 Maria Graziella Catalano1
Maria Graziella Catalano1
- 1 Department of Clinical Pathophysiology, University of Turin, Torino, Italy
- 2 Oncological Endocrinology, AUO San Giovanni Battista, Torino, Italy
At present no successful treatment is available for advanced thyroid cancer, which comprises poorly differentiated, anaplastic, and metastatic or recurrent differentiated thyroid cancer not responding to radioiodine. In the last few years, biologically targeted therapies for advanced thyroid carcinomas have been proposed on the basis of the recognition of key oncogenic mutations. Although the results of several phase II trials look promising, none of the patients treated had a complete response, and only a minority of them had a partial response, suggesting that the treatment is, at best, effective in stabilizing patients with progressive disease. “Epigenetic” refers to the study of heritable changes in gene expression that occur without any alteration in the primary DNA sequence. The epigenetic processes establish and maintain the global and local chromatin states that determine gene expression. Epigenetic abnormalities are present in almost all cancers and, together with genetic changes, drive tumor progression. Various genes involved in the control of cell proliferation and invasion (p16INK4A, RASSF1A, PTEN, Rap1GAP, TIMP3, DAPK, RARβ2, E-cadherin, and CITED1) as well as genes specific of thyroid differentiation (Na+/I− symport, TSH receptor, pendrin, SL5A8, and TTF-1) present aberrant methylation in thyroid cancer. This review deals with the most frequent epigenetic alterations in thyroid cancer and focuses on epigenetic therapy, whose goal is to target the chromatin in rapidly dividing tumor cells and potentially restore normal cell functions. Experimental data and clinical trials, especially using deacetylase inhibitors and demethylating agents, are discussed.
Introduction
Thyroid carcinoma is the most common endocrine malignancy worldwide, its incidence being approximately 1–5% of all cancers in females and less than 2% in males. Even though it is considered a relatively rare neoplasm, its incidence is progressively increasing (Kilfoy et al., 2009). Thyroid carcinomas arising from follicular epithelial cells (about 95% of all thyroid tumors) are traditionally classified as well-differentiated thyroid carcinomas (WDTC), including both papillary (PTC, 80%) and follicular types (FTC, 10–15%; Fassnacht et al., 2009), poorly differentiated thyroid carcinomas (PDTC; Patel and Shaha, 2006; Ghossein, 2009) and, finally, anaplastic thyroid carcinoma (ATC), accounting for 1–2% of thyroid malignancies. WDTC usually share slow growth rate and high percent of cure achieved by a combination of surgery, radioiodine ablation, and TSH-suppressive therapy. Poorly differentiated tumors, either from the beginning or loosing differentiation as disease progresses, as well as ATC, are generally resistant to conventional therapies, and no successful treatment is now available (Smith et al., 2009; Catalano et al., 2010; Sherman, 2011). Medullary thyroid carcinoma (MTC) represents a minority of thyroid cancers arising from para-follicular calcitonin producing cells. Neuroendocrine-derived MTC is not responsive to either radioiodine or TSH suppression and its treatment is mainly based on surgery for primary and regional metastatic disease. The outcome of patients with metastatic disease is similar to that of radioiodine-unresponsive WDTC (Roman et al., 2006).
The term advanced thyroid cancer refers to all thyroid tumors resistant to conventional therapies, and comprises PDTC, ATC, and metastatic or recurrent differentiated cancers which do not respond to radioiodine. Chemotherapy is generally taken into consideration only for patients with symptomatic or rapidly progressive metastatic disease unresponsive to or unsuitable for surgery, radioiodine, and external beam radiotherapy; but the efficacy of cytotoxic systemic chemotherapies for these tumors is fairly poor, response rates being around 25% or less (Sherman, 2010).
In the recent past, biologically targeted therapies for advanced thyroid carcinomas have been proposed on the basis of the recognition of key oncogenic mutations, but also of adaptation processes facilitating tumor growth, such as hypoxia-inducible angiogenesis or epigenetic modifications of chromosomal DNA and histones.
As far as oncogenes are concerned, RAS mutation may be an early step in thyroid tumorigenesis, as it is present with frequencies ranging from 24 to 53% in follicular adenomas (FA) and from 18 to 52% in FTC (Nikiforova et al., 2003a). In FTC, PAX8–PPARγ rearrangement was also reported (Kroll et al., 2000). RET/PTC rearrangement, distinctive of PTC, has not been identified in FA or FTC, supporting divergent tumor induction/progression (Wreesmann and Singh, 2008). The majority of PTC, in fact, are initiated by well characterized genetic events, involving single activating somatic mutation of BRAF or RAS, and translocations producing RET/PTC oncogenes (Fagin, 2004). BRAFV600E mutation, found in approximately 40% of PTC (Ciampi and Nikiforov, 2005), leads to constitutively active BRAF and subsequent activation of the RET/RAS/BRAF/MAPK signal transduction pathway and, in addition, the presence of BRAF mutations is associated with decreased expression of mRNAs for the sodium iodide symporter (NIS) and the TSH receptor, that are considered markers of thyroid differentiation (Durante et al., 2007; Figure 1). Mutated BRAF is also associated with the PTC to PDTC progression, since it makes thyroid cells sensitive to TGFβ-induced EMT (epithelial–mesenchymal transition; Knauf et al., 2011) and it is involved in the control of some ECM non-cellular components such as thrombospondin-1; it was reported that BRAFV600E knock-down caused a reduction of in TSP-1 expression and, as a consequence, of adhesion and migration/invasion of human thyroid cancer cells (Nucera et al., 2010, 2011). Moreover, treatment of mice carrying BRAFV600E PTC with BRAF inhibitors reduced tumor proliferative index and partially restored thyroid-specific gene expression (Chakravarty et al., 2011). Finally, in PTC the pro-angiogenic factor vascular endothelial growth factor (VEGF) expression correlates with a higher risk of metastasis and recurrence, a shorter disease-free survival, and BRAF mutation status (Lennard et al., 2001; Jo et al., 2006).
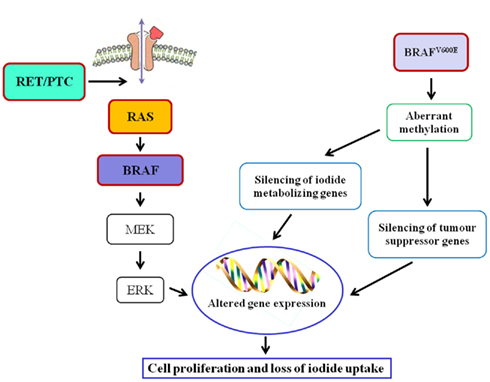
Figure 1. Main genetic alterations and related epigenetic modifications in thyroid cancer.
Poorly differentiated thyroid carcinomas and ATC have a more complex and less distinct gene profile and molecular data suggest that these aggressive forms may dedifferentiate from WDTC. For instance, the well-known mutations of BRAF and RAS occurring in PTC are present in more than 50% of PDTC (Ghossein, 2009). In about one-third of anaplastic carcinomas, probably those arisen from pre-existing PDTC, is present the BRAFV600E mutation (Nikiforova et al., 2003b). It was also suggested that ATC might develop from RAS-mutated FTC since RAS mutation was also detected in 6–50% of ATC (Smallridge et al., 2009). Further, in ATC a mutation of PI3KCA gene causing Akt and ERK activation was reported (Smallridge et al., 2009). Additional mutations frequently observed in ATC involve p53 and β-catenin. The tumor-suppressor gene p53 is fundamental for the progression from indolent to aggressive thyroid cancer. The inactivating p53 mutation, seldom detected in WDTC, is found in about 55% of PDTC and ATC (Smallridge et al., 2009). Membrane β-catenin expression is progressively reduced with loss of tumor differentiation, resulting in tumor invasiveness, and increasing metastatic potential (Garcia-Rostan et al., 2001).
