

 Rasmus Jorgensen3
Rasmus Jorgensen3- 1 Department of Incretin Biology, Novo Nordisk A/S, Gentofte, Denmark
- 2 Department of Biomedical Sciences, Faculty of Health Sciences, University of Copenhagen, Copenhagen, Denmark
- 3 Department of Type 2 Diabetes, Novo Nordisk A/S, Maaloev, Denmark
- 4 Diabetes Biology and Hagedorn Research Institute, Novo Nordisk A/S, Gentofte, Denmark
The superfamily of the seven transmembrane G-protein-coupled receptors (7TM/GPCRs) is the largest family of membrane-associated receptors. GPCRs are involved in the pathophysiology of numerous human diseases, and they constitute an estimated 30–40% of all drug targets. During the last two decades, GPCR oligomerization has been extensively studied using methods like bioluminescence resonance energy transfer (BRET) and today, receptor–receptor interactions within the GPCR superfamily is a well-established phenomenon. Evidence of the impact of GPCR oligomerization on, e.g., ligand binding, receptor expression, and signal transduction indicates the physiological and pharmacological importance of these receptor interactions. In contrast to the larger and more thoroughly studied GPCR subfamilies A and C, the B1 subfamily is small and comprises only 15 members, including, e.g., the secretin receptor, the glucagon receptor, and the receptors for parathyroid hormone (PTHR1 and PTHR2). The dysregulation of several family B1 receptors is involved in diseases, such as diabetes, chronic inflammation, and osteoporosis which underlines the pathophysiological importance of this GPCR subfamily. In spite of this, investigation of family B1 receptor oligomerization and especially its pharmacological importance is still at an early stage. Even though GPCR oligomerization is a well-established phenomenon, there is a need for more investigations providing a direct link between these interactions and receptor functionality in family B1 GPCRs. One example of the functional effects of GPCR oligomerization is the facilitation of allosterism including cooperativity in ligand binding to GPCRs. Here, we review the currently available data on family B1 GPCR homo- and heteromerization, mainly based on BRET investigations. Furthermore, we cover the functional influence of oligomerization on ligand binding as well as the link between oligomerization and binding cooperativity.
Introduction
The seven transmembrane G-protein-coupled receptors (7TM/GPCRs) comprise the largest family of membrane receptors. Through coupling to intracellular heterotrimeric G-proteins, the GPCRs mediate cellular responses to a diverse pallet of stimuli including photons, odorants, ions, nucleotides, lipids, neurotransmitters, proteases, and hormones (Bockaert and Pin, 1999). Based on structural and ligand binding criteria, several classification systems for GPCRs have been proposed, such as the A to F system (Kolakowski, 1994), the 1 to 5 system (Bockaert and Pin, 1999), and the GRAFS system (named after the first letters of the five families Glutamate, Rhodopsin, Adhesion, Frizzled, and Secretin; Fredriksson et al., 2003). In the Kolakowski system, the GPCRs are classified into six families, A–F, of which only the families A, B, and C are found in mammalian species (Kolakowski, 1994). These families share little or no inter-family sequence homology, even though the overall morphology of GPCRs is highly conserved (Vohra et al., 2007; Kenakin and Miller, 2010). Family A (rhodopsin-like receptors) is by far the largest family of GPCRs, and most receptors in this family are characterized by very short N- and C-termini. This family includes some of the most extensively studied GPCRs: rhodopsin, the β-adrenergic receptors (β-ARs), and the opioid receptors. Family C (metabotropic glutamate receptors, mGluRs) includes, e.g., the mGluRs, the Ca2+ sensing receptor (CaSR), and the receptors for γ-aminobutyric acid (GABABRs). This family is characterized by a long C-terminus as well as a very long N-terminal domain often containing a “Venus fly-trap” structure responsible for ligand binding (Kristiansen, 2004; Kenakin and Miller, 2010). Family B is subdivided into the subfamilies B1 (secretin-like receptor family), B2 [adhesion family, or “long amino terminus, family B” (LNB-7TM proteins)], and B3 (Methuselah-like family) as proposed by Harmar (2001). According to this classification, the human B1 family of GPCRs includes 15 receptors, all responding to peptide hormones: the secretin receptor (SECR), the glucagon receptor (GCGR), the glucagon-like peptide-1 and -2 receptors (GLP-1R and GLP-2R), the glucose-dependent insulinotropic polypeptide receptor (GIPR), the receptors for parathyroid hormone (PTHR1 and PTHR2), the receptors for vasoactive intestinal peptide (VPAC1 and VPAC2), the pituitary adenylate cyclase-activating peptide receptor (PAC1), growth hormone-releasing hormone receptor (GHRHR), the receptors for corticotrophin-releasing hormone (CRHR1 and CRHR2), the calcitonin receptor (CALCR), and the CALCR-like receptor (CRLR; Harmar, 2001).
