

- Division of Endocrinology and Diabetes, Department of Medicine 1, Friedrich-Alexander University Erlangen-Nuremberg, Erlangen, Germany
Craniopharyngiomas are slow growing benign tumors of the sellar and parasellar region with an overall incidence rate of approximately 1.3 per million. During adulthood there is a peak incidence between 40 and 44 years. There are two histopathological types, the adamantinomatous and the papillary type. The later type occurs almost exclusively in adult patients. The presenting symptoms develop over years and display a wide spectrum comprising visual, endocrine, hypothalamic, neurological, and neuropsychological manifestations. Currently, the main treatment option consists in surgical excision followed by radiation therapy in case of residual tumor. Whether gross total or partial resection should be preferred has to be balanced on an individual basis considering the extent of the tumor (e.g., hypothalamic invasion). Although the overall long-term survival is good it is often associated with substantial morbidity. Preexisting disorders are often permanent or even exacerbated by treatment. Endocrine disturbances need careful replacement and metabolic sequelae should be effectively treated. Regular follow-up by a multidisciplinary team is a prerequisite for optimal outcome of these patients.
Introduction
Craniopharyngiomas are rare benign tumors derived from cell remnants of Rathke’s pouch along a line from the nasopharynx to the diencephalon. The majority of these epithelial tumors occur in the sellar, para-, and suprasellar region (Jane and Laws, 2006; Karavitaki et al., 2006; Garnett et al., 2007). In the United States, craniopharyngiomas constitute approximately 2.5–3% of brain tumors with an incidence rate (IR) of 1.1–1.3 per million (Bunin et al., 1998; Jane and Laws, 2006; Karavitaki et al., 2006). In other parts of the world, like Japan or certain parts of Africa, IR seem to be higher (Stiller and Nectoux, 1994). A recent analysis calculated a worldwide overall IR of 1.34 in all ages (Nielsen et al., 2011). About half of the cases occur in adults with a peak IR between 40 and 44 years according to data from a Danish study (Nielsen et al., 2011). The male to female ratio is 1.1–1.4:1 (Nielsen et al., 2011). Although craniopharyngiomas are regarded as benign tumors they often infiltrate adjacent structures like the pituitary, hypothalamus, optic nerves, blood vessels, or the third ventricle thereby causing significant morbidity and mortality. As a consequence patients present with a wide range of symptoms which are often permanent or might even be exacerbated by therapy. Because of their infiltrative growth behavior and their high tendency for recurrence, the treatment is often challenging. In the present article we present an overview on craniopharyngiomas with special emphasis on patients with adult-onset of the disease.
Pathology and Pathogenesis
Craniopharyngiomas are divided in two main histological subtypes, the adamantinomatous and the papillary type, but transitional and mixed variants have also been reported (Weiner et al., 1994; Crotty et al., 1995; Louis et al., 2007). The adamantinomatous type may develop at all ages, whereas the papillary type almost exclusively occurs in adults comprising about 14–50% of the tumors in this age group (Adamson et al., 1990; Weiner et al., 1994; Karavitaki et al., 2006). The papillary type rarely presents with calcifications, is generally well-circumscribed and compared to the adamantinomatous type tumor infiltration of surrounding tissue is less frequent (Weiner et al., 1994; Crotty et al., 1995). Macroscopically craniopharyngiomas are predominantly cystic or mixed lesions although solid lesion might also occur. The tumor size varies from small, solid, and well-circumscribed tumors to large multilocular cysts invading the sella turcica and displacing adjacent cerebral structures (Crotty et al., 1995; Karavitaki et al., 2005). Most tumors are located in the sellar/parasellar region with suprasellar tumor mass extension. A smaller number of tumors are confined to the sella, or arise in the third ventricle or within the optic system (Van Effenterre and Boch, 2002).
The molecular pathogenesis of craniopharyngiomas remains widely unknown. In patients harboring the adamantinomatous variant mutations in the beta-catenin gene CTNNB1 resulting in degradation-resistant mutant forms of beta-catenin have been identified (Sekine et al., 2002; Kato et al., 2004; Buslei et al., 2005; Campanini et al., 2010). Studies in rodents showed that permanent activation of the Wnt signaling pathway, which leads to high expression levels of beta-catenin, causes formation of pituitary tumors that resemble the human adamantinomatous craniopharyngioma. Thus, reactivation of the Wnt signaling pathway may be one factor in the pathogenesis of craniopharyngiomas of the adamantinomatous type (Gaston-Massuet et al., 2011). The formation of cysts and their size appears to be associated with the expression of carbonic anhydrase IX, an enzyme causing fluid production. The specific tumor-associated regulating mechanisms, however, of this enzyme have not been clarified yet (Proescholdt et al., 2011).
Clinical Manifestation
The clinical manifestations are multiple and depend on the location of the tumor, its size, growth pattern, and relationship to adjacent cerebral structures (Table 1). As craniopharyngiomas are in general slowly growing, symptoms may develop gradually – a circumstance which may contribute to the reported delay of 1–2 years between symptom onset and diagnosis (Garnett et al., 2007). In adults the most common presenting clinical symptoms are visual field deficits and signs of hypopituitarism (Hoff and Patterson, 1972). In about 40–80% of the patients suprasellar tumor extension pressurizes the optic chiasm causing loss of visual acuity and visual field abnormalities (mostly asymmetric bitemporal hemianopsia; Crotty et al., 1995; Karavitaki et al., 2005, 2006). In a series of 78 adult patients 57% of the female patients reported about menstrual irregularities or amenorrhea and 28% complained about impaired sexual function (Karavitaki et al., 2005). Other symptoms like nausea and vomiting (26%), poor energy (32%), and lethargy (26%) are also frequent in the adult patient (Karavitaki et al., 2005). These symptoms are indicative for anterior pituitary dysfunction. Overall, growth hormone (GH) deficiency is most common, followed by gonadotropin, adrenocorticotropic hormone (ACTH), and thyroid stimulating hormone (TSH) deficiency, which are present in 86, 74, 58, and 42% of adult cases, respectively (Karavitaki et al., 2005). Compression of the pituitary stalk causes diabetes insipidus (DI) with polyuria and polydipsia in 17–38.5% of cases (Karavitaki et al., 2005; Mortini et al., 2011). Hypothalamic involvement may cause significant weight gain, which is a presenting symptom in 13–15.4% of adult patients (Karavitaki et al., 2005; Mortini et al., 2011). Severe headaches are also frequent (56%) and may be caused by raised intracranial pressure due to the tumor mass itself, to obstructive hydrocephalus resulting from compression of the third ventricle or due to leaked out cyst content which could lead to meningeal irritation (Karavitaki et al., 2006). Parasellar tumor extension with infiltration of the cavernous sinus could result in cranial nerve palsies with diplopia and paresis of ocular muscles. Involvement of the temporal lobe might trigger seizures and especially in the elderly, large tumors may cause deteriorating cognitive abilities and personality changes (Karavitaki et al., 2006).