As far as MTC is concerned, heritable germ-line activating mutations in RET are found in almost all familial cases and identical somatic mutations in sporadic disease. Activated RET mutant proteins also enhance MAPK signaling (Santoro et al., 1995).
The use of selective inhibitors of activated BRAF, RET, and RET/PTC kinases as well as of VEGF and VEGF receptor to treat advanced thyroid cancer is under thoughtful evaluation. To date, a number of clinical trials regarding tyrosine kinase and angiogenic factors inhibitors are in progress1 2.
Epigenetic Alterations
In the early 1940s, Conrad Hal Waddington coined the term epigenetics as “the causal interactions between genes and their products, which bring the phenotype into being” (Waddington, 1942). Currently, epigenetic refers to the study of heritable changes in gene expression that occur without any alteration in the primary DNA sequence (Sharma et al., 2010). The epigenetic processes establish and maintain the global and local condensed or decondensed chromatin states that determine gene expression. The continuous interplay of all these processes is today called “epigenome” – the epigenetic status that determines the way a single eukaryotic genome may manifest in different cell types and developmental stages and that, if aberrant, gives rise to cancer and other diseases. In fact, epigenetic abnormalities are present in almost all cancers and, together with genetic changes, drive tumor progression. Moreover, acting in concert with genetic changes, they play a role in the earliest steps of tumorigenesis (Feinberg et al., 2006), as also suggested by the growing list of tumor-suppressor genes that are often epigenetically silenced but rarely genetically mutated in the pre-invasive stages of many cancers (Jones and Baylin, 2007).
Epigenetic information that fulfills the requirement of heritability can be classified into three distinct types: DNA methylation, histone modifications, and non-coding RNAs.
In the present review, we will primarily discuss DNA methylation, and histone modifications (Figure 2), as drugs that target these epigenetic modifications are already at a clinical developmental stage.
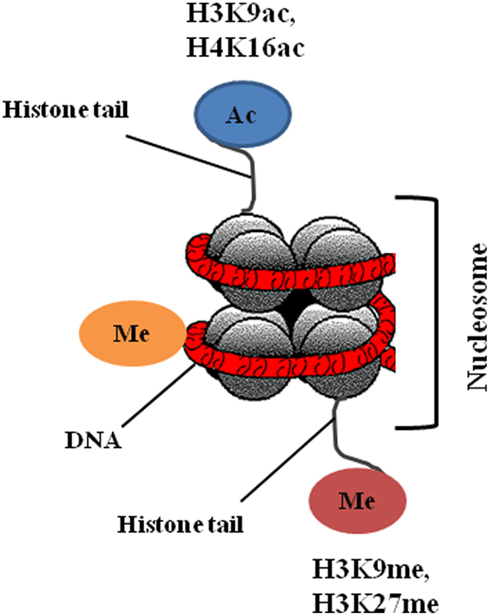
Figure 2. Nucleosome structure and main epigenetic alterations in cancer.
DNA methylation takes place within the CpG dinucleotides, and its consequence is the silencing of genes and non-coding genomic regions. There are three main DNA methyltransferases (DNMTs): DNMT1, which maintains the existing methylation patterns following DNA replication, and DNMT3A and DNMT3B, which target previously unmethylated CpGs. Cancer genome is typically characterized by global hypomethylation concomitant with hypermethylation of CpG islands in the promoters of genes that play important roles in regulating cell cycle, apoptosis, differentiation, and cell adhesion (Baylin and Herman, 2000).
Post-translational modifications of the N-terminal tails of histones include acetylation, methylation, phosphorylation, ubiquitination, SUMOylation, and ADP ribosylation. Histone modifications can lead to either gene activation or repression, depending upon which residues are modified and the type of modification (Chi et al., 2010). Overall, histone modifications affect chromatin conformation and consequently influence gene transcription, DNA repair and replication, and cell cycle checkpoints (Sawan et al., 2008). Histone acetylation and deacetylation cause activation and arrest of gene transcription, respectively, and the enzymes that catalyze these changes, histone acetyltransferases (HATs) and histone deacetylases (HDACs), can also target non-histone proteins such as p53, Hsp90, and α-tubulin (Sterner and Berger, 2000). There are four classes of HDACs: class I consists of HDAC 1, 2, 3, and 8; class II consists of HDAC 4, 5, 6, 7, 9, and 10; class III are sirtuins (SIRT 1–7); and class IV consisting of HDAC 11. Alterations in histone modifications significantly contribute to the onset and progression of cancer (Chi et al., 2010). The most common epigenetic modifications of histones in cancer are acetylation and methylation; reduction in monoacetylated H4-K16 and trimethylation of H4-K20 are general features of cancer cells (Fraga et al., 2005). Altered expression of HDACs has been reported in tumor samples; overexpression of class I HDACs has been reported in 5–40% of cancer tissues (Nakagawa et al., 2007), HDAC1 is overexpressed in prostate, gastric, colon, and breast carcinomas. HDAC2 is overexpressed in colorectal, cervical, and gastric cancer, whereas overexpression of HDAC6 is observed in breast cancer specimens (Ellis et al., 2009).
MicroRNAs (miRs), are small (19–25 nucleotides), non-coding RNA molecules representing the most recent class of molecules known to be involved in epigenetic regulation. They function as negative regulators of the expression of protein-encoding genes, involved in major processes such as development, apoptosis, cell proliferation, immune response, and hematopoiesis.
Epigenetic Modifications in Thyroid Cancer
Various genes involved in the control of cell proliferation and invasion as well as genes specific of thyroid differentiation are epigenetically silenced in thyroid cancer (Table 1). Cumulative epigenetic alterations play a role in the sequential progression from indolent WDTC to metastasizing carcinomas, through the spectrum of poorly differentiated to undifferentiated thyroid carcinoma.
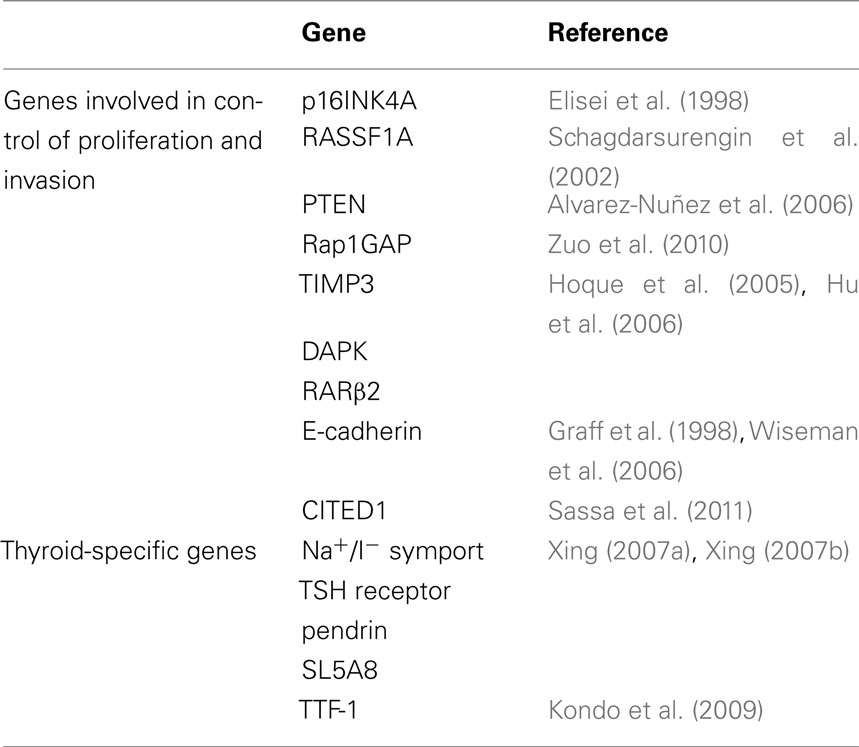
Table 1. Epigenetic modifications in thyroid cancer.