The natural ligands for family B1 GPCRs include secretin, glucagon, GLP-1 and GLP-2, glucose-dependent insulinotropic polypeptide (GIP), parathyroid hormone (PTH), vasoactive intestinal peptide (VIP), pituitary adenylate cyclase-activating peptide (PACAP), growth hormone-releasing hormone (GRH), corticotrophin-releasing hormone (CRH), and calcitonin. These peptide hormones and their receptors are currently of considerable interest to the pharmaceutical industry because they are involved in the pathophysiology of human diseases, such as, e.g., diabetes (glucagon, GLP-1, GIP; Knop et al., 2009; Bagger et al., 2011; D’Alessio, 2011), osteoporosis (calcitonin, PTH; de Paula and Rosen, 2010; Verhaar and Lems, 2010), inflammation, and neurodegeneration (VIP, PACAP; Gonzalez-Rey et al., 2005). For a full list of the involvement of family B1 GPCRs in diseases, see Table 1.
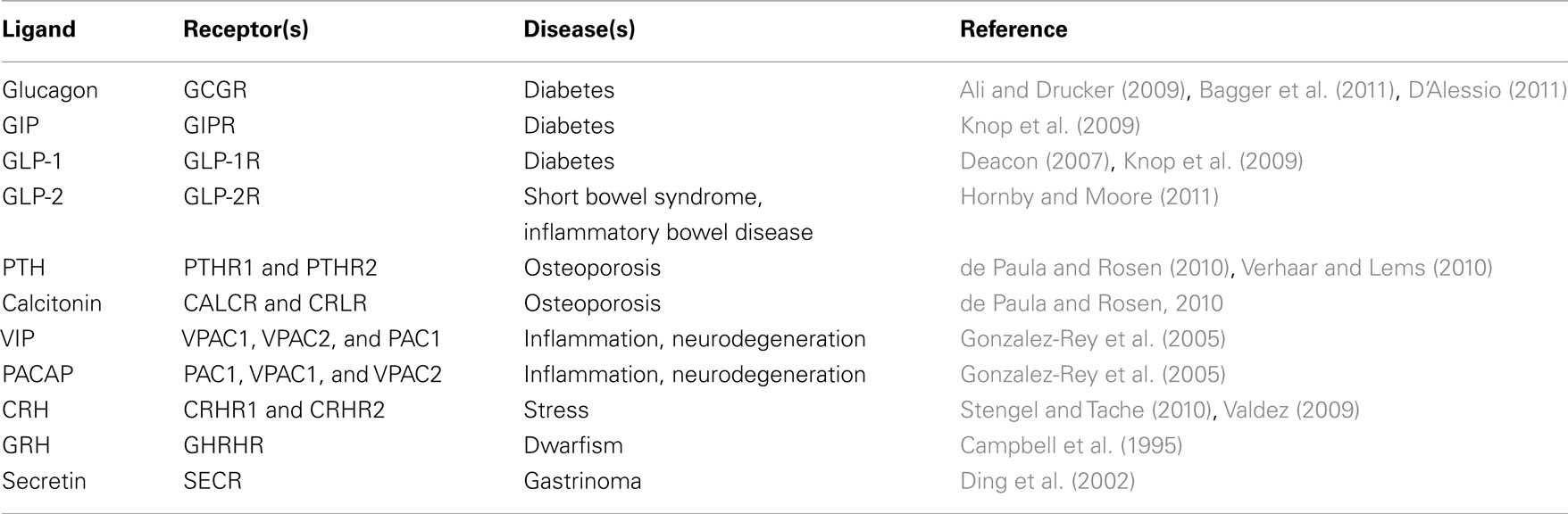
Table 1. Potential therapeutical interests of targeting family B1 GPCRs and/or their natural ligands in human diseases.
