

- 1 Department of Radiology, College of Medicine, Yonsei University, Seoul, Korea
- 2 Department of Pathology, Miller School of Medicine, University of Miami, Miami, FL, USA
Follicular cell-derived well-differentiated thyroid cancer, papillary (PTC) and follicular thyroid carcinomas comprise 95% of all thyroid malignancies. Familial follicular cell-derived well-differentiated thyroid cancers contribute 5% of cases. Such familial follicular cell-derived carcinomas or non-medullary thyroid carcinomas (NMTC) are divided into two clinical–pathological groups. The syndromic-associated group is composed of predominately non-thyroidal tumors and includes Pendred syndrome, Warner syndrome, Carney complex (CNC) type 1, PTEN-hamartoma tumor syndrome (PHTS; Cowden disease), and familial adenomatous polyposis (FAP)/Gardner syndrome. Other conditions with less established links to the development of follicular cell-derived tumors include ataxia–telangiectasia syndrome, McCune Albright syndrome, and Peutz–Jeghers syndrome. The final group encompasses syndromes typified by NMTC, as well as pure familial (f) PTC with or without oxyphilia, fPTC with multinodular goiter, and fPTC with papillary renal cell carcinoma. This heterogeneous group of diseases does not have the established genotype–phenotype correlations known as in the familial C-cell-derived tumors or medullary thyroid carcinomas (MTC). Clinicians should have the knowledge to identify the likelihood of a patient presenting with thyroid cancer having an additional underlying familial syndrome stemming from characteristics by examining morphological findings that would alert pathologists to recommend that patients undergo molecular genetic evaluation. This review discusses the clinical and pathological findings of patients with familial PTC, such as FAP, CNC, Werner syndrome, and Pendred syndrome, and the heterogeneous group of familial PTC.
Introduction
Malignant thyroid tumors account for 1% of all malignant tumors (American Cancer Society, 2007). The incidence of thyroid malignancy has increased more than that of any other cancer (Horner et al., 2009). Newly reported cases of thyroid cancer in the United States grew from 18,000 in 2000 to 35,000 in 2007 (Horner et al., 2009). This is likely due to advancements in diagnostic techniques and early detection of small thyroid nodules (microcarcinomas), which clinical meaning is uncertain (Zhu et al., 2009). Almost 95% of patients have well-differentiated cancer of follicular cell origin, including papillary (80–90%), and follicular (10–15%). Approximately 5% of patients have medullary thyroid cancer.
Advances in molecular genetics have also confirmed the presence of several familial cancer syndromes with non-medullary familial thyroid cancers, usually papillary or follicular cancers. These include familial adenomatous polyposis (FAP), Cowden syndrome (CS), Werner syndrome, Carney complex (CNC), and Pendred syndrome (Dotto and Nosé, 2008; Nosé, 2008; Vriens et al., 2009).
Familial follicular cell-derived tumors may account 5–15% of thyroid carcinoma cases. A classification of familial follicular cell thyroid cancer associated with hereditary syndromes (Dotto and Nosé, 2008; Nosé, 2008) is shown in Table 1. The inheritance, gene involved, and risks of familial cancer syndromes for developing thyroid cancer are also shown in Table 1.

Table 1. Hereditary tumor syndromes associated with thyroid cancer.
Familial Adenomatous Polyposis
Familial adenomatous polyposis is inherited as an autosomal dominant trait caused by a germline mutation in the adenomatous polyposis coli (APC) gene. The APC gene is a tumor suppressor gene on chromosome 5q21 (Spigelman et al., 1989; Luk, 1995; Cetta et al., 2000). Patients typically have thousands of adenomas that are primarily located in the colon and rectum. Virtually all patients will progress to colorectal cancer if FAP is not identified and treated surgically with proctocolectomy. Most patients also develop gastric polyps of the fundus, most of which do not progress to carcinoma. Polyps in the duodenum and periampullary region are adenomatous, with an increased risk for progression to cancer estimated to be 200 times greater than that of patients in the general population (Spigelman et al., 1989; Luk, 1995; Cetta et al., 2000).
Patients may have extraintestinal manifestations that include osteomas, dental abnormalities, epidermal cysts, desmoids tumors, congenital hypertrophy of the retinal pigment epithelium (CHRPE), hepatoblastoma, medulloblastoma, and thyroid cancers.