DNA Methylation
In thyroid cancer, aberrant methylation of tumor-suppressor genes is common. CDK inhibitors, such as p27KIP1 and p16INK4A, are commonly down-regulated; methylation of CpG islands in p16INK4A is detected in 30% of thyroid neoplasms (Elisei et al., 1998).
The tumor-suppressive Ras effector, RAS association domain family 1, splicing isoform A (RASSF1A) contains a Ras association domain, and plays a role in the regulation of cell cycle and apoptosis (Donninger et al., 2007). In the thyroid, RASSF1A promoter methylation is present in more than 30% of benign and malignant thyroid tumors. The high frequency of RASSF1A hypermethylation both in benign FA (33–44%) and the increased occurrence in FTC (70–100%) suggest that epigenetically silencing of RASSF1A is an early step in thyroid tumorigenesis (Schagdarsurengin et al., 2002).
PTEN, a phosphatase that terminates the PI3K/Akt pathway, has been found aberrantly methylated in about 50% of papillary carcinomas and almost 100% of follicular carcinomas and adenomas, suggesting that it may be involved in thyroid tumorigenesis (Alvarez-Nuñez et al., 2006).
Rap1GAP is a Rap1 GTPase-activating protein that inhibits the RAS superfamily protein Rap1 by facilitating hydrolysis of GTP to GDP. In human thyroid cancers, Rap1GAP expression is frequently lost or down-regulated secondary to promoter hypermethylation and/or loss of heterozygosity; Rap1GAP loss of expression correlates with tumor invasiveness (Zuo et al., 2010).
A close association between BRAF mutation and aberrant methylation of several tumor-suppressor genes in PTC, including the genes for tissue inhibitor of matrix metalloproteinase-3 (TIMP3), death-associated protein kinase (DAPK), and retinoic acid receptor β2 (RARβ2) has been reported (Hoque et al., 2005). This association correlated with high-risk clinicopathological characteristics of PTC, including extra-thyroid invasion, lymph node metastasis, and advanced disease stages (III and IV; Hu et al., 2006). TIMP3 suppresses tumor growth, angiogenesis, invasion, and metastasis both by preventing the interstitial matrix destruction promoted by matrix metalloproteinase (MMP)-3 and by blocking the binding of VEGF to the VEGF receptor (Anand-Apte et al., 1996). Therefore, methylation-mediated silencing of the TIMP3 gene may play a unique role in BRAF mutation-promoted invasiveness and progression of PTC.
As far as invasiveness is concerned, E-cadherin joins together with catenins to promote Ca2+-dependent, homotypic cell-to-cell adhesion and to establish normal epithelial tissue architecture. Disruption of the E-cadherin/catenin complex contributes to tumor metastasis, and decreased expression of E-cadherin is observed in advanced stage, poorly differentiated carcinomas, (Graff et al., 1998) and is associated with the transformation of differentiated into ATC (Wiseman et al., 2006).
Aberrant methylation also involves thyroid-specific genes such as the Na+/I− symporter (NIS), the promoter of the TSH receptor, the genes for the putative thyroid follicular cell apical iodide transport (pendrin and SCL5A8; Xing 2007a,b). Suppression of these thyroid iodide-metabolizing molecules results in the loss of cancer cells ability to concentrate iodine, rendering tumors insensitive to radioiodide therapy.
Hypomethylation of the CpGs in the promoter region of CITED1 (Cbp/p300 Interacting Transactivators with glutamic acid [E] and aspartic acid [D]-rich C-terminal domain) is associated with higher expression of CITED1 mRNA in PTC tissues (Sassa et al., 2011).
Histone Modifications
Unfortunately, little information about the histone modifications present in thyroid tumors and the relationship between such modifications and thyroid cancer behavior is at present available.
However, recently, Puppin et al. (2011) investigated whether global levels of acetylated histones are modified in thyroid cancer tissues. They show that levels of acetylated H3 at residue K18 are lower in undifferentiated cancers with respect to differentiated tumors, suggesting that acetylation is switched off in the thyroid tumor transition.
CpG hypermethylation in the promoter region of the thyroid transcription factor-1 (TTF-1), which is essential for thyroid organogenesis, concurrently with increased dimethyl-H3-K9 and decreased acetyl-H3-K9, has been observed in a subset of thyroid carcinoma cells that had lost TTF-1 expression (Kondo et al., 2009); moreover, it has recently been demonstrated that the enhancer of zeste homolog 2 (EZH2), a histone lysine methyltransferase belonging to the polycomb group protein family, is specifically overexpressed in ATC, and it directly contributes to transcriptional silencing of PAX8 gene and ATC differentiation (Borbone et al., 2011).
microRNA
Comparative analysis of miRs expression in normal thyroid tissue versus neoplastic tissue has shown that, in thyroid neoplasms, 32% of miRs were up-regulated and 38% were down-regulated; moreover, miRs expression profiles had substantial variability within specific tumor types (Nikiforova et al., 2008). Down-regulation of anti-invasive miR-200 and miR-30 families directs the EMT and invasive potential of ATCs (Braun et al., 2010). Recently it has been reported that two histone deacetylase inhibitors, trichostatin A and vorinostat, induced miR-129-5p overexpression, histone acetylation and cell death in papillary, and anaplastic cancer cell lines and in primary cultures of papillary thyroid cancer (Brest et al., 2011).
Epigenetic Drugs
Epigenetic therapy was conceived to activate genes abnormally silenced in cancer by epigenetic mechanisms. Epigenetic drugs are expected to target the two main mechanisms of epigenetic alterations, DNA methylation, and acetylation, controlling though the differentiation and proliferation of transformed cells. To date, different epigenetic drugs are in clinical trials for the treatment of advanced thyroid cancer (Table 2).
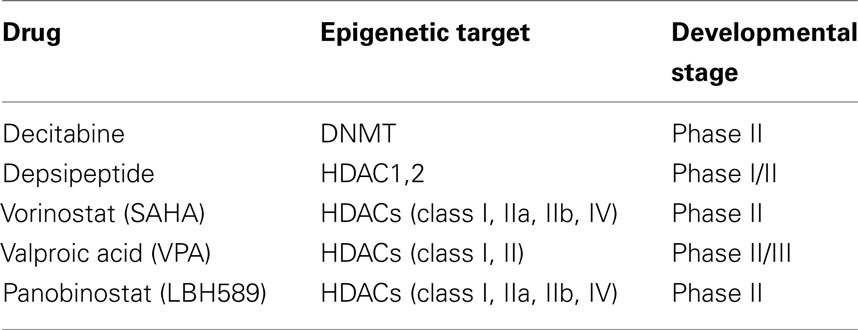
Table 2. Epigenetic drugs in clinical trials for the treatment of advanced thyroid cancer (www.clinicaltrials.gov and https://oss-sper-clin.agenziafarmaco.it).