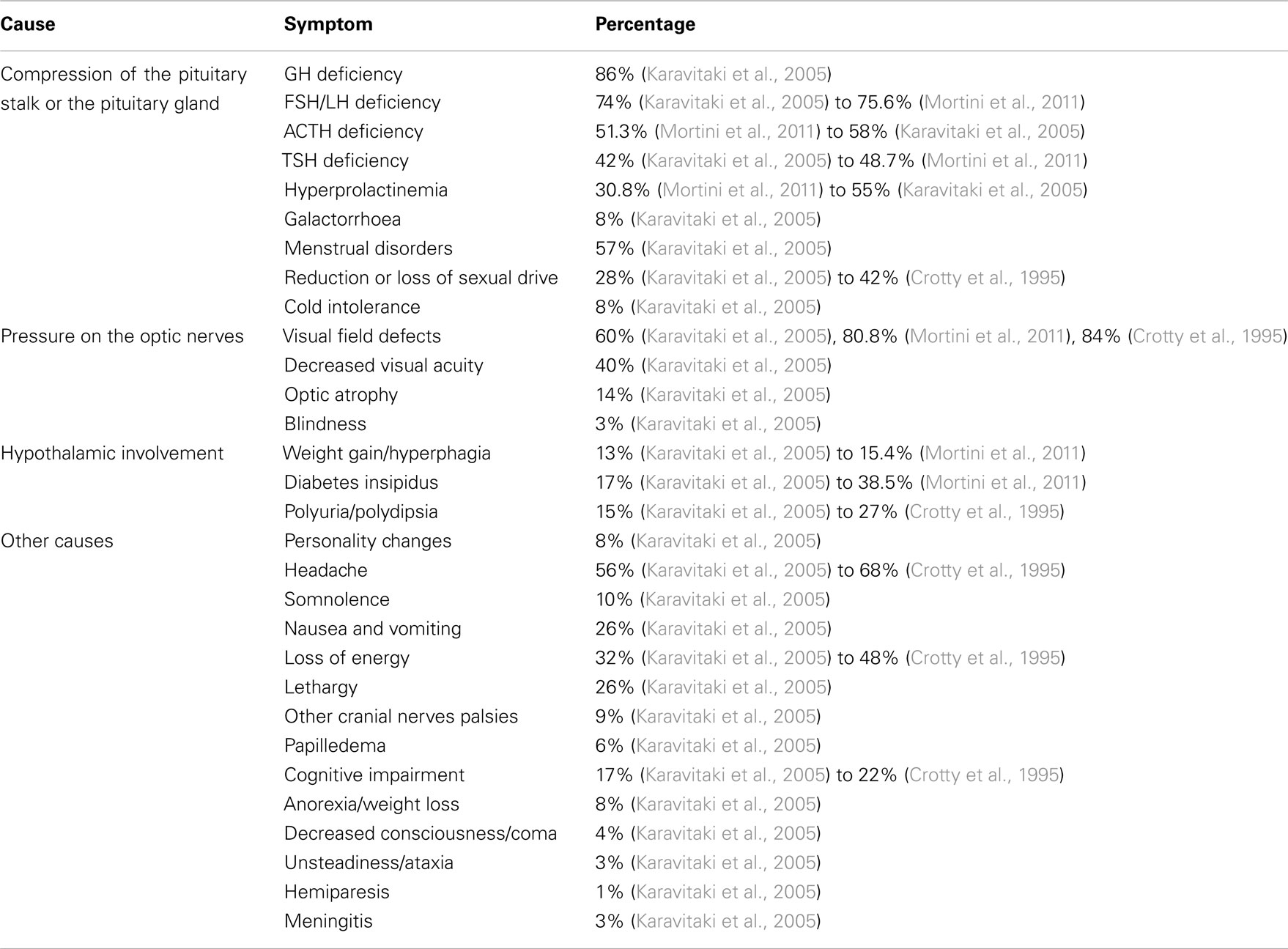
Table 1. Presenting symptoms and clinical manifestations of adults with craniopharyngioma.
Diagnosis
The suspicion of a craniopharyngioma is based on clinical and radiological findings. The diagnosis is finally proven by histology. A proposal for the initial diagnostic work-up in patients suspicious for craniopharyngioma is given in Table 2.
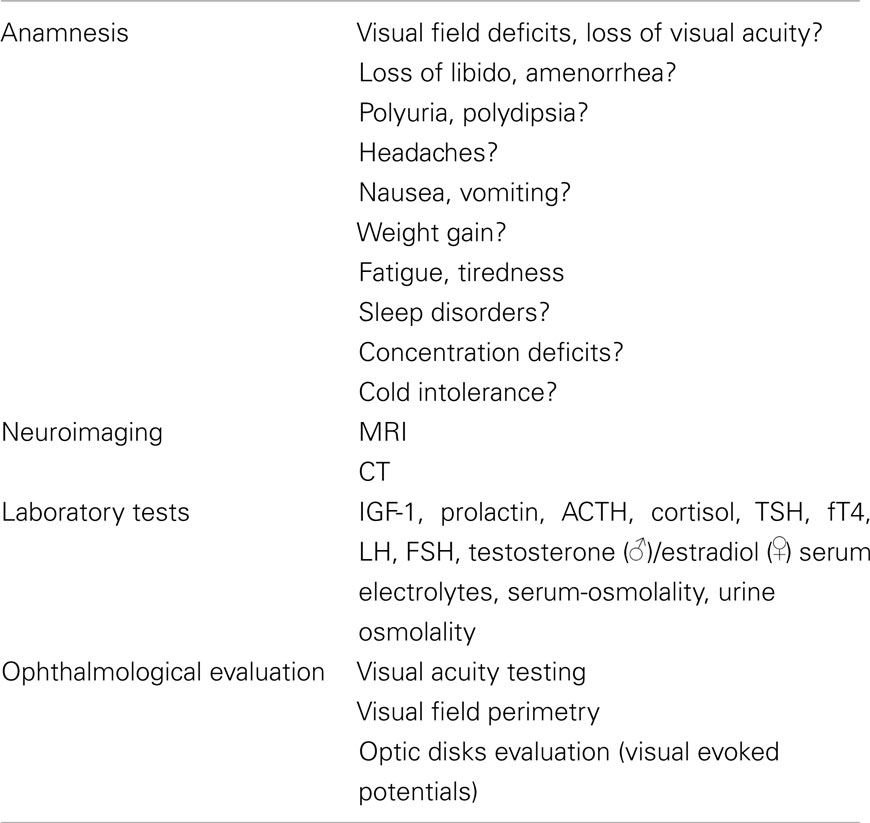
Table 2. Diagnostic work-up of patients with craniopharyngioma.