Structure and Binding Mechanism of Family B1 GPCRs
A common structural feature of the family B1 GPCRs is a relatively short C-terminus and a long N-terminal domain of 100–200 residues (George et al., 2002). The characteristic structure of the N-terminal region in family B1 includes two antiparallel β-sheets (β1, β2, β3, and β4), an N-terminal α-helix, and six cysteines that form three disulfide bridges. Furthermore, there is a conserved disulfide bridge between cysteine residues in extracellular loop 1 (ECL1) and ECL2 (Ulrich et al., 1998; Authier and Desbuquois, 2008; Kenakin and Miller, 2010). Like their receptors, the peptide ligands for the family B1 GPCRs are highly similar. They are all relatively short peptides of 27–44 amino acid residues and they are assumed to adopt an α-helical conformation during the contact with their respective receptors (Grace et al., 2004).
The N-terminal end of the receptor extracellular domain (ECD) constitutes the primary receptor–ligand interaction site that determines affinity and specificity through binding of the C-terminal portion of the ligand. This interaction constitutes the first step (step A) of ligand–receptor binding of family B1 in a model proposed by Hoare and Usdin(2001; see Figure 1). Subsequently, the ligand N-terminal portion interacts with the juxtamembrane domain of the receptor (step B) resulting in receptor activation (step C; Hoare and Usdin, 2001; Hoare, 2005; Tan et al., 2006).
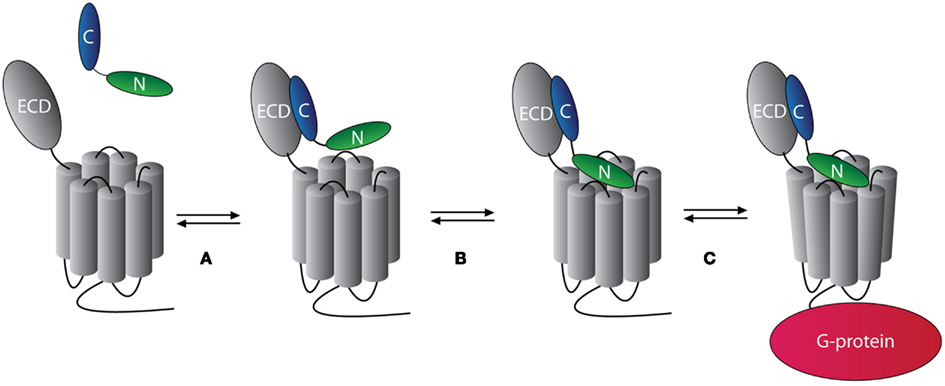
Figure 1. The two-step model of peptide ligand binding to family B1 GPCRs. The binding of one ligand to one family B1 GPCR occurs in two steps. In step (A) the C-terminal portion of the ligand (blue) binds to the N-terminal domain of the receptor with high affinity and specificity. In step (B), the N-terminal of the ligand (green) binds to the juxtamembrane domain of the receptor. This activates the receptor and mediates intracellular interaction with G-proteins [step (C)]. Modified from Hoare (2005).
