

- Instituto de Investigaciones Biomédicas “Alberto Sols”, Consejo Superior de Investigaciones Científicas, y Universidad Autónoma de Madrid, Madrid, Spain
Aberrant activation of Wnt signaling is involved in the development of several epithelial tumors. Wnt signaling includes two major types of pathways: (i) the canonical or Wnt/β-catenin pathway; and (ii) the non-canonical pathways, which do not involve β-catenin stabilization. Among these pathways, the Wnt/β-catenin pathway has received most attention during the past years for its critical role in cancer. A number of publications emphasize the role of the Wnt/β-catenin pathway in thyroid cancer. This pathway plays a crucial role in development and epithelial renewal, and components such as β-catenin and Axin are often mutated in thyroid cancer. Although it is accepted that altered Wnt signaling is a late event in thyroid cell transformation that affects anaplastic thyroid tumors, recent data suggest that it is also altered in papillary thyroid carcinoma (PTC) with RET/PTC mutations. Therefore, the purpose of this review is to summarize the main relevant data of Wnt signaling in thyroid cancer, with special emphasis on the Wnt/β-catenin pathway.
Introduction
Follicular epithelial thyroid carcinoma is the most common endocrine malignancy, and its incidence is rapidly rising in the world. Although thyroid cancer has a very good outcome, there are still several challenges that classical clinicopathological studies have not been able to resolve (Riesco-Eizaguirre and Santisteban, 2007). The most recent advances in thyroid cancer research derive from an increased understanding of the mechanisms that regulate thyroid cell differentiation and proliferation and the signal transduction pathways involved.
Differentiated thyroid cells form follicular structures surrounding a lumen and express a series of thyroid-specific transcription factors as well as other specific proteins (reviewed in De Felice and Di Lauro, 2004; Santisteban and Bernal, 2005). Thyroid proliferation and differentiation are finely regulated by Thyrotropin (thyroid stimulating hormone, TSH) that, after binding to its receptor (TSHR, a seven-transmembrane receptor coupling to G proteins), activates the cAMP/PKA cascade (Vassart and Dumont, 1992). Recently our group has shown that TSH can also act via PI3K/Akt (Zaballos et al., 2008). In addition, increasing evidence supports a role for other kinases, such as MAP kinase (MAPK), which are activated in response to TSH and lie downstream of cAMP (Dumaz and Marais, 2005).
The differentiated thyroid epithelial phenotype is characterized by structural and functional polarization of the cell surface into apical and basolateral domains, and the formation of junction complexes that mediate strong and tight intercellular adhesion. Cell polarization plays a central role in maintaining follicular structures and its alteration occurs in parallel with cell transformation. The presence of tight junctions in thyroid epithelial cells is thought to be essential for the integrity and maintenance of the follicular structure. Several molecules are involved in this process; of these, the occludins, claudins, and zonula occludens (ZO) proteins have been most intensively studied, as they play an important role in the maintenance of the epithelial cell phenotype. As occurs with others epithelial markers, their expression is reduced in thyroid cancer progression (Tzelepi et al., 2008). On the other hand, adherens junctions are mainly composed of cadherins. Among them, E-cadherin and Cadherin 16 are highly expressed in the thyroid (Cali et al., 2012). Both are members of the large superfamily of adhesion molecules and play a critical role in the establishment of cell polarity and firm contacts.
Adherens junctions have a similar structural organization as tight junctions: E-cadherins and Cadherin 16 form contacts with the catenins (α, β, and γ) and αB-crystallin respectively, and these latter proteins connect with the cystokeleton (Cali et al., 2012). The follicular structure is maintained by a series of proteins through which TSH and intercellular contact regulate adhesion of follicular cells to each other and to the extracellular matrix, and influence thyroid cell behavior. The extracellular matrix plays a role in the adhesion, proliferation, differentiation, and migration of thyroid follicular cells.
It has been reported that downregulation of cadherins takes place in carcinogenesis and is associated with tumor progression in a variety of human carcinomas (Strumane et al., 2004; Berx and van Roy, 2009). Therefore, decreased expression or loss of cadherins may lead to the development of epithelial tumors, invasion, and metastasis. In accordance with this, the loss of E-cadherin and Cadherin 16 expression in thyroid carcinoma has been correlated with a loss of differentiation and a poor prognosis (Brabant et al., 1993; Cali et al., 2012; de Cristofaro et al., 2012).
Thyroid Cancer Initiation and Progression
Thyroid cell proliferation requires the combined effects of TSH, acting via cAMP, and growth factors such IGF1, signaling primarily through MAPK and phosphatidylinositol-3-kinase (PI3K; Medina and Santisteban, 2000; Kimura et al., 2001). It is therefore well accepted that mutations in genes involved in these signaling pathways play prominent roles in the pathogenesis of thyroid neoplasia.
Currently there are two hypotheses to explain the onset of thyroid cancer, the classical multistep model and a theory based on cancer stem cells (CSCs).
The classical view considers thyroid carcinoma as a complication of a pre-existing follicular adenoma (FA) accumulating mutations that drive progression through a dedifferentiation process. Accordingly, a step model of thyroid carcinogenesis involving different defined types of thyroid tumor is well accepted (Figure 1). Thus, autonomously hyperfunctioning thyroid adenomas and FAs are benign tumors that rarely progress to malignancy. Papillary thyroid carcinoma (PTC) and follicular thyroid carcinoma (FTC) show follicular cell differentiation, and poorly differentiated carcinomas (PDC) have morphological and biological characteristics intermediate between well-differentiated and undifferentiated (anaplastic) thyroid carcinomas (ATC; reviewed in Riesco-Eizaguirre and Santisteban, 2007, and in Nikiforov and Nikiforova, 2011).
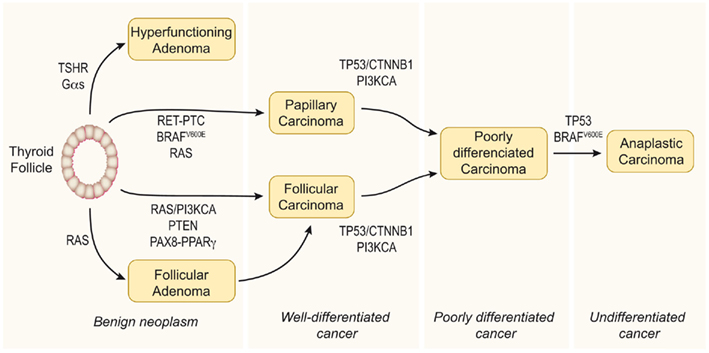
Figure 1. Step model of thyroid carcinogenesis. This model is based on histological and clinical features as well as on the degree of tumor differentiation. The well-differentiated thyroid follicular cell may give rise to both benign and malignant tumors. Autonomously hyperfunctioning thyroid adenomas are associated with activating mutations in the TSHR or Gαs genes (O’Sullivan et al., 1991; Parma et al., 1993). After gaining mutations in different oncogenes and tumor suppressor genes, the differentiated thyroid follicle can also give rise to well-differentiated papillary or follicular carcinomas, poorly differentiated carcinoma, and anaplastic carcinoma. The figure represents a schematic model showing the molecular events involved. Modified from Nikiforov and Nikiforova (2011).
The genetic events involved in tumor initiation have been identified in well-differentiated thyroid cancer. In PTC there are many data supporting a central role of mutations leading to constitutive activation of the MAPK pathway. Thus, the V600E mutation of BRAF, RET/PTC rearrangements, and RAS mutations have been clearly implicated in the pathogenesis of this disease. These three pathways are mutually exclusive and it is accepted that they are the cause of a significant proportion of PTCs. Similarly, there is increasing evidence that shows that cancer genes acting through the PI3K pathway (PI3KCA and PTEN) are involved in the pathogenesis of FTC. However, in this case more studies are needed to elucidate a possible causal effect of these genetic events. Furthermore, a PAX8/PPARγ rearrangement (Kroll et al., 2000) has been identified in a significant proportion of these tumors, although the mechanism of transformation induced by PAX8/PPARγ is still unclear.
