
 Tsuyoshi Miyahara
Tsuyoshi Miyahara- Department of Pharmacotherapy, Graduate School of Biomedical Sciences, Hiroshima University, Minami-ku, Hiroshima, Japan
Increasing evidence indicates that endoplasmic reticulum stress (ER stress) is involved in the development of metabolic syndrome. However, pharmacological treatments targeting ER stress are not well understood. In the present study, we found that fluvoxamine, a selective serotonin reuptake inhibitor used for depression, can attenuate ER stress-induced “leptin resistance,” i.e., insensitivity to the anti-obesity hormone leptin. Treatment with tunicamycin, an ER stress-inducing reagent, caused cell death which was significantly inhibited by fluvoxamine. Leptin activates JAK2–STAT3 signaling. ER stress caused an impairment of leptin-induced STAT3 phosphorylation which was reversed by fluvoxamine. Fluvoxamine would be a novel leptin-sensitizing drug, which targets ER stress.
Introduction
Obesity is associated with Type 2 diabetes, cardiovascular disease, and hypertension. However, the molecular mechanisms and pharmacological treatment of obesity are not well understood. Leptin is an anti-obesity hormone, identified in 1994 (Zhang et al., 1994). As it has an inhibitory effect on appetite, and stimulatory effect on energy metabolism, leptin was initially expected to be useful for treating obesity. However, recent evidence suggests that most obese patients are in a state of “leptin resistance,” and cannot adequately respond to circulating levels of leptin (Münzberg and Myers, 2005). These observations have led to the notion that “leptin resistance” is a major cause of obesity and elucidating the mechanisms responsible would be useful for treating obesity. We and others have recently proposed endoplasmic reticulum stress (ER stress) to be involved in the development of leptin resistance (Hosoi et al., 2008; Zhang et al., 2008; Hosoi and Ozawa, 2009, 2010; Milanski et al., 2009; Özcan et al., 2009; Won et al., 2009). The ER is involved in maintaining Ca2+ homeostasis, synthesizing lipids and steroids, and correcting the folding of newly made proteins. However, stress signals, which impair ER function, can lead to the accumulation of unfolded proteins, resulting in ER stress. In recent years, the involvement of ER stress in metabolic syndrome has been suggested. However, compounds targeting ER stress are an enigma. One interesting report found that the sigma-1 receptor (Sig-1R) can counteract the ER stress response, and that decreasing receptor numbers enhances apoptosis (Hayashi and Su, 2007). Sig-1R is an ER protein, first cloned in 1996 (Hanner et al., 1996). Sig-1R has been shown to play an important role in biological as well as pathological processes in the brain of patients with Alzheimer’s disease, depression, and stroke (Maurice and Su, 2009; Fishback et al., 2010). Interestingly, fluvoxamine, a selective serotonin reuptake inhibitor used to treat depression, has affinity forSig-1R (Narita et al., 1996). Moreover, fluvoxamine potentiated nerve growth factor-induced neurite outgrowth through Sig-1R (Takebayashi et al., 2002; Nishimura et al., 2008). These results suggest that fluvoxamine can reduce ER stress through Sig-1R. We hypothesized that fluvoxamine can reduce ER stress and the subsequent development of “leptin resistance.”
Materials and Methods
Materials and Reagents
Tunicamycin was obtained from Wako Pure Chemical Industries, Ltd. (Japan). Leptin and fluvoxamine were obtained from SIGMA (St. Louis, MO, USA).
Cell Culture
Human neuroblastoma SH-SY5Y cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% (v/v) heat-inactivated fetal calf serum at 37°C in humidified 5% CO2 and 95% air.
Generation of Ob–Rb Leptin Receptor-Transfected Cells
The human Ob–Rb leptin receptor construct, a gift from Genentech Inc., was transfected into SH-SY5Y and HEK293 cell using Lipofectamine Plus reagent (Life Technologies Inc.) according to the manufacturer’s instructions. Stable transfectants (SH-SY5Y–Ob–Rb and HEK293–Ob–Rb cells) were obtained by selection with the antibiotic G418 (Hosoi et al., 2006).