Recently, crystal structures of ligand-bound ECDs have been determined for several family B1 GPCRs, namely GIP (Parthier et al., 2007), PTHR1 (Pioszak and Xu, 2008), GLP-1R (Runge et al., 2008), CRHR1 and -2 (Pioszak et al., 2008; Pal et al., 2010), and CRLR (ter Haar et al., 2010). These structures support the binding model proposed by Hoare and Usdin (2001) in which one ligand binds to one receptor in a two-step process (Figure 1). However, the interaction between the N-terminal portion of the ligand and the juxtamembrane domain of the receptor may not be simplified into one single model. Based on studies on SECR, VPAC1, CALCR, and GLP-1R, an alternative model of receptor activation has been proposed, involving the exposure of a hidden epitope in the N-terminal domain of the receptor upon ligand binding that can act as an endogenous agonist ligand (Dong et al., 2006, 2008a,b).
GPCR Oligomerization
G-protein-coupled receptors were originally thought to be functional monomers, one receptor interacting with one heterotrimeric G-protein in a 1:1 stoichiometry. Today, however, the phenomenon of GPCR homo- and/or heteromerization is widely accepted, and several studies have revealed important functional roles of GPCR oligomerization. In general, is it difficult to distinguish between dimers and higher order oligomers of GPCRs experimentally. Thus, we will refer to GPCR–GPCR interactions as oligomers throughout this review unless the specific oligomeric state has been determined.
In family C of GPCRs, oligomerization is a fundamental theme; for example, the functional GABABR is a heteromer of the two protomers, GABAB1R and GABAB2R. The GABAB1R contains the ligand binding site but is dependent on heteromerization with the GABAB2R protomer for proper transport to the plasma membrane as well as for coupling to the G-protein signaling cascade (Galvez et al., 2001; Comps-Agrar et al., 2011). The crystal structure of the ECD of another family C GPCR, the mGluR, has revealed that this receptor must be expressed on the cell surface as a homodimer in order to be functional because two mGluR protomers in combination is needed to form the ligand binding cleft (Kunishima et al., 2000).
Within family A of GPCRs there are also several examples of functionality of oligomerization (Terrillon and Bouvier, 2004) as well as examples of the involvement of oligomerization in pathophysiological conditions. The possibility that GPCR heteromerization might play an important role in pharmacological diversity was first suggested by studies on the δ- and κ-opioid receptors. Co-expression of these receptors resulted in formation of heteromers with a very low affinity for either the δ- or the κ-selective ligand alone. However, when the two ligands were combined, high affinity was restored, suggesting the occurrence of positive cooperativity (Jordan and Devi, 1999). This is very interesting in the context of drug development because it might imply that selective compounds can be designed that target specific heteromers without affecting the individual protomers (George et al., 2002). Another example comes from the study of platelets from preeclamptic hypertensive women. This study revealed that an increase in the relative number of heteromers between the AT1 receptor for the vasopressor angiotensin II and the B2 receptor for the vasodepressor bradykinin compared to homomers of the respective receptors is involved in the pathophysiology of preeclampsia (AbdAlla et al., 2001). It should be noted though, that others have been unable to reproduce these notable findings in several cell lines (Hansen et al., 2009; See et al., 2011).
In line with these examples for family A and C GPCRs, oligomerization of family B1 GPCRs is likely to play an essential role in receptor function as well as in the pathophysiology of the diseases listed in Table 1. Except for the SECR, which has been extensively studied by Laurence Miller and his research group as described later, not many studies have addressed the functional issue of oligomerization in family B1 of GPCRs. However, given their importance in many severe diseases, including, e.g., diabetes which alone currently affects 346 million people worldwide (WHO, 2011), it is essential to clarify the interactions between these receptors with the prospect of developing new and better treatments in the future. This review will focus on the current status on homo- and heteromerization of family B1 GPCRs, especially based on bioluminescence resonance energy transfer (BRET) experiments as well as the possible functional link between receptor oligomerization and binding cooperativity.