Patients with FAP are at risk for developing papillary thyroid carcinoma (PTC). PTC is one of the extracolonic manifestations of FAP. Young women with FAP are at particular risk of developing thyroid cancer, with risk approximately 160 times higher than that of normal individuals, and PTC occurs with a frequency about 10 times that expected for sporadic PTC (Harach et al., 1994; Cameselle-Teijeiro and Chan, 1999; Soravia et al., 1999). Prevalence ranges from 2 to 12% of patients with FAP (Herraiz et al., 2007).
Thyroid carcinomas associated with FAP are typically bilateral and multifocal, with histological features different from sporadic tumors, with characteristic histopathological cribriform pattern with solid areas and a spindle cell component, and are most often associated with marked fibrosis (Figure 1). The characteristic cellular and nuclear findings of sporadic PTC as grooved, overlapping, and clear nuclei are absent in this subtype (Harach et al., 1994; Cameselle-Teijeiro and Chan, 1999; Soravia et al., 1999).
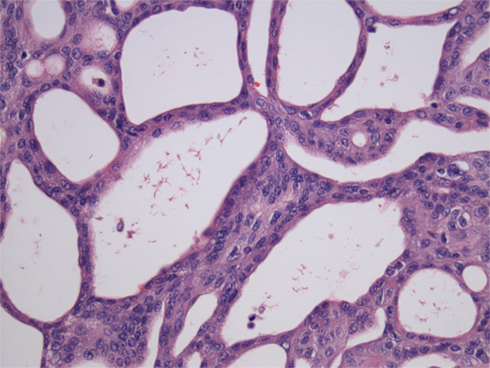
Figure 1. Cribriform-morular variant of PTC showing typical cribriform arrangement composed of fused follicles lined by tall cells and lumina lacking colloid (H&E).
The cribriform-morular variant of PTC (CMv-PTC) is a very rare subtype of PTC representing 0.1–0.2% of cases, or less than 1 in 500 cases of all papillary carcinoma cases (Harach et al., 1994; Cameselle-Teijeiro and Chan, 1999). The overall prognosis for CMv-PTC is similar to that of classical variants of PTC with less than 10% of cases demonstrating aggressive clinical behavior. Among patients with FAP who have synchronous PTC, over 90% of these cases have been reported to cribriform-morular variant. While not all CMv-PTC is associated with FAP, a significant proportion of cases are associated with FAP. Patients with cribriform-morular variant PTC should be evaluated for FAP. This form of PTC is typically bilateral, presents at a younger age, and is 10 times more common in female patients with FAP. The histology of CMv-PTC is characterized by cribriform, solid, and morular areas lacking typical nuclear features of PTC and CMv-PTC is associated with germline and somatic mutations in the APC and β-catenin genes. In contrast to conventional PTC, CMv-PTC rarely metastasizes and carries a benign prognosis. CMv-PTC is revealed by aberrant nuclear and cytoplasmic expression of β-catenin (β-catenin immunostaining is strong in cytoplasm and nuclei in the morular and cribriform areas, and it is only expressed in cell membrane of the non-tumoral follicular cells; Figure 2).
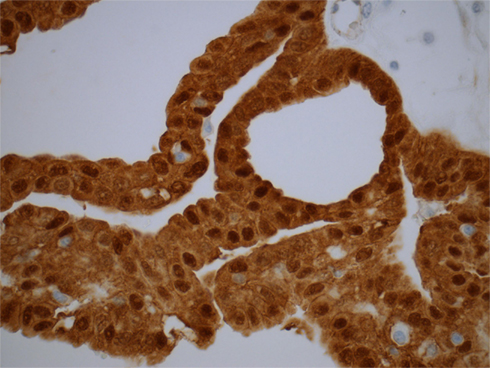
Figure 2. Immunostaining for β-catenin reveals an aberrant nuclear and cytoplasmic staining in the cribriform-morular variant of papillary thyroid carcinoma. The endothelial cells are negative.