Western Blot Analysis
Western blotting was performed as described previously (Hosoi et al., 2010a,b). Cells were washed with ice-cold PBS and lysed in a buffer containing 10 mM HEPES–NaOH (pH 7.5), 150 mM NaCl, 1 mM EGTA, 1 mM Na3VO4, 10 mM NaF, 10 μg/mL aprotinin, 10 μg/mL leupeptin, 1 mM phenylmethylsulfonyl fluoride (PMSF), and 1% NP-40 for 20 min. The lysate was centrifuged at 15,000 rpm for 20 min at 4°C, and the supernatant was collected. The samples were boiled with laemmli buffer for 3 min, fractionated by sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred at 4°C to nitrocellulose membranes. The membranes were incubated with anti-phospho STAT3 (Tyr705: Cell Signaling; 1:1,000), anti-STAT3 (Santa Cruz; 1:1,000), anti-KDEL (StressGen; 1:1,000), and anti-PARP (Santa Cruz; diluted to 1:1,000) antibodies followed by an anti-horseradish peroxidase-linked antibody. Peroxidase was detected by chemiluminescence using an enhanced chemiluminescence system.
RNAi Experiment
Transient transfections of siRNA were performed in HEK293-Ob–Rb cells. Lipofectamine RNAiMAX (Life technologies) was used to transfect siRNA according to the manufacturer’s directions. Opti-MEM1 medium was used for the transfection and the final concentrations of siRNA were 50 nM. We knocked down Sig-1R using the following siRNA sequence: human Sig-1R; 5′-CUC ACU AAC UGA GGC CUU UdTdT-3′. We used MISSION siRNA Universal Negative Control (SIGMA; SIC-001) for the control siRNA transfection. Cells were harvested 72 h after the transfection.
Lactate Dehydrogenase Leakage Assay
The viability of cells was estimated by the lactate dehydrogenase (LDH) leakage method using a cytotoxicity detection kit (Roche Molecular Biochemicals, Indianapolis, IN, USA) according to the manufacturer’s directions. LDH activity was measured as the optimal density at 492 nm.
Animals
ob/ob Mice were obtained from Japan SLC (Hamamatsu, Japan). Mice were maintained in a room at 22–24°C under a constant day-night rhythm and given food and water, ad libitum. All animal experiments were carried out in accordance with the NIH Guide for Care and Use of Laboratory Animals and approved by the animal care and use committee at Hiroshima University.
Measuring Food Intake
Nine weeks old female ob/ob mice were housed individually before this experiment. Three days after the isolation, saline (5 ml/kg) as a dummy was injected intraperitoneally (i.p.) for 3 days, and then we injected leptin (1 mg/kg, i.p.) and/or fluvoxamine (20 mg/kg). All treatments were started at 18:00. Measuring cumulative food intake was performed by measuring the weight of food at the specific time points.
Statistics
Results are expressed as the mean ± SE. The statistical analysis was performed using Student’s t-test.
Results
Fluvoxamine Attenuated Endoplasmic Reticulum Stress-Induced Cell Death
As fluvoxamine has affinity for Sig-1R (Narita et al., 1996), and Sig-1R is involved in ER stress (Hayashi and Su, 2007), we analyzed whether fluvoxamine could attenuate ER stress. Tunicamycin (Tm) was used to specifically induce ER stress. As shown in Figure 1, we observed an increase in GRP78 in tunicamycin-treated cells, indicating ER stress. In addition, tunicamycin treatment caused PARP cleavage (Figure 1) and cell death as assessed by the LDH leakage assay (Figure 2). The results suggest that tunicamycin induces ER stress-evoked apoptosis in human neuroblastoma SH-SY5Y cells. To evaluate the pharmacological properties of fluvoxamine, we next analyzed its effect on ER stress-induced cell death. Cells were pre-treated with fluvoxamine for 30 min and ER stress (tunicamycin)-induced cell death was analyzed. Treatment with fluvoxamine alone did not affect cell death (Figure 2). However, fluvoxamine dose-dependently attenuated ER stress-induced cell death (Figure 2). These results suggest that fluvoxamine can attenuate ER stress.