Oligomerization in the Family B1 of GPCRs
Methods Used to Study Oligomerization of GPCRs
Different approaches have been taken to study GPCR oligomerization over the last two decades. The first GPCR oligomer was indicated by the use of co-immunoprecipitation (Co-IP) by Hebert et al. (1996). This method was used extensively in early GPCR oligomerization studies. However, methods based on resonance energy transfer between two fluorescently/luminescently tagged receptors in living cells quickly gained traction. Today, the method which is being used by far the most in GPCR oligomerization studies is BRET. The principle of this method is relying on the energy transfer between an enzyme energy donor, Renilla luciferase (Rluc), and an energy acceptor usually in the form of either green fluorescent protein (GFP) or yellow fluorescent protein (YFP). In practice, an energy donor-tagged receptor is co-expressed with an energy acceptor-tagged receptor in a cell system. Upon receptor–receptor interaction, which brings the tags into a proximity of <100 Å, energy is transferred from the energy donor to the energy acceptor upon activation of the Rluc enzyme. This energy transfer results in a measurable acceptor emission signal, the BRET signal, reflecting receptor–receptor interactions (Pfleger and Eidne, 2006). The most convincing application of this method is a BRET saturation experiment. In this setup, a constant concentration of Rluc-tagged receptor is co-expressed with an increasing concentration of GFP-tagged receptor. For a specific receptor–receptor interaction, the BRET signal will increase with increasing amounts of energy acceptor molecules and produce a saturation curve. In contrast, a non-specific interaction will result in a linear curve, the so-called “bystander BRET,” resulting from random collisions of the tagged receptors, thereby providing a negative control. Such a negative control is required for validation of a specific interaction indicated in a BRET saturation experiment (Hamdan et al., 2006).
Fluorescence resonance energy transfer (FRET) is a method based on the same principle as BRET, however, FRET is relying on the energy transfer between two fluorescent proteins, e.g., YFP and cyan fluorescent protein (CFP). As in the case of BRET, FRET has been used in numerous studies on GPCR oligomerization (Ayoub and Pfleger, 2010). Recently, the FRET-principle was improved by including long-lived energy donors such as terbium and europium in the so-called time-resolved FRET (TR-FRET). This technique has the advantage of a lower background and thus a higher signal:noise ratio as compared to normal FRET (Albizu et al., 2010). In addition, FRET is also being used to study GPCR oligomerization by co-localization investigations using confocal microscopy, a principle referred to as morphological FRET (Harikumar et al., 2006).
Bimolecular fluorescence complementation (BiFC) and bimolecular luminescence complementation (BiLC) are two other recently developed fluorescence-based techniques employed in the study of GPCR oligomerization. These techniques apply splitting either a fluorescent or luminescent protein in two parts and tagging two receptors with a part each. Upon receptor–receptor interaction, the two parts of the energy emitting protein will come in close proximity and reassemble into a functional fluorescent or luminescent protein in a spontaneous reaction (Vidi et al., 2011).
All these different techniques have limitations and should in general be combined in order to obtain convincing evidence of GPCR oligomerization. It is, however, not the goal of this review to critically compare the benefits and limitations of these methods for which we refer to reviews on the subject (Milligan and Bouvier, 2005; Kaczor and Selent, 2011). In the following section, the use of these different techniques for studying oligomerization within the family B1 of GPCRs is reviewed.
Oligomerization of the SECR, the Prototypical Family B1 GPCR
Within the family B1 of GPCRs, the SECR is by far the most thoroughly studied receptor. In 1991, SECR was the first of the family B1 receptors to be cloned (Ishihara et al., 1991). Due to the similarity in sequences and structures of later characterized receptors of this family with the SECR, the B1 subfamily of GPCRs has been named the “secretin-like receptors.” Extensive studies on oligomerization properties of this prototypical family B1 GPCR have been carried out by the laboratory of Laurence Miller. SECR homomerization was indicated for the first time by morphological FRET in COS cells and further supported by energy transfer between Rluc- and GFP-tagged SECRs in BRET studies. Furthermore, this oligomerization was shown to be necessary for the functionality of the SECR since co-expression of a misspliced SECR with wild-type (WT) SECR had a dominant negative effect on receptor function (Ding et al., 2002). Importantly, secretin regulates growth-inhibitory effects through the SECR and the misspliced SECR has been found in gastrinoma. Thus heteromerization between the two, resulting in dominant negative activity of the misspliced SECR on the WT SECR, was speculated to facilitate tumor growth (Ding et al., 2002). This work on the SECR provided the first example of the functional and physiological relevance of oligomerization within the GPCR B1 family.