Deregulation of the p53 pathway seems to be an important second step leading to the progression of PDC/ATC (Fagin et al., 1993; Figures 1 and 2). In addition, activating mutations in the β-catenin gene (CTNNB1) have been described in ATC that occur late in thyroid tumor progression. Therefore, until the recent demonstration that RET/PTC stimulates the β-catenin pathway in the thyroid (Cassinelli et al., 2009; Castellone et al., 2009; Tartari et al., 2011), it was believed that β-catenin was not involved in thyroid tumor initiation. Compared with the well-documented RAS/RAF/MAPK pathway, the β-catenin signal in thyroid cancer is less well understood, and therefore in this review we will focus on updating the existing data and comment on future directions of research.
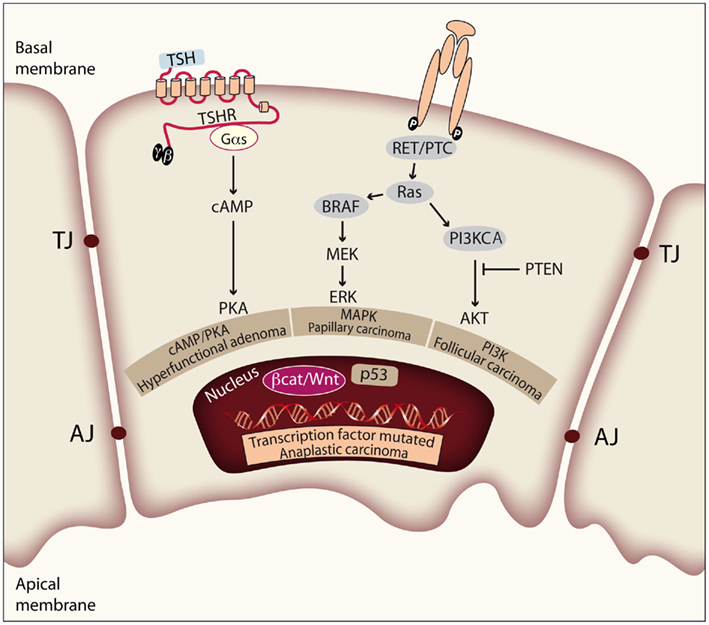
Figure 2. Genetic events involved in thyroid tumor initiation and progression. Thyroid cell proliferation depends on the combined activation of cAMP/PKA, MAPK, and PI3K pathways, induced by TSH and other growth factors. Mutations of effectors along these signaling pathways play prominent roles in the pathogenesis of thyroid neoplasia. The figure represents a thyroid epithelial cell with the basal and apical membrane, and shows the tight and adherens junctions (TJ and AJ) between cells in order to form follicular structures. Activating mutations in TSHR or Gαs lead to constitutive activation of the cAMP/PKA pathway and give rise to hyperfunctional adenomas. There is evidence that constitutive activation of the MAPK pathway is required for PTC initiation. Activation of the PI3K/Akt pathway is required for FTC initiation. Finally, deregulation of the p53 pathway and the Wnt/β-catenin pathway is observed in ATC and has more to do with tumor progression.
Recently, a new theory on the cancer initiation process was formulated, based on the existence of CSCs (Thomas et al., 2008; Lin, 2011). These CSCs are a small subpopulation (between 1 and 3.5%) of the cells with stem cell-like properties such as colony formation, self-renewal, and in vitro resistance to chemotherapy-induced apoptosis (Dick, 2008, 2009). In the case of the thyroid gland, the CSCs would derive from thyroid embryonic stem cells, intermediates in the differentiation pathway of thyroblasts or prothyrocytes, and accumulate mutations that lead to carcinogenesis. The CSC hypothesis assumes the presence of a hierarchy of embryonic cells in the thyroid gland that can give rise to different forms of thyroid cancer (reviewed in Lu et al., 2011).
The existence of CSCs in the thyroid is still a matter of controversy, but several lines of evidence support the model. Among them is the observation that it is uncommon that a benign adenoma evolves toward carcinoma; at present, it seems that most thyroid carcinomas are malignant from the onset; the original RET/PTC mutations seen in FTC and PTC are hardly seen in ATC. In addition, data from Chernobyl irradiation studies, in which the highest incidence of papillary thyroid cancer was among children rather than adults, strongly support the CSC theory. The establishment of ES cell cultures able to differentiate into thyrocytes may help clarifying these views (Arufe et al., 2006).
Wnt Signaling
Wnt proteins are a family of highly conserved secreted cysteine-rich glycoproteins, encoded by 19 genes in humans and 18 genes in mice. These proteins play important roles in embryonic development by controlling cell proliferation, cell fate specification, tissue patterning, and cell polarity. In adults, they contribute to tissue homeostasis by controlling proliferation, stem cell activation, and self-renewal. Signaling by Wnt proteins activates three different pathways: one canonical or β-catenin-dependent pathway and two non-canonical or β-catenin-independent pathways: Wnt/Ca2+ and planar cell polarity (PCP; Figure 3). The relevance of these pathways is also reflected by the fact that mutations in some elements of the Wnt pathways are often linked to human diseases like leukemia and other cancers, and type-II diabetes (Kikuchi and Yamamoto, 2008; MacDonald et al., 2009).
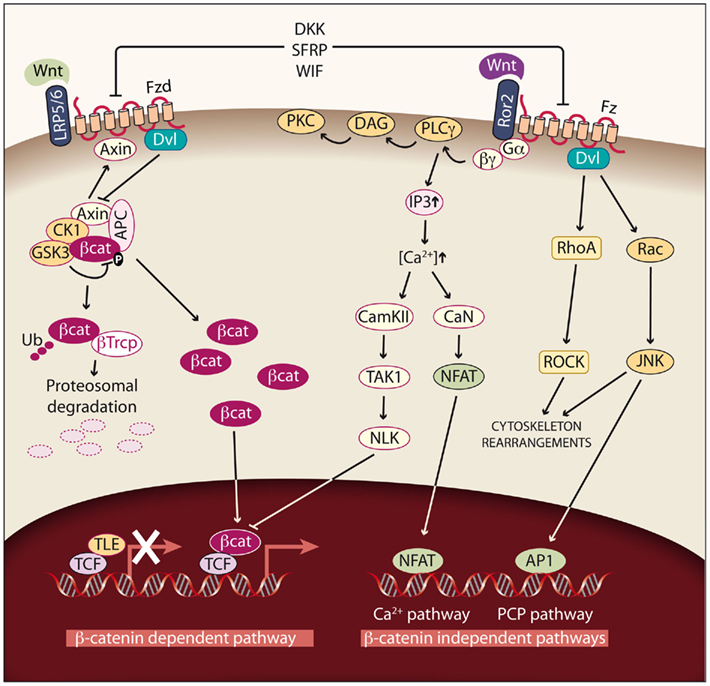
Figure 3. Wnt signaling pathways. Left: Canonical or Wnt/β-catenin dependent pathway. In cells not exposed to Wnt factors, cytoplasmic β-catenin is degraded, and TLE/Groucho proteins repress its target genes. If Wnt signaling is activated by binding of Wnt factors such as Wnt1, Wnt3, Wnt3A, Wnt7A, or Wnt10B to the Fzd receptor and LRP5/6 coreceptors, β-catenin degradation is reduced and the protein accumulates in the cytoplasm. As a consequence, β-catenin enters the nucleus, binds to TCF/LEF transcription factors and activates transcription. In this way, different processes such as proliferation are modulated. Right: Non-canonical or β-catenin-independent pathways. Binding of the non-canonical Wnt factors Wnt4, Wnt11, or Wnt5A to different Fzd receptors and the Ror2 coreceptor transduce signaling by two different pathways. The Ca2+ pathway promotes the activation of protein kinase C (PKC) via G-PLCγ and modulates cell adhesion and motility by activating calcium–calmodulin kinase (CamKII), and the phosphatase calcineurin (CaN). The planar cell polarity (PCP) pathway modulates cytoskeleton rearrangements through the activation of the small GTPases RhoA and Rac and their downstream effectors Rock and JNK.