The possibility of a microRNA-based therapy in cancer, using these small molecules as both targets and tools, suggests a new intriguing and promising therapy. Several preclinical studies underline this possibility; however, as to date no clinical trial is available, there will be no further discussion in the present review.
Demethylating Agents
During thyroid tumor progression, many specific thyroid genes (e.g., NIS and TSH receptor genes) are hypermethylated and, therefore silenced; demethylating agents might reverse the malignant cell phenotype. Indeed, it was reported that demethylating agents like decitabine are able to restore NIS and TSH-R expression in human thyroid carcinoma cell lines (Venkataraman et al., 1999; Xing et al., 2003; Provenzano et al., 2007; Tuncel et al., 2007). Furthermore, decitabine treatment suppressed the growth of undifferentiated and dedifferentiated thyroid carcinoma cells (Miasaki et al., 2008; Vivaldi et al., 2009).
At present, the methyltransferase inhibitors azacitidine and decitabine have been licensed for clinical therapy only in myelodysplastic syndrome, but new hypomethylating agents (zebularine and isothiocyanates) are in various stages of development for cancer therapy (Kurkjian et al., 2008). In particular, a phase II clinical trial is ongoing for treatment with decitabine of patients with metastatic thyroid cancers unresponsive to radioiodine (see text footnote 2). The results of the trial, whose primary objective is to determine whether decitabine can restore iodine 131 uptake in patients with metastatic papillary thyroid or follicular thyroid cancer, are expected for June 2012.
Histone Deacetylase Inhibitors
Histone acetylation and deacetylation are key events in controlling gene transcription; histone acetyltransferases (HATs) and histone deacetylases (HDACs; Kuo and Allis, 1998) catalyze these reactions and target also non-histone proteins, like transcription factors (Sterner and Berger, 2000).
HDAC inhibitors (DCI) are looked upon as very promising agents in tumor treatments since they, targeting multiple tumorigenic pathways, preferentially kill transformed cells, and are relatively non-toxic toward normal cells (Johnstone, 2002). Several structural classes of DCI have been identified, including short-chain fatty acids such as phenylbutyrate and valproic acid (VPA); cyclic tetrapeptides such as trapoxin A, cyclic peptides such as depsipeptide (FK228) and apicidin; benzamides such as MS27-275 and CI-994; hydroxamic acids such as suberoylanilide hydroxamic acid (SAHA), oxamflatin, trichostatin A, and the more recently developed pan-inhibitors LAQ824, PXD101, and LBH589 (Fuino et al., 2003; Plumb et al., 2003; Atadja, 2009).
In thyroid cancer, depsipeptide, SAHA, valproic acid, and panobinostat are the main DCI that are under investigation.
Depsipeptide
Depsipeptide (FK228 or FR901228) is a cyclic peptide that inhibits histone deacetylase activity at nanomolar concentrations (Johnstone, 2002). In SW-1736 cells it stimulates the expression of NIS, restores iodide uptake (Kitazono et al., 2001), and sensitizes cells to doxorubicin (Kitazono et al., 2002). The drug used alone inhibits the growth of a primary culture from a metastatic BRAF V600E papillary tumor; while in combination with paclitaxel, lovastatin, or gefitinib has synergic effects (Copland et al., 2006). p53 gene therapy associated with depsipeptide inhibits the growth of anaplastic FRO cell line (Imanishi et al., 2002).
There are currently two clinical studies at the National Cancer Institute (see text footnote 2). The first is a phase I trial examining the safety and tolerability of depsipeptide in patients with solid tumors, including thyroid cancer, which has recently been completed; the second is a phase II trial determining the anti-tumor activity of the drug in patients with progressive recurrent and/or metastatic non-MTCs that are refractory to radioactive iodine. No conclusive data from these studies are available at the moment.
Vorinostat
Vorinostat (SAHA) is a hydroxamic acid that, binding directly to the HDAC catalytic site, inhibits DAC enzymatic activity (Finnin et al., 1999). SAHA induces growth arrest and caspase-mediated apoptosis in anaplastic thyroid cancer FRO cells (Mitsiades et al., 2005). Moreover it sensitizes cells to doxorubicin and inhibits the proliferation of FRO anaplastic tumors grown in nude mice (Luong et al., 2006).
In a phase I study on vorinostat in advanced cancers, six patients with thyroid cancer were recruited; one patient with advanced papillary tumor exhibited a partial response, while one papillary thyroid patient showed improvement in the RAI scan post-therapy (Kelly et al., 2005). Successively, a phase II trial was undertaken to determine whether SAHA was active in patients with metastatic thyroid cancers who did not respond to standard therapy, but unfortunately, it concluded that 200 mg of vorinostat twice a day was not a useful treatment for advanced thyroid cancer (Woyach et al., 2009).
Valproic acid
Valproic acid is a short-chain fatty acid which has been used for a long time in epilepsy and mood disorder treatment (Blaheta et al., 2005). More recently its activity as DCI (Göttlicher et al., 2001; Phiel et al., 2001) as well as its anti-proliferative and pro-differentiating properties in a variety of hematologic and solid tumors (Duenas-Gonzalez et al., 2008) were reported.
In the last decade, our laboratory was very interested in studying the anti-tumor effects of VPA on undifferentiated thyroid cancer cells. In 2004 we demonstrated that valproic acid induces NIS gene expression, NIS membrane localization, and iodide accumulation in poorly differentiated thyroid cancer cells (Fortunati et al., 2004). We also showed (Catalano et al., 2005) that the drug is highly effective in suppressing the growth of poorly differentiated thyroid cancer cell lines, inducing apoptosis and cell cycle. In agreement with our results, Shen et al. (2005) demonstrated that, also in metastatic follicular cell lines, VPA causes significant growth inhibition. Unfortunately, in ATC cells, the induction of NIS mRNA is not followed by a change in iodide uptake (Fortunati et al., 2004) and VPA does not induce apoptosis (Catalano et al., 2006). Taken together, these data indicate that VPA alone is not effective in anaplastic thyroid cancer; however, we demonstrated that it enhances the cytotoxicity of doxorubicin in ATC cells and that the sensitizing effect increased apoptosis and doxorubicin-induced G2 cell cycle arrest (Catalano et al., 2006). Afterward, it has been reported that some cell lines employed in those in vitro studies are of non-thyroid origin (Schweppe et al., 2008); thus, results obtained using these cell lines need to be regarded with caution. However in the same studies, cells of assessed thyroid origin showed a similar behavior.
