce responding on the lever associated with the training drug, indicating similar discriminative stimulus effects. Threshold and magnitude of response can be dissociated in drug discrimination data by potency (effective dose, ED50) and efficacy (maximum responding on the ethanol lever), respectively. Discrimination of the stimulus effects of drugs can be trained in humans using the same operant techniques. Drugs that positively modulate GABAA receptors, including ethanol, and produce similar discriminative stimulus effects also tend to produce common subjective effects in humans (Kelly et al., 2003).
Studies in non-human primates indicate that the discriminative stimulus effects of a wide range of ethanol doses (1.0–2.0 g/kg) are mediated largely by GABAA receptors (Grant et al., 2000; Vivian et al., 2002; Helms et al., 2009). In contrast, the discriminative stimulus effects of higher doses of ethanol (2.0 g/kg) appear to involve specific subtypes of GABAA receptors (i.e., zolpidem-sensitive α1-containing; Helms et al., 2008). The discriminative stimulus effects of 5 mg/kg pregnanolone are also mediated by positive modulation of GABAA receptors. For example, in rats trained to discriminate pregnanolone, high levels of responding on the pregnanolone-appropriate lever occurred after administration of benzodiazepines and barbiturates, which are positive modulators of GABAA receptors. In contrast, the direct agonists muscimol and gaboxadol produced low levels of pregnanolone-appropriate responding (Engel et al., 2001). Thus, the discriminative stimulus effects produced by GABAA receptor agonists and positive modulators are distinct. Consistent with this conclusion, in animals trained to discriminate ethanol, the direct agonists muscimol and gaboxadol do not produce ethanol-like effects (cynomolgus monkeys, Grant et al., 2000; mice, Shelton and Grant, 2002). The activity of gaboxadol as an agonist rather than a positive modulator may account for its lack of ethanol-like stimulus effects. On the other hand, if infused directly into the nucleus accumbens or amygdala, muscimol can substitute for the discriminative stimulus effects of peripherally administered ethanol in rats (Hodge and Cox, 1998). Taken together, these data suggest that, although neurosteroids can directly activate GABA currents at high concentrations (Hosie et al., 2006), ethanol-like discriminative stimulus effects occur at lower, positive-modulatory concentrations. Second, interactions among specific brain regions may contribute to the discriminative stimulus effects of ethanol. For example, Hodge et al. (2001) found that allopregnanolone substituted for the discriminative stimulus effects of systemic ethanol when administered into the nucleus accumbens but not the hippocampus (CA1). Thus, hippocampal CA1 neurons that are sensitive to neurosteroids may not be involved in ethanol-like discriminative stimulus effects.
The GABAA receptor subtypes at which neuroactive steroids produce discriminative stimulus effects similar to ethanol have not been extensively investigated. In one study, δ subunit knockout mice learned to discriminate 1.5 g/kg ethanol, and showed similar substitution of neuroactive steroids (pregnanolone and alphaxalone) for the discriminative stimulus effects of ethanol (Shannon et al., 2004). Thus, δ subunits do not appear to be necessary for the ethanol-like discriminative stimulus effects of neuroactive steroids. Zolpidem is an imidazopyridine with selectivity for α1-containing GABAA receptors (Lüddens et al., 1994), which combine with γ subunits and are purported to be primarily synaptic, although co-assembly with δ subunits and mediation of tonic currents has been observed in interneurons of the hippocampus (Glykys et al., 2007). Predominantly, however, α1-containing receptors are synaptic and therefore mediate phasic GABA neurotransmission. In drug discrimination procedures, zolpidem substituted for the discriminative stimulus effects of pregnanolone in rats (Engel et al., 2001) but not mice (Shannon et al., 2005a,b), and no data exist for primates. Zolpidem partially substituted for ethanol in rats (Sanger et al., 1999) but not mice (Shannon et al., 2004) and completely substituted for 2.0 g/kg ethanol in cynomolgus monkeys (Helms et al., 2008). Overall, these data suggest that α1 subunits could mediate the discriminative stimulus effects of both ethanol and neuroactive steroids under some conditions. However, whether this subunit is involved in the ethanol-like effects of neuroactive steroids has not been studied. Finally, using the 5α-reductase inhibitor finasteride, the subjective effects of ethanol (0.7–0.8 g/kg, BEC, 50–70 mg/dl) were attenuated in humans, but only in individuals homozygous for the A (but not the G) allele at the GABAA receptor α2 subunit gene (Pierucci-Lagha et al., 2005). These data suggest that the subjective effects of ethanol are enhanced by neuroactive steroids in a select population carrying a polymorphism in the α2 subunit of the GABAA receptor. The role of α2 subunits in the discriminative stimulus effects of ethanol and neuroactive steroids has not been explored due to a lack of drugs with appropriate selectivity.
The GABAA receptors mediating the ethanol-like effects of neuroactive steroids have been studied using pharmacological antagonism. Ro15-4513 is a partial inverse agonist with high affinity for receptors containing α4, α5, or α6 subunits and lower affinity for α1, α2, or α3 subunits. This drug is celebrated as a competitive antagonist of the electrophysiological and behavioral effects of ethanol (Suzdak et al., 1986; Wallner and Olsen, 2008). In drug discrimination studies, we found that Ro15-4513 antagonized the substitution of allopregnanolone and pregnanolone for the discriminative stimulus effects of ethanol (Figure 2). Antagonism of pregnanolone substitution for ethanol was observed in 6/6 monkeys trained to discriminate 1.0 g/kg ethanol, and in 3/5 monkeys trained to discriminate 2.0 g/kg ethanol. Ro15-4513 antagonized allopregnanolone substitution for ethanol in 3/6 monkeys trained to discriminate 1.0 g/kg ethanol, and 4/5 monkeys trained to discriminate 2.0 g/kg ethanol. Among the monkeys showing antagonism, Ro15-4513 resulted in decreased potency, but not efficacy, of these neuroactive steroids to substitute for ethanol. In addition, according to an apparent pA2 analysis (Rowlett and Woolverton, 1996), the estimated affinity of Ro15-4513 for the receptor population mediating substitution of pregnanolone (pKB of 5.2–7.0) and allopregnanolone (pKB of 6.1–8.9) overlapped with zolpidem (pKB of 6.3–8.0). These affinity estimates suggest that pregnanolone and allopregnanolone act at benzodiazepine-sensitive and -insensitive GABAA receptors to produce discriminative stimulus effects similar to ethanol. Because the γ subunit is obligatory for benzodiazepine sensitivity and this subunit is primarily synaptic (Möhler, 2006), these data suggest that the ethanol-like discriminative stimulus effects of neuroactive steroids are mediated by synaptically located GABAA receptors that regulate phasic inhibition.
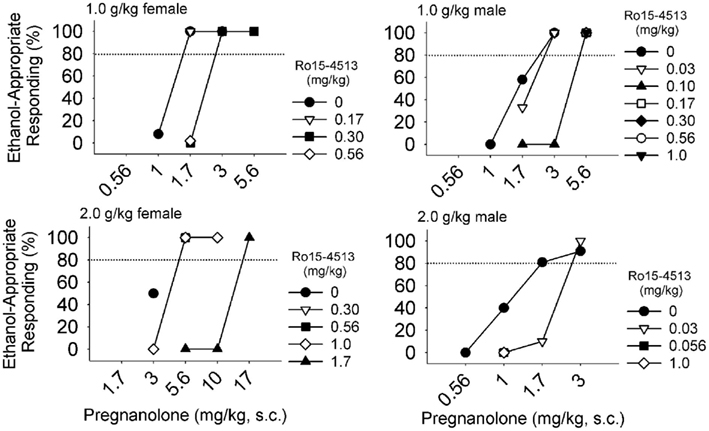
Figure 2. Among male (n = 6) and female (n = 6) cynomolgus macaques that were trained to discriminate 1.0 or 2.0 g/kg ethanol, pregnanolone produced ethanol-like discriminative stimulus effects in all monkeys, which were antagonized when Ro15-4513 (symbols indicate doses in mg/kg, i.m.) was administered after pregnanolone, 5 min before the levers extended into the chamber. Shifts in the dose-response curves of four monkeys are shown.
In contrast to the ethanol-like effects of positive modulators of GABAA receptors, neuroactive steroids that negatively modulate GABAA receptors, including epipregnanolone and epiallopregnanolone sulfate, might antagonize the discriminative stimulus effects of ethanol. Our preliminary data indicate that epipregnanolone (0, 10, 17, 30 mg/kg, s.c.) and epiallopregnanolone (0, 17, 30 mg/kg, s.c.) administered 30 min prior to ethanol (0.25–2.5 g/kg, i.g.) blocked (≤20% ethanol-appropriate) discriminative effects in only a subset of cynomolgus monkeys. Specifically, epipregnanolone blocked the discriminative stimulus effects of ethanol in 1/6 of monkeys trained to discriminate 1.0 g/kg ethanol, and in 2/6 monkeys trained to discriminate 2.0 g/kg ethanol. In another study in rats, administration of epipregnanolone (10–20 mg/kg, i.p.) decreased responses for ethanol measured over 30 min from 40 to 20 (O’Dell et al., 2005). Although preliminary, these data suggest that neuroactive steroid negative modulators could block the subjective effects of ethanol and consequently responding for ethanol, and thus could be therapeutic targets. However, these data are also consistent with the wide individual differences in the substitution patterns of neuroactive steroids (e.g., Bowen et al., 1999; Grant et al., 2008a), the causes of which are currently unknown. Some possible sources of individual differences in the modulation of ethanol effects by neuroactive steroids could be basal hormone concentration, polymorphisms in GABAA receptor subunits, basal activity of steroidogenic enzymes, reproductive state, or the effects of stress on HPA axis function.
Many studies have indicated that physiological concentrations of the neuroactive steroids allopregnanolone and pregnanolone substitute for the discriminative stimulus effects of ethanol (Grant et al., 1996, 1997, 2008a; Bowen et al., 1999). These findings suggest that endogenous variation in neuroactive steroids could influence sensitivity to ethanol. Indeed, the potency of 1.0 g/kg ethanol (Grant et al., 1997) but not 2.0 g/kg ethanol (Green et al., 1999) to produce discriminative stimulus effects was greater during the luteal compared to the follicular phase of the menstrual cycle in monkeys. During the luteal phase, peak progesterone concentration is associated with greater circulating concentrations of progesterone-derived neuroactive steroids, including allopregnanolone and pregnanolone (Genazzani et al., 1998; Hill et al., 2005). Although self-reported subjective effects of low ethanol doses (up to 0.6 g/kg) did not vary across a menstrual cycle in women (Holdstock and de Wit, 2000), sensitivity to the benzodiazepine triazolam was greater during women’s luteal compared to the follicular phase when measured using a drug discrimination procedure (Kelly et al., 2001). Despite the absence of menstrual cycles in males, male and female cynomolgus monkeys show only minor differences in the ethanol-like discriminative stimulus effects of neuroactive steroids. For example, the potency of androsterone to produce ethanol-like stimulus effects is on average lower in males compared to females (Grant et al., 2008b). In humans, subjective effects associated with varying plasma concentrations of neuroactive steroids could contribute to regulation of ethanol consumption. For example, change in plasma neuroactive steroid concentration after consuming 0.80 g/kg ethanol (11:00 am) correlated significantly with subjective effects of ethanol including “liking” (pregnenolone, r = 0.53; allopregnanolone, r = 0.50) and “want more alcohol” (r = 0.51; Pierucci-Lagha et al., 2006). These data further suggest that neuroactive steroids produced as a result of ethanol self-administration could subsequently regulate intake. By extension, the effects of ethanol may vary with other endogenous (e.g., stress: Droogleever Fortuyn et al., 2004) or disease states (e.g., premenstrual dysphoric disorder, Bäckström et al., 2011) associated with altered concentration of, or sensitivity to, GABAergic neuroactive steroids. Such endogenous conditions could influence the risk of alcohol abuse.