Canonical Wnt/β-Catenin Pathway
The most extensively studied Wnt pathway is the Wnt/β-catenin pathway, due to its important role in cancer initiation and progression (Reya and Clevers, 2005; see Figure 3, left). In the absence of Wnt, β-catenin, the central component of the pathway, is localized in adherens junctions bound to E-cadherin. The unbound cytoplasmic protein is constantly degraded by a protein complex called destruction complex, which is composed of the scaffold proteins Axin and adenomatous polyposis coli (APC), and the kinases casein kinase 1 (CK1) and glycogen synthase kinase 3 beta (GSK3β; Rubinfeld et al., 1996). CK1 and GSK3β sequentially phosphorylate β-catenin, resulting in β-catenin being recognized and ubiquitinated by the β-Trcp ubiquitin ligase, followed by proteasomal degradation. With low levels of free cytoplasmic β-catenin, the transcription factors T-cell factor/lymphoid enhancer factors (TCF/LEF) function as transcriptional repressors by recruiting corepresors of the TLE/Groucho family.
The Wnt pathway is activated when a protein of the family, such as Wnt1, Wnt3, Wnt3a, Wnt7A, or Wnt10B, binds to a frizzled receptor (Fzd) and the LDL-Receptor-related protein coreceptor (LRP5/6). Formation of a Wnt–Fzd–LRP complex induces the binding of the cytoplasmic protein Disheveled (Dvl) to Fzd and the LRP phosphorylation-dependent recruitment of Axin to the membrane. Recruitment of Axin, which is the limiting component of the destruction complex, promotes the release of β-catenin, and its accumulation in cytoplasm and nuclei. In the nuclei, β-catenin displaces TLE/Groucho corepressors and recruits coactivators, activating expression of Wnt target genes. The meanings of the acronyms used for the different members of the Wnt pathway are given in Table 1; see http://www.stanford.edu/group/nusselab/cgi-bin/wnt/ for more details.
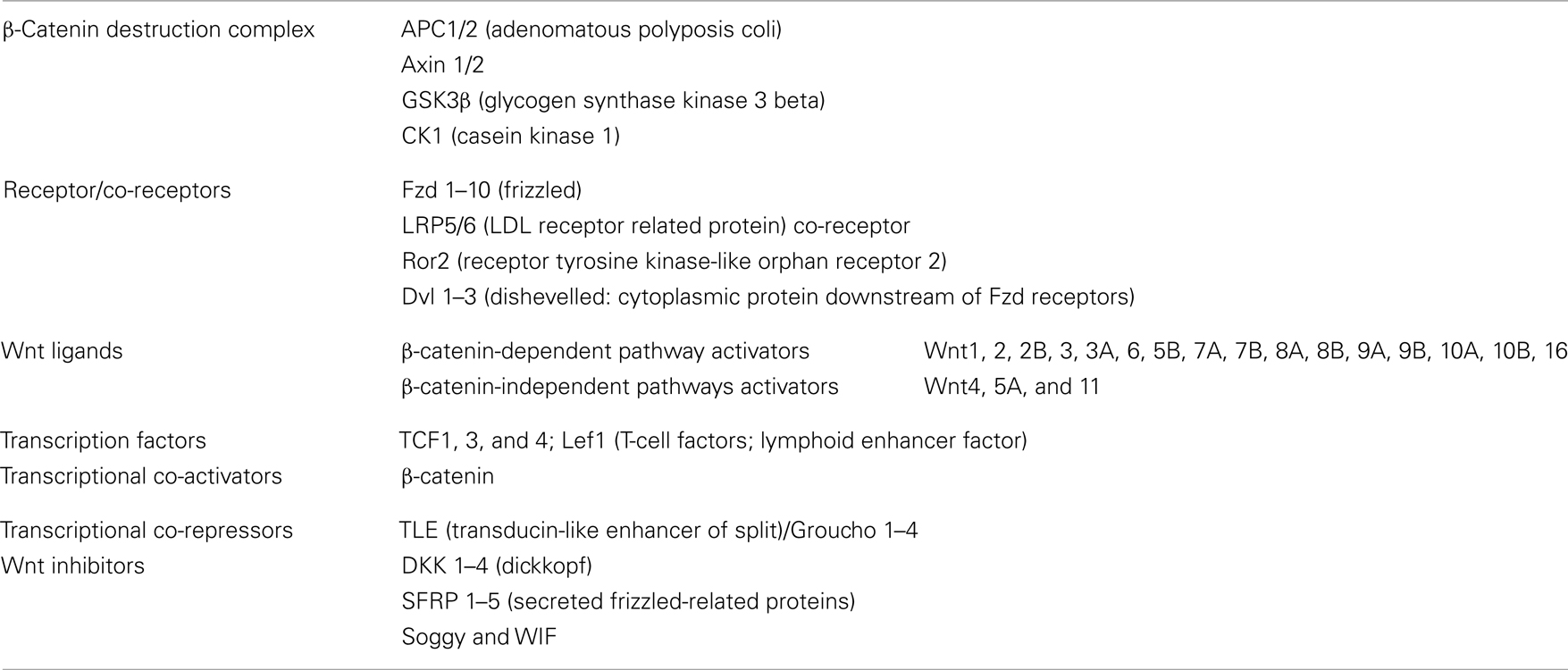
Table 1. Members of the Wnt pathway grouped according to function.
The most important genes regulated are those related to proliferation, such as Cyclin D1 and c-Myc (He et al., 1998; Tetsu and McCormick, 1999), which are over-expressed in most β-catenin-dependent tumors.
Non-Canonical Wnt Pathways
There are two main β-catenin-independent pathways (see Figure 3, right). The activation of these pathways is mediated by Wnt factors such Wnt4, Wnt11, and Wnt5A in a tissue and context specific way. The first pathway is the PCP pathway. This pathway can be triggered through several of the Fzd receptors and it activates the small GTP-binding proteins RhoA and Rac and their downstream effectors Rho-kinase and JNK. This non-canonical pathway is involved in tissue polarity, cell migration, and cytoskeleton organization. It has been described in Drosophila, and although its relevance in mammals has not been fully demonstrated, there is important evidence pointing to its existence in these vertebrates (Wada and Okamoto, 2009). The second pathway is the Ca2+ pathway, which can be activated by the interaction with two Fzd receptors (Fzd 2 and 7) or with Ror2 (receptor tyrosine kinase-like orphan receptor 2). The activation of this pathway leads to an activation of PKC and to increased intracellular Ca2+ levels, which activates CaMKII and calcineurin (CaN) and regulates cell migration and proliferation (Nishita et al., 2010).
Wnt Inhibitors
Activation of the Wnt pathway is highly regulated, and there are two families of antagonists: DKK and secreted frizzled-related proteins (SFRP). The Dickkopf (DKK) family is a group of four soluble proteins that bind to LRP and thus prevent the formation of the Wnt–Fzd complex. The SFRP are soluble glycoproteins that inhibit the Wnt pathway by sequestering Wnt proteins, thus preventing their interaction with Fzd receptors. There are other Wnt inhibitors like WIF or Soggy1 (Filipovich et al., 2011; Figure 3). The physiological role of these proteins is to inhibit the Wnt pathway in a time and tissue-specific manner both during development and in adult tissues. The silencing of DKK and SFRP genes has been described in several tumor types, contributing to the activation of the canonical pathway (Aguilera et al., 2006).
Wnt and Thyroid Cancer
Normal thyroid cells express several Fzd, Dvl, and Wnt proteins and have a functional destruction complex (Helmbrecht et al., 2001). In addition, TSH-dependent over-expression of Wnt1 (Kim et al., 2007) and inhibition of GSK3β by adenoviral-interference (Chen et al., 2010) lead to an increase of rat and human thyroid cell proliferation. These data suggest the existence of a functional Wnt pathway, relevant for the proliferation of thyroid cells.
Given the hypothetical role of this pathway in proliferation, it is not surprising that over-activation of the Wnt/β-catenin pathway promotes tumor growth in the thyroid, as in other Wnt-dependent tissues like skin or colon.
Classically, activation of the Wnt pathway in thyroid cancer has been related with ATCs, as a second mutational event involved in the progression from a well-differentiated to a poorly or undifferentiated (anaplastic) and more aggressive thyroid carcinoma. However, as commented above, recent data correlate this pathway also with early stages of thyroid carcinogenesis.
Wnt in Poorly Differentiated and Anaplastic Thyroid Carcinoma
Mutations of several components of the Wnt pathway have been described in ATC (Table 2). The most frequent mutations are those in the scaffold proteins APC and Axin, as well as in β-catenin (Polakis, 2007). In tumors such as colon tumors mutations most frequently affect the APC gene, but in the thyroid the mutations appear mainly in β-catenin and Axin.