In line with these findings, it has been reported that VPA, in combination with the highest concentration of doxorubicin that does not induce KAT-18 cell death, efficiently induced apoptosis in KAT-18 cells (Kim et al., 2009). Since some DCI that target HDAC6 (Zhang et al., 2003), like VPA, can also have α-tubulin as a substrate and tubulin acetylation is linked to microtubule dynamics and stability (Matsuyama et al., 2002), the effect of VPA used together with paclitaxel, a microtubule-targeting drug, were evaluated. The addition of VPA to paclitaxel significantly enhanced the cytostatic and cytotoxic effects of paclitaxel in ATC cells (Catalano et al., 2007). Beside the in vitro studies, one case of successful treatment of ATC with a combination of oral VPA, cisplatin and doxorubicin, plus external and intra-operative radiation, and surgery has been reported the patient being alive and disease-free 2 years after diagnosis (Noguchi et al., 2009). A phase II trial of VPA in patients with advanced thyroid cancers of follicular origin is active and currently recruiting patients at the NCI (see text footnote 2). Moreover, an Italian multicenter phase II/III clinical trial in patients with undifferentiated thyroid cancer treated with a combination of VPA and paclitaxel is ongoing3.
Panobinostat
Panobinostat (LBH589) is a hydroxamic acid with potent inhibitory activity at low nanomolar concentrations against all classes of HDAC enzymes (pan-DAC inhibitor; Atadja, 2009). In vitro treatment with LBH589 of three ATC cell lines (BHT-101, CAL-62, and 8305C) resulted in impairment of cell viability, inhibition of colony formation, cell cycle arrest, and apoptosis induction. LBH589 markedly determined microtubule stabilization as evidenced by tubulin acetylation and increased tubulin polymerization. The cytotoxic properties of LBH589 were confirmed in a SCID xenograft model implanted with CAL-62 cells (Catalano et al., 2011).
A phase II trial of LBH589 in patients with metastatic MTC and radioactive iodine resistant differentiated thyroid cancer is now recruiting participants at the NCI (see text footnote 2).
Concluding Remarks
Epigenetic drugs, that are expected to target the two main mechanisms of epigenetic alterations, DNA methylation, and acetylation, are regarded with increasing interest by both endocrinologists and oncologists. Definitive results from clinical trials will ultimately clarify the real effectiveness of epigenetic drugs used alone for treatment of advanced thyroid cancer. Nowadays, trials that have been concluded showed that only few patients have a complete response, suggesting that the treatment is, at best, effective in stabilizing the progressive disease.
The complexity of crosstalk between protein signaling pathways implies that a single signaling inhibitor can only produce modest effects, and suggests that multiple pathways need to be inhibited in order to substantially affect thyroid cancer growth. In this direction, epigenetic drugs used in combination with other target molecules might significantly increase the response rate to treatment in advanced thyroid cancer, either by relaxing the chromatin structure and making it more accessible to the effect of a DNA targeting drug, or by acting synergistically with anti-mitotic drugs.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
Alvarez-Nuñez, F., Bussaglia, E., Mauricio, D., Ybarra, J., Vilar, M., Lerma, E., de Leiva, A., and Matias-Guiu, X. (2006). Thyroid Neoplasia Study Group. PTEN promoter methylation in sporadic thyroid carcinomas. Thyroid 16, 17–23.
Anand-Apte, B., Bao, L., Smith, R., Iwata, K., Olsen, B. R., Zetter, B., and Apte, S. S. (1996). A review of tissue inhibitor of metalloproteinases-3 (TIMP-3) and experimental analysis of its effect on primary tumor growth. Biochem. Cell Biol. 74, 853–862.
Atadja, P. (2009). Development of the pan-DAC inhibitor panobinostat (LBH589): successes and challenges. Cancer Lett. 280, 233–241.
Baylin, S. B., and Herman, J. G. (2000). DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet. 16, 168–174.
Blaheta, R. A., Michaelis, M., Driever, P. H., and Cinatl, J. Jr. (2005). Evolving anticancer drug valproic acid: insights into the mechanism and clinical studies. Med. Res. Rev. 25, 383–397.
Borbone, E., Troncone, G., Ferraro, A., Jasencakova, Z., Stojic, L., Esposito, F., Hornig, N., Fusco, A., and Orlando, V. (2011). Enhancer of zeste homolog 2 overexpression has a role in the development of anaplastic thyroid carcinomas. J. Clin. Endocrinol. Metab. 96, 1029–1038.
Braun, J., Hoang-Vu, C., Dralle, H., and Hüttelmaier, S. (2010). Downregulation of microRNAs directs the EMT and invasive potential of anaplastic thyroid carcinomas. Oncogene 29, 4237–4244.
Brest, P., Lassalle, S., Hofman, V., Bordone, O., Gavric Tanga, V., Bonnetaud, C., Moreilhon, C., Rios, G., Santini, J., Barbry, P., Svanborg, C., Mograbi, B., Mari, B., and Hofman, P. (2011). MiR-129-5p is required for histone deacetylase inhibitor-induced cell death in thyroid cancer cells. Endocr. Relat. Cancer 18, 711–719.
Catalano, M. G., Fortunati, N., Pugliese, M., Costantino, L., Poli, R., Bosco, O., and Boccuzzi, G. (2005). Valproic acid induces apoptosis and cell cycle arrest in poorly differentiated thyroid cancer cells. J. Clin. Endocrinol. Metab. 90, 1383–1389.
Catalano, M. G., Fortunati, N., Pugliese, M., Poli, R., Bosco, O., Mastrocola, R., Aragno, M., and Boccuzzi, G. (2006). Valproic acid, a histone deacetylase inhibitor, enhances sensitivity to doxorubicin in anaplastic thyroid cancer cells. J. Endocrinol. 191, 465–472.
Catalano, M. G., Poli, R., Pugliese, M., Fortunati, N., and Boccuzzi, G. (2007). Valproic acid enhances tubulin acetylation and apoptotic activity of paclitaxel on anaplastic thyroid cancer cell lines. Endocr. Relat. Cancer 14, 839–845.
Catalano, M. G., Poli, R., Pugliese, M., Fortunati, N., and Boccuzzi, G. (2010). Emerging molecular therapies of advanced thyroid cancer. Mol. Aspects Med. 31, 215–226.
Catalano, M. G., Pugliese, M., Gargantini, E., Grange, C., Bussolati, B., Asioli, S., Bosco, O., Poli, R., Compagnone, A., Bandino, A., Mainini, F., Fortunati, N., and Boccuzzi, G. (2011). Cytotoxic activity of the histone deacetylase inhibitor panobinostat (LBH589) in anaplastic thyroid cancer in vitro and in vivo. Int. J. Cancer 130, 694–704.
Chakravarty, D., Santos, E., Ryder, M., Knauf, J. A., Liao, X. H., West, B. L., Bollag, G., Kolesnick, R., Thin, T. H., Rosen, N., Zanzonico, P., Larson, S. M., Refetoff, S., Ghossein, R., and Fagin, J. A. (2011). Small-molecule MAPK inhibitors restore radioiodine incorporation in mouse thyroid cancers with conditional BRAF activation. J. Clin. Invest. 121, 4700–4711.
Chi, P., Allis, C. D., and Wang, G. G. (2010). Covalent histone modifications–miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer 10, 457–459.
Ciampi, R., and Nikiforov, Y. E. (2005). Alterations of the BRAF gene in thyroid tumors. Endocr. Pathol. 16, 163–172.
Copland, J. A., Marlow, L. A., Williams, S. F., Grebe, S. K., Gumz, M. L., Maples, W. J., Silverman, V. E., and Smallridge, R. C. (2006). Molecular diagnosis of a BRAF papillary thyroid carcinoma with multiple chromosome abnormalities and rare adrenal and hypothalamic metastases. Thyroid 16, 1293–1302.