Using BRET, one cannot readily distinguish between dimers and higher order oligomers. Thus, in order to investigate the oligomeric state of the SECR, Millers group in 2008 used a combination of BiFC and BRET. The results of this study showed energy transfer between two but not three of the fluorescently/luminescently tagged SECRs, indicating that the SECR forms dimers but not higher order oligomers (Harikumar et al., 2008a).
The interaction site between two SECR protomers within a dimer has also been investigated by Miller’s group. By competing SECR dimer interactions measured by BRET with the co-expression of isolated SECR transmembrane (TM) sections, the interaction site of a SECR dimer was mapped to TM4 (Lisenbee and Miller, 2006). Further, by the use of mutational studies, the exact interaction site was localized to two specific residues in the lipid-exposed face of TM4, namely Gly243 and Ile247 (Harikumar et al., 2007). Subsequent mutations of these sites enabled comparative studies on ligand binding and signaling on receptor dimers versus monomers. These studies showed that SECR monomers have similar ligand binding properties as SECR dimers. In contrast, the potency for cAMP production was decreased for the monomer, indicating that the dimeric state of the SECR is required for correct G-protein coupling (Harikumar et al., 2007). Thus, dimerization is important for optimal signaling of the SECR, thereby adding evidence to the physiological and pharmacological importance of family B1 GPCR oligomerization.
Finally, an extensive study using BRET to detect heteromerization between the SECR and other members of the family B1 GPCRs was published in 2008 (Harikumar et al., 2008b). Interestingly, this study revealed a basic constitutive interaction between SECR and almost all other tested family B1 GPCRs, including GLP-1R, GLP-2R, GHRHR, VPAC1 and -2, PTHR1 and -2, and CRLR. This indicates a possible broad communication between receptors of the family B1 GPCRs. The only receptor of this study that turned out not to interact with the SECR was the CALCR. The absence of heteromerization between these receptors was suggested to be caused by the presence of two different residues in the CALCR TM4 (shown to be the dimer interaction site for SECR) not present in any other of the tested family B1 GPCRs (Harikumar et al., 2008b; for further details, see below).
Oligomerization of the Glucagon Receptor Family of B1 GPCRs
The GCGR family branch of the family B1 GPCRs consists of the structurally related GLP-1R, GCGR, and GIPR, which are all involved in the intricate control of blood-glucose levels (Holst et al., 2011), as well as the GLP-2R.
Homo- and heteromerization studies within this subfamily have been carried out by Schelshorn et al. (2011) using the BRET technique. The most interesting finding of this study was the association of the GLP-1R and the GIPR in a heteromer complex induced by stimulation with GLP-1. This is the first indication of ligand-induced oligomerization in the family B1 GPCRs. The effect of GLP-1 on the GLP-1R/GIPR heteromer assembly seemed to be reversed by GIP stimulation, which led to dissociation of the heteromer complex. This finding was validated by BRET saturation experiments, negative controls, as well as BRET kinetic experiments where the GLP-1-induced heteromerization was found to be very fast occurring within 30 s after ligand addition. Furthermore, a functional role of the GLP-1R/GIPR heteromer was indicated. Upon co-expression of the GLP-1R with the GIPR, GLP-1R signaling was altered and the β-arrestin recruitment was reduced (Schelshorn et al., 2011). These findings indicate a functional interaction between the receptor targets of the two important incretin hormones that together regulate post-prandial blood-glucose levels by potentiating insulin secretion from the pancreatic β-cells (Holst et al., 2009).