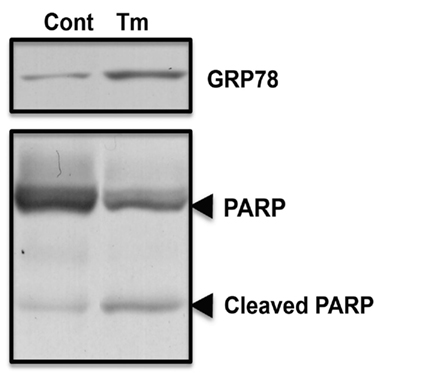
Figure 1. Specific inducer of ER stress, tunicamycin, evoked GRP78 expression, and PARP cleavage. SH-SY5Y cells were stimulated with tunicamycin (Tm: 0.5 μg/mL) for 24 h. Levels of GRP78 expression and PARP cleavage were analyzed by Western blotting.
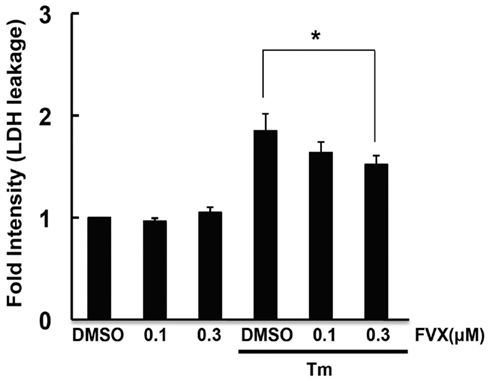
Figure 2. Fluvoxamine attenuated ER stress-induced cell death. SH-SY5Y cells were pre-incubated with fluvoxamine (FVX: 0.1 μ M, 0.3 μ M) for 1 h and then stimulated with tunicamycin (Tm: 0.5 μg/mL) for 36 h. LDH activity was measured as an indicator of cytotoxicity. *P < 0.05.
Fluvoxamine Attenuated Endoplasmic Reticulum Stress-Induced Leptin Resistance
We and others had recently reported the involvement of ER stress in leptin resistance (Hosoi et al., 2008; Zhang et al., 2008; Milanski et al., 2009; Özcan et al., 2009; Won et al., 2009). As fluvoxamine attenuated ER stress, we next examined whether it can reduce ER stress-induced leptin resistance. Consistent with a previous report (Hosoi et al., 2008), leptin-induced STAT3 phosphorylation was inhibited in ER stressed SH-SY5Y cells stably transfected with the Ob–Rb leptin receptor (Figure 3). Thus, we next analyzed whether fluvoxamine could reverse the ER stress-induced impairment of leptin’s signaling. As shown in Figure 3, we observed a dose-dependent recovery of STAT3 phosphorylation in fluvoxamine-treated cells. Furthermore, we found that knocking down endogenous Sig-1R by siRNA reduced leptin-induced STAT3 phosphorylation (Figure 4). The results suggest that Sig-1R might be involved in downstream signal of leptin’s action. Overall, these results suggest that fluvoxamine could attenuate leptin resistance.
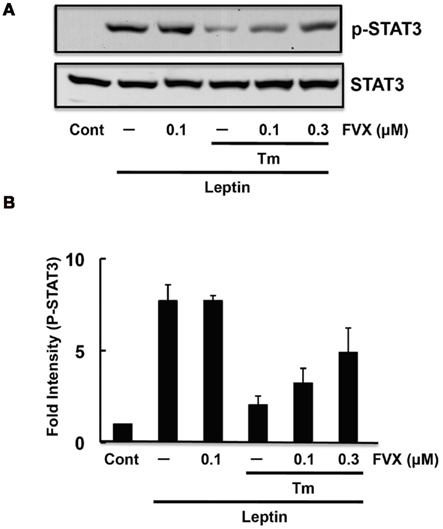
Figure 3. Fluvoxamine restored ER stress-induced leptin resistance. (A) SH-SY5Y–Ob–Rb cells were pre-treated with fluvoxamine (FVX: 0.1 μM) for 1 h and then treated with tunicamycin (Tm: 0.5 μg/mL). Four hours after the tunicamycin treatment, the cells were stimulated with leptin (Lep: 0.5 μg/mL) for 15 min and the phosphorylation status of STAT3 was analyzed. Western blotting was performed using specific antibodies for phospho STAT3 and STAT3. (B) Densitometric analysis of STAT3 phosphorylation using image analyzing software.