The Role of Neuroactive Steroids in Chronic Ethanol Effects and Ethanol Withdrawal
Although naïve individuals differ in sensitivity to the acute effects of ethanol, a much greater variability in sensitivity to ethanol occurs within individuals as a result of repeated administration. Neuroadaptation to chronic ethanol involves decreased sensitivity to GABA and changes in GABAA receptor subunit expression and function. These neuroadaptations have additional pharmacological consequences including decreased sensitivity to benzodiazepines and increased sensitivity to some neuroactive steroids (reviewed by Biggio et al., 2007). Changes in subunit expression after ethanol or progesterone withdrawal differ between brain areas (cerebral cortex, cerebellum, and hippocampus are most often studied), but a relatively consistent result is decreased expression of α1 and δ subunits and increased expression of α4 and α6 subunits (Mhatre and Ticku, 1992; Rani and Ticku, 2006). Electrophysiological studies in cultured neurons have confirmed that the changes in GABAA receptor subunits after chronic ethanol treatment and withdrawal are associated with changes in receptor function (reviewed by Kumar et al., 2009). In a report by Shen et al. (2011), endocytosis of GABAA receptors containing α4 and δ subunits was shown to be increased by ethanol treatment in cultured hippocampal neurons. In another model, rats are administered high doses of ethanol intermittently (chronic intermittent ethanol, CIE: 5 g/kg every other day for 5 days, then 6 g/kg/day for 55 days) and then brain slices are prepared for electrophysiology. The CIE treatment results in the decreased efficacy of several GABAergic sedative/hypnotic drugs (e.g., ethanol, zolpidem, gaboxadol) to potentiate tonic currents in hippocampal slices (Liang et al., 2004, 2009). At the same time, these drugs showed greater efficacy to potentiate phasic (synaptic) currents after CIE. This result was related to decreased α1 subunits at synaptic sites, decreased α4, β, and δ subunit protein at extrasynaptic sites, and increased α4, β, and γ2 subunits at synaptic sites (Liang et al., 2004, 2009). Notably, similar to the effects of withdrawal from chronic ethanol, increased α4 subunit expression is observed in the hippocampus after chronic allopregnanolone and withdrawal (reviewed by Smith et al., 2007). Overall, these data suggest that the neuroadaptations to chronic ethanol and neuroactive steroids are similar, possibly to maintain homeostasis of inhibitory neurotransmission. Regardless of the mechanisms by which it is accomplished, i.e., direct receptor action by neuroactive steroids or increased GABA release by ethanol, positive modulation of GABAA receptors appears to be a common effect of ethanol and exogenous neuroactive steroids that can result in homeostatic molecular adaptations to restore GABAergic tone. Such homeostatic adaptations result in withdrawal following the removal of neurosteroids or ethanol.
The great complexity of results from in vitro studies, in which differences are commonly found between brain areas, suggests that in vivo pharmacological studies could inform changes in GABAA receptor function after chronic ethanol and withdrawal. For example, 10 mg/kg alphaxalone induced sleep, as measured by a loss of righting reflex. After CIE, however, the duration of sleep induced by alphaxalone was decreased, indicating cross-tolerance (Cagetti et al., 2003). In contrast, the efficacy of alphaxalone to increase time spent on the open arms in the elevated plus maze (purportedly an anxiolytic effect) was increased after CIE (Cagetti et al., 2004). In the same study, rats exposed to CIE were sensitized to the convulsant effect of pentylenetetrazol, as 20–25 mg/kg induced seizures in this group compared to 40 mg/kg in control rats. This treatment also resulted in greater sensitivity to the anticonvulsant effects of alphaxalone. Likewise, the anticonvulsant efficacy and potency of allopregnanolone (Devaud et al., 1995) and THDOC (Devaud et al., 1996) was increased by chronic ethanol treatment in rats (liquid diet, 10–12 g/kg/day for 14 days) and mice (72 h ethanol vapor inhalation, Finn et al., 2000). These data indicate that chronic ethanol and withdrawal are associated with increased sensitivity, rather than cross-tolerance, to the anticonvulsant and possibly the anxiolytic effects of GABAergic neuroactive steroids. Thus, data from the limited studies conducted so far suggest that adaptations to chronic ethanol and withdrawal alter the effects of neuroactive steroids in a behavior-specific manner likely related to the GABAA receptor subunits expressed in the brain regions mediating the behavior that is being studied.
Neuroactive Steroid Regulation of the HPA Axis and Interactions with Ethanol
Similar to the negative feedback of the HPA axis by glucocorticoids, GABA is a primary regulator of hypothalamic responses to stress. Activity of the HPA axis, beginning with secretion of CRH, is inhibited by GABA (Calogero et al., 1988). Decreased GABAergic activity during acute stress (Biggio et al., 1990) results in stimulation of the HPA axis and increased brain and plasma concentrations of neuroactive steroids, at least in rodents (Purdy et al., 1991). A variety of stressors have been shown to increase plasma and/or brain neuroactive steroids to concentrations that modulate GABAA receptors in vitro (Belelli et al., 2002). For example, swim stress (22°C, 10 min) in rats increased allopregnanolone in plasma (<1 ng/ml to 2.5 ng/ml after 70 min) and brain (cerebral cortex, 2.5 ng/ml to 5 ng/ml after 10 min; hypothalamus, <2 ng/ml to 4.5 ng/ml after 40 min; Purdy et al., 1991). Similarly, plasma allopregnanolone increased from 1.0 to 1.2 nmol/l during oral defense for a Ph.D. degree (Droogleever Fortuyn et al., 2004). Increased neuroactive steroids are proposed to dampen the physiological impact of the stress and mediate restoration of homeostasis of the HPA axis (Finn and Purdy, 2007). For example, allopregnanolone decreased anxiety in response to heat stress and administration of CRH (Patchev et al., 1994, 1996). A recent report indicated that physiological concentrations of allopregnanolone, THDOC, and pregnenolone in cell culture inhibited CRH reporter gene activity and forskolin-stimulated transcription (Budziszewska et al., 2010). Overall, these data are consistent with the idea that endogenous GABAergic neuroactive steroids exert negative feedback on stress responses. Chronic stress has been proposed to result in adaptations of the HPA axis that increase risk for alcohol abuse (Lovallo et al., 2000; Biggio et al., 2007). Neuroactive steroids may be a target for amelioration of HPA axis abnormalities.
The interaction between neuroactive steroids, ethanol, and chronic stress has been extensively investigated using the social isolation model in which rodents are housed individually after weaning. Compared to group-housed rats, socially isolated rats had lower cortical and plasma concentrations of pregnenolone, progesterone, THDOC and allopregnanolone, and 32% greater basal concentration of plasma corticosterone (Serra et al., 2000). After acute ethanol (1.0 g/kg), however, socially isolated rats had greater concentrations of plasma and cerebral cortex pregnenolone, progesterone, allopregnanolone, and THDOC compared to group-housed rats (Serra et al., 2003). The chronic stress of social isolation therefore down-regulated the basal concentration of neurosteroids, perhaps lowering inhibitory control of the HPA axis, resulting in greater corticosterone. Furthermore, the efficacy of ethanol to increase neuroactive steroids was greater following social isolation, perhaps related to suppressed basal concentrations. In contrast, following treatment with chronic ethanol (8–10 g/kg/day for 7 days), the efficacy of ethanol to increase plasma concentrations of neuroactive steroids was lower compared to control diet (Boyd et al., 2010a). Insofar as stress substitutes for, and has additive effects toward, social anxiety induced by ethanol withdrawal (5 days ethanol diet, restraint stress; Breese et al., 2004), one hypothesis is that ethanol withdrawal may have effects similar to stress, and potentiate the increase in neuroactive steroid concentration after acute ethanol. To our knowledge, the effects of ethanol withdrawal on the efficacy of acute ethanol to alter concentrations of neuroactive steroids have not been studied.
Sanna et al. (2011) hypothesized that, in socially isolated mice, ethanol drinking would normalize neurosteroid concentrations. Data supporting this hypothesis would suggest that increased neuroactive steroids resulting from consumption of ethanol might contribute to reinforcement of ethanol drinking. This hypothesis was previously investigated by Janak et al. (1998), showing that 3 mg/kg allopregnanolone slightly increased ethanol self-administration from 0.30 to 0.43 g/kg/30 min session. However, Sanna et al. (2011) reported that hippocampal allopregnanolone concentration did not differ between mice consuming ethanol (group-housed, 3.5 g/kg/2 h session; socially isolated, 4.25 g/kg/2 h session) or water for 6 weeks, and remained lower in socially isolated (11–13 ng/g) compared to group-housed mice (15–17 ng/g). Thus, ethanol consumption did not normalize hippocampal (Sanna et al., 2011) or cerebral cortical (rats, Pisu et al., 2011) neurosteroids that were suppressed by the chronic stress of social isolation. However, there may be differences between voluntary and experimenter-administered ethanol. The proceedings of the 2008 Volterra symposium (Morrow et al., 2009) reported that Biggio’s lab observed a two-fold increase in cortical allopregnanolone in socially isolated rats exposed to intermittent ethanol vapor for 8 days (BEC, 205 mg/dl at sacrifice; 91% increase in group-housed rats). It may be that compensatory processes induced by ethanol-conditioned stimuli during voluntary drinking maintain neurosteroid homeostasis, similar to neurosteroid regulation by the anticipation of food (Pisu et al., 2006).
Changes in the expression of GABAA receptor subunits during chronic stress or ethanol appear to alter GABAA receptor-mediated tonic currents. As a result, changes in the ability of neuroactive steroids to modulate tonic currents might influence the threshold for HPA axis stimulation. In support of this idea, socially isolated rats showed greater expression of α4 and δ subunits in the hippocampus compared to group-housed rats (Serra et al., 2006), and greater enhancement of tonic current by gaboxadol (Sanna et al., 2011). Furthermore, δ subunit mRNA expression and protein was attenuated by ethanol consumption in socially isolated mice, in which basal expression was 50% greater than group-housed mice (Sanna et al., 2011). Whereas social isolation resulted in approximately 10% greater α4 subunit protein compared to group-housed mice drinking water, this difference was absent when the mice drank ethanol, suggesting that ethanol consumption opposed the effects of stress on α4 subunit expression and peptide up-regulation (Sanna et al., 2011). Ethanol consumption in socially isolated mice also resulted in electrophysiological effects of gaboxadol more similar to group-housed mice, consistent with regulation of α4δ subunits. Additional research is needed, as the combination of chronic ethanol and associated social stress in humans could have unique pharmacological consequences that are significant for the treatment of alcoholism.
Changes in GABAA receptor subunit expression in relation to ethanol consumption have been studied using a primate model. By using non-human primates, chronic alcohol intake can be characterized in the absence of confounding variables present in human post-mortem studies (e.g., reliable measures of intake, multi-drug histories, nutrition). Studies using monkeys model important aspects of human alcohol consumption, including alcoholic-like patterns of intake with high (>100 mg/dl) BECs (Grant et al., 2008b). After more than 18 months of ethanol self-administration, previous studies found marked alterations in GABAA and NMDA receptor subunit mRNA expression in the dorsal lateral prefrontal cortex (DLPFC) and the orbital frontal (OFC) but not in anterior cingulate cortex (ACC; Hemby et al., 2006; Acosta et al., 2010). Specifically, the GABAA receptor subunits α2, α4, β1, β3, γ1, γ2, γ3, were decreased in the OFC, whereas only α1, α2, γ1, and δ were decreased in the DLPFC. These data suggest that GABAA receptor adaptation in response to chronic ethanol drinking differed between cortical regions of the primate brain, and only the DLPFC showed changes in the δ subunit. Likewise, the only change observed with respect to α subunits was lowered expression, including a decrease in the α4 subtype within the OFC. The significance of these findings with regard to ethanol and neuroactive steroid pharmacology and effects on the HPA axis requires further investigation. However, Porcu et al. (2006) found that the synthetic glucocorticoid dexamethasone suppressed plasma concentrations of deoxycorticosterone, and that the efficacy of dexamethasone was negatively correlated with subsequent ethanol self-administration using the primate model. Thus, lower glucocorticoid modulation of neuroactive steroid precursors may be a risk factor for heavy drinking, particularly among individuals with GABAA receptor subunit polymorphisms associated with greater ethanol-like effects of neuroactive steroids (Pierucci-Lagha et al., 2005).