As with other familial non-medullary thyroid carcinoma (FNMTC) syndromes, the low incidence in FAP patients suggests that PTC occurs primarily as a result of a susceptibility gene. Somatic mutations of RET–PTC1 and RET–PTC3 have been identified. Investigators have also identified differences in the location of APC germline mutations in FAP patients with and without PTC (Cetta et al., 2000). They found that 13/15 (87%) patients with FAP-associated PTC had germline mutations and that 12 of these patients had mutations in the genomic region associated with CHRPE and in the mutation cluster region in the 5′ region of exon 15. This led to a recommendation that thyroid screening begin early (age 15 years) in patients or kindred with CHRPE and for patients with exon 15 mutations in the 5′ region (Cetta et al., 2000).
PTEN-Hamartoma Tumor Syndrome
Cowden syndrome is an autosomal dominant disorder caused by a germline mutation in PTEN (phosphatase and tensin homolog, deleted on chromosome 10) and characterized by the development of multiple hamartomas and carcinomas of the thyroid, breast, and uterus. PTEN is a tumor suppressor gene located on 10q23.3. Two-thirds of CS patients develop thyroid tumors including multinodular goiter, multiple adenomatous nodules, follicular adenoma, follicular carcinoma and, less frequently, PTC (Parisi et al., 2001; Zambrano et al., 2004). The majority of thyroid lesions occurring in PHTS are of follicular origin and are characteristically multicentric, bilateral, benign, and malignant thyroid lesions. Adenomatous nodules seen in PHTS are bilateral, more numerous, and distinct from age-related nodules with only a scant atrophic rim of normal thyroid follicles (Nosé, 2010).
Follicular carcinoma is a major criteria and an important feature in PTEN-hamartoma tumor syndrome (PHTS). These tumors are more frequently multicentric, and progress from a preexisting follicular adenoma. However, PTC is rarely associated with this entity. Multiple adenomatous nodules in a background of lymphocytic thyroiditis and/or C-cell hyperplasia are distinctive findings in this syndrome. Pathologists should notify clinicians of the possibility of PHTS (Dotto and Nosé, 2008).
The diagnosis of a thyroid lesion usually precedes the diagnosis of PHTS by many years. In some patients, a work-up for PHTS is triggered by unusual pathologic findings in the thyroid of a young patient, most specifically, multiple adenomatous nodules.
The current diagnostic criteria for CS include follicular carcinoma (present in 10–15%) as a major criterion, while multinodular goiter (including multiple adenomatous nodules) and follicular adenomas are minor criteria (present in 50–67%).
Adenomatous nodules are unusually numerous in these cases reaching over 100 nodules, are not encapsulated, homogeneous, firm, yellow-tan, lack gelatinous colloid, and do not exhibit secondary changes. Histologically, the adenomatous nodules in PHTS are solid, cellular, and composed of small follicles lacking abundant colloid, side by side with others. Some nodules may have a thin, discontinuous rim of fibrous tissue simulating a capsule (Nosé, 2011). On immunohistochemical staining, 63% of CS cases showed complete loss of PTEN expression and 38% showed heterogeneous loss of PTEN (Figure 3). All intact PTEN expression cases were non-CS patients (Barletta et al., 2011).
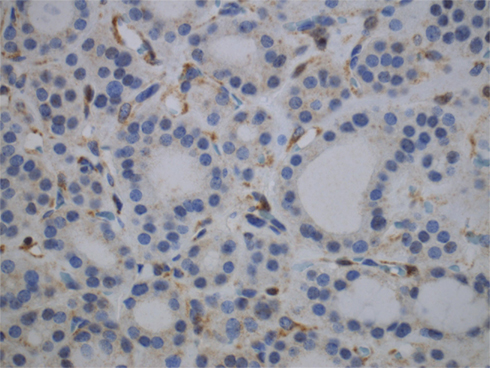
Figure 3. Immunohistochemistry for PTEN shows loss of PTEN expression in an adenomatous nodule in a PTEN-hamartoma tumor syndrome (Cowden’s disease) patient. Note positivity in the endothelial cells.
The high incidence of thyroid pathology in patients with CS warrants routine thyroid screening with ultrasonography and a low threshold for recommending thyroidectomy.
Carney Complex
The CNC is a dominantly inherited syndrome characterized by spotty skin pigmentation, endocrine overactivity, and myxomas. The syndrome was first described in 1985 as “myxomas, spotty pigmentation, and endocrine overactivity” (Carney et al., 1985).