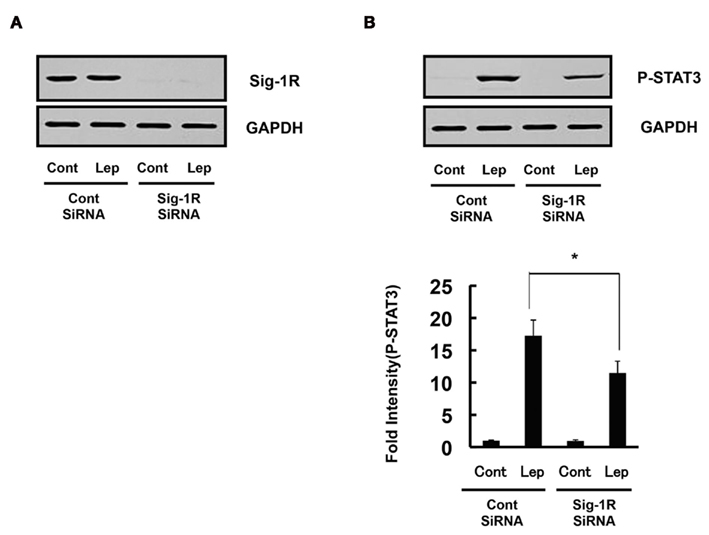
Figure 4. Knocking down sigma-1 receptor attenuated leptin’s signal. (A) Western blot analysis of endogenous sigma-1 receptor (Sig-1R) expression in lysates of HEK293-Ob–Rb cells transfected with siRNA (50 nM) directed at Sig-1R or the control sequence. Sig-1R siRNA reduced the expression of Sig-1R compared with control siRNA. (B) Sig-1R siRNA decreased leptin-induced STAT3 phosphorylation compared with control siRNA. HEK293-Ob–Rb cells were transfected with 50 nM siRNA and treated with leptin (0.5 μg/mL) for 15 min. Western blot analysis was then performed. *P < 0.05 compared with control siRNA.
To further evaluate the effect of fluvoxamine on anti-obesity property of leptin’s action, we next investigated in vivo function of this drug. In the present study, we used ob/ob mice to evaluate the pharmacological action. The mouse is used for obesity model, which has no functional leptin due to the mutation of ob gene. Moreover, the mouse has been shown to cause ER stress (Özcan et al., 2004). Therefore, we investigated whether fluvoxamine can enhance leptin’s action by measuring food intake. Treatment with leptin (1 mg/kg/day) reduced food intake. On the other hand, treatment with fluvoxamine alone slightly increased food intake (Saline vs. fluvoxamine: 30.3 ± 1.7 vs. 37.5 ± 2.8 g, cumulative food intake of 6 days, respectively). However, efficiency of leptin’s action in inhibiting food intake was significantly decreased in fluvoxamine + leptin-treated mice compared with leptin-treatment alone (Figure 5). Therefore, these results suggest that fluvoxamine can improve leptin’s action in ob/ob mice model.
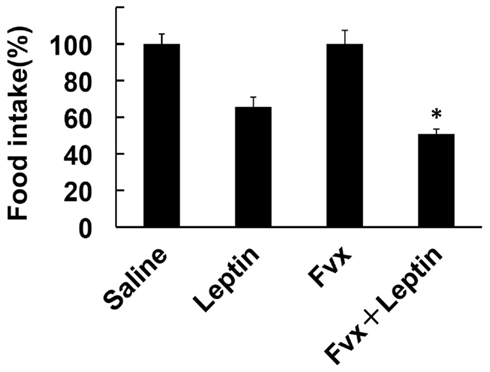
Figure 5. Effect of fluvoxamine on leptin-induced food intake. ob/ob Mice were treated with fluvoxamine (Fvx) along with leptin and analyzed food intake. The effect of leptin on food intake were presented as percentage of saline vs. leptin or fluvoxamine vs. fluvoxamine + leptin. Fluvoxamine significantly enhanced the efficiency of the leptin’s action on inhibiting food intake. *P < 0.05 compared with leptin-treatment.