Most adult patients present with visual field deficits and signs of hypopituitarism (Hoff and Patterson, 1972). Visual field and visual acuity assessment is therefore important to determine any deficit and to establish baseline values. In addition, an examination of the optic disks is required to rule out papilledema and visual evoked potentials are recommended. An endocrine evaluation should include the assessment of anterior and posterior pituitary functions. Signs and symptoms indicative for hypopituitarism including DI should be asked for. Basal ACTH and cortisol, TSH and free T4, LH and testosterone in men and FSH and estradiol in women, IGF-1 and prolactin as well as serum electrolytes, serum, and urine osmolality are determined. Further endocrine testings should be performed according to the results and the individual clinical situation.
Cranial computer tomography (CT) and/or magnetic resonance imaging (MRI) with and without contrast enhancement are part of the neuroradiological evaluation of craniopharyngiomas. Calcifications and bony structures are best evaluated on CT, while the delineation of tumor size and the involvement of neighboring structures can be assessed most accurately by MRI. The MRI scan is therefore the method of choice in order to prepare a surgical approach (Garnett et al., 2007). According to a recent study, there are no significant differences in neuroradiological characteristics of craniopharyngiomas between children and adults with the exception of lower rates of tumor calcification in adult patients (Mortini et al., 2011). In some studies hydrocephalus occurred less frequently in adults than in children, however other authors reported no differences in this regard (Van Effenterre and Boch, 2002; Karavitaki et al., 2005). Although several attempts have been made, no reliable discrimination between the two histological subtypes is possible based on neuroradiological criteria (Crotty et al., 1995; Eldevik et al., 1996; Sartoretti-Schefer et al., 1997). In some cases the presence of a craniopharyngioma is suggested by an incidental finding of a mass lesion on neuroimaging scans. The differential diagnosis comprises other sellar and parasellar tumors like Rathke’s cleft cysts and other non-malignant cysts, pituitary adenomas, optic and hypothalamic gliomas, meningiomas, teratomas, germinomas, lymphomas, metastases, and infiltrative diseases, like sarcoidosis or systemic histiocytosis (Garnett et al., 2007).
Treatment
Prospective and controlled studies comparing different treatment strategies in adult patients are missing. Most data come from retrospective analyses of mixed populations of patients with childhood- and adult-onset disease. There are no evidence-based guidelines or a clear consensus for best treatment of primary or recurrent craniopharyngiomas in adults. Like in children a radical approach with complete tumor resection and potential cure has to be balanced with a more conservative approach to avoid substantial treatment-associated long-term morbidity. Therefore, optimal treatment should always be individualized taking into account, e.g., the patient’s symptoms, age, tumor localization, and extension. Endocrine and metabolic complications should be treated prior to any tumor-directed therapy such as surgery. This is especially important in case of adrenal or thyroidal insufficiency, as well as DI. It is generally agreed that a multidisciplinary team comprising endocrinologists, neurosurgeons, radiotherapists, and ophthalmologists is essential for optimized treatment of patients of any age (Garnett et al., 2007; Garre and Cama, 2007; Honegger and Tatagiba, 2008).
Surgery
Surgery is the first-line approach to the treatment of craniopharyngiomas. Complete surgical resection is the goal of initial therapy and because of major advances in neurosurgical techniques, it is increasingly feasible. However, due to their size, their often irregular margins and intense peritumoral gliosis with adherence to vital neighboring neurovascular structures this is often difficult and potentially dangerous. According to the literature complete tumor removal by radical surgery can be achieved in 18–84% of selected childhood and adult cases (Weiner et al., 1994; Fahlbusch et al., 1999; Van Effenterre and Boch, 2002; Karavitaki et al., 2005, 2006). Aggressive surgery with gross total resection (GTR) of the tumor, however, may result in significant and devastating peri- and postoperative morbidity especially after resection of tumors invading the hypothalamus (Rutka, 2002). If complete tumor removal is unlikely to be achieved or too hazardous, subtotal, or partial resection (PR) of the tumor, e.g., to reduce pressure effects on adjacent structures like the optic pathways and/or to re-establish circulation of the cerebrospinal fluid with or without subsequent radiotherapy is an alternative approach (Habrand et al., 1999; Cama et al., 2006). In any case the benefits of surgery must be balanced against the risks of treatment-related morbidity. For pediatric patients a preoperative grading system has been proposed according to the degree of hypothalamic involvement (Puget et al., 2007). Using this classification a significant relationship between the preoperative tumor grade and post-surgical morbidity could be demonstrated. GTR should only be attempted in patients without hypothalamic involvement (grade 0) or with a distorted or elevated but still visible hypothalamus (grade 1). In grade 2 craniopharyngiomas (hypothalamic structures not discernible) a subtotal resection leaving the hypothalamic part is recommended (Puget et al., 2007). Similar studies in adults are lacking. Nevertheless it appears reasonable to follow these recommendations established in pediatric patients. For craniopharyngiomas with major cystic portions stereotactic cyst decompression is another treatment option. This can be used for acute symptomatic therapy, prior to definite craniopharyngioma resection, or may be recommended as intermittent therapy whenever a total cyst excision is not possible (Fahlbusch et al., 1999; Honegger and Tatagiba, 2008).