Donninger, H., Vos, M. D., and Clark, G. J. (2007). The RASSF1A tumor suppressor. J. Cell Sci. 120, 3163–3172.
Duenas-Gonzalez, A., Candelaria, M., Perez-Plascencia, C., Perez-Cardenas, E., de la Cruz-Hernandez, E., and Herrera, L. A. (2008). Valproic acid as epigenetic cancer drug: preclinical, clinical and transcriptional effects on solid tumors. Cancer Treat. Rev. 34, 206–222.
Durante, C., Puxeddu, E., Ferretti, E., Morisi, R., Moretti, S., Bruno, R., Barbi, F., Avenia, N., Scipioni, A., Verrienti, A., Tosi, E., Cavaliere, A., Gulino, A., Filetti, S., and Russo, D. (2007). BRAF mutations in papillary thyroid carcinomas inhibit genes involved in iodine metabolism. J. Clin. Endocrinol. Metab. 92, 2840–2843.
Elisei, R., Shiohara, M., Koeffler, H. P., and Fagin, J. A. (1998). Genetic and epigenetic alterations of the cyclin-dependent kinase inhibitors p15INK4b and p16INK4a in human thyroid carcinoma cell lines and primary thyroid carcinomas. Cancer 83, 2185–2193.
Ellis, L., Atadja, P. W., and Johnstone, R. W. (2009). Epigenetics in cancer: targeting chromatin modifications. Mol. Cancer. Ther. 8, 1409–1420.
Fagin, J. A. (2004). How thyroid tumors start and why it matters: kinase mutants as targets for solid cancer pharmacotherapy. J. Endocrinol. 183, 249–256.
Fassnacht, M., Kreissl, M. C., Weismann, D., and Allolio, B. (2009). New targets and therapeutic approaches for endocrine malignancies. Pharmacol. Ther. 123, 117–141.
Feinberg, A. P., Ohlsson, R., and Henikoff, S. (2006). The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 7, 21–33.
Finnin, M. S., Donigian, J. R., Cohen, A., Richon, V. M., Rifkind, R. A., Marks, P. A., Breslow, R., and Pavletich, N. P. (1999). Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 401, 188–193.
Fortunati, N., Catalano, M. G., Arena, K., Brignardello, E., Piovesan, A., and Boccuzzi, G. (2004). Valproic acid induces the expression of the Na+/I- symporter and iodine uptake in poorly differentiated thyroid cancer cells. J. Clin. Endocrinol. Metab. 89, 1006–1009.
Fraga, M. F., Ballestar, E., Villar-Garea, A., Boix-Chornet, M., Espada, J., Schotta, G., Bonaldi, T., Haydon, C., Ropero, S., Petrie, K., Iyer, N. G., Pérez-Rosado, A., Calvo, E., Lopez, J. A., Cano, A., Calasanz, M. J., Colomer, D., Piris, M. A., Ahn, N., Imhof, A., Caldas, C., Jenuwein, T., and Esteller, M. (2005). Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 37, 391–400.
Fuino, L., Bali, P., Wittmann, S., Donapaty, S., Guo, F., Yamaguchi, H., Wang, H. G., Atadja, P., and Bhalla, K. (2003). Histone deacetylase inhibitor LAQ824 down-regulates Her-2 and sensitizes human breast cancer cells to trastuzumab, taxotere, gemcitabine, and epothilone B. Mol. Cancer Ther. 2, 971–984.
Garcia-Rostan, G., Camp, R. L., Herrero, A., Carcangiu, M. L., Rimm, D. L., and Tallini, G. (2001). Beta-catenin dysregulation in thyroid neoplasms: down-regulation, aberrant nuclear expression, and CTNNB1 exon 3 mutations are markers for aggressive tumor phenotypes and poor prognosis. Am. J. Pathol. 158, 987–996.
Ghossein, R. (2009). Problems and controversies in the histopathology of thyroid carcinomas of follicular cell origin. Arch. Pathol. Lab. Med. 133, 683–691.
Göttlicher, M., Minucci, S., Zhu, P., Krämer, O. H., Schimpf, A., Giavara, S., Sleeman, J. P., Lo Coco, F., Nervi, C., Pelicci, P. G., and Heinzel, T. (2001). Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 17, 6969–6978.
Graff, J. R., Greenberg, V. E., Herman, J. G., Westra, W. H., Boghaert, E. R., Ain, K. B., Saji, M., Zeiger, M. A., Zimmer, S. G., and Baylin, S. B. (1998). Distinct patterns of E-cadherin CpG island methylation in papillary, follicular, Hurthle’s cell, and poorly differentiated human thyroid carcinoma. Cancer Res. 58, 2063–2066.
Hoque, M. O., Rosenbaum, E., Westra, W. H., Xing, M., Ladenson, P., Zeiger, M. A., Sidransky, D., and Umbricht, C. B. (2005). Quantitative assessment of promoter methylation profiles in thyroid neoplasms. J. Clin. Endocrinol. Metab. 90, 4011–4018.
Hu, S., Liu, D. X., Tufano, R. P., Carson, K. A., Rosenbaum, E., Cohen, Y., Holt, E. H., Kiseljak-Vassiliades, K., Rhoden, K. J., Tolaney, S., Condouris, S., Tallini, G., Westra, W. H., Umbricht, C. B., Zeiger, M. A., Califano, J. A., Vasko, V., and Xing, M. (2006). Association of aberrant methylation of tumor suppressor genes with tumor aggressiveness and BRAF mutation in papillary thyroid cancer. Int. J. Cancer. 119, 2322–2329.
Imanishi, R., Ohtsuru, A., Iwamatsu, M., Iioka, T., Namba, H., Seto, S., Yano, K., and Yamashita, S. (2002). A histone deacetylase inhibitor enhances killing of undifferentiated thyroid carcinoma cells by p53 gene therapy. J. Clin. Endocrinol. Metab. 87, 4821–4824.
Jo, Y. S., Li, S., Song, J. H., Kwon, K. H., Lee, J. C., Rha, S. Y., Lee, H. J., Sul, J. Y., Kweon, G. R., Ro, H. K., Kim, J. M., and Shong, M. (2006). Influence of the BRAF V600E mutation on expression of vascular endothelial growth factor in papillary thyroid cancer. J. Clin. Endocrinol. Metab. 91, 3667–3670.
Johnstone, R. W. (2002). Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat. Rev. Drug Discov. 1, 287–299.
Kelly, W. K., O’Connor, O. A., Krug, L. M., Chiao, J. H., Heaney, M., Curley, T., MacGregore-Cortelli, B., Tong, W., Secrist, J. P., Schwartz, L., Richardson, S., Chu, E., Olgac, S., Marks, P. A., Scher, H., and Richon, V. M. (2005). Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J. Clin. Oncol. 23, 3923–3931.
Kilfoy, B. A., Zheng, T., Holford, T. R., Han, X., Ward, M. H., Sjodin, A., Zhang, Y., Bai, Y., Zhu, C., Guo, G. L., Rothman, N., and Zhang, Y. (2009). International patterns and trends in thyroid cancer incidence, 1973–2002. Cancer Causes Control 20, 525–531.
Kim, T. H., Yoo, Y. H., Kang, D. Y., Suh, H., Park, M. K., Park, K. J., and Kim, S. H. (2009). Efficacy on anaplastic thyroid carcinoma of valproic acid alone or in combination with doxorubicin, a synthetic chenodeoxycholic acid derivative, or lactacystin. Int. J. Oncol. 34, 1353–1362.