Inhibition of the HPA axis by GABAergic transmission is difficult to reconcile with the ability of ethanol, a positive modulator of GABAA receptors, to activate the HPA axis and increase neuroactive steroids in rodents. One possibility is that activation of the sympathetic nervous system related to ethanol administration (restraint, needle poke) interacts with the effects of ethanol to increase neuroactive steroids. Barbaccia et al. (1994) reported that CO2 inhalation increased neuroactive steroid precursors (pregnenolone, progesterone) in rat brain, but that the magnitude of increase was reduced after experience with handling, the experimental context, and the containment apparatus, twice daily for 5 days. A majority of studies using rats currently include 1–2 weeks of acclimation to the housing conditions, but few include daily handling. We measured plasma pregnenolone, progesterone, ACTH, and deoxycorticosterone in female rats that were extremely acclimated to the procedures and found no difference between rats administered 1.0 g/kg ethanol or water (i.g.; Helms et al., 2010). These rats had >1 year daily handling and were gavaged 5–7 days/week in which 1.0 g/kg ethanol was administered on about half the days. Another explanation was recently suggested by Boyd et al. (2010a). Compared to rats fed an isocaloric control diet, rats fed an ethanol liquid diet for 7 days (8–10 g/kg/day) showed blunted plasma ACTH, progesterone, and allopregnanolone when injected with 2.0 g/kg ethanol. These data suggest tolerance develops to ethanol induction of neuroactive steroids. However, because administering ACTH restored the efficacy of ethanol to increase neuroactive steroids, adrenal responsiveness was not impaired by chronic ethanol. Instead, chronic exposure to ethanol also may alter steroidogenesis. For example, injection of pregnenolone (50 mg/kg, i.p.) increased allopregnanolone in the cortex of alcohol-preferring P rats, but only if they had been trained to self-administer ethanol (Besheer et al., 2010).
The Role of Endogenous Neuroactive Steroids in Sex Differences in the Effects of Ethanol
Greater alcoholism risk among males is a robust observation in humans (Chan et al., 2007; Grant et al., 2007) and non-human primates (Vivian et al., 2002), suggesting that this sex difference is caused directly, at least in part, by biological rather social and behavioral differences. This sex difference is opposite in adult rodents and has been related to gonadal hormones (Vetter-O’Hagen et al., 2009; Vetter-O’Hagen and Spear, 2011), as testosterone appears to protect adult male rodents against heavy ethanol intake. Although contradictory results have been obtained in studies of both species, the results of human studies generally contrast with rodent findings to suggest that testosterone is a risk factor for heavy ethanol drinking. Testosterone is particularly important in the development of male-specific maturation in rodents and primates (Gupta et al., 1975; Sato et al., 2008). Although present in adult females, the concentration of circulating testosterone is much greater in adult male rodents (e.g., Pluchino et al., 2009) and primates (e.g., Muller et al., 2011), in whom it is linked to aggressive and reproductive behavior (Sato et al., 2008). Stålenheim et al. (1998) reported that testosterone was associated with aggressive and antisocial behavior, which were more prevalent in alcoholic men with a family history of alcoholism. Among adolescent males, individuals with ≥3 symptoms of alcohol abuse had greater testosterone (n = 13, 0.16 nmol/l) than individuals with ≤2 symptoms (n = 451, 0.11 nmol/l). A significant positive correlation was found between total alcohol abuse symptoms and testosterone after adjusting for pubertal development (Eriksson et al., 2005). In another study using premenopausal females, testosterone was elevated among individuals with early onset alcohol abuse compared to controls, in contrast to decreased estradiol, progesterone, androstenedione, and sex hormone binding globulin (Pettersson et al., 1990). Consistent with a positive correlation between alcohol drinking and circulating androgens, a more recent study of post-menopausal women reported a significant association between testosterone and alcohol intake, which had a narrow range around 1.5 drinks per day (Wayne et al., 2008). In contradiction, however, Onland-Moret et al. (2005) reported that testosterone concentrations were similar amongst post-menopausal women who drank >2.5 drinks per day and those who abstained, although the androgen DHEA sulfate was elevated amongst the drinkers. In another correlative study, alcohol-preferring rats had greater baseline testosterone compared to alcohol non-preferring rats (Apter and Eriksson, 2003), which is inconsistent with the idea that testosterone protects against heavy alcohol drinking in male rodents. Few studies have tested the hypothesis that testosterone is causally related to ethanol drinking, and these have produced contradictory findings. First, among castrated male albino rats, those treated with testosterone more rapidly acquired a greater preference for ethanol compared to those treated with estradiol, progesterone, or oil control solution (Lakoza and Barkov, 1980). On the other hand, castrated male Sprague-Dawley rats showed greater ethanol intake (2.2 g/kg/2 h) compared to sham rats (1.4 g/kg/2 h), which was decreased on average by testosterone replacement (1.75 g/kg/2 h; Vetter-O’Hagen et al., 2011). Many procedural differences could account for these discrepant findings, including “alcoholization” by Lakoza and Barkov (1980) in which ethanol was administered intraperitoneally for 2 weeks prior to access to ethanol, and the use of a sweetened solution by Vetter-O’Hagen et al. (2011). Overall, a majority of studies suggest that testosterone influences the risk for heavy drinking, although obviously more work is needed to determine a causal mechanism. In terms of neurosteroids, one possible mechanism is metabolism to androsterone.
Androsterone, as reviewed above, has discriminative stimulus effects similar to ethanol (monkeys, Grant et al., 2008a) and pregnanolone (rats, Engel et al., 2001), and therefore could increase sensitivity to ethanol via additive pharmacological effects. Consistent with the distinct pharmacological effects of isomers of neuroactive steroids, 3α,5α-androsterone, but not 3β,5α-androsterone, is a low-potency but efficacious anticonvulsant (Kaminski et al., 2005). In one study, male volunteers, who may have been social drinkers, had high levels of circulating androsterone in the form of a glucuronide or sulfated conjugate (androsterone glucuronide, 45 ng/ml; Hsing et al., 2007), compared to 97 pg/ml for the non-conjugated form of androsterone (Porcu et al., 2010). Fabre et al. (1973) reported that both androsterone isomers (3α,5α- and 3α,5β-androsterone) were present in lower concentrations among actively drinking (mean BEC, 148 mg/dl; 5α, 0.43 mg/24 h; 5β, 0.29 mg/14 h) compared to abstinent alcoholics (5α, 0.95 mg/24 h; 5β, 0.71 mg/24 h) or controls (5α, 1.4 mg/24 h; 5β, 0.63 mg/24 h). To our knowledge, glucuronide or sulfate conjugates of androgens have not been measured in heavy drinkers. If the older results of Fabre et al. (1973) are confirmed, lower levels of androgens in alcoholics could indicate that the role of androgens in alcoholism is primarily as a risk factor and not a regulator of ethanol sensitivity or consummatory behavior, although once again, only correlative data is available.
Although present in males, progesterone concentration is two-fold greater in reproductively fertile women (Genazzani et al., 1998). Progesterone mediates reproductive behavior related to pregnancy that supports fetal development such as increased consummatory behavior (hyperphagia, Douglas et al., 2007). Females of old-world primates (including humans) have menstrual cycles that occur over approximately 28–30 days. Following a 10- to 12-day follicular phase, a rapid rise in luteinizing hormone and estrogen precedes ovulation at mid-cycle (Hotchkiss and Knobil, 1994). In contrast, rodents have a 4-day estrus cycle during which rising estradiol precedes a surge of luteinizing hormone that coincides with the onset of proestrus and induces ovulation. Rodent ovaries produce progesterone and progesterone-derived neuroactive steroids (Holzbauer, 1975) such that circulating levels rise throughout proestrus. Approximately 12 h after the luteinizing hormone surge, ovulation coincides with peak progesterone which declines throughout estrus (Goldman et al., 2007). In primates, progesterone mediates development of the uterine epithelium prior to menses, but in rodents, progesterone mediates the cellular composition of the vaginal epithelium.
The duration of exposure to endogenous neuroactive steroids across the menstrual or estrus cycle may influence subsequent effects of ethanol. For example, declining progesterone in the latter half of the primate luteal phase after prolonged (about 1 week) progesterone exposure could enhance the aversive effects of ethanol withdrawal. Compared to vehicle, twice daily injection of progesterone (5 mg/kg) to rats maintained on a chronic ethanol diet increased anxiety during ethanol withdrawal measured using elevated plus maze (Sharma et al., 2007). Exposure to progesterone for several days altered the pharmacological activity of allopregnanolone, and GABAA receptor subunit expression (i.e., increased α4 and δ, decreased α1 subunits; reviewed by Smith et al., 2007; Maguire and Mody, 2009). Indeed, tolerance to endogenous neuroactive steroids during the menstrual cycle and during pregnancy correlated with changes in GABAA receptor subunit conformation and function (Turkmen et al., 2011). Differences between rodents and primates in the time-course of endogenous progesterone concentration during reproductive cycles suggests that primate studies will provide important translational data about the influence of fluctuations in endogenous neuroactive steroids on sensitivity to ethanol, including withdrawal from chronic ethanol.
Consistent with sex differences in ethanol drinking and alcoholism susceptibility, males and females differ in symptoms of ethanol withdrawal (reviewed by Finn et al., 2010). For example, compared to males, female rats are susceptible to seizures for fewer days after ethanol withdrawal (Alele and Devaud, 2007). Furthermore, withdrawal symptoms are ameliorated by GABAergic neurosteroids differentially in male and female rodents (e.g., Devaud et al., 1998). In one study, rats’ amplitude of startle to an auditory stimulus was slightly enhanced during withdrawal from ethanol compared to withdrawal from a dextrose diet, but only in male rats, not sham or ovariectomized female rats. This effect was attenuated by progesterone (Reilly et al., 2009). In female mice, the magnitude of handling-induced convulsions during ethanol withdrawal was increased by finasteride, suggesting modulation by 5α-reduced neuroactive steroids. In contrast, finasteride decreased the magnitude of handling-induced convulsions in males (Gorin-Meyer et al., 2007). Sex differences in anticonvulsant effects of endogenous neurosteroids could relate to basal steroid concentrations. In male mice, removal of endogenous neurosteroids by adrenalectomy increased seizure magnitude during acute ethanol withdrawal, with no additional effect of gonadectomy. In contrast, in female mice, both adrenalectomy and gonadectomy were necessary to potentiate handling-induced convulsions during ethanol withdrawal. In both sexes, treatment with progesterone or deoxycorticosterone decreased the severity of convulsions (Kaufman et al., 2010). The absence of an effect of gonadectomy in males suggests that testosterone and its neuroactive metabolites have little impact on seizure susceptibility during ethanol withdrawal (Finn et al., 2010), or perhaps de novo testosterone synthesis in the brain could be up-regulated to compensate for gonadectomy.
Withdrawal seizures in alcoholics have a relatively low incidence, even among humans undergoing alcoholism treatment (8–16%; Caetano et al., 1998). A more commonly reported withdrawal syndrome is anxiety, often comorbid with alcohol dependence in humans (Kushner et al., 2005). Repeated high doses of ethanol followed by withdrawal from ethanol decrease the threshold for seizures similar to kindling, in which seizure threshold is reduced by repeated sub-threshold electrical activity (reviewed by Breese et al., 2005; Rogawski, 2005). Kia et al. (2011) reported that daily electrical stimulation of the basolateral amygdala, which elicited convulsions, also eliminated the potentiation of tonic currents by THDOC in pyramidal neurons of the piriform cortex. Thus, kindling resulting in basal neural excitation was associated with decreased efficacy of a neuroactive steroid to enhance tonic currents. These in vitro data are reminiscent of the reports of cross-tolerance to alphaxalone-induced sleep after CIE, which includes daily withdrawal (Cagetti et al., 2003). Basal neurosteroid concentrations may regulate seizure threshold via modulation of tonic currents, and changes in neurosteroid concentrations in alcoholics that undergo abstinence could represent attempts to maintain homeostasis of neural excitability.
Ethanol and Neuroactive Steroid Effects in Relation to Changes in GABA Neurotransmission during Maturation
The expression of GABAA receptor subunits is developmentally regulated. However, the effect of ethanol and neuroactive steroids on different components of GABA neurotransmission has rarely been investigated at different maturational stages. In the cerebellum and thalamus of rodents, the δ subunit and associated α subunits (α4/α6) are not expressed at birth (developmentally equivalent to humans in the third trimester of pregnancy), but they are progressively expressed over the first month of life (roughly peaking at puberty). Accordingly, in those brain regions, tonic GABAA currents, mediated by α4/α6δ containing GABAA receptors, first appear at ~15 days, and get progressively larger until adulthood (Brickley et al., 1996; Wall and Usowicz, 1997; Peden et al., 2008). In contrast, tonic GABAA currents mediated by α4δ subunits are present at birth in the hippocampus (Holter et al., 2010). Thus, the role of extrasynaptic GABAA receptors and tonic inhibition in mediating neurosteroid and ethanol actions may be broadly developmentally regulated in some brain regions (cerebellum/thalamus) but not in others (hippocampus). In addition to developmental changes in GABAA receptors, there are developmental changes in the transporters that establish the Cl− gradient which is what makes GABAA receptors inhibitory in the adult (Wang and Kriegstein, 2009). Accordingly, in the early post-natal period in rodents (third trimester in humans) GABAA receptors are excitatory and play a crucial role in maturation of the nervous system (Eilers et al., 2001; Wang and Kriegstein, 2009). Thus, neurosteroid and ethanol enhancement of GABAA receptors in the human fetus is likely to be excitatory, and may interfere with proper brain development. In the case of ethanol, such interference may contribute to the brain damage underlying fetal alcohol syndrome (Costa et al., 2000; Galindo et al., 2005).