Carney complex is defined by the association of multiple endocrine neoplasia and cardiocutaneous manifestations. Patients previously characterized as having LAMB (lentigines, atrial myxoma, mucocutaneous myxoma, blue nevi) or NAME (nevi, atrial myxoma, myxoid neurofibroma, ephelide) could be considered to have CNC. Numerous organs may be involved in CNC and manifestations vary greatly among patients. Skin pigmentation anomalies include lentigines and blue nevi. Myxomas can occur at multiple sites such as the heart, skin or soft tissue, external auditory canal, and breast. Cardiac myxomas can develop in any cardiac chamber and may be multiple. These patients may also present with schwannomas and testicular tumors (Stratakis et al., 1997, 1998).
The most common endocrine gland involvements are growth hormone (GH)-secreting pituitary adenomas (acromegaly), thyroid tumors, and adrenocorticotropic hormone (ACTH)-independent Cushing’s syndrome due to primary pigmented nodular adrenocortical disease (PPNAD).
Most cases of this autosomal dominant condition are classified as type 1 and are associated with a mutation to the protein kinase A regulatory subunit type 1-alpha (PRKAR1-α) gene. Type 2 patients have been confirmed to have a mutation on chromosome 2p16, which may be a regulator of genomic stability (Stratakis et al., 1997; Matyakhina et al., 2003; Pan et al., 2010).
The risk of thyroid cancer is low, and the presence of thyroid nodules is very common. The thyroid is multinodular with multiple adenomatous nodules and follicular adenomas. Both PTC and follicular thyroid carcinomas (FTC) are present in about 15% of patients with CNC. A recent review of CNC in 53 patients of 12 kindred found clinically significant thyroid disease in 11% of patients (Stratakis et al., 1997).
Werner Syndrome
Werner syndrome is a rare premature aging syndrome (progeroid) that typically begins in the third decade. The clinical presentation includes an elderly appearance with thin skin, wrinkles, alopecia, and muscle atrophy in proportion to the patient’s age. The patients with this syndrome have short stature secondary to an absent pubertal growth period (Nehlin et al., 2000; Muftuoglu et al., 2008). Patients also present with age-related disorders such as diabetes, osteoporosis, cataracts, peripheral vascular disease, or different types of malignant tumors. Malignancy and cardiac disease are the most common causes of death in these patients, who have a median life expectancy of 54 years (Nehlin et al., 2000; Muftuoglu et al., 2008).
It is an autosomal recessive disease that is caused by mutations in the WRN gene on chromosome 8p11–p12. This WRN gene encodes a protein that is both a RecQ helicase and exonuclease. It is important in DNA repair and replication.
Patients present at a younger age and have approximately a threefold increased risk for follicular carcinoma and six times increased risk for developing anaplastic thyroid carcinoma (Ishikawa et al., 1999).
The overall incidence of thyroid cancer in Japanese patients with Werner syndrome is 18% (Ishikawa et al., 1999). The risk of PTC is increased, specifically in Caucasian populations. This high prevalence of thyroid cancer in Werner syndrome supports routine thyroid screening in patients with this disorder.
Familial Non-Medullary Thyroid Carcinoma Syndrome
Familial non-medullary thyroid carcinoma syndrome is diagnosed when three or more family members have non-medullary thyroid cancer in the absence of other known associated syndromes. Statistical estimates suggest that a grouping of two family members with non-medullary thyroid carcinomas (NMTC) could represent the concurrence of sporadic tumors, but thyroid tumors in three or more members in kindred, or the diagnosis of PTC in men and children, is more suggestive of a familial predisposition (Malchoff et al., 1999; Musholt et al., 2000; Hemminki et al., 2005; Sturgeon and Clark, 2005; Charkes, 2006). The search for a genetic susceptibility locus for FNMTC started about a decade ago.
Familial non-medullary thyroid carcinoma is now recognized as a distinct clinical entity and accounts for up to 10.5% of all follicular cell origin thyroid carcinomas. FNMTC has been associated with the presence of multiple benign nodules, to behave in a more aggressive clinical behavior, and to have a worse prognosis than sporadic non-medullary thyroid cancer. Individuals with FNMTC have an increased risk of multifocal disease, local invasion, and increased local or regional recurrence, lymph node metastases, and intraglandular dissemination. FNMTC is an independent predictor of shorter disease-free survival (Malchoff et al., 1999; Alsanea et al., 2000; Musholt et al., 2000; Alsanea and Clark, 2001; Hemminki et al., 2005; Sturgeon and Clark, 2005; Charkes, 2006; Mazeh et al., 2012).