Radiotherapy
Radiotherapy (RT) is a therapeutic option in patients for whom surgery is contraindicated, with residual tumor tissue after subtotal or partial surgical resection, as well as in patients with recurrent disease. Advances in RT techniques allow higher treatment precision with less long-term toxicity by focusing the ionizing radiation on the tumor and limiting the exposure of adjacent tissues to a minimum. Techniques used for the treatment of craniopharyngiomas include stereotactic fractionated radiotherapy, radiosurgery, intensity modulated radiation therapy, and proton beam therapy (Fitzek et al., 2006; Minniti et al., 2009; Niranjan et al., 2010). For fractionated conformal RT schedules the probability of local tumor control is best with radiation doses above 54–55 Gy (Regine et al., 1993) but less than 61 Gy to lower the risk of radiation-associated side-effects such as visual impairment, pituitary deficiency, impaired cognitive function or development of secondary malignancies (Cavazzuti et al., 1983; Merchant et al., 2006). Application of radiosurgery with delivery of a single high-dose radiation to the tumor is an attractive technique but restricted to smaller tumor volumes and requires sufficient safety margins of adjacent critical structures. Although reports published so far are promising (Minniti et al., 2009; Iwata et al., 2012), further studies are needed to define its role in the prevention of tumor recurrence and its potential long-term adverse effects. Intracavitary irradiation with stereotactically guided instillation of radioisotopes is another approach to treat mono- or multicystic tumors (Voges et al., 1997; Barriger et al., 2011).
Other Treatments
Other treatment options for predominantly cystic craniopharyngiomas comprise intracystic chemotherapy or immunological therapy with bleomycin or interferon alpha, respectively (Steinbok and Hukin, 2010). The experience regarding these treatments is mainly restricted to pediatric patients. In general, these options were used for temporary tumor control with the expectation for further therapies like surgery or radiotherapy. Systemic chemotherapy is generally regarded as ineffective (Karavitaki et al., 2006).
Recurrent Disease
Recurrence of craniopharyngiomas remains a problem which highly impacts on the long-term prognosis of the patients. In a recent study with 106 patients including 78 adults, 24.5% experienced a relapse within a median follow-up of 83 months (Mortini et al., 2011). The risk of recurrence appears highest in the first 3 years after surgery and then tends to reach a plateau (Fahlbusch et al., 1999; Karavitaki et al., 2005). After GTR the risk of tumor recurrence has been reported to range between 0 and 26% (Maira et al., 1995; Fahlbusch et al., 1999; Duff et al., 2000). This is significantly lower than in patients with subtotal or partial tumor resection in whom tumor recurrence occurs in 25–100% of the patients (Fahlbusch et al., 1999; Duff et al., 2000; Karavitaki et al., 2005). In these patients adjuvant RT significantly improves the tumor control rates with recurrence rates ranging from 10 to 63% at 10-years follow-up (Karavitaki et al., 2006). Thus, patients with subtotal resection without subsequent RT have the highest risk of relapse (Duff et al., 2000; Karavitaki et al., 2005). In patients treated with RT alone tumor growth or recurrence was observed in 0–23% of the patients at 10 years follow-up (Hetelekidis et al., 1993; Rajan et al., 1993). The data about the prognostic significance of age at diagnosis, gender, tumor size, location, or consistency, as well as the histological subtype are inconsistent (Karavitaki et al., 2005, 2006; Mortini et al., 2011), but in general these factors do not seem to affect the risk of recurrence (Karavitaki et al., 2006).
Treatment of Recurrent Disease
Recurrent disease is difficult to treat. Because of scars and adhesions due to previous surgery or radiation, the surgical success rates for recurrent disease are significantly lower than for primary surgery and peri- and postoperative morbidity and mortality is significantly increased (Yasargil et al., 1990; Wisoff, 1994; Fahlbusch et al., 1999; Karavitaki et al., 2005). Therefore, the severity of clinical symptoms has to be taken into account and it has been suggested that repeated surgery should only be performed when acute pressure effects occur (Karavitaki et al., 2006). In patients not previously treated with RT irradiation is another option either instead of or following a second surgery, and it appears that RT is equally effective in the control of recurrent tumors as for primary treatment (Jose et al., 1992). Further options comprise cyst controlling procedures like aspiration, intracystic irradiation or application of bleomycin, salvage surgery in case of life-threatening solid lesions, or radiosurgery. In any case treatment decisions for recurrent disease should be made on an individualized basis.
Disease and Treatment Complications
Endocrine
Anterior pituitary dysfunction and DI are common in adult patients with craniopharyngiomas and the majority of adult craniopharyngioma patients present with signs and symptoms of hypopituitarism (Paja et al., 1995; Honegger et al., 1999; Karavitaki et al., 2005). Neuroendocrine dysfunction may worsen upon treatment. In a recent study by Mortini et al. (2011) 82.3, 75.9, 72.7, and 66.7% of patients with normal baseline values for GH, ACTH, TSH, and gonadotropins developed a new deficiency of the respective pituitary axis after surgery. Post-surgical onset of DI was observed in 69.6% of their patients. The risk for new hormone deficiencies appears to be lower after transsphenoidal operation (Honegger et al., 1999; Mortini et al., 2011). In contrast to pituitary adenomas, recovery of preexisting pituitary dysfunction after surgery is rare (Webb et al., 1999; Mortini et al., 2011). Although symptomatic DI appears to occur more frequent in surgically treated patients (Hetelekidis et al., 1993; Karavitaki et al., 2006), long-term endocrine morbidity in general seems not to be affected by the type of therapy (Weiner et al., 1994; Habrand et al., 1999; Karavitaki et al., 2005). Most of the patients chronically suffer from partial or complete hypopituitarism as well as DI, with approximately 80% requiring the substitution of more than two hormones (Kendall-Taylor et al., 2005; Verhelst et al., 2005). Concerning GH substitution, observational studies suggest that GH replacement does not increase the risk of tumor recurrence (Abs et al., 1999). Because of the often complex endocrine morbidity lifelong surveillance by an endocrinologist is required.
Visual
In adults impaired visual function is common at first presentation (Crotty et al., 1995; Karavitaki et al., 2005, 2006). After surgical decompression visual deficits often improve, but may also remain unchanged or even become worse (Fahlbusch et al., 1999; Van Effenterre and Boch, 2002; Mortini et al., 2011). In a series of 173 patients treated with RT only or after limited surgery, improvement in visual field defects or in visual acuity was observed in 36 and 30%, respectively after a median follow-up of 12 years (Rajan et al., 1993). In about one-third of the patients pretreatment visual deficits deteriorated (Rajan et al., 1993). No radiation optic neuropathy developed by applying accurate RT techniques and doses below the tolerance limit of the optic system (Harris and Levene, 1976; Rajan et al., 1993). Karavitaki et al. (2005) reported in their series of patients with long-term follow-up about major visual field defects in 49 and 72% of the patients after 10 and 20 years, respectively. This reflects treatment-associated deterioration of visual function on the one hand and on the other hand the natural course of the disease with a high risk for recurrence. Risk factors for adverse visual outcome are the presence of visual symptoms at diagnosis, irradiation dose above 2 Gy per day, and a PR of the tumor, probably because of the increased risk of recurrence (Harris and Levene, 1976; Karavitaki et al., 2005, 2006). Long-term follow-up by an ophthalmologist is recommended.