As well as the broad developmental changes in the GABAergic system that likely affect the impact of neurosteroids and ethanol, there are specific developmental changes in the GABAergic system that may influence responses to neurosteroids and ethanol. Specifically, GABA neurotransmission appears to be distinct during puberty (post-natal days 35–45 in mice), which may contribute to the vulnerability of adolescents to alcohol abuse and dependence (Grant and Dawson, 1997). Shen et al. (2010) reported 300 and 700% greater α4 and δ subunit expression, respectively, on dendritic spine (not shaft) membranes of CA1 pyramidal cells in pubertal compared to pre-pubertal and adult female mice. Additionally, whereas allopregnanolone potentiated GABA-induced currents and decreased anxiety in adult rodents (Finn and Purdy, 2007), allopregnanolone increased anxiety and decreased tonic GABA-induced currents (CA1 pyramidal cells) in female mice during puberty. This differential effect of allopregnanolone was related to a basal outward Cl− current at α4βδ receptors during puberty, as opposed to inward during adulthood (Shen et al., 2007). Pyramidal cells of pubertal mice were more sensitive to currents induced by the α4 and δ subunit-selective agonist gaboxadol, consistent with the unique subunit expression occurring during puberty. Allopregnanolone (30 nM) facilitated long-term potentiation and spatial learning in pubertal mice, but decreased long-term potentiation in pre-pubertal mice (Shen et al., 2010). A pattern of ethanol exposure in rats meant to model binge drinking in adolescence (3 g/kg/day, 8 days of injections with two intervening saline injections) heightened HPA axis reactivity to stress (acute ethanol) as adults (Przybycien-Szymanska et al., 2011). Ethanol exposure during the sensitive pubertal period could disrupt the maturation of stress response systems including neurosteroid regulation of homeostasis, perhaps altering ethanol sensitivity, and increasing risk of alcoholism.
Future Directions
An area of research needing attention is the contribution of neurosteroids to ethanol effects on sleep, an essential process regulating mood and responses to stress that is disrupted in alcoholism (Brower, 2003). A key brain structure involved in sleep is the ventrolateral preoptic area of the hypothalamus, which sends galanin and GABA projections to brainstem arousal nuclei to maintain sleep, with reverse inhibitory projections maintaining wakefulness (Harrison, 2007). During the first half of a sleep bout, high doses of ethanol decrease the latency to sleep onset, increase slow-wave sleep and decrease rapid eye movement (REM) sleep (young adult women; Williams et al., 1983), whereas the opposite effects are observed in the last half of a sleep bout (Feige et al., 2006). Sleep deprivation interacts with ethanol to potentiate daytime sleepiness and performance impairments due to ethanol (Roehrs and Roth, 2001). Both alcoholic men and women were reported to have more REM sleep, and less slow-wave sleep, compared to controls, with altered electroencephalograms during non-REM sleep (Gann et al., 2001; Colrain et al., 2010). A recent study found that moderately intoxicating doses of ethanol are associated with greater sleep disruption in women compared to men (Arnedt et al., 2011). It is tempting to speculate that neuroactive steroids could be a factor in sex differences in ethanol-induced disruption of sleep. Only a few studies, however, have investigated neuroactive steroid influences on sleep. Pregnenolone sulfate (47.5 mg/kg) increased REM sleep in rats without affecting slow-wave sleep or wakefulness (Darnaudéry et al., 1999). Administration of 15 mg/kg allopregnanolone per day for 5 days to rats dramatically altered the timing of sleep stages including decreased non-REM episodes, decreased latency to non-REM sleep, and increased latency to REM sleep (Damianisch et al., 2001). The effects of acute allopregnanolone and THDOC are similar, both decreasing sleep latency and increasing the duration of transition between non-REM and REM sleep (Müller-Preuss et al., 2002). Likewise, allopregnanolone (7.5 and 15 mg/kg) decreased the latency to sleep and increased the time in transition between non-REM and REM sleep (rats, Lancel et al., 1997). Pregnenolone sulfate regulated the percentage of REM sleep, but not non-REM or wakefulness, when infused into the pedunculopontine tegmentum, a component of the reticular formation (Darbra et al., 2004). In contrast, up to 100 mg/kg dehydroepiandrosterone sulfate does not affect sleep cycles, but affects electroencephalograms associated with non-REM sleep (Schiffelholz et al., 2000). Additional studies are needed to determine whether neurosteroid mechanisms are involved in ethanol influences on sleep, and whether neuroactive steroids could ameliorate sleep deficits in alcoholics.
Summary
Investigation of the effects of neuroactive steroids has come a long way since the first realizations of their shared receptor mechanisms with ethanol. These common receptor mechanisms (e.g., GABAA, NMDA receptors) mediate neurotransmission in ubiquitous neural pathways regulating excitation and inhibition across many brain areas. The diversity of neuronal types regulated by GABA and glutamate and expressing receptors for these neurotransmitters accounts for the multiple behavioral effects of ethanol and their modulation by neuroactive steroids. Neuroactive steroids appear to be able to modify many of the behaviors affected by ethanol along a continuum broadly bounded by excitation (anxiety or vigilance, convulsions, perhaps impulsivity) and inhibition (anxiolysis, decreased seizure threshold, sedation, ataxia). The endogenous role of neuroactive steroids in regulating these behaviors provides a window into the abnormalities observed in alcoholics. The strongest candidates for the endogenous ligands acting at the receptor systems mediating ethanol effects are neuroactive steroids, although not all of the receptor systems were covered in this review (e.g., 5-hydroxytryptamine receptors, sigma receptors). Additional evidence for neuroactive steroids as endogenous alcohols comes from their common metabolic substrates (Figure 1). Although we have learned much, it has only been less than 30 years since Harrison and Simmond’s (1984) study of the anesthetic actions of alphaxalone, and plenty of work remains to be done. For example, clarifying the functions of neuroactive steroids in maturation is likely to reveal the mechanisms for abnormalities induced by prenatal alcohol. It has been 20 years since the initial studies linking neuroactive steroids to the HPA axis (e.g., Purdy et al., 1991). The HPA axis may serve as a bridge between peripheral organs, the brain, and ultimately behavior to for optimal energy utilization depending on environmental conditions. Neuroactive steroids may be a central mechanism by which these environmental conditions are signaled to the brain, HPA axis, and the rest of the body. The significance of neuroactive steroids for a variety of physiological and behavioral processes affected by ethanol suggests that they should continue to be a focus of research for alcoholism therapies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Acosta, G., Hasenkamp, W., Daunais, J. B., Friedman, D. P., Grant, K. A., and Hemby, S. E. (2010). Ethanol self-administration modulation of NMDA receptor subunit and related synaptic protein mRNA expression in prefrontal cortical fields in cynomolgus monkeys. Brain Res. 1318, 144–154.
Alele, P. E., and Devaud, L. L. (2007). Sex differences in steroid modulation of ethanol withdrawal in male and female rats. J. Pharmacol. Exp. Ther. 320, 427–436.
Allan, A. M., and Harris, R. A. (1986). Gamma-aminobutyric acid and alcohol actions: neurochemical studies of long sleep and short sleep mice. Life Sci. 39, 2005–2015.
Apter, S. J., and Eriksson, C. J. P. (2003). The effect of alcohol on testosterone concentrations in alcohol-preferring and non-preferring rat lines. Alcohol. Clin. Exp. Res. 27, 1190–1193.
Arnedt, J. T., Rohsenow, D. J., Almeida, A. B., Hunt, S. K., Gokhale, M., Gottlieb, D. J., and Howland, J. (2011). Sleep following alcohol intoxication in healthy, young adults: effects of sex and family history of alcoholism. Alcohol. Clin. Exp. Res. 35, 870–878.
Ator, N. A., Grant, K. A., Purdy, R. H., Paul, S. M., and Griffiths, R. R. (1993). Drug discrimination analysis of endogenous neuroactive steroids in rats. Eur. J. Pharmacol. 241, 237–243.
Bäckström, T., Haage, D., Löfgren, M., Johansson, I. M., Strömberg, J., Nyberg, S., Andréen, L., Ossewaarde, L., Van Wingen, G. A., Turkmen, S., and Bengtsson, S. K. (2011). Paradoxical effects of GABA-A modulators may explain sex steroid induced negative mood symptoms in some persons. Neuroscience 191, 46–54.
Barbaccia, M. L., Roscetti, G., Trabucchi, M., Cuccheddu, T., Concas, A., and Biggio, G. (1994). Neurosteroids in the brain of handling-habituated and naïve rats: effects of CO2 inhalation. Eur. J. Pharmacol. 261, 317–320.
Belelli, D., Casula, A., Ling, A., and Lambert, J. J. (2002). The influence of subunit composition on the interaction of neurosteroids with GABAA receptors. Neuropharmacology 43, 651–661.
Belelli, D., Herd, M. B., Mitchell, E. A., Peden, D. R., Vardy, A. W., Gentet, L., and Lambert, J. J. (2006). Neuroactive steroids and inhibitory neurotransmission: mechanisms of action and physiological relevance. Neuroscience 138, 821–829.
Besheer, J., Lindsay, T. G., O’Buckley, T. K., Hodge, C. W., and Morrow, A. L. (2010). Pregnenolone and ganaxolone reduce operant ethanol self-administration in alcohol-preferring P rats. Alcohol. Clin. Exp. Res. 34, 2044–2052.
Biggio, G., Concas, A., Corda, M. G., Giorgi, O., Sanna, E., and Serra, M. (1990). GABAergic and dopaminergic transmission in the rat cerebral cortex: effect of stress, anxiolytic and anxiogenic drugs. Pharmacol. Ther. 48, 121–142.
Biggio, G., Concas, A., Follesa, P., Sanna, E., and Serra, M. (2007). Stress, ethanol, and neuroactive steroids. Pharmacol. Ther. 116, 140–171.
Borghese, C. M., and Harris, R. A. (2007). Studies of ethanol actions on recombinant delta-containing gamma-aminobutyric acid type A receptors yield contradictory results. Alcohol 41, 155–162.
Borghese, C. M., Stórustovu, S. Í., Ebert, B., Herd, M. B., Belelli, D., Lambert, J. J., Marshall, G., Wafford, K. A., and Harris, R. A. (2006). The δ subunit of γ-aminobutyric acid type A receptors does not confer sensitivity to low concentrations of ethanol. J. Pharmacol. Exp. Ther. 316, 1360–1368.
Botta, P., Radcliffe, R. A., Carta, M., Mameli, M., Daly, E., Floyd, K. L., Deitrich, R. A., and Valenzuela, C. F. (2007a). Modulation of GABAA receptors in cerebellar granule neurons by ethanol: a review of genetic and electrophysiological studies. Alcohol 41, 187–199.
Botta, P., Mameli, M., Floyd, K. L., Radcliffe, R. A., and Valenzuela, C. F. (2007b). Ethanol sensitivity of GABAergic currents in cerebellar granule neurons is not increased by a single amino acid change (R100Q) in the alpha6 GABAA receptor subunit. J. Pharmacol. Exp. Ther. 323, 684–691.
Bowen, C. A., Purdy, R. H., and Grant, K. A. (1999). Ethanol-like discriminative stimulus effects of endogenous neuroactive steroids: effects of ethanol training dose and dosing procedure. J. Pharmacol. Exp. Ther. 289, 405–411.
Boyd, K. N., Kumar, S., O’Buckley, T. K., and Morrow, A. L. (2010a). Chronic ethanol exposure produces tolerance to elevations in neuroactive steroids: mechanisms and reversal by exogenous ethanol. J. Neurochem. 115, 142–152.