In addition to the GLP-1R/GIPR heteromer, Schelshorn et al. (2011) also investigated homo- and heteromerization of the other members of the GCGR family B1 GPCRs. Homomerization of the GLP-1R as well as the GIPR was claimed to be found in BRET saturation experiments. Furthermore, stimulation of these homomers with GLP-1 and GIP, respectively, was found to slightly reduce the BRET signal indicating either a reduced affinity or conformational changes in the receptor homomers upon ligand binding. However, these results were not compared with a negative control nor were there any statistical evaluation supporting this theory in the publication. We have performed similar BRET saturation experiments on GLP-1R homomerization showing a saturation curve but no effect of GLP-1 stimulation (see Figure 2A; Roed, 2011). Several attempts to validate this specific receptor–receptor interaction by negative controls were, however, unsuccessful. Thus, it still remains to be firmly established whether GLP-1R homomerization occurs.
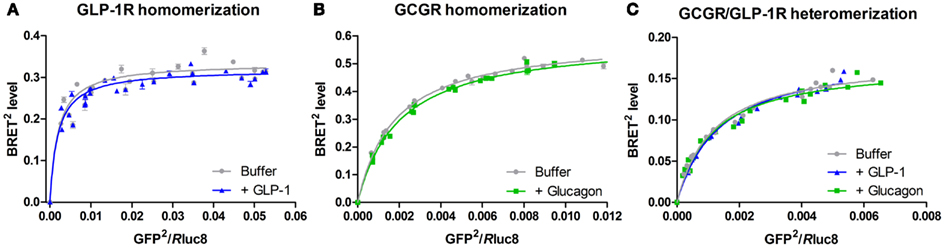
Figure 2. Bioluminescence resonance energy transfer saturation studies on homo- and heteromerization of GLP-1R and GCGR. These BRET studies were performed as BRET2 saturation assays, which uses a spectrally improved form of the luciferase enzyme substrate (coelenterazine), DeepBlueC, as well as a modified form of the energy acceptor, GFP2 (Pfleger and Eidne, 2006). In addition, a mutationally improved energy donor, Rluc8, with increased stability and quantum yield was used (De et al., 2007). HEK293 cells were transiently co-transfected with a constant amount of either GCGR-Rluc8 or GLP-1R-Rluc8 and an increasing amount of either GCGR-GFP2 or GLP-1R-GFP2 and incubated for 48 h at 37°C and 5% CO2. Subsequently, the cells were harvested, diluted to a concentration of 1 × 106 cells/ml, and stimulated with either 105 nM GLP-1, 83.5 nM glucagon, or buffer (negative control) for 5 min at room temperature. The BRET2 signal was measured upon addition of the Rluc8 substrate, DeepBlueC in a Mithras plate reader. All data are plotted as the BRET2 level as a function of the GFP2/Rluc8 ratio and fitted to a one-site specific binding model in GraphPad Prism. A saturating curve indicates a specific interaction between the Rluc- and GFP2-tagged receptor. The data represent mean ± SD of three independent experiments carried out in quadruplicates. (A) GLP-1R-Rluc8/GLP-1R-GFP2 homomerization in the presence (blue) or the absence (gray) of 105 nM GLP-1 (Roed, 2011). (B) GCGR-Rluc8/GCGR-GFP2 homomerization in the presence (green) or the absence (gray) of 83.5 nM glucagon (Orgaard, 2011). (C) GCGR-Rluc8/GLP-1R-GFP2 heteromerization in the presence of either 105 nM GLP-1 (blue), 83.5 nM glucagon (green), or buffer (gray) (Orgaard, 2011).
Schelshorn et al. (2011) also indicated homomerization of the GCGR as well as the GLP-2R by BRET studies. However, these results were not validated by BRET saturation experiments nor by negative controls. Figure 2B (Orgaard, 2011) shows our own results: a BRET saturation experiment with the GCGR in the presence or the absence of glucagon. A saturation curve was observed indicating homomerization of the GCGR with no effect of ligand addition. However, again, attempts to validate these data by negative controls were unsuccessful.