Discussion
In recent years, obesity has become a serious health concern in industrialized countries. It is thus important to evaluate the mechanisms and pharmacological treatment of obesity, although effective treatments have yet to be established. In the present study, we discovered a pharmacological strategy targeting “leptin resistance,” a major cause of obesity. We found that fluvoxamine attenuated leptin resistance caused by ER stress. Using ob/ob mice as obesity model, we found that fluvoxamine can slightly increase responsiveness of leptin’s action. Although the pharmacological property of fluvoxamine would be weak, our finding would point another novel type of anti-obesity drugs, which would be a lead compound for the more efficient treatments.
Fluvoxamine would affect leptin resistance by attenuating ER stress, because it inhibited ER stress-induced cell death. Fluvoxamine is a selective serotonin (5-HT: 5-hydroxtryptamine) reuptake inhibitor (SSRI) used for depression. The involvement of serotonin in the inhibition of obesity has been suggested in studies using several agents which activate the brain’s serotonin system (Guy-Grand et al., 1989; Jackson et al., 1997; Lucas et al., 1998). Fluvoxamine attenuated food intake in food-restricted hyperphagic rats (Shinozaki et al., 2008) and during rebound hyperphagia induced by a time-restricted feeding schedule in rats (Inoue et al., 1997). These pharmacological effects of fluvoxamine would be mediated through the serotonin system. However, in addition to its pharmacological effect as a SSRI, fluvoxamine possesses high affinity for Sig-1R, an ER protein involved in ER stress (Hayashi and Su, 2007). We found that fluvoxamine can attenuate ER stress. Thus, fluvoxamine would have a novel pharmacological action, targeting ER stress-induced leptin resistance. At present, the mechanisms how fluvoxamine attenuated ER stress and leptin resistance are enigma but our results suggest it would be mediated through Sig-1R because knocking down Sig-R1 reduced leptin-induced signal. Interestingly, Sig-R1 is expressed in several hypothalamic areas involved in food intake such as paraventricular and arcuate nuclei (Alonso et al., 2000). Therefore, it is of interest to examine whether there exist a physiological link between Sig-R1 and leptin’s action. It is unknown whether physiological function of hypothalamic Sig-R1 is impaired in obesity, which would need future analysis.
Interestingly, clinical studies indicate a possible association between obesity and depression (Faith et al., 2002; Stunkard et al., 2003). It has been reported that obese patients are more likely to have depressive disorders than non-obese people (Simon et al., 2006). Considering the link between leptin and depression, and that leptin has an anti-depressive effect in mice models (Lu et al., 2006; Lu, 2007), it is possible that depression is linked with leptin resistance, and the pharmacological effects of fluvoxamine in attenuating leptin resistance would contribute to the therapeutic action against depression.
Conclusion
Our observation suggest that fluvoxamine has unique pharmacological properties in attenuating leptin resistance. It is of interest to investigate whether fluvoxamine can also attenuate leptin resistance evoked by physiological stimuli-induced leptin resistance. Clearly, further studies are required to determine whether the fluvoxamine has leptin-sensitizing effect in human levels to conclude our hypothesis.
Our present study suggest that fluvoxamine would contribute to attenuate pathophysiology of leptin resistance, which may provide basic information for treating obesity as well as depressive disorders.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by Grants-in-aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology, Japan; Takeda Science Foundation; Nestle Nutrition Council; Tokyo Biochemical Research Foundation; and The Nakajima Foundation.
References
Alonso, G., Phan, V., Guillemain, I., Saunier, M., Legrand, A., Anoal, M., and Maurice, T. (2000). Immunocytochemical localization of the sigma(1) receptor in the adult rat central nervous system. Neuroscience 9 7, 155–170.