Hypothalamic and Metabolic
Tumor- or treatment-related damage of the ventromedial hypothalamus may lead to the impairment of mechanisms controlling satiety, hunger, and energy expenditure resulting in severe obesity (Daousi et al., 2005). Hypothalamic obesity is the most common manifestation of hypothalamic complications. At presentation about 15% of adult patients complain about excessive weight gain or are obese (Karavitaki et al., 2005; Mortini et al., 2011). During long-time follow-up excessive weight gain has been reported in up to 67% of patients after surgery with and without adjuvant RT (Hoffman et al., 1992; Karavitaki et al., 2005). Hypothalamic obesity is often associated with disastrous metabolic and psychological consequences leading to severe morbidity, impaired quality of life, and reduced life expectancy (Karavitaki et al., 2006). Features of the metabolic syndrome like abdominal obesity, dyslipidemia, hyperinsulinemia caused by insulin resistance, and elevated blood pressure are commonly seen in patients with craniopharyngiomas (Kendall-Taylor et al., 2005; Pereira et al., 2005; Holmer et al., 2009). In a retrospective analysis from the KIMS database patients with adult-onset disease were more obese, had a greater waist circumference, and had higher cholesterol and triglycerides levels with similar HDL- and LDL-cholesterol when compared to patients with childhood disease onset (Kendall-Taylor et al., 2005). Together, these metabolic alterations increase the risk of hypertension, diabetes, and atherosclerosis, which all lead to cardiovascular disease and mortality from vascular events (Van Gaal et al., 2006). Thus, the necessity for treatment is vital but treatment of hypothalamic obesity is difficult and patients need to comply with dietary and behavioral modifications comprising regular physical activities, require anti obesity drugs, or even bariatric surgery (Karavitaki et al., 2006). DI with an absent sense of thirst is another hypothalamic complication resulting in serious water and electrolyte imbalances, which was observed in 19% of adult patients after surgery with and without adjuvant RT (Smith et al., 2004). Sleep disorders and increased daytime somnolence caused by disruption of the circadian rhythm occur in up to one-third of adult craniopharyngioma patients (van der Klaauw et al., 2008). Impaired thermoregulation with hyper- or hypothermia has also been observed in adults (Lipton et al., 1981; Griffiths et al., 1988). Risk factors for hypothalamic complications are preexisting disorders at diagnosis, young age at presentation of symptoms, hypothalamic invasion, tumor size, multiple operations due to recurrence, and hypothalamic radiation with doses above 51 Gy (de Vile et al., 1996; Lustig et al., 2003; Poretti et al., 2004).
Neuropsychological and Cognitive Dysfunction
Behavioral problems and deteriorated cognitive functions are relatively common, which contribute to decreased academic and occupational performance, difficulties in maintaining family, and social relationships, resulting in a loss in quality of life (Cavazzuti et al., 1983; Karavitaki et al., 2006).
Others
In patients who received RT vascular damage like aneurysms and secondary brain tumors may occur (Enchev et al., 2009; Liu et al., 2009).
Long-Term Prognosis and Mortality
Control of tumor growth and the disease- or treatment-related complications determine the long-term prognosis. Overall survival rates reported range from 89 to 94% at 5-years and from 85 to 90% at 10-years follow-up (Van Effenterre and Boch, 2002; Karavitaki et al., 2005). Nevertheless, overall mortality appears three to five times higher than those of the general population (Sherlock et al., 2010). Currently, it is unclear which therapeutic strategy (e.g., GTR or subtotal or PR followed by radiotherapy) is associated with a better survival (Karavitaki et al., 2005; Sherlock et al., 2010) and whether the histological subtype is of relevance (Weiner et al., 1994). Recurrent disease, however, is significantly associated with increased morbidity and mortality, resulting in lower 10-year survival rates (29–70%; Karavitaki et al., 2005; Honegger and Tatagiba, 2008). Apart from mortality caused by the tumor itself or by surgical treatment, mortality from cardio- and cerebrovascular as well as respiratory causes is increased (Bulow et al., 1998; Tomlinson et al., 2001). It is therefore essential to optimize the treatment of any endocrine and metabolic sequelae of the disease in order to reduce mortality in this high risk patient population.
Conclusion
Although craniopharyngiomas are generally benign their location, size, and tendency to infiltrate adjacent cerebral structures makes their management rather demanding and may lead to sometimes devastating complications. According to the data available adult- and childhood-onset craniopharyngiomas behave similar in many aspects. Treatment decisions for primary or recurrent disease need to consider long-term tumor control as well as disease and treatment-related morbidity and they are often reached on an individualized basis. Surgery is the initial treatment approach, which should remove as much tumor as safely possible, while avoiding severe treatment-induced complications. There is some controversy as to whether a more radical surgical approach with gross total tumor resection should be aimed at or a more limited approach with reducing the tumor mass followed by radiotherapy in order to minimize long-term morbidity. As some patients with subtotal or partial tumor resection may have stable disease and since RT may cause long-term complications it is currently unclear, whether all patients with residual tumor should receive immediate postoperative RT. Likewise it is unknown which therapeutic strategy is best in patients with tumor recurrence despite RT. Posttreatment follow-up should include neuroimaging with MRI, visual assessment, and monitoring of endocrine functions and metabolic status. Endocrine and metabolic alterations need to be treated adequately. There is no doubt that for optimal results a lifelong surveillance of an experienced multidisciplinary team is essential.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Emerging Field Initiative “Neurotrition” of the Friedrich-Alexander University Erlangen-Nuremberg, Germany. We thank Dr. Josefine Römmler for critical reading of the manuscript and her valuable comments.
References
Abs, R., Bengtsson, B. A., Hernberg-Stahl, E., Monson, J. P., Tauber, J. P., Wilton, P., and Wuster, C. (1999). GH replacement in 1034 growth hormone deficient hypopituitary adults: demographic and clinical characteristics, dosing and safety. Clin. Endocrinol. (Oxf.) 50, 703–713.