The genetic inheritance of FNMTC remains unknown, but it is believed to be autosomal dominant with incomplete penetrance and variable expressivity. Genetic analyses of large FNMTC kindreds not only support the hypothesis of an inherited genetic predisposition to FNMTC, but also represent the first steps in identification of the putative susceptibility genes (Lesueur et al., 1999; McKay et al., 1999; Malchoff et al., 2000; Bevan et al., 2001; Harach, 2001; Xing, 2005). Six potential regions for harboring an FNMTC gene have been identified: MNG1 (14q32), thyroid carcinoma with oxyphilia (TCO; 19p13.2), fPTC/papillary renal neoplasia (PRN; 1q21), NMTC1 (2q21), FTEN (8p23.1–p22), and the telomere–telomerase complex. Linkage analyses have identified three different chromosomal regions that may harbor an FNMTC susceptibility gene (Nosé, 2010).
The systematic use of an algorithmic approach will help in the understanding of familial history and findings, and the patient’s clinical history associated with the pathology findings to suggest a familial disease. The clinical features and relative frequency of different forms of familial thyroid cancer have been discussed and presented recently (Vriens et al., 2009).
Familial tumor syndromes characterized by a predominance of non-medullary thyroid carcinoma are listed below and summarized in Table 2.
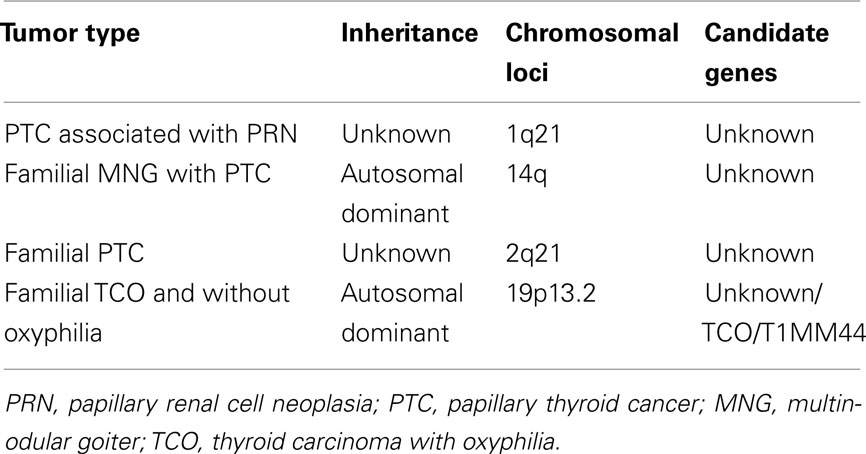
Table 2. Familial tumor syndrome characterized by a predominance of non-medullary thyroid carcinoma.
Familial Papillary Thyroid Carcinoma
Papillary thyroid carcinoma in FNMTC has a well-documented predisposition to multicentric disease, bilateral disease, local invasion, extrathyroidal extension, lymph node metastases, recurrence, and specific histology.
The background thyroid may show lymphocytic thyroiditis, multinodular hyperplasia, and multiple adenomatous nodules. Sporadic PTC has BRAF mutation in approximately 40% of cases. However, no BRAF mutation was reported in a group of 40 patients with FNMTC as germline mutation or a susceptibility genetic event for FNMTC (Xing, 2005).
The fPTC enriched in TCO has been mapped to chromosomal region 19p13, and FNMTC without oxyphilia has also been mapped to 19p13.31. Tumor-specific loss of heterozygosity is found in sporadic FTC with and without oxyphilia at both 19p13 and 2q21.32 (Lesueur et al., 1999; Musholt et al., 2000).
FNMTC Type 1 Syndrome
The FNMTC type 1 syndrome (chromosomal region 2q21) is characterized by PTC without any distinguishing pathologic features and without an obvious increase in frequency of non-thyroidal neoplasms in kindred members (McKay et al., 2001).
fPTC Associated with Renal Papillary Neoplasia
The fPTC associated with renal papillary neoplasia presents with the usual classical variant of PTC and with no special features. The PRN syndrome (fPTC/PRN), mapped to chromosomal region 1q21, includes not only PTC and the expected benign thyroid nodules, but also PRN and possibly other malignancies as well (Malchoff et al., 2000).