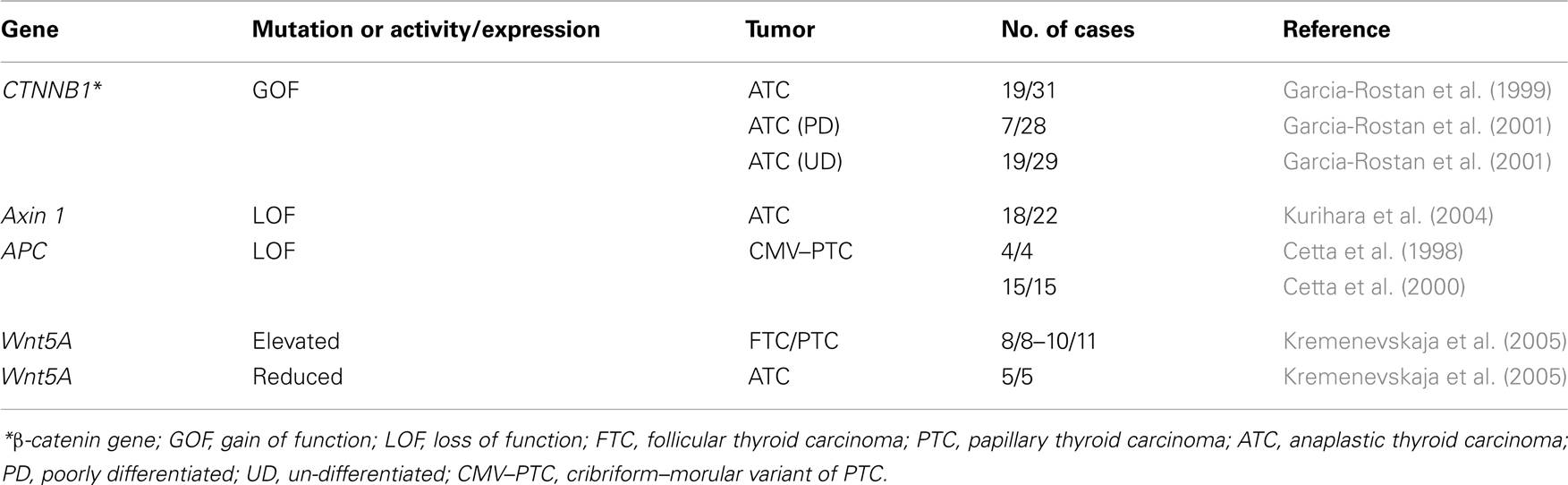
Table 2. Events linked with aberrant activation of Wnt signaling in thyroid cancer.
In ATC, three different studies found nuclear localization of β-catenin in 40–60% of the samples analyzed, highlighting the role of the Wnt pathway in this type of tumor (Garcia-Rostan et al., 1999, 2001; Kurihara et al., 2004). In these studies, performed in three different human populations, mutations in the β-catenin and Axin1 genes were described. In two of them, all β-catenin mutations found were in the conserved Ser and Thr residues phosphorylated by CK1 and GSK3β, which results in a constitutive stabilization of the protein and successive accumulation in both cytoplasm and nucleus. The presence of nuclear β-catenin correlated with a higher proliferation and a loss of tumor differentiation, and therefore with a poor prognosis (Garcia-Rostan et al., 1999, 2001). Despite the role of β-catenin in proliferation, these latter two studies together with another one (Cerrato et al., 1998) also described a percentage of anaplastic tumors in which the β-catenin expression is absent.
These data can be interpreted to mean that there are distinct subtypes of tumors in ATC with different, mutually exclusive mutations. Thus, one subtype would carry the Wnt/β-catenin pathway mutation, while the other would carry the p53 mutation (Fagin et al., 1993) or an activated PI3K/Akt pathway, e.g., through a mutation in PI3KCA (Garcia-Rostan et al., 2005); these three are the most frequent mutations found in ATC. It would be of great interest to establish whether mutations in oncogenes or tumor suppressor genes are mutually exclusive in order to better classify anaplastic thyroid tumors.
By contrast, the study by Kurihara et al. (2004) described a low percentage of β-catenin mutations, while more than 50% of the tumors carried mutations in the functional domain of Axin1. The different mutations found in the Axin1 gene affect the domains for interaction with APC, β-catenin and Dvl, and the G-protein regulatory domain, and thus affect the role of Axin as a negative regulator of β-catenin. The difference in data regarding β-catenin and Axin mutations may be due to the different genetic backgrounds of the populations analyzed.
Wnt in Well-Differentiated Thyroid Carcinoma
It is well accepted that there is a correlation between the subcellular localization of β-catenin and cancer progression (Garcia-Rostan et al., 2001). In hyperfunctioning adenomas and FTC, β-catenin is localized in the plasma membrane, as it is in the normal thyroid gland. In PTC, there is an accumulation of β-catenin in the cytoplasm, and in poorly and undifferentiated carcinoma β-catenin is translocated to the nuclei due to mutations in the β-catenin gene as well as in other genes of the Wnt pathway (Table 3). We already discussed the role of β-catenin in ATC, but the role and the molecular mechanisms that lead to the cytoplasmic stabilization of β-catenin have not been established. Some groups have correlated cytoplasmic β-catenin in PTC with higher levels of cyclin D1 (Ishigaki et al., 2002; Meirmanov et al., 2003; Rezk et al., 2004; Zhang et al., 2011) and increased proliferation. Nevertheless, at present there are no in vitro results that corroborate this hypothesis and no relationship between cytoplasmic β-catenin and transcriptional activity has been found. More studies are needed to demonstrate a direct link between cytoplasmic β-catenin and cyclin D1 expression in order to correlate both events with proliferation.
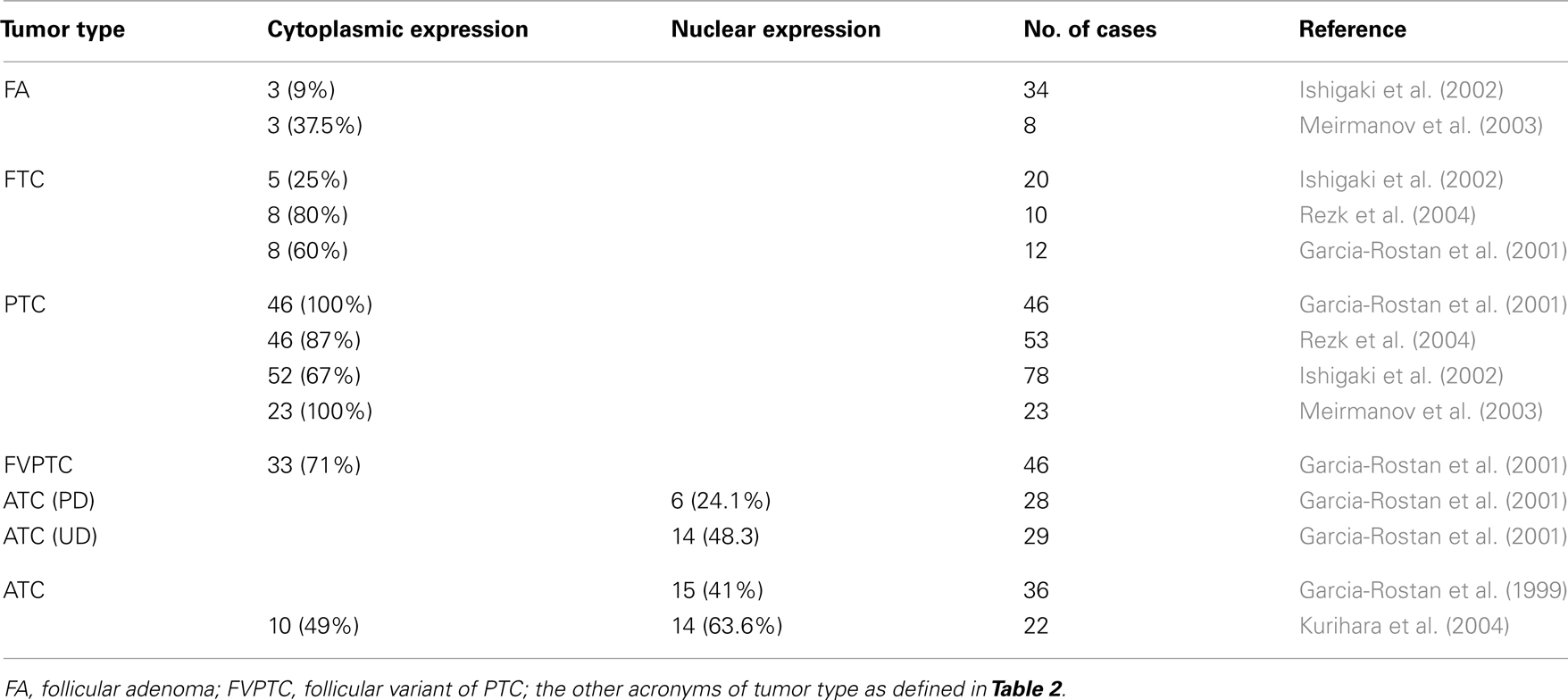
Table 3. Localization of β-catenin in thyroid carcinoma.