Adamson, T. E., Wiestler, O. D., Kleihues, P., and Yasargil, M. G. (1990). Correlation of clinical and pathological features in surgically treated craniopharyngiomas. J. Neurosurg. 73, 12–17.
Barriger, R. B., Chang, A., Lo, S. S., Timmerman, R. D., DesRosiers, C., Boaz, J. C., and Fakiris, A. J. (2011). Phosphorus-32 therapy for cystic craniopharyngiomas. Radiother. Oncol. 98, 207–212.
Bulow, B., Attewell, R., Hagmar, L., Malmstrom, P., Nordstrom, C. H., and Erfurth, E. M. (1998). Postoperative prognosis in craniopharyngioma with respect to cardiovascular mortality, survival, and tumor recurrence. J. Clin. Endocrinol. Metab. 83, 3897–3904.
Bunin, G. R., Surawicz, T. S., Witman, P. A., Preston-Martin, S., Davis, F., and Bruner, J. M. (1998). The descriptive epidemiology of craniopharyngioma. J. Neurosurg. 89, 547–551.
Buslei, R., Nolde, M., Hofmann, B., Meissner, S., Eyupoglu, I. Y., Siebzehnrubl, F., Hahnen, E., Kreutzer, J., and Fahlbusch, R. (2005). Common mutations of beta-catenin in adamantinomatous craniopharyngiomas but not in other tumours originating from the sellar region. Acta Neuropathol. 109, 589–597.
Cama, A., Ravegnani, M., Piatelli, G., Rossi, A., Gandolfo, C., and Garre, M. L. (2006). Conservative surgical approach in treatment strategy of craniopharyngioma: experience at a single institution in Italy. J. Pediatr. Endocrinol. Metab. 19(Suppl. 1), 337–340.
Campanini, M. L., Colli, L. M., Paixao, B. M., Cabral, T. P., Amaral, F. C., Machado, H. R., Neder, L. S., Saggioro, F., Moreira, A. C., Antonini, S. R., and de Castro, M. (2010). CTNNB1 gene mutations, pituitary transcription factors, and MicroRNA expression involvement in the pathogenesis of adamantinomatous craniopharyngiomas. Horm. Cancer 1, 187–196.
Cavazzuti, V., Fischer, E. G., Welch, K., Belli, J. A., and Winston, K. R. (1983). Neurological and psychophysiological sequelae following different treatments of craniopharyngioma in children. J. Neurosurg. 59, 409–417.
Crotty, T. B., Scheithauer, B. W., Young, W. F. Jr., Davis, D. H., Shaw, E. G., Miller, G. M., and Burger, P. C. (1995). Papillary craniopharyngioma: a clinicopathological study of 48 cases. J. Neurosurg. 83, 206–214.
Daousi, C., Dunn, A. J., Foy, P. M., MacFarlane, I. A., and Pinkney, J. H. (2005). Endocrine and neuroanatomic features associated with weight gain and obesity in adult patients with hypothalamic damage. Am. J. Med. 118, 45–50.
de Vile, C. J., Grant, D. B., Hayward, R. D., Kendall, B. E., Neville, B. G., and Stanhope, R. (1996). Obesity in childhood craniopharyngioma: relation to post-operative hypothalamic damage shown by magnetic resonance imaging. J. Clin. Endocrinol. Metab. 81, 2734–2737.
Duff, J. M., Mayer, F. B., Ilstrup, D. M., Laws, E. R. Jr., Scleck, C. D., and Scheithauer, B. W. (2000). Long-term outcomes for surgically resected craniopharyngiomas. Neurosurgery 46, 291–302.
Eldevik, O. P., Blaivas, M., Gabrielsen, T. O., Hald, J. K., and Chandler, W. F. (1996). Craniopharyngioma: radiologic and histologic findings and recurrence. AJNR Am. J. Neuroradiol. 17, 1427–1439.
Enchev, Y., Ferdinandov, D., Kounin, G., Encheva, E., and Bussarsky, V. (2009). Radiation-induced gliomas following radiotherapy for craniopharyngiomas: a case report and review of the literature. Clin. Neurol. Neurosurg. 111, 591–596.
Fahlbusch, R., Honegger, J., Paulus, W., Huk, W., and Buchfelder, M. (1999). Surgical treatment of craniopharyngiomas: experience with 168 patients. J. Neurosurg. 90, 237–250.
Fitzek, M. M., Linggood, R. M., Adams, J., and Munzenrider, J. E. (2006). Combined proton and photon irradiation for craniopharyngioma: long-term results of the early cohort of patients treated at Harvard Cyclotron Laboratory and Massachusetts General Hospital. Int. J. Radiat. Oncol. Biol. Phys. 64, 1348–1354.
Garnett, M. R., Puget, S., Grill, J., and Sainte-Rose, C. (2007). Craniopharyngioma. Orphanet. J. Rare Dis. 2, 18.
Garre, M. L., and Cama, A. (2007). Craniopharyngioma: modern concepts in pathogenesis and treatment. Curr. Opin. Pediatr. 19, 471–479.
Gaston-Massuet, C., Andoniadou, C. L., Signore, M., Jayakody, S. A., Charolidi, N., Kyeyune, R., Vernay, B., Jacques, T. S., Taketo, M. M., Le Tissier, P., Dattani, M. T., and Martinez-Barbera, J. P. (2011). Increased Wingless (Wnt) signaling in pituitary progenitor/stem cells gives rise to pituitary tumors in mice and humans. Proc. Natl. Acad. Sci. U.S.A. 108, 11482–11487.
Griffiths, A. P., Henderson, M., Penn, N. D., and Tindall, H. (1988). Haematological, neurological and psychiatric complications of chronic hypothermia following surgery for craniopharyngioma. Postgrad. Med. J. 64, 617–620.
Habrand, J. L., Ganry, O., Couanet, D., Rouxel, V., Levy-Piedbois, C., Pierre-Kahn, A., and Kalifa, C. (1999). The role of radiation therapy in the management of craniopharyngioma: a 25-year experience and review of the literature. Int. J. Radiat. Oncol. Biol. Phys. 44, 255–263.
Harris, J. R., and Levene, M. B. (1976). Visual complications following irradiation for pituitary adenomas and craniopharyngiomas. Radiology 120, 167–171.
Hetelekidis, S., Barnes, P. D., Tao, M. L., Fischer, E. G., Schneider, L., Scott, R. M., and Tarbell, N. J. (1993). 20-year experience in childhood craniopharyngioma. Int. J. Radiat. Oncol. Biol. Phys. 27, 189–195.