Boyd, K. N., Kumar, S., O’Buckley, T. K., Porcu, P., and Morrow, A. L. (2010b). Ethanol induction of steroidogenesis in rat adrenal and brain is dependent upon pituitary ACTH release and de novo adrenal StAR synthesis. J. Neurochem. 112, 784–796.
Breese, G. R., Knapp, D. J., and Overstreet, D. J. (2004). Stress sensitization of ethanol withdrawal-induced reduction in social interaction: inhibition by CRF-1 and benzodiazepine receptor antagonists and a 5-HT1A-receptor agonist. Neuropsychopharmacology 29, 470–482.
Breese, G. R., Overstreet, D. H., and Knapp, D. J. (2005). Conceptual framework for the etiology of alcoholism: a “kindling”/stress hypothesis. Psychopharmacology (Berl.) 178, 367–380.
Brickley, S. G., Cull-Candy, S. G., and Farrant, M. (1996). Development of a tonic form of synaptic inhibition in rat cerebellar granule cells resulting from persistent activation of GABAA receptors. J. Physiol. (Lond.) 497, 753–759.
Budziszewska, B., Zając, A., Basta-Kaim, A., Leśkiewicz, M., Steczkowska, M., Lasoń, W., and Kaciński, M. (2010). Effect of neurosteroids on the human corticotropin-releasing hormone gene. Pharmacol. Rep. 62, 1030–1040.
Caetano, R., Clark, C. L., and Greenfield, T. K. (1998). Prevalence, trends, and incidence of alcohol withdrawal symptoms: analysis of general population and clinical samples. Alcohol Health Res. World 22, 73–80.
Cagetti, E., Liang, J., Spigelman, I., and Olsen, R. W. (2003). Withdrawal from chronic intermittent ethanol treatment changes subunit composition, reduces synaptic function, and decreases behavioral responses to positive allosteric modulators of GABAA receptors. Mol. Pharmacol. 63, 53–64.
Cagetti, E., Liang, J., Spigelman, I., and Olsen, R. W. (2004). Chronic intermittent ethanol (CIE) administration in rats decreases levels of neurosteroids in hippocampus, accompanied by altered behavioral responses to neurosteroids and memory function. Neuropharmacology 46, 570–579.
Calogero, A. E., Gallucci, W. T., Chrousos, G. P., and Gold, P. W. (1988). Interaction between GABAergic neurotransmission and rat hypothalamic corticotropin-releasing hormone secretion in vitro. Brain Res. 463, 28–36.
Carta, M., Mameli, M., and Valenzuela, C. F. (2004). Alcohol enhances GABAergic transmission to cerebellar granule cells via an increase in Golgi cell excitability. J. Neurosci. 24, 3746–3751.
Cavelier, P., Hamann, M., Rossi, D., Mobbs, P., and Attwell, D. (2005). Tonic excitation and inhibition of neurons: ambient transmitter sources and computational consequences. Prog. Biophys. Mol. Biol. 87, 3–16.
Chan, K. K., Neighbors, C., Gilson, M., Larimer, M. E., and Marlatt, G. A. (2007). Epidemiological trends in drinking by age and gender: providing normative feedback to adults. Addict. Behav. 32, 967–976.
Chisari, M., Eisenman, L. N., Covey, D. F., Mennerick, S., and Zorumski, C. F. (2010). The sticky issue of neurosteroids and GABAA receptors. Trends Neurosci. 33, 299–306.
Choi, D. S., Wei, W., Deitchman, J. K., Kharazia, V. N., Lesscher, H. M., McMahon, T., Wang, D., Qi, Z. H., Sieghart, W., Zhang, C., Shokat, K. M., Modi, I., and Messing, R. O. (2008). Protein kinase Cdelta regulates ethanol intoxication and enhancement of GABA-stimulated tonic current. J. Neurosci. 28, 11890–11899.
Colrain, I. M., Turlington, S., and Baker, F. C. (2010). Impact of alcoholism on sleep architecture and EEG power spectra in men and women. Sleep 32, 1341–1352.
Costa, E. T., Savage, D. D., and Valenzuela, C. F. (2000). A review of the effects of prenatal or early postnatal ethanol exposure on brain ligand-gated ion channels. Alcohol. Clin. Exp. Res. 24, 706–715.
Crabb, D. W., Matsumoto, M., Chang, D., and You, M. (2004). Overview of the role of alcohol dehydrogenase and aldehyde dehydrogenase and their variants in the genesis of alcohol-related pathology. Proc. Nutr. Soc. 63, 49–63.
Criswell, H. E., Ming, Z., Kelm, M. K., and Breese, G. R. (2008). Brain regional differences in the effect of ethanol on GABA release from presynaptic terminals. J. Pharmacol. Exp. Ther. 326, 596–603.
Damianisch, K., Rupprecht, R., and Lancel, M. (2001). The influence of subchronic administration of the neurosteroid allopregnanolone on sleep in the rat. Neuropsychopharmacology 25, 576–584.
Darbra, S., George, O., Bouyer, J.-J., Piazza, P.-V., Le Moal, M., and Mayo, W. (2004). Sleep–wake states and cortical synchronization control by pregnenolone sulfate into the pedunculopontine nucleus. J. Neurosci. Res. 76, 742–747.
Darbra, S., Mòdol, L., and Pallarès, M. (2011). Allopregnanolone infused into the dorsal (CA1) hippocampus increases prepulse inhibition of startle response in Wistar rats. Psychoneuroendocrinology. doi: 10.1016/j.psyneuen.2011.08.002
Darbra, S., and Pallarès, M. (2010). Alterations in neonatal neurosteroids affect exploration during adolescence and prepulse inhibition in adulthood. Psychoneuroendocrinology 35, 525–535.
Darnaudéry, M., Bouyer, J.-J., Pallarés, M., Le Moal, M., and Mayo, W. (1999). The promnesic neurosteroid pregnenolone sulfate increases paradoxical sleep in rats. Brain Res. 818, 492–498.
Devaud, L. L., Fritschy, J.-M., and Morrow, A. L. (1998). Influence of gender on chronic ethanol-induced alterations in GABAA receptors in rats. Brain Res. 796, 222–230.
Devaud, L. L., Purdy, R. H., Finn, D. A., and Morrow, A. L. (1996). Sensitization of γ-aminobutyric acid A receptors to neuroactive steroids in rats during ethanol withdrawal. J. Pharmacol. Exp. Ther. 278, 510–517.
Devaud, L. L., Purdy, R. H., and Morrow, A. L. (1995). The neurosteroid, 3alpha-hydroxy-5 alpha-pregnan-20-one, protects against bicuculline-induced seizures during ethanol withdrawal in rats. Alcohol. Clin. Exp. Res. 19, 350–355.
Douglas, A. J., Johnstone, L. E., and Leng, G. (2007). Neuroendocrine mechanisms of change in food intake during pregnancy: a potential role for brain oxytocin. Physiol. Behav. 91, 352–365.
Droogleever Fortuyn, H. A., van Broekhoven, F., Span, P. N., Bäckström, T., Zitman, F. G., and Verkes, R. J. (2004). Effects of PhD examination stress on allopregnanolone and cortisol plasma levels and peripheral benzodiazepine receptor density. Psychoneuroendocrinology 29, 1341–1344.
Eilers, J., Plant, T. D., Marandi, N., and Konnerth, A. (2001). GABA-mediated Ca2+ signalling in developing rat cerebellar Purkinje neurones. J. Physiol. (Lond.) 536, 429–437.
Engel, S. R., Purdy, R. H., and Grant, K. A. (2001). Characterization of discriminative stimulus effects of the neuroactive steroid pregnenolone. J. Pharmacol. Exp. Ther. 297, 498–495.
Eriksson, C. J. P., Kaprio, J., Pulkkinen, L., and Rose, R. J. (2005). Testosterone and alcohol use among adolescent male twins: testing between-family associations in within-family comparisons. Behav. Genet. 35, 359–368.
Fabre, L. F. Jr., Pasco, P. J., Liegel, J. M., and Farmer, R. W. (1973). Abnormal testosterone excretion in men alcoholics. Q. J. Stud. Alcohol. 34, 57–63.
Farrant, M., and Kaila, K. (2007). The cellular, molecular and ionic basis of GABAA receptor signaling. Prog. Brain Res. 160, 59–87.
Farrant, M., and Nusser, Z. (2005). Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat. Rev. Neurosci. 6, 215–229.
Feige, B., Gann, H., Brueck, R., Hornyak, M., Litsch, S., Hohagen, F., and Riemann, D. (2006). Effects of alcohol on polysomnographically recorded sleep in healthy subjects. Alcohol. Clin. Exp. Res. 30, 1527–1537.
Finn, D. A., Beckley, E. H., Kaufman, K. R., and Ford, M. M. (2010). Manipulation of GABAergic steroids: sex differences in the effects on ethanol drinking- and withdrawal-related behaviors. Horm. Behav. 57, 12–22.
Finn, D. A., Gallaher, E. J., and Crabbe, J. C. (2000). Differential change in neuroactive steroid sensitivity during ethanol withdrawal. J. Pharmacol. Exp. Ther. 292, 394–405.
Finn, D. A., and Purdy, R. H. (2007). “Neuroactive steroids in anxiety and stress,” in Handbook of Contemporary Neuropharmacology, eds D. R. Sibley, I. Hanin, M. Kuhar, and P. Skolnick (Hoboken: John Wiley and Sons), 133–176.
Galindo, R., Zamudio, P. A., and Valenzuela, C. F. (2005). Alcohol is a potent stimulant of immature neuronal networks: implications for fetal alcohol spectrum disorder. J. Neurochem. 94, 1500–1511.
Gann, H., Feige, B., Hohagen, F., van Calker, D., Geiss, D., and Dieter, R. (2001). Sleep and the cholinergic rapid eye movement sleep induction test in patients with primary alcohol dependence. Biol. Psychiatry 50, 383–390.
Genazzani, A. R., Petraglia, F., Bernardi, F., Casarosa, E., Salvestroni, C., Tonetti, A., Nappi, R. E., Luisi, S., Palumbo, M., Purdy, R. H., and Luisi, M. (1998). Circulating levels of allopregnanolone in humans: gender, age, and endocrine influences. J. Clin. Endocrinol. Metab. 83, 2099–2103.
Gibbs, T. T., Russek, S. J., and Farb, D. H. (2006). Sulfated steroids as endogenous neuromodulators. Pharmacol. Biochem. Behav. 84, 555–567.
Glykys, J., and Mody, I. (2007a). Activation of GABAA receptors: views from outside the synaptic cleft. Neuron 56, 763–770.
Glykys, J., and Mody, I. (2007b). The main source of ambient GABA responsible for tonic inhibition in the mouse hippocampus. J. Physiol. (Lond.) 582, 1163–1178.
Glykys, J., Peng, Z., Chandra, D., Homanics, G. E., Houser, C. R., and Mody, I. (2007). A new naturally occurring GABAA receptor subunit partnership with high sensitivity to ethanol. Nat. Neurosci. 10, 40–48.
Goldman, J. M., Murr, A. S., and Cooper, R. L. (2007). The rodent estrous cycle: characterization n of vaginal cytology and its utility in toxicological studies. Birth Defects Res. B Dev. Reprod. Toxicol. 80, 84–97.
Gorin-Meyer, R., Wiren, K. M., Tanchuck, M. A., Long, S. L., Yoneyama, N., and Finna, D. A. (2007). Sex differences in the effect of finasteride on acute ethanol withdrawal severity in C57BL/6J and DBA/2J mice. Neuroscience 146, 1302–1315.
Grant, B. F., and Dawson, D. A. (1997). Age at onset of alcohol use and its association with DSM-IV alcohol abuse and dependence: results from the National Longitudinal Alcohol Epidemiologic Survey. J. Subst. Abuse 9, 103–110.
Grant, B. F., Harford, T. C., Muthén, B. O., Yi, H.-Y., Hasin, D. S., and Stinson, F. S. (2007). DSM-IV alcohol dependence and abuse: further evidence of validity in the general population. Drug Alcohol Depend. 86, 154–166.
Grant, K. A., Azarov, A., Bowen, C. A., Mirkis, S., and Purdy, R. H. (1996). Ethanol-like discriminative stimulus effects of the neurosteroid 3a-hydroxy-5a-pregnan-20-one in female Macaca fascicularis monkeys. Psychopharmacology (Berl.) 124, 340–346.