Familial Multinodular Goiter Syndrome
In familial multinodular goiter syndrome, which is mapped to 14q, some patients may develop an associated PTC (Bakhsh et al., 2006).
Conclusion
Mutations in patients with FNMTC syndromes have not been well defined as in medullary thyroid carcinoma (MTC). Most patients with these familial syndromes may have susceptibility genes that increase the risk of thyroid cancer. Thyroid cancer in many of these patients has been characterized as more aggressive than sporadic thyroid cancer, with a predisposition for lymph node metastasis, extrathyroidal invasion, and a younger age of onset. Most patients with a familial syndrome and FNMTC will have PTC, suggesting that a specific gene for PTC may also be present. In many cases, patients have a known familial syndrome that has a defined risk for thyroid cancer. These patients can be followed closely and the thyroid can be identified at an earlier stage (Nosé, 2010). In a few cases, thyroid cancer may be the initial presentation of an underlying familial syndrome.
The distinct thyroid pathology in some of these syndromes should alert the pathologist of a possible familial cancer syndrome. Knowledge of FNMTC and its histopathology can allow the pathologist to diagnose familial tumor syndrome. Awareness and screening of FNMTC will permit earlier detection, proper treatment, and improved outcomes for patients and their families.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alsanea, O., Wada, N., Ain, K., Wong, M., Taylor, K., Ituarte, P. H., Treseler, P. A., Weier, H. U., Freimer, N., Siperstein, A. E., Duh, Q. Y., Takami, H., and Clark, O. H. (2000). Is familial nonmedullary thyroid carcinoma more aggressive than sporadic thyroid cancer? A multicenter series. Surgery 128, 1043–1050; discussion 1050–1051.
American Cancer Society. (2007). Cancer Facts and Figures 2007. Atlanta, GA: American Cancer Society.
Bakhsh, A., Kirov, G., Gregory, J. W., Williams, E. D., and Ludgate, M. (2006). A new form of familial multi-nodular goitre with progression to differentiated thyroid cancer. Endocr. Relat. Cancer 13, 475–483.
Barletta, J. A., Bellizzi, A. M., and Hornick, J. L. (2011). Immunohistochemical staining of thyroidectomy specimens for PTEN can aid in the identification of patients with Cowden syndrome. Am. J. Surg. Pathol. 35, 1505–1511.
Bevan, S., Pal, T., Greenberg, C. R., Green, H., Wixey, J., Bignell, G., Narod, S. A., Foulkes, W. D., Stratton, M. R., and Houlston, R. S. (2001). A comprehensive analysis of MNG1, TCO1, fPTC, PTEN, TSHR, and TRKA in familial nonmedullary thyroid cancer: confirmation of linkage to TCO1. J. Clin. Endocrinol. Metab. 86, 3701–3704.
Cameselle-Teijeiro, J., and Chan, J. K. (1999). Cribriform-morular variant of papillary carcinoma: a distinctive variant representing the sporadic counterpart of familial adenomatous polyposis-associated thyroid carcinoma? Mod. Pathol. 12, 400–411.
Carney, J. A., Gordon, H., Carpenter, P. C., Shenoy, B. V., and Go, V. L. (1985). The complex of myxomas, spotty pigmentation, and endocrine overactivity. Medicine (Baltimore) 64, 270–283.
Cetta, F., Montalto, G., Gori, M., Curia, M., Cama, A., and Olschwang, S. (2000). Germline mutations of the APC gene in patients with FAP associated thyroid carcinoma. Results from a European cooperative study. J. Clin. Endocrinol. Metab. 85, 286–292.
Charkes, N. D. (2006). On the prevalence of familial nonmedullary thyroid cancer in multiple affected kindreds. Thyroid 16, 181–186.
Dotto, J., and Nosé, V. (2008). Familial thyroid carcinoma a diagnostic algorithm. Adv. Anat. Pathol. 15, 332–349.
Harach, H. R., Williams, G. T., and Williams, E. D. (1994). Familial adenomatous polyposis associated thyroid carcinoma: a distinct type of follicular cell neoplasm. Histopathology 25, 549–561.