Faith, M. S., Matz, P. E., and Jorge, M. A. (2002). Obesity-depression associations in the population. J. Psychosom. Res. 53, 935–942.
Fishback, J. A., Robson, M. J., Xu, Y. T., and Matsumoto, R. R. (2010). Sigma receptors: potential targets for a new class of antidepressant drug. Pharmacol. Ther. 127, 271–282.
Guy-Grand, B., Apfelbaum, M., Crepaldi, G., Gries, A., Lefebvre, P., and Turner, P. (1989). International trial of long-term dexfenfluramine in obesity. Lancet 2, 1142–1145.
Hanner, M., Moebius, F. F., Flandorfer, A., Knaus, H. G., Striessnig, J., Kempner, E., and Glossmann, H. (1996). Purification, molecular cloning, and expression of the mammalian sigma1-binding site. Proc. Natl. Acad. Sci. U.S.A. 93, 8072–8077.
Hayashi, T., and Su, T. P. (2007). Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 131, 596–610.
Hosoi, T., Kume, A., Otani, K., Oba, T., and Ozawa, K. (2010a). A unique modulator of endoplasmic reticulum stress-signalling pathways: the novel pharmacological properties of amiloride in glial cells. Br. J. Pharmacol. 159, 428–437.
Hosoi, T., Matsunami, N., Nagahama, T., Okuma, Y., Ozawa, K., Takizawa, T., and Nomura, Y. (2006). 2-Aminopurine inhibits leptin receptor signal transduction. Eur. J. Pharmacol. 553, 61–66.
Hosoi, T., and Ozawa, K. (2009). Possible involvement of endoplasmic reticulum stress in obesity associated with leptin resistance. J. Med. Invest. 56, 296–298.
Hosoi, T., and Ozawa, K. (2010). Endoplasmic reticulum stress in disease: mechanisms and therapeutic opportunities. Clin. Sci. (Lond). 1180.118, 19–29.
Hosoi, T., Sasaki, M., Miyahara, T., Hashimoto, C., Matsuo, S., Yoshii, M., and Ozawa, K. (2008). Endoplasmic reticulum stress induces leptin resistance. Mol. Pharmacol. 74, 1610–1619.
Hosoi, T., Tamubo, T., Horie, N., Okuma, Y., Nomura, Y., and Ozawa, K. (2010b). TEK/Tie2 is a novel gene involved in endoplasmic reticulum stress. J. Pharmacol. Sci. 114, 230–233.
Inoue, K., Kiriike, N., Fujisaki, Y., Kurioka, M., and Yamagami, S. (1997). Effects of fluvoxamine on food intake during rebound hyperphagia in rats. Physiol. Behav. 61, 603–608.
Jackson, H. C., Needham, A. M., Hutchins, L. J., Mazurkiewicz, S. E., and Heal, D. J. (1997). Comparison of the effects of sibutramine and other monoamine reuptake inhibitors on food intake in the rat. Br. J. Pharmacol. 121, 1758–1762.
Lu, X. Y. (2007). The leptin hypothesis of depression: a potential link between mood disorders and obesity? Curr. Opin. Pharmacol. 7, 648–652.
Lu, X. Y., Kim, C. S., Frazer, A., and Zhang, W. (2006). Leptin: a potential novel antidepressant. Proc. Natl. Acad. Sci. U.S.A. 103, 1593–1598.
Lucas, J. J., Yamamoto, A., Scearce-Levie, K., Saudou, F., and Hen, R. (1998). Absence of fenfluramine-induced anorexia and reduced c-Fos induction in the hypothalamus and central amygdaloid complex of serotonin 1B receptor knock-out mice. J. Neurosci. 18, 5537–5544.
Maurice, T., and Su, T. P. (2009). The pharmacology of sigma-1 receptors. Pharmacol. Ther. 124, 195–206.