Grant, K. A., Azarov, A., Shively, C. A., and Purdy, R. H. (1997). Discriminative stimulus effects of ethanol and 3α-hydroxy-5α-pregnan-20-one in relation to menstrual cycle phase in cynomolgus monkeys (Macaca fascicularis). Psychopharmacology (Berl.) 130, 59–68.
Grant, K. A., Helms, C. M., Rogers, L. S. M., and Purdy, R. H. (2008a). Neuroactive steroid stereospecificity of ethanol-like discriminative stimulus effects in monkeys. J. Pharmacol. Exp. Ther. 326, 354–361.
Grant, K. A., Leng, X., Green, H. L., Szeliga, K. T., Rogers, L. S. M., and Gonzales, S. W. (2008b). Drinking typography established by schedule induction predicts chronic heavy drinking in a monkey model of ethanol self-administration. Alcohol. Clin. Exp. Res. 32, 1824–1838.
Grant, K. A., Waters, C. A., Green-Jordan, K., Azarov, A., and Szeliga, K. T. (2000). Characterization of the discriminative stimulus effects of GABAA receptor ligands in Macaca fascicularis monkeys under different ethanol training conditions. Psychopharmacology (Berl.) 152, 181–188.
Green, K. L., Azarov, A. V., Szeliga, K. T., Purdy, R. H., and Grant, K. A. (1999). The influence of menstrual cycle phase on sensitivity to ethanol-like discriminative stimulus effects of GABAA-positive modulators. Pharmacol. Biochem. Behav. 64, 379–383.
Gupta, D., Rager, K., Attanasio, A., Klemm, W., and Eichner, M. (1975). Sex steroid hormones during multiphase pubertal developments. J. Steroid Biochem. 6, 859–868.
Haage, D., Druzin, M., and Johansson, S. (2002). Allopregnanolone modulates spontaneous GABA release via presynaptic Cl- permeability in rat preoptic nerve terminals. Brain Res. 958, 405–413.
Hamann, M., Rossi, D. J., and Attwell, D. (2002). Tonic and spillover inhibition of granule cells control information flow through cerebellar cortex. Neuron 33, 625–633.
Hanchar, H. J., Dodson, P. D., Olsen, R. W., Otis, T. S., and Wallner, M. (2005). Alcohol-induced motor impairment caused by increased extrasynaptic GABA(A) receptor activity. Nat. Neurosci. 8, 339–345.
Hanchar, H. J., Wallner, M., and Olsen, R. W. (2004). Alcohol effects on gamma-aminobutyric acid type A receptors: are extrasynaptic receptors the answer? Life Sci. 76, 1–8.
Harrison, N. L. (2007). Mechanisms of sleep induction by GABAA receptor agonists. J. Clin. Psychiatry 68, 6–12.
Harrison, N. L., and Simmonds, M. A. (1984). Modulation of the GABA receptor complex by a steroid anaesthetic. Brain Res. 323, 287–292.
Helms, C. M., Buckert, A. D., and Grant, K. A. (2010). Effect of Ovariectomy on the Receptor Mechanisms Contributing to the Discriminative Stimulus Effects of Ethanol. Anaheim, CA: Experimental Biology – American Society for Pharmacology and Experimental Therapeutics C116.767.6.
Helms, C. M., Rogers, L. S. M., and Grant, K. A. (2009). Antagonism of the ethanol-like discriminative stimulus effects of ethanol, pentobarbital, and midazolam in cynomolgus monkeys reveals involvement of specific GABAA receptor subtypes. J. Pharmacol. Exp. Ther. 331, 142–152.
Helms, C. M., Rogers, L. S. M., Waters, C. A., and Grant, K. A. (2008). Zolpidem generalization and antagonism in male and female cynomolgus monkeys trained to discriminate 1.0 or 2.0 g/kg ethanol. Alcohol. Clin. Exp. Res. 32, 1197–1206.
Hemby, S. E., O’Connor, J. A., Acosta, G., Floyd, D., Anderson, N., McCool, B. A., Friedman, D., and Grant, K. A. (2006). Ethanol-induced regulation of GABAA subunit mRNAs in prefrontal fields of cynomolgus monkeys. Alcohol. Clin. Exp. Res. 30, 1978–1985.
Herd, M. B., Foister, N., Chandra, D., Peden, D. R., Homanics, G. E., Brown, V. J., Balfour, D. J. K., Lambert, J. J., and Belelli, D. (2009). Inhibition of thalamic excitability by THIP: a selective role for δ-GABAA receptors. Eur. J. Neurosci. 29, 1177–1187.
Hill, M., Popov, P., Havlíková, H., Kancheva, L., Vrbíková, J., Kancheva, R., Pouzar, V., Čcerný, I., and Stárka, L. (2005). Altered profiles of serum neuroactive steroids in premenopausal women treated for alcohol addiction. Steroids 70, 515–524.
Hodge, C. W., and Cox, A. A. (1998). The discriminative stimulus effects of ethanol are mediated by NMDA and GABAA receptors in specific limbic brain regions. Psychopharmacology (Berl.) 139, 95–107.
Hodge, C. W., Nannini, M. A., Olive, M. F., Kelley, S. P., and Mehmert, K. K. (2001). Allopregnanolone and pentobarbital infused into the nucleus accumbens substitute for the discriminative stimulus effects of ethanol. Alcohol. Clin. Exp. Res. 25, 1441–1447.
Holdstock, L., and de Wit, H. (2000). Effects of ethanol at four phases of the menstrual cycle. Psychopharmacology (Berl.) 150, 374–382.
Holter, N. I., Zylla, M. M., Zuber, N., Bruehl, C., and Draguhn, A. (2010). Tonic GABAergic control of mouse dentate granule cells during postnatal development. Eur. J. Neurosci. 32, 1300–1309.
Holzbauer, M. (1975). Physiological variations in the ovarian production of 5α-pregnane derivatives with sedative properties in the rat. J. Steroid Biochem. 6, 1307–1310.
Hosie, A. M., Clarke, L., da Silva, H., and Smart, T. G. (2009). Conserved site for neurosteroid modulation of GABAA receptors. Neuropharmacology 56, 149–154.
Hosie, A. M., Wilkins, M. E., da Silva, H. M. A., and Smart, T. G. (2006). Endogenous neurosteroids regulate GABAA receptors through two discrete transmembrane sites. Nature 444, 486–489.
Hotchkiss, J., and Knobil, E. (1994). “The menstrual cycle and its neuroendocrine control,” in The Physiology of Reproduction, 2nd Edn, eds E. Knobil, and J. D. Neill (New York: Raven Press), 711–749.
Hsing, A. W., Stanczyk, F. Z., Bélanger Schroeder, P., Chang, L., Falk, R. T., and Fears, T. R. (2007). Reproducibility of serum sex steroid assays in men by RIA and mass spectrometry. Cancer Epidemiol. Biomarkers Prev. 16, 1004–1008.
Huguenard, J. R., and McCormick, D. A. (2007). Thalamic synchrony and dynamic regulation of global forebrain oscillations. Trends Neurosci. 30, 350–356.
Janak, P. H., Redfern, J. E. M., and Samson, H. H. (1998). The reinforcing effects of ethanol are altered by the endogenous neurosteroid, allopregnanolone. Alcohol. Clin. Exp. Res. 22, 1106–1112.
Jia, F., Chandra, D., Homanics, G. E., and Harrison, N. L. (2008). Ethanol modulates synaptic and extrasynaptic GABAA receptors in the thalamus. J. Pharmacol. Exp. Ther. 326, 475–482.
Kaminski, R. M., Marini, H., Kim, W.-J., and Rogawski, M. A. (2005). Anticonvulsant activity of androsterone and etiocholanolone. Epilepsia 46, 819–827.
Kask, K., Bäckström, T., Lundgren, P., and Sundström Poromaa, I. (2009). Allopregnanolone has no effect on startle response and prepulse inhibition of startle response in patients with premenstrual dysphoric disorder or healthy controls. Pharmacol. Biochem. Behav. 92, 608–613.
Kaufman, K. R., Tanchuck, M. A., Strong, M. N., and Finn, D. A. (2010). Replacement with GABAergic steroid precursors restores the acute ethanol withdrawal profile in adrenalectomy/gonadectomy mice. Neuroscience 166, 5–14.
Kelley, A. E., Baldo, B. A., Pratt, W. E., and Will, M. J. (2005). Corticostriatal-hypothalamic circuitry and food motivation: integration of energy, action and reward. Physiol. Behav. 86, 773–795.
Kelly, T. H., Fredenburg, A. M., Emurian, C. S., Martin, C. A., Hays, L. R., and Rush, C. R. (2001). Discriminative stimulus effects of triazolam: influence of menstrual cycle phase. Drug Alcohol Depend. 63, S3–S178.
Kelly, T. H., Stoops, W. W., Perry, A. S., Prendergast, M. A., and Rush, C. R. (2003). Clinical neuropharmacology of drugs of abuse: a comparison of drug-discrimination and subject-report measures. Behav. Cogn. Neurosci. Rev. 2, 227–260.
Kelm, M. K., Criswell, H. E., and Breese, G. R. (2008). The role of protein kinase A in the ethanol-induced increase in spontaneous GABA release onto cerebellar Purkinje neurons. J. Neurophysiol. 100, 3417–3428.
Kelm, M. K., Criswell, H. E., and Breese, G. R. (2011). Ethanol-enhanced GABA release: a focus on G protein-coupled receptors. Brain Res. Rev. 65, 113–123.
Kia, A. K., Ribeiro, F., Nelson, R., Gavrilovici, C., Ferguson, S. S. G., and Poulter, M. O. (2011). Kindling alters neurosteroid- induced modulation of phasic and tonic GABAA receptor-mediated currents: role of phosphorylation. J. Neurochem. 116, 1043–1056.
Krause, J. E., and Karavolas, H. J. (1980). Subcellular location of hypothalamic progesterone metabolizing enzymes and evidence for distinct NADH- and NADPH-linked 3α-hydroxysteroid oxidoreductase activities. J. Steroid Biochem. 13, 271–280.
Kumar, S., Porcu, P., Werner, D. F., Matthews, D. B., Diaz-Granados, J. L., Helfand, R. S., and Morrow, A. L. (2009). The role of GABAA receptors in the acute and chronic effects of ethanol: a decade of progress. Psychopharmacology (Berl.) 205, 529–564.
Kushner, M. G., Abrams, K., Thuras, P., Hanson, K. L., Brekke, M., and Sletten, S. (2005). Follow-up study of anxiety disorder and alcohol dependence in comorbid alcoholism treatment patients. Alcohol. Clin. Exp. Res. 29, 1432–1443.
Lakoza, G. N., and Barkov, N. K. (1980). The role of testosterone in the development of experimental alcoholism. Bull. Narc. 32, 41–48.
Lancel, M., Faulhaber, J., Schiffelholz, T., Romeo, E., Michele, F. D., Holsboer, F., and Rupprecht, R. (1997). Allopregnanolone affects sleep in a benzodiazepine-like fashion. J. Pharmacol. Exp. Ther. 282, 1213–1218.
Lavoie, H. A., and King, S. R. (2009). Transcriptional regulation of steroidogenic genes: STARD1, CYP11A1 and HSD3B. Exp. Biol. Med. 234, 880–907.
Lee, M., Schwab, C., and McGeer, P. L. (2011). Astrocytes are GABAergic cells that modulate microglial activity. Glia 59, 152–165.
Lee, S., Yoon, B. E., Berglund, K., Oh, S. J., Park, H., Shin, H. S., Augustine, G. J., and Lee, C. J. (2010). Channel-mediated tonic GABA release from glia. Science 330, 790–796.
Lee, S., Selvage, D., Hansen, K., and Rivier, C. (2004). Site of action of acute alcohol administration in stimulating the rat hypothalamic-pituitary-adrenal axis: comparison between the effect of systemic and intracerebroventricular injection of this drug on pituitary and hypothalamic responses. Endocrinology 145, 4470–4479.
Liang, J., Cagetti, E., Olsen, R. W., and Spigelman, I. (2004). Altered pharmacology of synaptic and extrasynaptic GABAA receptors on CA2 hippocampal neurons is consistent with subunit changes in a model of alcohol withdrawal and dependence. J. Pharmacol. Exp. Ther. 310, 1234–1245.