Hemminki, K., Eng, C., and Chen, B. (2005). Familial risks for nonmedullary thyroid cancer. J. Clin. Endocrinol. Metab. 90, 5747–5753.
Herraiz, M., Barbesino, G., Faquin, W., Chan-Smutko, G., Patel, D., Shannon, K., Daniels, G., and Chung, D. (2007). Prevalence of thyroid cancer familial adenomatous polyposis syndrome and the role of screening ultrasound examinations. Clin. Gastroenterol. Hepatol. 5, 367–373.
Horner, M. J., Ries, L., Krapcho, M., Neyman, N., Aminou, R., Howlader, N., Altekruse, S., Feuer, E., Huang, L., Mariotto, A., Miller, B., Lewis, D., Eisner, M., Stinchcomb, D., and Edwards, B. (eds). (2009). SEER Cancer Statistics Review, 1975–2006. Bethesda, MD: National Cancer Institute.
Ishikawa, Y., Sugano, H., Matsumoto, T., Furuichi, Y., Miller, R., and Goto, M. (1999). Unusual features of thyroid carcinomas in Japanese patients with Werner syndrome and possible genotype-phenotype relations to cell type and race. Cancer Sci. 85, 1345–1352.
Lesueur, F., Stark, M., Tocco, T., Ayadi, H., Delisle, M. J., Goldgar, D. E., Schlumberger, M., Romeo, G., and Canzian, F. (1999). Genetic heterogeneity in familial nonmedullary thyroid carcinoma:exclusion of linkage to RET, MNG1, and TCO in 56 families. NMTC Consortium. J. Clin. Endocrinol. Metab. 84, 2157–2162.
Luk, G. (1995). Diagnosis and therapy of hereditary polyposis syndromes. Gastroenterologist 3, 153–167.
Malchoff, C. D., Sarfarazi, M., Tendler, B., Forouhar, F., Whalen, G., Joshi, V., Arnold, A., and Malchoff, D. M. (2000). Papillary thyroid carcinoma associated with papillary renal neoplasia: genetic linkage analysis of a distinct heritable tumor syndrome. J. Clin. Endocrinol. Metab. 85, 1758–1764.
Malchoff, C. D., Sarfarazi, M., Tendler, B., Forouhar, F., Whalen, G., and Malchoff, D. M. (1999). Familial papillary thyroid carcinoma is genetically distinct from familial adenomatous polyposis coli. Thyroid 9, 247–252.
Matyakhina, L., Pack, S., Kirschner, L., Pak, E., Mannan, P., Jaikumar, J., Taymans, S., Sandrini, F., Carney, J., and Stratakis, C. (2003). Chromosome 2 (2p16) abnormalities in Carney complex tumours. J. Med. Genet. 40, 268–277.
Mazeh, H., Benavidez, J., Poehls, J. L., Youngwirth, L., Chen, H., and Sippel, R. S. (2012). In patients with thyroid cancer of follicular cell origin, a family history of nonmedullary thyroid cancer in one first-degree relative is associated with more aggressive disease. Thyroid. 22, 3–8.
McKay, J. D., Lesueur, F., Jonard, L., Pastore, A., Williamson, J., Hoffman, L., Burgess, J., Duffield, A., Papotti, M., Stark, M., Sobol, H., Maes, B., Murat, A., Kääriäinen, H., Bertholon-Grégoire, M., Zini, M., Rossing, M. A., Toubert, M. E., Bonichon, F., Cavarec, M., Bernard, A. M., Boneu, A., Leprat, F., Haas, O., Lasset, C., Schlumberger, M., Canzian, F., Goldgar, D. E., and Romeo, G. (2001). Localization of a susceptibility gene for familial nonmedullary thyroid carcinoma to chromosome 2q21. Am. J. Hum. Genet. 69, 440–446.
McKay, J. D., Williamson, J., Lesueur, F., Stark, M., Duffield, A., Canzian, F., Romeo, G., and Hoffman, L. (1999). At least three genes account for familial papillary thyroid carcinoma: TCO and MNG1 excluded as susceptibility loci from a large Tasmanian family. Eur. J. Endocrinol. 141, 122–125.
Muftuoglu, M., Oshima, J., von Kobbe, C., Cheng, W., Leistritz, D., and Bohr, V. (2008). The clinical characteristics of Werner syndrome: molecular and biochemical diagnosis. Hum. Genet. 124, 369–377.