Milanski, M., Degasperi, G., Coope, A., Morari, J., Denis, R., Cintra, D. E., Tsukumo, D. M., Anhe, G., Amaral, M. E., Takahashi, H. K., Curi, R., Oliveira, H. C., Carvalheira, J. B., Bordin, S., Saad, M. J., and Velloso, L. A. (2009). Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. J. Neurosci. 29, 359–370.
Münzberg, H., and Myers, M. G. Jr. (2005). Molecular and anatomical determinants of central leptin resistance. Nat. Neurosci. 8, 566–570.
Narita, N., Hashimoto, K., Tomitaka, S., and Minabe, Y. (1996). Interactions of selective serotonin reuptake inhibitors with subtypes of sigma receptors in rat brain. Eur. J. Pharmacol. 307, 117–119.
Nishimura, T., Ishima, T., Iyo, M., and Hashimoto, K. (2008). Potentiation of nerve growth factor-induced neurite outgrowth by fluvoxamine: role of sigma-1 receptors, IP3 receptors and cellular signaling pathways. PLoS ONE 3, e2558. doi:10.1371/journal.pone.0002558
Özcan, L., Ergin, A. S., Lu, A., Chung, J., Sarkar, S., Nie, D., Myers, M. G. Jr., and Özcan, U. (2009). Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 9, 35–51.
Özcan, U., Cao, Q., Yilmaz, E., Lee, A. H., Iwakoshi, N. N., Ozdelen, E., Tuncman, G., Görgün, C., Glimcher, L. H., and Hotamisligil, G. S. (2004). Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306, 457–461.
Shinozaki, T., Kimura, M., Hosoyamada, M., and Shibasaki, T. (2008). Fluvoxamine inhibits weight gain and food intake in food restricted hyperphagic Wistar rats. Biol. Pharm. Bull. 31, 2250–2254.
Simon, G. E., Von Korff, M., Saunders, K., Miglioretti, D. L., Crane, P. K., van Belle, G., and Kessler, R. C. (2006). Association between obesity and psychiatric disorders in the US adult population. Arch. Gen. Psychiatry 63, 824–830.
Stunkard, A. J., Faith, M. S., and Allison, K. C. (2003). Depression and obesity. Biol. Psychiatry 54, 330–337.
Takebayashi, M., Hayashi, T., and Su, T. P. (2002). Nerve growth factor-induced neurite sprouting in PC12 cells involves sigma-1 receptors: implications for antidepressants. J. Pharmacol. Exp. Ther. 303, 1227–1237.
Won, J. C., Jang, P. G., Namkoong, C., Koh, E. H., Kim, S. K., Park, J. Y., Lee, K. U., and Kim, M. S. (2009). Central administration of an endoplasmic reticulum stress inducer inhibits the anorexigenic effects of leptin and insulin. Obesity (Silver Spring) 170.17, 1861–1865.
Zhang, X., Zhang, G., Zhang, H., Karin, M., Bai, H., and Cai, D. (2008). Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 135, 61–73.
Keywords: leptin, fluvoxamine, endoplasmic reticulum stress, STAT3, obesity, depression
Citation: Hosoi T, Miyahara T, Kayano T, Yokoyama S and Ozawa K (2012) Fluvoxamine attenuated endoplasmic reticulum stress-induced leptin resistance. Front. Endocrin. 3:12. doi: 10.3389/fendo.2012.00012
Received: 16 April 2011;
Accepted: 12 January 2012;
Published online: 30 January 2012.
Edited by:
Arthur Donny Strosberg, The Scripps Research Organization, USAReviewed by:
Donghai Wu, Guangzhou Institute of Biomedicine and Health, ChinaTimo Dirk Mueller, University of Cincinnati Metabolic Diseases Institute, USA
Copyright: © 2012 Hosoi, Miyahara, Kayano, Yokoyama and Ozawa. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Toru Hosoi and Koichiro Ozawa, Department of Pharmacotherapy, Graduate School of Biomedical Sciences, Hiroshima University, 1-2-3 Kasumi, Minami-ku, Hiroshima 734-8553, Japan. e-mail:dG9ydWhAaGlyb3NoaW1hLXUuYWMuanA=;b3phd2FrQGhpcm9zaGltYS11LmFjLmpw