Kitazono, M., Bates, S., Fok, P., Fojo, T., and Blagosklonny, M. V. (2002). The histone deacetylase inhibitor FR901228 (desipeptide) restores expression and function of pseudo-null p53. Cancer Biol. Ther. 1, 665–668.
Kitazono, M., Robey, R., Zhan, Z., Sarlis, N. J., Skarulis, M. C., Aikou, T., Bates, S., and Fojo, T. (2001). Low concentrations of the histone deacetylase inhibitor, depsipeptide (FR901228), increase expression of the Na(+)/I(-) symporter and iodine accumulation in poorly differentiated thyroid carcinoma cells. J. Clin. Endocrinol. Metab. 86, 3430–3435.
Knauf, J. A., Sartor, M. A., Medvedovic, M., Lundsmith, E., Ryder, M., Salzano, M., Nikiforov, Y. E., Giordano, T. J., Ghossein, R. A., and Fagin, J. A. (2011). Progression of BRAF-induced thyroid cancer is associated with epithelial-mesenchymal transition requiring concomitant MAP kinase and TGFβ signaling. Oncogene 30, 3153–3162.
Kondo, T., Nakazawa, T., Ma, D., Niu, D., Mochizuki, K., Kawasaki, T., Nakamura, N., Yamane, T., Kobayashi, M., and Katoh, R. (2009). Epigenetic silencing of TTF-1/NKX2-1 through DNA hypermethylation and histone H3 modulation in thyroid carcinomas. Lab. Invest. 89, 791–799.
Kroll, T. G., Sarraf, P., Pecciarini, L., Chen, C. J., Mueller, E., Spiegelman, B. M., and Fletcher, J. A. (2000). PAX8-PPARgamma1 fusion oncogene in human thyroid carcinoma [corrected]. Science 289, 1357–1360.
Kuo, M. H., and Allis, C. D. (1998). Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays 20, 615–626.
Kurkjian, C., Kummar, S., and Murgo, A. J. (2008). DNA methylation: its role in cancer development and therapy. Curr. Probl. Cancer 32, 187–235.
Lennard, C. M., Patel, A., Wilson, J., Reinhardt, B., Tuman, C., Fenton, C., Blair, E., Francis, G. L., and Tuttle, R. M. (2001). Intensity of vascular endothelial growth factor expression is associated with increased risk of recurrence and decreased disease-free survival in papillary thyroid cancer. Surgery 129, 552–558.
Luong, Q. T., O’Kelly, J., Braunstein, G. D., Hershman, J. M., and Koeffler, H. P. (2006). Antitumor activity of suberoylanilide hydroxamic acid against thyroid cancer cell lines in vitro and in vivo. Clin. Cancer Res. 12, 5570–5577.
Matsuyama, A., Shimazu, T., Sumida, Y., Saito, A., Yoshimatsu, Y., Seigneurin-Berny, D., Osada, H., Komatsu, Y., Nishino, N., Khochbin, S., Horinouchi, S., and Yoshida, M. (2002). In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J. 21, 6820–6831.
Miasaki, F. Y., Vivaldi, A., Ciampi, R., Agate, L., Collecchi, P., Capodanno, A., Pinchera, A., and Elisei, R. (2008). Retinoic acid receptor beta2 re-expression and growth inhibition in thyroid carcinoma cell lines after 5-aza-2′-deoxycytidine treatment. J. Endocrinol. Invest. 31, 724–730.
Mitsiades, C. S., Poulaki, V., McMullan, C., Negri, J., Fanourakis, G., Goudopoulou, A., Richon, V. M., Marks, P. A., and Mitsiades, N. (2005). Novel histone deacetylase inhibitors in the treatment of thyroid cancer. Clin. Cancer Res. 11, 3958–3965.
Nakagawa, M., Oda, Y., Eguchi, T., Aishima, S., Yao, T., Hosoi, F., Basaki, Y., Ono, M., Kuwano, M., Tanaka, M., and Tsuneyoshi, M. (2007). Expression profile of class I histone deacetylases in human cancer tissues. Oncol. Rep. 18, 769–774.
Nikiforova, M. N., Lynch, R. A., Biddinger, P. W., Alexander, E. K., Dorn, G. W. II, Tallini, G., Kroll, T. G., and Nikiforov, Y. E. (2003a). RAS point mutations and PAX8-PPAR gamma rearrangement in thyroid tumors: evidence for distinct molecular pathways in thyroid follicular carcinoma. J. Clin. Endocrinol. Metab. 88, 2318–2326.
Nikiforova, M. N., Kimura, E. T., Gandhi, M., Biddinger, P. W., Knauf, J. A., Basolo, F., Zhu, Z., Giannini, R., Salvatore, G., Fusco, A., Santoro, M., Fagin, J. A., and Nikiforov, Y. E. (2003b). BRAF mutations in thyroid tumors are restricted to papillary carcinomas and anaplastic or poorly differentiated carcinomas arising from papillary carcinomas. J. Clin. Endocrinol. Metab. 88, 5399–5404.
Nikiforova, M. N., Tseng, G. C., Steward, D., Diorio, D., and Nikiforov, Y. E. (2008). MicroRNA expression profiling of thyroid tumors: biological significance and 550 diagnostic utility. J. Clin. Endocrinol. Metab. 93, 1600–1608.
Noguchi, H., Yamashita, H., Murakami, T., Hirai, K., Noguchi, Y., Maruta, J., Yokoi, T., and Noguchi, S. (2009). Successful treatment of anaplastic thyroid carcinoma with a combination of oral valproic acid, chemotherapy, radiation and surgery. Endocr. J. 56, 245–249.
Nucera, C., Lawler, J., and Parangi, S. (2011). BRAF(V600E) and microenvironment in thyroid cancer: a functional link to drive cancer progression. Cancer Res. 71, 2417–2422.
Nucera, C., Porrello, A., Antonello, Z. A., Mekel, M., Nehs, M. A., Giordano, T. J., Gerald, D., Benjamin, L. E., Priolo, C., Puxeddu, E., Finn, S., Jarzab, B., Hodin, R. A., Pontecorvi, A., Nose, V., Lawler, J., and Parangi, S. (2010). B-Raf(V600E) and thrombospondin-1 promote thyroid cancer progression. Proc. Natl. Acad. Sci. U.S.A. 107, 10649–10654.
Patel, K. N., and Shaha, A. R. (2006). Poorly differentiated and anaplastic thyroid cancer. Cancer Control 13, 119–128.
Phiel, C. J., Zhang, F., Huang, E. Y., Guenther, M. G., Lazar, M. A., and Klein, P. S. (2001). Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J. Biol. Chem. 276, 36734–36741.
Plumb, J. A., Finn, P. W., Williams, R. J., Bandara, M. J., Romero, M. R., Watkins, C. J., La Thangue, N. B., and Brown, R. (2003). Pharmacodynamic response and inhibition of growth of human tumor xenografts by the novel histone deacetylase inhibitor PXD101. Mol. Cancer Ther. 2, 721–728.
Provenzano, M. J., Fitzgerald, M. P., Krager, K., and Domann, F. E. (2007). Increase iodine uptake in thyroid carcinoma after treatment with sodium butyrate and decitabine (5-Aza-dC). Otolaryngol. Head Neck Surg. 137, 722–728.