Hoff, J. T., and Patterson, R. H. Jr. (1972). Craniopharyngiomas in children and adults. J. Neurosurg. 36, 299–302.
Hoffman, H. J., De Silva, M., Humphreys, R. P., Drake, J. M., Smith, M. L., and Blaser, S. I. (1992). Aggressive surgical management of craniopharyngiomas in children. J. Neurosurg. 76, 47–52.
Holmer, H., Ekman, B., Bjork, J., Nordstom, C. H., Popovic, V., Siversson, A., and Erfurth, E. M. (2009). Hypothalamic involvement predicts cardiovascular risk in adults with childhood onset craniopharyngioma on long-term GH therapy. Eur. J. Endocrinol. 161, 671–679.
Honegger, J., Buchfelder, M., and Fahlbusch, R. (1999). Surgical treatment of craniopharyngiomas: endocrinological results. J. Neurosurg. 90, 251–257.
Iwata, H., Tatewaki, K., Inoue, M., Yokota, N., Baba, Y., Nomura, R., Shibamoto, Y., and Sato, K. (2012). Single and hypofractionated stereotactic radiotherapy with CyberKnife for craniopharyngioma. J. Neurooncol. 106, 571–577.
Jose, C. C., Rajan, B., Ashley, S., Marsh, H., and Brada, M. (1992). Radiotherapy for the treatment of recurrent craniopharyngioma. Clin. Oncol. (R. Coll. Radiol.) 4, 287–289.
Karavitaki, N., Brufani, C., Warner, J. T., Adams, C. B., Richards, P., Ansorge, O., Shine, B., Turner, H. E., and Wass, J. A. (2005). Craniopharyngiomas in children and adults: systematic analysis of 121 cases with long-term follow-up. Clin. Endocrinol. (Oxf.) 62, 397–409.
Karavitaki, N., Cudlip, S., Adams, C. B., and Wass, J. A. (2006). Craniopharyngiomas. Endocr. Rev. 27, 371–397.
Kato, K., Nakatani, Y., Kanno, H., Inayama, Y., Ijiri, R., Nagahara, N., Miyake, T., Tanaka, M., Ito, Y., Aida, N., Tachibana, K., Sekido, K., and Tanaka, Y. (2004). Possible linkage between specific histological structures and aberrant reactivation of the Wnt pathway in adamantinomatous craniopharyngioma. J. Pathol. 203, 814–821.
Kendall-Taylor, P., Jonsson, P. J., Abs, R., Erfurth, E. M., Koltowska-Haggstrom, M., Price, D. A., and Verhelst, J. (2005). The clinical, metabolic and endocrine features and the quality of life in adults with childhood-onset craniopharyngioma compared with adult-onset craniopharyngioma. Eur. J. Endocrinol. 152, 557–567.
Lipton, J. M., Rosenstein, J., and Sklar, F. H. (1981). Thermoregulatory disorders after removal of a craniopharyngioma from the third cerebral ventricle. Brain Res. Bull. 7, 369–373.
Liu, A. K., Bagrosky, B., Fenton, L. Z., Gaspar, L. E., Handler, M. H., McNatt, S. A., and Foreman, N. K. (2009). Vascular abnormalities in pediatric craniopharyngioma patients treated with radiation therapy. Pediatr. Blood Cancer 52, 227–230.
Louis, D. N., Ohgaki, H., Wiestler, O. D., Cavenee, W. K., Burger, P. C., Jouvet, A., Scheithauer, B. W., and Kleihues, P. (2007). The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 114, 97–109.
Lustig, R. H., Post, S. R., Srivannaboon, K., Rose, S. R., Danish, R. K., Burghen, G. A., Xiong, X., Wu, S., and Merchant, T. E. (2003). Risk factors for the development of obesity in children surviving brain tumors. J. Clin. Endocrinol. Metab. 88, 611–616.
Maira, G., Anile, C., Rossi, G. F., and Colosimo, C. (1995). Surgical treatment of craniopharyngiomas: an evaluation of the transsphenoidal and pterional approaches. Neurosurgery 36, 715–724.
Merchant, T. E., Kiehna, E. N., Kun, L. E., Mulhern, R. K., Li, C., Xiong, X., Boop, F. A., and Sanford, R. A. (2006). Phase II trial of conformal radiation therapy for pediatric patients with craniopharyngioma and correlation of surgical factors and radiation dosimetry with change in cognitive function. J. Neurosurg. 104, 94–102.
Minniti, G., Esposito, V., Amichetti, M., and Enrici, R. M. (2009). The role of fractionated radiotherapy and radiosurgery in the management of patients with craniopharyngioma. Neurosurg. Rev. 32, 125–132.
Mortini, P., Losa, M., Pozzobon, G., Barzaghi, R., Riva, M., Acerno, S., Angius, D., Weber, G., Chiumello, G., and Giovanelli, M. (2011). Neurosurgical treatment of craniopharyngioma in adults and children: early and long-term results in a large case series. J. Neurosurg. 114, 1350–1359.
Nielsen, E. H., Feldt-Rasmussen, U., Poulsgaard, L., Kristensen, L. O., Astrup, J., Jorgensen, J. O., Bjerre, P., Andersen, M., Andersen, C., Jorgensen, J., Lindholm, J., and Laurberg, P. (2011). Incidence of craniopharyngioma in Denmark (n = 189) and estimated world incidence of craniopharyngioma in children and adults. J. Neurooncol. 104, 755–763.
Niranjan, A., Kano, H., Mathieu, D., Kondziolka, D., Flickinger, J. C., and Lunsford, L. D. (2010). Radiosurgery for craniopharyngioma. Int. J. Radiat. Oncol. Biol. Phys. 78, 64–71.
Paja, M., Lucas, T., Garcia-Uria, J., Salame, F., Barcelo, B., and Estrada, J. (1995). Hypothalamic-pituitary dysfunction in patients with craniopharyngioma. Clin. Endocrinol. (Oxf.) 42, 467–473.
Pereira, A. M., Schmid, E. M., Schutte, P. J., Voormolen, J. H., Biermasz, N. R., van Thiel, S. W., Corssmit, E. P., Smit, J. W., Roelfsema, F., and Romijn, J. A. (2005). High prevalence of long-term cardiovascular, neurological and psychosocial morbidity after treatment for craniopharyngioma. Clin. Endocrinol. (Oxf.) 62, 197–204.