E-cadherin is a transmembrane protein that mediates cell–cell adhesion in a Ca2+-dependent manner. It interacts through its cytoplasmic domain with β-catenin and the actin cytoskeleton, controlling cell migration and cell polarity (Hulsken et al., 1994). In normal thyroid cells, E-cadherin is expressed in the basolateral membrane and its downregulation, by promoter methylation or by activation of oncogenes such as BRAF, has been implicated in the induction of the epithelial mesenchymal transition (EMT) in follicular, papillary, and anaplastic thyroid tumor cells (Brabant et al., 1993; Graff et al., 1998; Riesco-Eizaguirre et al., 2009). As E-cadherin keeps β-catenin bound to the cell membrane, the presence of cytoplasmic β-catenin could be merely a consequence of the loss of E-cadherin expression, although this correlation has not yet been demonstrated.
Supporting the role of β-catenin in earlier stages of tumor progression, recent publications describe the involvement of β-catenin in RET/PTC-induced proliferation (Cassinelli et al., 2009; Castellone et al., 2009; Tartari et al., 2011). These studies show a RET/PTC-dependent stabilization of β-catenin by phosphorylation of a residue outside the GSK3β Ser/Thr domain. This stabilization, together with an Akt–MAPK-dependent inhibition of GSK3β, leads to an increase of β-catenin in the nuclei where it is able to interact with the transcription factors TCF/LEF and CREB binding to the cyclin D1 promoter. In consequence, DNA synthesis and cell proliferation are induced. This is another way of Wnt/β-catenin pathway regulation, by post-translational modifications, that points to a participation of this pathway in the first steps of thyroid cell transformation. Unfortunately, the authors did not correlate their results with the localization of β-catenin in RET/PTC-carrying thyroid tumors.
Data obtained from the TRβPV/PV mouse model of FTC that harbors a dominant negative mutation (PV) of the thyroid hormone-beta receptor (TRβ), also support the notion that β-catenin could contribute to thyroid carcinogenesis (Guigon et al., 2008; Lu et al., 2011). Thus, in the FTC tumors of these mice thyroid hormone and its receptors seem to modulate the Wnt/β-catenin pathways in two ways. First, the TRβ acts as negative regulator of β-catenin in a T3-dependent manner (Guigon et al., 2008). In the absence of ligand, TRβ binds to β-catenin and stabilizes it in the cytoplasm and the nuclei allowing β-catenin to operate as a transcriptional activator promoting cell proliferation. Binding of T3 to TRβ weakens the physical interaction between β-catenin and TRβ and allows the uncomplexed β-catenin to be targeted for proteasomal degradation. In the thyroid tumors developed in the transgenic TRβPV/PV mice, β-catenin is constitutively stabilized through its binding to the mutant TRβ, because the mutated TRβ cannot bind T3. In this way, β-catenin activates the expression of its target genes cyclin D1, myc, and MT1-MMP (matrix metalloproteinase), and promotes tumor growth and progression. The second mechanism involves a non-genomic action: T3 (elevated in the TRβPV/PV mice) is able to increase the PTEN/PI3K/Akt pathway through the αvβ3 receptor, leading to the phosphorylation of β-catenin, which increases its stability and transcriptional activity and promotes cell proliferation (Lu et al., 2011). Overall, these results show a new mechanism of activation of the Wnt/β-catenin pathway, which through the stabilization of β-catenin could be promoting cancer progression in the thyroid gland.
All the above evidence points to a role of β-catenin in well-differentiated thyroid carcinomas, but more in vitro and in vivo evidence is needed and the molecular mechanism remains to be resolved.
Non-Canonical Wnt Pathways in Thyroid Carcinomas
Wnt5A is an activator of the non-canonical Wnt pathways. Due to its roles in planar polarity and cell migration and invasiveness, Wnt5A has been implicated in several human cancers, but, as occurs in normal tissues, the roles of Wnt5A in cancer are tissue and receptor specific. In osteosarcoma as well as in prostate and renal cell carcinomas, Wnt5A acting through the Ror2 receptor is involved in matrix metalloprotease expression, enhancing cell migration and invasiveness of these cells, and consequently it is a poor prognosis factor for these types of tumors. By contrast, in colon and thyroid tumors Wnt5A acts as a tumor suppressor, highlighting the variety of roles of this Wnt member (McDonald and Silver, 2009).
While Wnt5A is expressed in FA, PTC, and FTC, no expression has been detected in ATC or in the normal thyroid (Kremenevskaja et al., 2005). In well-differentiated cancer cells, Wnt5A acts as a tumor suppressor by inhibiting both Wnt/β-catenin-dependent proliferation in a Ca2+/CaMKII dependent manner, and migration and invasiveness. In this way, it promotes a mesenchymal epithelial transition (MET) by the induction of cadherin expression and the re-localization of β-catenin from the nuclei to the membrane. Wnt5A expression is lost in anaplastic carcinoma, leading to a more aggressive tumor in which proliferation, migration, and invasiveness are enhanced.
Wnt in Familial Syndromes
Sporadic mutation of the APC gene is less frequent in thyroid tumors, but there is a high frequency of PTC in several syndromes carrying APC mutations, such as familial adenomatous polyposis (FAP), Gardner’s syndrome, and Turcot’s syndrome (Soravia et al., 1999). FAP arises as a consequence of germinal mutations in one allele of the APC gene. Patients develop mainly colonic polyps and non-colonic malignancies, the most frequent being PTC. These PTCs have characteristic histological structures and are called the Cribriform–Morular Variant of PTC (CMV of PTC), because of their morula-like structure. Such structures are frequent in other tumors and correlate with nuclear β-catenin and activation of the Wnt/β-catenin pathway. Contrary to colorectal neoplasms that exhibit a loss of heterozygosity in the APC gene, the CMV of PTC maintains heterozygosity and seems to need other gene alterations to originate a tumor. RET–PTC rearrangements have been described as a frequent event in CMV PTC-FAP tumors, further suggesting that APC mutation alone is not sufficient as a tumor initiator (Cetta et al., 1998, 2000).
Mutations in the phosphorylation sites of β-catenin are also found in the CMV of PTC, where nuclear localization of β-catenin correlates with poorly or undifferentiated tumors. In one study it was suggested that a mutation in exon 3 of the β-catenin gene, CTNNB1, could be an early molecular event in the CMV of PTC, although mutations in other genes frequently altered in PTC, such as RET, RAS, or BRAF were not studied (Xu et al., 2003).
Cancer Stem Cells and the Wnt Pathway
Wnt proteins contribute to the homeostasis of several tissues of epithelial origin, like intestine and skin. This is because activation of the Wnt pathway is absolutely required for driving the stem cell/progenitor compartment. This pathway is altered in thyroid malignancies, together with other stem cell-regulating pathways such as Hedgehog and Notch signaling, which supports the CSC model (reviewed in Derwahl, 2011).