Puppin, C., Passon, N., Lavarone, E., Di Loreto, C., Frasca, F., Vella, V., Vigneri, R., and Damante, G. (2011). Levels of histone acetylation in thyroid tumors. Biochem. Biophys. Res. Commun. 411, 679–83.
Roman, S., Lin, R., and Sosa, J. A. (2006). Prognosis of medullary thyroid carcinoma: demographic, clinical, and pathologic predictors of survival in 1252 cases. Cancer 107, 2134–2142.
Santoro, M., Carlomagno, F., Romano, A., Bottaro, D. P., Dathan, N. A., Grieco, M., Fusco, A., Vecchio, G., Matoskova, B., Kraus, M. H., et al.. (1995). Activation of RET as a dominant transforming gene by germline mutations of MEN2A and MEN2B. Science 267, 381–383.
Sassa, M., Hayashi, Y., Watanabe, R., Kikumori, T., Imai, T., Kurebayashi, J., Kiuchi, T., and Murata, Y. (2011). Aberrant promoter methylation in overexpression of CITED1 in papillary thyroid cancer. Thyroid 21, 511–517.
Sawan, C., Vaissière, T., Murr, R., and Herceg, Z. (2008). Epigenetic drivers and genetic passengers on the road to cancer. Mutat. Res. 642, 1–13.
Schagdarsurengin, U., Gimm, O., Hoang-Vu, C., Dralle, H., Pfeifer, G. P., and Dammann, R. (2002). Frequent epigenetic silencing of the CpG island promoter of RASSF1A in thyroid carcinoma. Cancer Res. 62, 3698–3701.
Schweppe, R. E., Klopper, J. P., Korch, C., Pugazhenthi, U., Benezra, M., Knauf, J. A., Fagin, J. A., Marlow, L. A., Copland, J. A., Smallridge, R. C., and Haugen, B. R. (2008). Deoxyribonucleic acid profiling analysis of 40 human thyroid cancer cell lines reveals cross-contamination resulting in cell line redundancy and misidentification. J. Clin. Endocrinol. Metab. 93, 4331–4341.
Shen, W. T., Wong, T. S., Chung, W. Y., Wong, M. G., Kebebew, E., Duh, Q. Y., and Clark, O. H. (2005). Valproic acid inhibits growth, induces apoptosis, and modulates apoptosis-regulatory and differentiation gene expression in human thyroid cancer cells. Surgery 138, 979–984.
Sherman, S. I. (2010). Cytotoxic chemotherapy for differentiated thyroid carcinoma. Clin. Oncol. (R. Coll. Radiol.) 22, 464–468.
Smallridge, R. C., Marlow, L. A., and Copland, J. A. (2009). Anaplastic thyroid cancer: molecular pathogenesis and emerging therapies. Endocr. Relat. Cancer 16, 17–44.
Smith, B. D., Smith, G. L., Hurria, A., Hortobagyi, G. N., and Buchholz, T. A. (2009). Future of cancer incidence in the United States: burdens upon an aging, changing nation. J. Clin. Oncol. 27, 2758–2765.
Sterner, D. E., and Berger, S. L. (2000). Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 64, 435–459.
Tuncel, M., Aydin, D., Yaman, E., Tazebay, U. H., Güç, D., Dogan, A. L., Tasbasan, B., and Ugur, O. (2007). The comparative effects of gene modulators on thyroid specific genes and radioiodine uptake. Cancer Biother. Radiopharm 22, 281–288.
Venkataraman, G. M., Yatin, M., Marcinek, R., and Ain, K. B. (1999). Restoration of iodide uptake in dedifferentiated thyroid carcinoma: relationship to human NaC/IK symporter gene methylation status. J. Clin. Endocrinol. Metab. 84, 2449–2457.
Vivaldi, A., Miasaki, F. Y., Ciampi, R., Agate, L., Collecchi, P., Capodanno, A., Pinchera, A., and Elisei, R. (2009). Re-differentiation of thyroid carcinoma cell lines treated with 5-Aza-20-deoxycytidine and retinoic acid. Mol. Cell. Endocrinol. 307, 142–148.
Wiseman, S. M., Masoudi, H., Niblock, P., Turbin, D., Rajput, A., Hay, J., Filipenko, D., Huntsman, D., and Gilks, B. (2006). Derangement of the E-cadherin/catenin complex is involved in transformation of differentiated to anaplastic thyroid carcinoma. Am. J. Surg. 191, 581–587.
Woyach, J. A., Kloos, R. T., Ringel, M. D., Arbogast, D., Collamore, M., Zwiebel, J. A., Grever, M., Villalona-Calero, M., and Shah, M. H. (2009). Lack of therapeutic effect of the histone deacetylase inhibitor vorinostat in patients with metastatic radioiodine-refractory thyroid carcinoma. J. Clin. Endocrinol. Metab. 94, 164–170.
Wreesmann, V. B., and Singh, B. (2008). Clinical impact of molecular analysis on thyroid cancer management. Surg. Oncol. Clin. N. Am. 17, 1–35.
Xing, M. (2007a). BRAF mutation in papillary thyroid cancer: pathogenic role, molecular bases, and clinical implications. Endocr. Rev. 28, 742–762.
Xing, M., Westra, W. H., Tufano, R. P., Cohen, Y., Rosenbaum, E., Rhoden, K. J., Carson, K. A., Vasko, V., Larin, A., Tallini, G., Tolaney, S., Holt, E. H., Hui, P., Umbricht, C. B., Basaria, S., Ewertz, M., Tufaro, A. P., Califano, J. A., Ringel, M. D., Zeiger, M. A., Sidransky, D., and Ladenson, P. W. (2003). Methylation of the thyroid-stimulating hormone receptor gene in epithelial thyroid tumors: a marker of malignancy and a cause of gene silencing. Cancer Res. 63, 2316–2321.
Zhang, Y., Li, N., Caron, C., Matthias, G., Hess, D., Khochbin, S., and Matthias, P. (2003). HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 22, 1168–1179.
Zuo, H., Gandhi, M., Edreira, M. M., Hochbaum, D., Nimgaonkar, V. L., Zhang, P., Dipaola, J., Evdokimova, V., Altschuler, D. L., and Nikiforov, Y. E. (2010). Downregulation of Rap1GAP through epigenetic silencing and loss of heterozygosity promotes invasion and progression of thyroid tumors. Cancer Res. 70, 1389–1397.
Keywords: advanced thyroid cancer, anaplastic thyroid cancer, target therapy, epigenetic, histone deacetylase inhibitors, demethylating agents
Citation: Catalano MG, Fortunati N and Boccuzzi G (2012) Epigenetics modifications and therapeutic prospects in human thyroid cancer. Front. Endocrin. 3:40. doi: 10.3389/fendo.2012.00040
Received: 09 December 2011; Accepted: 28 February 2012;
Published online: 19 March 2012.
Edited by:
Carmelo Nucera, Beth Israel Deaconess Medical Center/Harvard Medical School, USAReviewed by:
Jeffrey Knauf, Memorial Sloan-Kettering Cancer Center, USADario Giuffrida, Instituto Oncologico Del Mediterraneo, Italy
Copyright: © 2012 Catalano, Fortunati and Boccuzzi. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Giuseppe Boccuzzi, Dipartimento di Fisiopatologia Clinica, University of Turin, Via Genova 3, 10126 Torino, Italy. e-mail:Z2l1c2VwcGUuYm9jY3V6emlAdW5pdG8uaXQ=