Liang, J., Spigelman, I., and Olsen, R. W. (2009). Tolerance to sedative/hypnotic action of GABAergic drugs correlates with tolerance to potentiation of extrasynaptic tonic currents of alcohol-dependent rats. J. Neurophysiol. 102, 224–233.
Lovallo, W. R., Dickensheets, S. L., Myers, D. A., Thomas, T. L., and Nixon, S. J. (2000). Blunted stress cortisol response in abstinent alcoholic and polysubstance-abusing men. Alcohol. Clin. Exp. Res. 24, 651–658.
Lovinger, D. M., White, G., and Weight, F. F. (1989). Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science 243, 1721–1724.
Lüddens, H., Seeburg, P. H., and Korpi, E. R. (1994). Impact of β and γ variants on ligand-binding properties of γ-aminobutyric acid type A receptors. Mol. Pharmacol. 45, 810–814.
Maguire, J., and Mody, I. (2009). Steroid hormone fluctuations and GABAAR plasticity. Psychoneuroendocrinology 342, S84–S90.
Majewska, M. D., Harrison, N. L., Schwartz, R. D., Barker, J. L., and Paul, S. M. (1986). Steroid hormone metabolites are barbiturate-like modulators of the GABA receptor. Science 232, 1004–1007.
Meera, P., Wallner, M., and Otis, T. S. (2011). Molecular basis for the high THIP/gaboxadol sensitivity of extrasynaptic GABAA receptors. J. Neurophysiol. 106, 2057–2064.
Meyer, D. A., Carta, M., Partridge, L. D., Covey, D. F., and Valenzuela, C. F. (2002). Neurosteroids enhance spontaneous glutamate release in hippocampal neurons. J. Biol. Chem. 277, 28725–28732.
Mhatre, M. C., and Ticku, M. K. (1992). Chronic ethanol administration alters γ-aminobutyric acid A receptor gene expression. Mol. Pharmacol. 42, 415–422.
Mihalek, R. M., Banerjee, P. K., Korpi, E. R., Quinlan, J. J., Firestone, L. L., Mi, Z.-P., Lagenaur, C., Tretter, V., Sieghart, W., Anagnostaras, S. G., Sage, J. R., Fanselow, M. S., Guidotti, A., Spigelman, I., Li, Z., DeLorey, T. M., Olsen, R. W., and Homanics, G. E. (1999). Attenuated sensitivity to neuroactive steroids in gamma-aminobutyrate type A receptor delta subunit knockout mice. Proc. Natl. Acad. Sci. U.S.A. 96, 12905–12910.
Mody, I. (2008). Extrasynaptic GABAA receptors in the crosshairs of hormones and ethanol. Neurochem. Int. 52, 60–64.
Morrow, A. L., Biggio, G., Serra, M., Becker, H. C., Lopez, M. F., Porcu, P., Alward, S. E., and O’Buckley, T. K. (2009). The role of neuroactive steroids in ethanol/stress interactions: proceedings of symposium VII at the Volterra conference on alcohol and stress, May 2008. Alcohol 43, 521–530.
Morrow, A. L., Porcu, P., Boyd, K. N., and Grant, K. A. (2006). Hypothalamic-pituitary-adrenal axis modulation of GABAergic neuroactive steroids influences ethanol sensitivity and drinking behavior. Dialogues Clin. Neurosci. 8, 463–477.
Muller, D. C., Giles, G. G., Bassett, J., Morris, H. A., Manning, J. T., Hopper, J. L., English, D. R., and Severi, G. (2011). Second to fourth digit ratio (2D:4D) and concentrations of circulating sex hormones in adulthood. Reprod. Biol. Endocrinol. 9, 57–69.
Müller-Preuss, P., Rupprecht, R., and Lancel, M. (2002). The effects of the neuroactive steroid 3α,5α-THDOC on sleep in the rat. Neuroreport 13, 487–490.
Nie, H., Rewal, M., Gill, M., Ron, D., and Janak, P. H. (2011). Extrasynaptic δ-containing GABAA receptors in the nucleus accumbens dorsomedial shell contribute to alcohol intake. Proc. Natl. Acad. Sci. U.S.A. 108, 4459–4464.
O’Dell, L. E., Purdy, R. H., Covey, D. F., Richardson, H. N., Roberto, M., and Koob, G. F. (2005). Epipregnanolone and a novel synthetic neuroactive steroid reduce alcohol self-administration in rats. Pharmacol. Biochem. Behav. 81, 543–550.
Olsen, R. W., Hanchar, H. J., Meera, P., and Wallner, M. (2007). GABAA receptor subtypes: the “one glass of wine” receptors. Alcohol 41, 201–209.
Onland-Moret, N. C., Peeters, P. H. M., van der Schouw, Y. T., Grobbee, D. E., and van Gils, C. H. (2005). Alcohol and endogenous sex steroid levels in postmenopausal women: a cross-sectional study. J. Clin. Endocrinol. Metab. 90, 1414–1419.
Park-Chung, M., Malayev, A., Purdy, R. H., Gibbs, T. T., and Farb, D. H. (1999). Sulfated and unsulfated steroids modulate γ-aminobutyric acid A receptor function through distinct sites. Brain Res. 830, 72–87.
Park-Chung, M., Wu, F.-S., Purdy, R. H., Malayev, A. A., Gibbs, T. T., and Farb, D. H. (1997). Distinct sites for inverse modulation of N-methyl-D-aspartate receptors by sulfated steroids. Mol. Pharmacol. 52, 1113–1123.
Patchev, V. K., Hassan, A. H., Holsboer, D. F., and Almeida, O. F. (1996). The neurosteroid tetrahydroprogesterone attenuates the endocrine response to stress and exerts glucocorticoid-like effects on vasopressin gene transcription in the rat hypothalamus. Neuropsychopharmacology 15, 533–540.
Patchev, V. K., Shoaib, M., Holsboer, F., and Almeida, O. F. (1994). The neurosteroid tetrahydroprogesterone counteracts corticotropin-releasing hormone-induced anxiety and alters the release and gene expression of corticotropin-releasing hormone in the rat hypothalamus. Neuroscience 62, 265–271.
Peden, D. R., Petitjean, C. M., Herd, M. B., Durakoglugil, M. S., Rosahl, T. W., Wafford, K., Homanics, G. E., Belelli, D., Fritschy, J. M., and Lambert, J. J. (2008). Developmental maturation of synaptic and extrasynaptic GABAA receptors in mouse thalamic ventrobasal neurones. J. Physiol. (Lond.) 586, 965–987.
Pettersson, P., Ellsinger, B. M., Sjoberg, C., and Bjorntorp, P. (1990). Fat distribution and steroid hormones in women with alcohol abuse. J. Intern. Med. 228, 311–316.
Pierucci-Lagha, A., Covault, J., Feinn, R., Khisti, R. T., Morrow, A. L., Marx, C. E., Shampine, L. J., and Kranzler, H. R. (2006). Subjective effects and changes in steroid hormone concentrations in humans following acute consumption of alcohol. Psychopharmacology (Berl.) 186, 451–461.
Pierucci-Lagha, A., Covault, J., Feinn, R., Nellissery, M., Hernandez-Avila, C., Oncken, C., Morrow, A. L., and Kranzler, H. R. (2005). GABRA2 alleles moderate the subjective effects of alcohol, which are attenuated by finasteride. Neuropsychopharmacology 30, 1193–1203.
Pisu, M. G., Floris, I., Maciocco, E., Serra, M., and Biggio, G. (2006). Anticipation and consumption of food each increase the concentration of neuroactive steroids in rat brain and plasma. Pharmacol. Biochem. Behav. 85, 76–81.
Pisu, M. G., Mostallino, M. C., Dore, R., Maciocco, E., Secci, P. P., and Serra, M. (2011). Effects of voluntary ethanol consumption on emotional state and stress responsiveness in socially isolated rats. Eur. Neuropsychopharmacol. 21, 414–425.
Pluchino, N., Ninni, F., Casarosa, E., Giannini, A., Merlini, S., Cubeddu, A., Luisi, M., Cela, V., and Genazzani, A. R. (2009). Sex differences in brain and plasma β-endorphin content following testosterone, dihydrotestosterone and estradiol administration to gonadectomized rats. Neuroendocrinology 89, 411–423.
Porcu, P., Grant, K. A., Green, H. L., Rogers, L. S. M., and Morrow, A. L. (2006). Hypothalamic-pituitary-adrenal axis and ethanol modulation of deoxycorticosterone levels in cynomolgus monkeys. Psychopharmacology (Berl.) 186, 293–301.
Porcu, P., O’Buckley, T. K., Alward, S. E., Song, S. C., Grant, K. A., de Wit, H., and Morrow, A. L. (2010). Differential effects of ethanol on serum GABAergic 3α,5α/3α,5β neuroactive steroids in mice, rats, cynomolgus monkeys and humans. Alcohol. Clin. Exp. Res. 34, 432–442.
Porcu, P., O’Buckley, T. K., Morrow, A. L., and Adinoff, B. (2008). Differential hypothalamic-pituitary-adrenal activation of the neuroactive steroids pregnenolone sulfate and deoxycorticosterone in healthy controls and alcohol-dependent subjects. Psychoneuroendocrinology 33, 214–226.
Porcu, P., Sogliano, C., Ibba, C., Piredda, M., Tocco, S., Marra, C., Purdy, R. H., Biggio, G., and Concas, A. (2004). Failure of γ-hydroxybutyric acid both to increase neuroactive steroid concentration in adrenalectomized-orchiectomized rats and to induce tolerance to its steroidogenic effect in intact animals. Brain Res. 1012, 160–168.
Przybycien-Szymanska, M. M., Mott, N. N., Paul, C. R., Gillespie, R. A., and Pak, T. R. (2011). Binge-pattern alcohol exposure during puberty induces long-term changes in HPA axis reactivity. PLoS ONE 6, e18350. doi: 10.1371/journal.pone.0018350
Puia, G., Santi, M., Vicini, S., Pritchett, D. B., Purdy, R. H., Paul, S. M., Seeburg, P. H., and Costa, E. (1990). Neurosteroids act on recombinant human GABAA receptors. Neuron 4, 759–765.
Purdy, R. H., Morrow, A. L., Moore, P. H. Jr., and Paul, S. M. (1991). Stress-induced elevations of γ-aminobutyric acid type A receptor-active steroids in the rat brain. Proc. Natl. Acad. Sci. U.S.A. 88, 4553–4557.
Ramaker, M. J., Ford, M. M., Fretwell, A. M., and Finn, D. A. (2011). Alteration of ethanol drinking in mice via modulation of the GABA(A) receptor with ganaxolone, finasteride, and gaboxadol. Alcohol. Clin. Exp. Res. 35, 1994–2007.
Rani, C. S. S., and Ticku, M. K. (2006). Comparison of chronic ethanol and chronic intermittent ethanol treatments on the expression of GABAA and NMDA receptor subunits. Alcohol 38, 89–97.
Reilly, W., Koirala, B., and Devaud, L. L. (2009). Sex differences in acoustic startle responses and seizure thresholds between ethanol-withdrawn male and female rats. Alcohol Alcohol. 44, 561–566.
Reith, C. A., and Sillar, K. T. (1997). Pre- and postsynaptic modulation of spinal GABAergic neurotransmission by the neurosteroid, 5β-pregnan-3α-ol-20-one. Brain Res. 770, 202–212.
Roehrs, T., and Roth, T. (2001). Sleep, sleepiness, and alcohol use. Alcohol Res. Health 25, 101–109.
Rogawski, M. A. (2005). Update on the neurobiology of alcohol withdrawal seizures. Epilepsy Curr. 5, 225–230.
Rossi, D. J., and Hamann, M. (1998). Spillover-mediated transmission at inhibitory synapses promoted by high affinity alpha6 subunit GABA(A) receptors and glomerular geometry. Neuron 20, 783–795.
Rossi, D. J., Hamann, M., and Attwell, D. (2003). Multiple modes of GABAergic inhibition of rat cerebellar granule cells. J. Physiol. (Lond.) 548, 97–110.
Rowlett, J. K., and Woolverton, W. L. (1996). Assessment of benzodiazepine receptor heterogeneity in vivo: apparent pA2 and pKB analyses from behavioral studies. Psychopharmacology (Berl.) 128, 1–16.
Sanger, D. J., Griebel, G., Perrault, G., Claustre, Y., and Schoemaker, H. (1999). Discriminative stimulus effects of drugs acting at GABAA receptors: differential profiles and receptor selectivity. Pharmacol. Biochem. Behav. 64, 269–273.