In fact, it is well accepted that the Wnt/β-catenin pathway is also necessary for the maintenance of CSCs. Thus, in colon the inappropriate activation of this pathway by APC or β-catenin gene mutations promotes the growth of tumor cells exhibiting a stem cell-like expression profile (Vermeulen et al., 2010). In breast cancer, the Wnt pathway is upregulated in CSCs by Wnt ligands secreted by the tumor microenviroment (Malanchi et al., 2011). Concerning thyroid CSCs there are still few data, but increasing evidence supports the existence of CSCs and the role of the Wnt pathway in this gland. Recent data demonstrate that CSCs from different types of thyroid carcinomas (PTC, FTC, and ATC), have different properties: CSCs isolated from ATC are the most aggressive and tumorogenic, followed by CSCs from PTC and FTC (Todaro et al., 2010). These data demonstrate that the malignancy of the CSCs correlates with the characteristics of the tumor and could reconcile the multistep process of thyroid carcinogenesis with the CSC hypothesis (Derwahl, 2011). Interestingly, the data reported by Todaro et al. showed constitutive activation of cMet, Akt, and β-catenin, together with downregulation of E-cadherin, in CSCs derived from the most undifferentiated thyroid tumors. This correlated with a higher migration capacity and metastatic rate. Although the above results are very promising, further studies are needed to evaluate the activation of the Wnt pathway and its role in CSC stemness or maintenance.
The Wnt Pathway as a Target for Thyroid Cancer Treatment
Thyroid cancer has, in general terms, a very good outcome as radioiodine treatment is a very effective therapy. However, there are still some critical challenges that the classical clinicopathological approach has not been able to solve, and in some poorly differentiated radioiodine-resistant tumors and in ATC treatment options are limited.
In the past years, new drugs such as tyrosine kinase inhibitors or MAPK inhibitors proved to be quite efficient. The findings regarding the involvement of the Wnt pathway in thyroid cancer, its crosstalk with thyrosine kinase receptors such as RET and the involvement of β-catenin and Axin in ATC suggest that this pathway may be a potential therapeutic target. Current therapies with tyrosine kinase inhibitors such as Imatinid or Vandetanib seem to work in part by inhibiting the Wnt/β-catenin pathway. Treatment with Imatinid of anaplastic human cells, positive for the tyrosine kinase c-abl, induced a decrease in cell proliferation and invasiveness by reducing nuclear β-catenin and increasing β-catenin/E-cadherin binding to the plasma membrane. Imatinid attenuated TCF activity, which in turn reduced expression of its target gene cyclin D1 leading to cell growth arrest (Rao et al., 2006). Treatment with Vandetanib of papillary TPC1 cells carrying a RET/PTC rearrangement also stabilized β-catenin in the plasma membrane, decreasing the expression of β-catenin target genes such as c-myc and cyclin D1, and decreasing cell growth and migration (Tartari et al., 2011). These results underscore the importance of Wnt pathway activation in thyroid cancer progression.
Interestingly, non-steroidal anti-inflammatory drugs such as Sundilac, which target the Wnt/β-catenin pathway, have been used in colon cancer treatment (Rice et al., 2003). Sundilac also reduces β-catenin expression, which is accompanied by a decrease in cell growth in human PTC cell lines overexpressing BRAFV600E but not RET/PTC3 (Cho et al., 2010).
Finally, a conditionally replicative adenovirus harbouring the E1A and E1B expression under the control of TCF response elements has been reported. These constructs replicate specifically in cells with an active Wnt/β-catenin pathway. This therapeutic approach has been used in xenograft tumors in nude mice developed from several thyroid cancer cell lines with good results regarding tumor size reduction, and should be further developed in the future (Abbosh et al., 2007).
Concluding Remarks and Future Perspectives
It has become evident that the Wnt pathways are involved in thyroid cancer progression.
Wnt/β-catenin-independent pathways appear to act as tumor suppressors, because downregulation of Wnt5A, an activator of the non-canonical pathways, is needed to enhance the migratory and invasive capacity of thyroid tumor cells. The Wnt/β-catenin-dependent pathway, on the other hand, seems to be involved in the proliferation of normal thyroid cells, which is a highly controlled process.
In the earliest stages of progression of tumors such as FA or well-differentiated FTC and PTC, β-catenin remains mainly attached to the cell membrane, but begins to appear in the cytoplasm. In these early stages, tumors carrying RET/PTC rearrangements proliferate in a β-catenin-dependent way. Although in cell culture this enhanced proliferation correlates with nuclear accumulation of β-catenin and high expression of the cyclin D1 and c-Myc genes, in tumor samples no such correlations were found and β-catenin was visible only in the cytoplasm. Further studies are needed to understand the correlation between cytoplasmic β-catenin, the expression of its cell cycle target genes, and the role of β-catenin in these tumors.
In poorly and undifferentiated carcinomas, β-catenin is found in the nuclei, and mutations in this gene or in other genes of the pathway such as Axin1 induce the constitutive activation of the canonical Wnt pathway, which triggers an increase in proliferation.
In conclusion, β-catenin has a direct role in the proliferation of poorly and undifferentiated thyroid tumor cells, but more studies are needed to establish the role of β-catenin in earlier stages of thyroid tumor progression.
The use of animal models like APCMin or ΔNβ-catenin mice, which have a constitutively activated Wnt/β-catenin pathway, could be of great interest for better understanding the mechanism by which the Wnt pathway promotes tumor growth in thyroid cancer.
The Wnt/β-catenin pathway is involved in the transformation of a large number of tumors, and for this reason in the past few years several groups have been looking for specific inhibitors of this pathway. These inhibitors act through several mechanisms: increasing the stability of the destruction complex (Huang et al., 2009) in order to decrease cytoplasmic and nuclear β-catenin levels, or disrupting the interaction between β-catenin and the transcription factors TCF/LEF (Lepourcelet et al., 2004) abolishing the transcription of cell cycle genes. The development of these inhibitors could contribute to the treatment of thyroid carcinomas, especially some anaplastic carcinomas, which show a clear activation of the Wnt/β-catenin pathway and have a poor prognosis due to their aggressiveness and the loss of differentiation markers.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Dr. Ronald Hartong for criticisms and linguistic assistance. We acknowledge the support of Grants BFU-2010-16025 from the Dirección General de Proyectos de Investigación; RD06/0020/0060 from FIS, Instituto de Salud Carlos III, and S2011/BMD-2328 TIRONET project from the Comunidad de Madrid (Spain).
References
Abbosh, P. H., Li, X., Li, L., Gardner, T. A., Kao, C., and Nephew, K. P. (2007). A conditionally replicative, Wnt/beta-catenin pathway-based adenovirus therapy for anaplastic thyroid cancer. Cancer Gene Ther. 14, 399–408.
Aguilera, O., Fraga, M. F., Ballestar, E., Paz, M. F., Herranz, M., Espada, J., Garcia, J. M., Munoz, A., Esteller, M., and Gonzalez-Sancho, J. M. (2006). Epigenetic inactivation of the Wnt antagonist DICKKOPF-1 (DKK-1) gene in human colorectal cancer. Oncogene 25, 4116–4121.
Arufe, M. C., Lu, M., Kubo, A., Keller, G., Davies, T. F., and Lin, R. Y. (2006). Directed differentiation of mouse embryonic stem cells into thyroid follicular cells. Endocrinology 147, 3007–3015.
Berx, G., and van Roy, F. (2009). Involvement of members of the cadherin superfamily in cancer. Cold Spring Harb. Perspect. Biol. 1, a003129.
Brabant, G., Hoang-Vu, C., Cetin, Y., Dralle, H., Scheumann, G., Molne, J., Hansson, G., Jansson, S., Ericson, L. E., and Nilsson, M. (1993). E-cadherin: a differentiation marker in thyroid malignancies. Cancer Res. 53, 4987–4993.
Cali, G., Gentile, F., Mogavero, S., Pallante, P., Nitsch, R., Ciancia, G., Ferraro, A., Fusco, A., and Nitsch, L. (2012). CDH16/Ksp-cadherin is expressed in the developing thyroid gland and is strongly down-regulated in thyroid carcinomas. Endocrinology 153, 522–534.
Cassinelli, G., Favini, E., Degl’Innocenti, D., Salvi, A., De Petro, G., Pierotti, M. A., Zunino, F., Borrello, M. G., and Lanzi, C. (2009). RET/PTC1-driven neoplastic transformation and proinvasive phenotype of human thyrocytes involve Met induction and beta-catenin nuclear translocation. Neoplasia 11, 10–21.
Castellone, M. D., De Falco, V., Rao, D. M., Bellelli, R., Muthu, M., Basolo, F., Fusco, A., Gutkind, J. S., and Santoro, M. (2009). The beta-catenin axis integrates multiple signals downstream from RET/papillary thyroid carcinoma leading to cell proliferation. Cancer Res. 69, 1867–1876.