Musholt, T. J., Musholt, P. B., Petrich, T., Oetting, G., Knapp, W. H., and Klempnauer, J. (2000). Familial papillary thyroid carcinoma: genetics, criteria for diagnosis, clinical features, and surgical treatment. World J. Surg. 24, 1409–1417.
Nehlin, J., Skovgaard, G., and Bohr, V. (2000). The Werner syndrome. A model for the study of human aging. Ann. N. Y. Acad. Sci. 908, 167–179.
Nosé, V. (2010). Familial follicular cell tumors: classification and morphological characteristics. Endocr. Pathol. 21, 219–226.
Pan, L., Peng, L., Jean-Gilles, J., Zhang, X., Wieczorek, R., Jain, S., Levine, V., Osman, I., Prieto, V. G., and Lee, P. (2010). Novel PRKAR1A gene mutations in Carney complex. Int. J. Clin. Exp. Pathol. 3, 545–548.
Parisi, M. A., Dinulos, M. B., Leppig, K. A., Sybert, V. P., Eng, C., and Hudgins, L. (2001). The spectrum and evolution of phenotypic findings in PTEN mutation positive cases of Bannayan-Riley-Ruvalcaba syndrome. J. Med. Genet. 38, 52–58.
Soravia, C., Sugg, S., Berk, T., Mitri, A., Cheng, H., Gallinger, S., Cohen, Z., Asa, S., and Bapat, B. (1999). Familial adenomatous polyposis-associated thyroid cancer: a clinical, pathological, and molecular genetics study. Am. J. Pathol. 154, 127–135.
Spigelman, A., Williams, C., Talbot, I., Domizio, P., and Phillips, R. (1989). Upper gastrointestinal cancer in patients with familial adenomatous polyposis. Lancet 2, 783–785.
Stratakis, C. A., Courcoutsakis, N. A., Abati, A., Filie, A., Doppman, J. L., Carney, J. A., and Shawker, T. (1997). Thyroid gland abnormalities in patients with the syndrome of spotty skin pigmentation, myxomas, endocrine overactivity, and schwannomas (Carney complex). J. Clin. Endocrinol. Metab. 82, 2037–2043.
Stratakis, C. A., Kirschner, L. S., and Carney, J. A. (1998). Carney complex: diagnosis and management of the complex of spotty skin pigmentation, myxomas, endocrine overactivity, and schwannomas. Am. J. Med. Genet. 80, 183–185.
Vriens, M., Suh, I., Moses, W., and Kebebew, E. (2009). Clinical features and genetic predisposition to hereditary nonmedullary thyroid cancer. Thyroid 19, 1343–1349.
Xing, M. (2005). The T1799A BRAF mutation is not a germline mutation in familial nonmedullary thyroid cancer. Clin. Endocrinol. (Oxf.) 63, 263–266.
Zambrano, E., Holm, I., Glickman, J., Huang, S., Perez-Atayde, A., Kozakewich, H. P., Shamberger, R. C., and Nosé, V. (2004). Abnormal distribution and hyperplasia of thyroid C-cells in PTEN-associated diseases. Endocr. Pathol. 15, 55–64.
Keywords: familial thyroid carcinoma, familial adenomatous polyposis, Cowden syndrome, Carney complex, Pendred syndrome, Werner syndrome, familial papillary thyroid carcinoma
Citation: Son EJ and Nosé V (2012) Familial follicular cell-derived thyroid carcinoma. Front. Endocrin. 3:61. doi: 10.3389/fendo.2012.00061
Received: 01 February 2012; Accepted: 13 April 2012;
Published online: 03 May 2012.
Edited by:
Carmelo Nucera, Beth Israel Deaconess Medical Center, USAReviewed by:
Ronald Cohen, University of Chicago, USAVasyl Vasko, Uniformed Services University of the Health Sciences, USA
Marco Centanni, Sapienza University of Rome, Italy
Copyright: © 2012 Son and Nosé. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Vânia Nosé, Miller School of Medicine, University of Miami, Clinical Research Building, (R-5), 1120 N.W., 14 Street, Suite 1411, Miami, FL 33136, USA. e-mail:dm5vc2VAbWVkLm1pYW1pLmVkdQ==