Poretti, A., Grotzer, M. A., Ribi, K., Schonle, E., and Boltshauser, E. (2004). Outcome of craniopharyngioma in children: long-term complications and quality of life. Dev. Med. Child Neurol. 46, 220–229.
Proescholdt, M., Merrill, M., Stoerr, E. M., Lohmeier, A., Dietmaier, W., and Brawanski, A. (2011). Expression of carbonic anhydrase IX in craniopharyngiomas. J. Neurosurg. 115, 796–801.
Puget, S., Garnett, M., Wray, A., Grill, J., Habrand, J. L., Bodaert, N., Zerah, M., Bezerra, M., Renier, D., Pierre-Kahn, A., and Sainte-Rose, C. (2007). Pediatric craniopharyngiomas: classification and treatment according to the degree of hypothalamic involvement. J. Neurosurg. 106, 3–12.
Rajan, B., Ashley, S., Gorman, C., Jose, C. C., Horwich, A., Bloom, H. J., Marsh, H., and Brada, M. (1993). Craniopharyngioma – a long-term results following limited surgery and radiotherapy. Radiother. Oncol. 26, 1–10.
Regine, W. F., Mohiuddin, M., and Kramer, S. (1993). Long-term results of pediatric and adult craniopharyngiomas treated with combined surgery and radiation. Radiother. Oncol. 27, 13–21.
Sartoretti-Schefer, S., Wichmann, W., Aguzzi, A., and Valavanis, A. (1997). MR differentiation of adamantinous and squamous-papillary craniopharyngiomas. AJNR Am. J. Neuroradiol. 18, 77–87.
Sekine, S., Shibata, T., Kokubu, A., Morishita, Y., Noguchi, M., Nakanishi, Y., Sakamoto, M., and Hirohashi, S. (2002). Craniopharyngiomas of adamantinomatous type harbor beta-catenin gene mutations. Am. J. Pathol. 161, 1997–2001.
Sherlock, M., Ayuk, J., Tomlinson, J. W., Toogood, A. A., Aragon-Alonso, A., Sheppard, M. C., Bates, A. S., and Stewart, P. M. (2010). Mortality in patients with pituitary disease. Endocr. Rev. 31, 301–342.
Smith, D., Finucane, F., Phillips, J., Baylis, P. H., Finucane, J., Tormey, W., and Thompson, C. J. (2004). Abnormal regulation of thirst and vasopressin secretion following surgery for craniopharyngioma. Clin. Endocrinol. (Oxf.) 61, 273–279.
Steinbok, P., and Hukin, J. (2010). Intracystic treatments for craniopharyngioma. Neurosurg. Focus 28, E13.
Stiller, C. A., and Nectoux, J. (1994). International incidence of childhood brain and spinal tumours. Int. J. Epidemiol. 23, 458–464.
Tomlinson, J. W., Holden, N., Hills, R. K., Wheatley, K., Clayton, R. N., Bates, A. S., Sheppard, M. C., and Stewart, P. M. (2001). Association between premature mortality and hypopituitarism. West Midlands Prospective Hypopituitary Study Group. Lancet 357, 425–431.
van der Klaauw, A. A., Biermasz, N. R., Pereira, A. M., van Kralingen, K. W., Dekkers, O. M., Rabe, K. F., Smit, J. W., and Romijn, J. A. (2008). Patients cured from craniopharyngioma or nonfunctioning pituitary macroadenoma (NFMA) suffer similarly from increased daytime somnolence despite normal sleep patterns compared to healthy controls. Clin. Endocrinol. (Oxf.) 69, 769–774.
Van Effenterre, R., and Boch, A. L. (2002). Craniopharyngioma in adults and children: a study of 122 surgical cases. J. Neurosurg. 97, 3–11.
Van Gaal, L. F., Mertens, I. L., and De Block, C. E. (2006). Mechanisms linking obesity with cardiovascular disease. Nature 444, 875–880.
Verhelst, J., Kendall-Taylor, P., Erfurth, E. M., Price, D. A., Geffner, M., Koltowska-Haggstrom, M., Jonsson, P. J., Wilton, P., and Abs, R. (2005). Baseline characteristics and response to 2 years of growth hormone (GH) replacement of hypopituitary patients with GH deficiency due to adult-onset craniopharyngioma in comparison with patients with nonfunctioning pituitary adenoma: data from KIMS (Pfizer International Metabolic Database). J. Clin. Endocrinol. Metab. 90, 4636–4643.
Voges, J., Sturm, V., Lehrke, R., Treuer, H., Gauss, C., and Berthold, F. (1997). Cystic craniopharyngioma: long-term results after intracavitary irradiation with stereotactically applied colloidal beta-emitting radioactive sources. Neurosurgery 40, 263–269.
Webb, S. M., Rigla, M., Wagner, A., Oliver, B., and Bartumeus, F. (1999). Recovery of hypopituitarism after neurosurgical treatment of pituitary adenomas. J. Clin. Endocrinol. Metab. 84, 3696–3700.
Weiner, H. L., Wisoff, J. H., Rosenberg, M. E., Kupersmith, M. J., Cohen, H., Zagzag, D., Shiminski-Maher, T., Flamm, E. S., Epstein, F. J., and Miller, D. C. (1994). Craniopharyngiomas: a clinicopathological analysis of factors predictive of recurrence and functional outcome. Neurosurgery 35, 1001–1010; discussion 1010–1011.
Wisoff, J. H. (1994). Surgical management of recurrent craniopharyngiomas. Pediatr. Neurosurg. 21(Suppl. 1), 108–113.
Keywords: craniopharyngioma, adults, treatment, diagnosis, complications
Citation: Zoicas F and Schöfl C (2012) Craniopharyngioma in adults. Front. Endocrin. 3:46. doi: 10.3389/fendo.2012.00046
Received: 29 February 2012; Accepted: 11 March 2012;
Published online: 29 March 2012.
Edited by:
Hermann Lothar Mueller, Klinikum Oldenburg GmbH, GermanyReviewed by:
Vera Popovic-Brkic, Clinical Center and School of Medicine University Belgrade, SerbiaJörg Flitsch, University Hospital Hamburg-Eppendorf, Germany
Copyright: © 2012 Zoicas and Schöfl. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Christof Schöfl, Division of Endocrinology and Diabetes, Department of Medicine 1, Friedrich-Alexander University Erlangen-Nuremberg, Ulmenweg 18, 91054 Erlangen, Germany. e-mail:Y2hyaXN0b2Yuc2Nob2VmbEB1ay1lcmxhbmdlbi5kZQ==