Cerrato, A., Fulciniti, F., Avallone, A., Benincasa, G., Palombini, L., and Grieco, M. (1998). Beta- and gamma-catenin expression in thyroid carcinomas. J. Pathol. 185, 267–272.
Cetta, F., Chiappetta, G., Melillo, R. M., Petracci, M., Montalto, G., Santoro, M., and Fusco, A. (1998). The ret/ptc1 oncogene is activated in familial adenomatous polyposis-associated thyroid papillary carcinomas. J. Clin. Endocrinol. Metab. 83, 1003–1006.
Cetta, F., Montalto, G., Gori, M., Curia, M. C., Cama, A., and Olschwang, S. (2000). Germline mutations of the APC gene in patients with familial adenomatous polyposis-associated thyroid carcinoma: results from a European cooperative study. J. Clin. Endocrinol. Metab. 85, 286–292.
Chen, G., Jiang, Q., You, Z., Yao, J., Mou, L., Lin, X., Shen, X., You, T., Lin, Q., Wen, J., and Lin, L. (2010). Regulation of GSK-3 beta in the proliferation and apoptosis of human thyrocytes investigated using a GSK-3 beta-targeting RNAi adenovirus expression vector: involvement the Wnt/beta-catenin pathway. Mol. Biol. Rep. 37, 2773–2779.
Cho, N. L., Lin, C. I., Whang, E. E., Carothers, A. M., Moore, F. D. Jr., and Ruan, D. T. (2010). Sulindac reverses aberrant expression and localization of beta-catenin in papillary thyroid cancer cells with the BRAFV600E mutation. Thyroid 20, 615–622.
de Cristofaro, T., Di Palma, T., Fichera, I., Lucci, V., Parrillo, L., De Felice, M., and Zannini, M. (2012). An essential role for pax8 in the transcriptional regulation of cadherin-16 in thyroid cells. Mol. Endocrinol. 26, 67–78.
De Felice, M., and Di Lauro, R. (2004). Thyroid development and its disorders: genetics and molecular mechanisms. Endocr. Rev. 25, 722–746.
Dumaz, N., and Marais, R. (2005). Integrating signals between cAMP and the RAS/RAF/MEK/ERK signalling pathways. Based on the anniversary prize of the Gesellschaft fur Biochemie und Molekularbiologie lecture delivered on 5 July 2003 at the Special FEBS Meeting in Brussels. FEBS J. 272, 3491–504.
Fagin, J. A., Matsuo, K., Karmakar, A., Chen, D. L., Tang, S. H., and Koeffler, H. P. (1993). High prevalence of mutations of the p53 gene in poorly differentiated human thyroid carcinomas. J. Clin. Invest. 91, 179–184.
Filipovich, A., Gehrke, I., Poll-Wolbeck, S. J., and Kreuzer, K. A. (2011). Physiological inhibitors of Wnt signaling. Eur. J. Haematol. 86, 453–465.
Garcia-Rostan, G., Camp, R. L., Herrero, A., Carcangiu, M. L., Rimm, D. L., and Tallini, G. (2001). Beta-catenin dysregulation in thyroid neoplasms: down-regulation, aberrant nuclear expression, and CTNNB1 exon 3 mutations are markers for aggressive tumor phenotypes and poor prognosis. Am. J. Pathol. 158, 987–996.
Garcia-Rostan, G., Costa, A. M., Pereira-Castro, I., Salvatore, G., Hernandez, R., Hermsem, M. J., Herrero, A., Fusco, A., Cameselle-Teijeiro, J., and Santoro, M. (2005). Mutation of the PIK3CA gene in anaplastic thyroid cancer. Cancer Res. 65, 10199–10207.
Garcia-Rostan, G., Tallini, G., Herrero, A., D’Aquila, T. G., Carcangiu, M. L., and Rimm, D. L. (1999). Frequent mutation and nuclear localization of beta-catenin in anaplastic thyroid carcinoma. Cancer Res. 59, 1811–1815.
Graff, J. R., Greenberg, V. E., Herman, J. G., Westra, W. H., Boghaert, E. R., Ain, K. B., Saji, M., Zeiger, M. A., Zimmer, S. G., and Baylin, S. B. (1998). Distinct patterns of E-cadherin CpG island methylation in papillary, follicular, Hurthle’s cell, and poorly differentiated human thyroid carcinoma. Cancer Res. 58, 2063–2066.
Guigon, C. J., Zhao, L., Lu, C., Willingham, M. C., and Cheng, S. Y. (2008). Regulation of beta-catenin by a novel nongenomic action of thyroid hormone beta receptor. Mol. Cell. Biol. 28, 4598–4608.
He, T. C., Sparks, A. B., Rago, C., Hermeking, H., Zawel, L., da Costa, L. T., Morin, P. J., Vogelstein, B., and Kinzler, K. W. (1998). Identification of c-MYC as a target of the APC pathway. Science 281, 1509–1512.
Helmbrecht, K., Kispert, A., von Wasielewski, R., and Brabant, G. (2001). Identification of a Wnt/beta-catenin signaling pathway in human thyroid cells. Endocrinology 142, 5261–5266.
Huang, S. M., Mishina, Y. M., Liu, S., Cheung, A., Stegmeier, F., Michaud, G. A., Charlat, O., Wiellette, E., Zhang, Y., Wiessner, S., Hild, M., Shi, X., Wilson, C. J., Mickanin, C., Myer, V., Fazal, A., Tomlinson, R., Serluca, F., Shao, W., Cheng, H., Shultz, M., Rau, C., Schirle, M., Schlegl, J., Ghidelli, S., Fawell, S., Lu, C., Curtis, D., Kirschner, M. W., Lengauer, C., Finan, P. M., Tallarico, J. A., Bouwmeester, T., Porter, J. A., Bauer, A., and Cong, F. (2009). Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 461, 614–620.
Hulsken, J., Birchmeier, W., and Behrens, J. (1994). E-cadherin and APC compete for the interaction with beta-catenin and the cytoskeleton. J. Cell Biol. 127(Pt 2), 2061–2069.
Ishigaki, K., Namba, H., Nakashima, M., Nakayama, T., Mitsutake, N., Hayashi, T., Maeda, S., Ichinose, M., Kanematsu, T., and Yamashita, S. (2002). Aberrant localization of beta-catenin correlates with overexpression of its target gene in human papillary thyroid cancer. J. Clin. Endocrinol. Metab. 87, 3433–3440.
Kikuchi, A., and Yamamoto, H. (2008). Tumor formation due to abnormalities in the beta-catenin-independent pathway of Wnt signaling. Cancer Sci. 99, 202–208.
Kim, W. B., Lewis, C. J., McCall, K. D., Malgor, R., Kohn, A. D., Moon, R. T., and Kohn, L. D. (2007). Overexpression of Wnt-1 in thyrocytes enhances cellular growth but suppresses transcription of the thyroperoxidase gene via different signaling mechanisms. J. Endocrinol. 193, 93–106.
Kimura, T., Van Keymeulen, A., Golstein, J., Fusco, A., Dumont, J. E., and Roger, P. P. (2001). Regulation of thyroid cell proliferation by TSH and other factors: a critical evaluation of in vitro models. Endocr. Rev. 22, 631–656.
Kremenevskaja, N., von Wasielewski, R., Rao, A. S., Schofl, C., Andersson, T., and Brabant, G. (2005). Wnt-5a has tumor suppressor activity in thyroid carcinoma. Oncogene 24, 2144–2154.
Kroll, T. G., Sarraf, P., Pecciarini, L., Chen, C. J., Mueller, E., Spiegelman, B. M., and Fletcher, J. A. (2000). PAX8-PPARgamma1 fusion oncogene in human thyroid carcinoma [corrected]. Science 289, 1357–1360.
Kurihara, T., Ikeda, S., Ishizaki, Y., Fujimori, M., Tokumoto, N., Hirata, Y., Ozaki, S., Okajima, M., Sugino, K., and Asahara, T. (2004). Immunohistochemical and sequencing analyses of the Wnt signaling components in Japanese anaplastic thyroid cancers. Thyroid 14, 1020–1029.