Sanna, E., Talani, G., Busonero, F., Pisu, M. G., Purdy, R. H., Serra, M., and Biggio, G. (2004). Brain steroidogenesis mediates ethanol modulation of GABAA receptor activity in rat hippocampus. J. Neurosci. 24, 6521–6530.
Sanna, E., Talani, G., Obili, N., Mascia, M. P., Mostallino, M. C., Secci, P. P., Pisu, M. G., Biggio, F., Utzeri, C., Olla, P., Biggio, G., and Follesa, P. (2011). Voluntary ethanol consumption induced by social isolation reverses the increase of α4/δ GABAA receptor gene expression and function in the hippocampus of C57BL/6J mice. Front. Neurosci. 5:15. doi: 10.3389/fnins.2011.00015
Sato, S. M., Schulz, K. M., Sisk, C. L., and Wood, R. I. (2008). Adolescents and androgens, receptors and rewards. Horm. Behav. 53, 647–658.
Saxena, N. C., and Macdonald, R. L. (1994). Assembly of GABAA receptor subunits: role of the delta subunit. J. Neurosci. 14, 7077–7086.
Saxena, N. C., and Macdonald, R. L. (1996). Properties of putative cerebellar gamma-aminobutyric acid A receptor isoforms. Mol. Pharmacol. 49, 567–579.
Schiffelholz, T., Holsboer, F., and Lancel, M. (2000). High doses of systemic DHEA-sulfate do not affect sleep structure and elicit moderate changes in non-REM sleep EEG in rats. Physiol. Behav. 69, 399–404.
Serra, M., Mostallino, M. C., Talani, G., Pisu, M. G., Carta, M., Mura, M. L., Floris, I., Maciocco, E., Sann, E., and Biggio, G. (2006). Social isolation-induced increase in α4 and δ subunit gene expression in associated with a greater efficacy of ethanol on steroidogenesis and GABAA receptor function. J. Neurochem. 98, 122–133.
Serra, M., Pisu, M. G., Floris, I., Cara, V., Purdy, R. H., and Biggio, G. (2003). Social isolation-induced increase in the sensitivity of rats to the steroidogenic effect of ethanol. J. Neurochem. 85, 257–263.
Serra, M., Pisu, M. G., Littera, M., Papi, G., Sanna, E., Tuveri, F., Usala, L., Purdy, R. H., and Biggio, G. (2000). Social isolation-induced decreases in both the abundance of neuroactive steroids and GABAA receptor function in rat brain. J. Neurochem. 75, 732–740.
Shannon, E. E., Porcu, P., Purdy, R. H., and Grant, K. A. (2005a). Characterization of the discriminative stimulus effects of the neuroactive steroid pregnanolone in DBA/2J and C57BL/6J inbred mice. J. Pharmacol. Exp. Ther. 314, 675–685.
Shannon, E. E., Purdy, R. H., and Grant, K. A. (2005b). Discriminative stimulus effects of 5.6 mg/kg pregnanolone in DBA/2J and C57BL/6J inbred mice. Alcohol 37, 35–45.
Shannon, E. E., Shelton, K. L., Vivian, J. A., Yount, I., Morgan, A. R., Homanics, G. E., and Grant, K. A. (2004). Discriminative stimulus effects of ethanol in mice lacking the γ-aminobutyric acid type A receptor δ subunit. Alcohol. Clin. Exp. Res. 28, 906–913.
Sharma, A. N., Chopde, C. T., Hirani, K., Kokare, D. M., and Ugale, R. R. (2007). Chronic progesterone treatment augments while dehydroepiandrosterone sulphate prevents tolerance to ethanol anxiolysis and withdrawal anxiety in rats. Eur. J. Pharmacol. 567, 211–222.
Shelton, K. L., and Grant, K. A. (2002). Discriminative stimulus effects of ethanol in C57BL/6J and DBA/2J inbred mice. Alcohol. Clin. Exp. Res. 26, 747–757.
Shen, H., Gong, Q. H., Aoki, C., Yuan, M., Ruderman, Y., Dattilo, M., Williams, K., and Smith, S. S. (2007). Reversal of neurosteroid effects at α4β2δ GABAA receptors triggers anxiety at puberty. Nat. Neurosci. 10, 469–477.
Shen, H., Sabaliauskas, N., Sherpa, A., Fenton, A. A., Stelzer, A., Aoki, C., and Smith, S. S. (2010). A critical role for α4βδ GABAA receptors in shaping learning deficits at puberty in mice. Science 327, 1515–1518.
Shen, Y., Lindemeyer, A. K., Spigelman, I., Sieghart, W., Olsen, W., and Liang, J. (2011). Plasticity of GABAA receptors after ethanol pre-exposure in cultured hippocampal neurons. J. Pharmacol. Exp. Ther. 79, 432–442.
Slawecki, C. J., Purdy, R. H., and Ehlers, C. L. (2005). Electrophysiological effects of allopregnanolone in rats with a history of ethanol exposure. Alcohol. Clin. Exp. Res. 29, 66–74.
Slawecki, C. J., Walpole, T., Purdy, R. H., and Ehlers, C. L. (2000). Comparison of the neurophysiological effects of allopregnanolone and ethanol in rats. Psychopharmacology (Berl.) 149, 351–359.
Smith, S. S., Shen, H., Gong, Q. H., and Zhou, X. (2007). Neurosteroid regulation of GABAA receptors: focus on the α4 and δ subunits. Pharmacol. Ther. 116, 58–76.
Stålenheim, E. G., Eriksson, E., von Knorring, L., and Wide, L. (1998). Testosterone as a biological marker in psychopathology. Psychiatry Res 77, 79–88.
Stell, B. M., Brickley, S. G., Tang, C. Y., Farrant, M., and Mody, I. (2003). Neuroactive steroids reduce neuronal excitability by selectively enhancing tonic inhibition mediated by δ subunit- containing GABAA receptors. Proc. Natl. Acad. Sci. U.S.A. 100, 14439–14444.
Stocco, D. M., and Clark, B. J. (1996). Regulation of the acute production of steroids in steroidogenic cells. Endocr. Rev. 17, 221–244.
Suzdak, P. D., Glowa, J. R., Crawley, J. N., Schwartz, R. D., Skolnick, P., and Paul, S. M. (1986). A selective imidazobenzodiazepine antagonist of ethanol in the rat. Science 234, 1243–1247.
Turkmen, S., Backstrom, T., Wahlstrom, G., Andreen, L., and Johansson, I. M. (2011). Tolerance to allopregnanolone with focus on the GABA-A receptor. Br. J. Pharmacol. 162, 311–327.
Valenzuela, C. F., Partridge, L. D., Mameli, M., and Meyer, D. A. (2007). Modulation of glutamatergic transmission by sulfated steroids: role in fetal alcohol spectrum disorder. Brain Res. Rev. 57, 506–519.
VanDoren, M. J., Matthews, D. B., Janis, G. C., Grobin, A. C., Devaud, L. L., and Morrow, A. L. (2000). Neuroactive steroid 3α-hydroxy-5α-pregnan-20-one modulates electrophysiological and behavioral action of ethanol. J. Neurosci. 20, 1982–1989.
Vetter-O’Hagen, C. S., Sanders, K. W., and Spear, L. P. (2011). Evidence for suppressant effects of testosterone on sex-typical ethanol intake in male Sprague-Dawley rats. Behav. Brain Res. 224, 403–407.
Vetter-O’Hagen, C. S., and Spear, L. P. (2011). The effects of gonadectomy on age- and sex-typical patterns of ethanol consumption in Sprague-Dawley rats. Alcohol. Clin. Exp. Res. 35, 2039–2049.
Vetter-O’Hagen, C. S., Varlinskaya, E., and Spear, L. (2009). Sex differences in ethanol intake and sensitivity to aversive effects during adolescence and adulthood. Alcohol Alcohol. 44, 547–554.
Vivian, J. A., Waters, C. A., Szeliga, K. T., Jordan, K., and Grant, K. A. (2002). Characterization of the discriminative stimulus effects of N-methyl-D-aspartate ligands under different ethanol training conditions in the cynomolgus monkey (Macaca fascicularis). Psychopharmacology (Berl.) 162, 273–281.
Wall, M. J., and Usowicz, M. M. (1997). Development of action potential-dependent and independent spontaneous GABAA receptor-mediated currents in granule cells of postnatal rat cerebellum. Eur. J. Neurosci. 9, 533–548.
Wallner, M., and Olsen, R. W. (2008). Physiology and pharmacology of alcohol: the imidazobenzodiazepine alcohol antagonist site on subtypes of GABAA receptors as an opportunity for drug development? Br. J. Pharmacol. 154, 288–298.
Wang, D. D., and Kriegstein, A. R. (2009). Defining the role of GABA in cortical development. J. Physiol. (Lond.) 587, 1873–1879.
Wang, M., He, Y., Eisenman, L. N., Fields, C., Zeng, C.-M., Mathews, J., Benz, A., Fu, T., Zorumski, E., Steinbach, J. H., Covey, D. F., Zorumski, C. F., and Mennerick, S. (2002). 3β-hydroxypregnane steroids are pregnenolone sulfate-like GABAA receptor antagonists. J. Neurosci. 22, 3366–3375.
Wayne, S., Neuhouser, M. L., Ulrich, C. M., Koprowski, C., Wiggins, C., Baumgartner, K. B., Bernstein, L., Baumgartner, R. N., Gilliland, F., McTiernan, A., and Ballard-Barbash, R. (2008). Association between alcohol intake and serum sex hormones and peptides differs by tamoxifen use in breast cancer survivors. Cancer Epidemiol. Biomarkers Prev. 17, 3224–3332.
Wei, W., Faria, L. C., and Mody, I. (2004). Low ethanol concentrations selectively augment the tonic inhibition mediated by δ subunit-containing GABAA receptors in hippocampal neurons. J. Neurosci. 24, 8379–8382.
Weitzman, E. D., Fukushima, D., Nogeire, C., Roffwarg, H., Gallagher, T. F., and Hellman, L. (1971). Twenty-four hour pattern of the episodic secretion of cortisol in normal subjects. J. Clin. Endocrinol. Metab. 33, 14–22.
Williams, D. L., MacLean, A. W., and Cairns, J. (1983). Dose-response effects of ethanol on the sleep of young women. J. Stud. Alcohol 44, 515–523.
Wisden, W., Laurie, D. J., Monyer, H., and Seeburg, P. H. (1992). The distribution of 13 GABAA receptor subunit mRNAs in the rat brain. I. Telencephalon, diencephalon, mesencephalon. J. Neurosci. 12, 1040–1062.
Wohlfarth, K. M., Bianchi, M. T., and Macdonald, R. L. (2002). Enhanced neurosteroid potentiation of ternary GABA(A) receptors containing the delta subunit. J. Neurosci. 22, 1541–1549.
Woolfrey, K. M., Musisca, N. J., Hunt, P. S., and Burk, J. A. (2005). Early postnatal ethanol administration does not affect prepulse inhibition in rats. Physiol. Behav. 84, 747–752.
Wu, F. S., Gibbs, T. T., and Farb, D. H. (1991). Pregnenolone sulfate: a positive allosteric modulator at the N-methyl-D-aspartate receptor. Mol. Pharmacol. 40, 333–336.
Zamudio-Bulcock, P. A., and Valenzuela, C. F. (2011). Pregnenolone sulfate increases glutamate release at neonatal climbing fiber-to-purkinje cell synapses. Neuroscience 175, 24–36.
Keywords: ethanol, neurosteroids, behavioral pharmacology, drug discrimination, GABAA receptors
Citation: Helms CM, Rossi DJ and Grant KA (2012) Neurosteroid influences on sensitivity to ethanol. Front. Endocrin. 3:10. doi: 10.3389/fendo.2012.00010
Received: 29 September 2011; Paper pending published: 30 October 2011;
Accepted: 11 January 2012; Published online: 31 January 2012.
Edited by:
Hubert Vaudry, University of Rouen, FranceReviewed by:
Charlotte A. Cornil, University of Liege, BelgiumGiovanni Biggio, University of Cagliari, Italy
Copyright: © 2012 Helms, Rossi and Grant. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Christa M. Helms, Division of Neuroscience, Oregon National Primate Research Center, L-584, 505 North-West 185th Avenue, Beaverton, OR 97006, USA. e-mail:aGVsbXNjQG9oc3UuZWR1