Lepourcelet, M., Chen, Y. N., France, D. S., Wang, H., Crews, P., Petersen, F., Bruseo, C., Wood, A. W., and Shivdasani, R. A. (2004). Small-molecule antagonists of the oncogenic Tcf/beta-catenin protein complex. Cancer Cell 5, 91–102.
Lu, C., Zhu, X., Willingham, M. C., and Cheng, S. Y. (2011). Activation of tumor cell proliferation by thyroid hormone in a mouse model of follicular thyroid carcinoma. Oncogene. doi: 10.1038/onc.2011.390. [Epub ahead of print].
MacDonald, B. T., Tamai, K., and He, X. (2009). Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9–26.
Malanchi, I., Santamaria-Martinez, A., Susanto, E., Peng, H., Lehr, H. A., Delaloye, J. F., and Huelsken, J. (2011). Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 481, 85–89.
McDonald, S. L., and Silver, A. (2009). The opposing roles of Wnt-5a in cancer. Br. J. Cancer 101, 209–214.
Medina, D. L., and Santisteban, P. (2000). Thyrotropin-dependent proliferation of in vitro rat thyroid cell systems. Eur. J. Endocrinol. 143, 161–178.
Meirmanov, S., Nakashima, M., Kondo, H., Matsufuji, R., Takamura, N., Ishigaki, K., Ito, M., Prouglo, Y., Yamashita, S., and Sekine, I. (2003). Correlation of cytoplasmic beta-catenin and cyclin D1 overexpression during thyroid carcinogenesis around Semipalatinsk nuclear test site. Thyroid 13, 537–545.
Nikiforov, Y. E., and Nikiforova, M. N. (2011). Molecular genetics and diagnosis of thyroid cancer. Nat. Rev. Endocrinol. 7, 569–580.
Nishita, M., Enomoto, M., Yamagata, K., and Minami, Y. (2010). Cell/tissue-tropic functions of Wnt5a signaling in normal and cancer cells. Trends Cell Biol. 20, 346–354.
O’Sullivan, C., Barton, C. M., Staddon, S. L., Brown, C. L., and Lemoine, N. R. (1991). Activating point mutations of the gsp oncogene in human thyroid adenomas. Mol. Carcinog. 4, 345–349.
Parma, J., Duprez, L., Van Sande, J., Cochaux, P., Gervy, C., Mockel, J., Dumont, J., and Vassart, G. (1993). Somatic mutations in the thyrotropin receptor gene cause hyperfunctioning thyroid adenomas. Nature 365, 649–651.
Rao, A. S., Kremenevskaja, N., von Wasielewski, R., Jakubcakova, V., Kant, S., Resch, J., and Brabant, G. (2006). Wnt/beta-catenin signaling mediates antineoplastic effects of imatinib mesylate (gleevec) in anaplastic thyroid cancer. J. Clin. Endocrinol. Metab. 91, 159–168.
Rezk, S., Brynes, R. K., Nelson, V., Thein, M., Patwardhan, N., Fischer, A., and Khan, A. (2004). Beta-catenin expression in thyroid follicular lesions: potential role in nuclear envelope changes in papillary carcinomas. Endocr. Pathol. 15, 329–337.
Rice, P. L., Kelloff, J., Sullivan, H., Driggers, L. J., Beard, K. S., Kuwada, S., Piazza, G., and Ahnen, D. J. (2003). Sulindac metabolites induce caspase- and proteasome-dependent degradation of beta-catenin protein in human colon cancer cells. Mol. Cancer Ther. 2, 885–892.
Riesco-Eizaguirre, G., Rodriguez, I., De la Vieja, A., Costamagna, E., Carrasco, N., Nistal, M., and Santisteban, P. (2009). The BRAFV600E oncogene induces transforming growth factor beta secretion leading to sodium iodide symporter repression and increased malignancy in thyroid cancer. Cancer Res. 69, 8317–8325.
Riesco-Eizaguirre, G., and Santisteban, P. (2007). Molecular biology of thyroid cancer initiation. Clin. Transl. Oncol. 9, 686–693.
Rubinfeld, B., Albert, I., Porfiri, E., Fiol, C., Munemitsu, S., and Polakis, P. (1996). Binding of GSK3beta to the APC-beta-catenin complex and regulation of complex assembly. Science 272, 1023–1026.
Santisteban, P., and Bernal, J. (2005). Thyroid development and effect on the nervous system. Rev. Endocr. Metab. Disord. 6, 217–228.
Soravia, C., Sugg, S. L., Berk, T., Mitri, A., Cheng, H., Gallinger, S., Cohen, Z., Asa, S. L., and Bapat, B. V. (1999). Familial adenomatous polyposis-associated thyroid cancer: a clinical, pathological, and molecular genetics study. Am. J. Pathol. 154, 127–135.
Tartari, C. J., Donadoni, C., Manieri, E., Mologni, L., Mina, P. D., Villa, A., and Gambacorti-Passerini, C. (2011). Dissection of the RET/beta-catenin interaction in the TPC1 thyroid cancer cell line. Am. J. Cancer Res. 1, 716–725.
Tetsu, O., and McCormick, F. (1999). Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 398, 422–426.
Thomas, D., Friedman, S., and Lin, R. Y. (2008). Thyroid stem cells: lessons from normal development and thyroid cancer. Endocr. Relat. Cancer 15, 51–58.
Todaro, M., Iovino, F., Eterno, V., Cammareri, P., Gambara, G., Espina, V., Gulotta, G., Dieli, F., Giordano, S., De Maria, R., and Stassi, G. (2010). Tumorigenic and metastatic activity of human thyroid cancer stem cells. Cancer Res. 70, 8874–8885.
Tzelepi, V. N., Tsamandas, A. C., Vlotinou, H. D., Vagianos, C. E., and Scopa, C. D. (2008). Tight junctions in thyroid carcinogenesis: diverse expression of claudin-1, claudin-4, claudin-7 and occludin in thyroid neoplasms. Mod. Pathol. 21, 22–30.
Vassart, G., and Dumont, J. E. (1992). The thyrotropin receptor and the regulation of thyrocyte function and growth. Endocr. Rev. 13, 596–611.
Vermeulen, L., De Sousa, E. M. F., van der Heijden, M., Cameron, K., de Jong, J. H., Borovski, T., Tuynman, J. B., Todaro, M., Merz, C., Rodermond, H., Sprick, M. R., Kemper, K., Richel, D. J., Stassi, G., and Medema, J. P. (2010). Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 12, 468–476.
Wada, H., and Okamoto, H. (2009). Roles of planar cell polarity pathway genes for neural migration and differentiation. Dev. Growth Differ. 51, 233–240.
Xu, B., Yoshimoto, K., Miyauchi, A., Kuma, S., Mizusawa, N., Hirokawa, M., and Sano, T. (2003). Cribriform-morular variant of papillary thyroid carcinoma: a pathological and molecular genetic study with evidence of frequent somatic mutations in exon 3 of the beta-catenin gene. J. Pathol. 199, 58–67.
Zaballos, M. A., Garcia, B., and Santisteban, P. (2008). Gbetagamma dimers released in response to thyrotropin activate phosphoinositide 3-kinase and regulate gene expression in thyroid cells. Mol. Endocrinol. 22, 1183–1199.
Keywords: Wnt pathway, signaling network, thyroid cancer, proliferation, differentiation
Citation: Sastre-Perona A and Santisteban P (2012) Role of the Wnt pathway in thyroid cancer. Front. Endocrin. 3:31. doi: 10.3389/fendo.2012.00031
Received: 01 December 2011; Accepted: 09 February 2012;
Published online: 29 February 2012.
Edited by:
Carmelo Nucera, Beth Israel Deaconess Medical Center/Harvard Medical School, USAReviewed by:
Ettore Ciro Degli Uberti, University of Ferrara, ItalyRoberta Malaguarnera, University “Magna Graecia” of Catanzaro, Italy
Marialuisa Appetecchia, Regina Elena National Cancer Institute IRCCS, Italy
Copyright: © 2012 Sastre-Perona and Santisteban. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Pilar Santisteban, Instituto de Investigaciones Biomédicas, Consejo Superior de Investigaciones Científicas, y Universidad Autónoma de Madrid, C/Arturo Duperier 4, 28029 Madrid, Spain. e-mail:cHNhbnRpc3RlYmFuQGlpYi51YW0uZXM=