Finally, heteromerization for all possible interaction pairs of the structurally related receptors of the GCGR family was investigated by Schelshorn et al. (2011), by BRET studies testing both possible combinations of Rluc/YFP- or YFP/Rluc-tagged receptor pairs (e.g., both GLP-1R-Rluc/GCGR-YFP and GLP-1R-YFP/GCGR-Rluc). A small but significant reducing effect of GLP-1 on GLP-1R/GCGR heteromerization was observed as well as a small and significant reducing effect of both glucagon and GLP-2 on GCGR/GLP-2R heteromerization. These results were, however, not reproducible upon shifting from an Rluc/YFP-tagged to an YFP/Rluc-tagged receptor pair. Such differences in the BRET signal upon a shift in the tagging of the receptor pair can result from (1) differences in receptor expression levels influencing the ratio of formed homomers:heteromers or (2) changes in the relative orientation between the two tags known to affect the BRET signal (Ayoub and Pfleger, 2010). Further studies supporting either of the two possibilities were not carried out and thus no conclusion on the effect of ligand stimulation of GLP-1R/GCGR as well as GCGR/GLP-2R heteromers can be drawn from this study. We have performed BRET saturation experiments on cells co-expressing GLP-1R and GCGR resulting in saturation curves (see Figure 2C; Orgaard, 2011). Here, no change in the saturation curve was observed upon addition of either GLP-1 or glucagon.
Thus, within the GCGR subfamily an example of the physiological importance of family B1 GPCR oligomerization has been found for the GLP-1R/GIPR heteromer. BRET experiments indicating homo- and heteromerization of the other members of the GCGR subfamily have also been attempted. However, no firm conclusions can be drawn without further investigations including proper negative controls.
Oligomerization of the “Non-Prototypical” and “Non-Glucagon Receptor Family” B1 GPCRs
In addition to the prototypical SECR and the glucagon subfamily receptors, oligomerization among almost all other family B1 GPCRs has now been reported.
An interesting case is the PTHR1. Crystal structures of the isolated ECD of this receptor in its ligand-bound state showed a monomeric receptor with ligand binding in a 1:1 stoichiometry (Pioszak and Xu, 2008; Pioszak et al., 2009). However, in 2010 the crystal structure of the unbound PTHR1 ECD was published, surprisingly showing a dimerization between two ECDs (Pioszak et al., 2010). Interestingly, this dimerization was mediated by the receptor ECD C-termini taking up α-helical structures, similar to the helix of the ligand, PTH. The receptor C-termini bound to the binding site of the opposing PTHR1 protomer in the absence of ligand. This cross-linking is possible due to the sequence similarity between the receptor ECD C-terminal and PTH. The oligomerization of the full-length PTHR1 in the absence of PTH was confirmed by BRET saturation studies as well as morphological FRET in living cells. As predicted from the presence of receptor ECD dimerization only in the ligand unbound state found by crystallography, PTH stimulation of the receptors in BRET studies resulted in dissociation of PTHR1 oligomers. In agreement with receptor monomerization upon ligand binding and activation, the monomeric PTHR1 was found to be sufficient for coupling and activation of G-proteins. In addition, it was shown that PTHR1 oligomerization did not influence either receptor surface expression or ligand binding and signaling (Pioszak et al., 2010). Hence, PTHR1 provides an interesting example of family B1 GPCR oligomerization where constitutive presumably dimeric receptors on the cell surface are disrupted upon ligand binding, and, therefore, likely does not influence the functionality of the receptor. Interestingly, stimulation with PTH has also been shown to disrupt interactions between the SECR and both the PTHR1 and the PTHR2 (Harikumar et al., 2008b) confirming the homomer disruption effects of PTH and the full functionality of a monomeric PTHR. The effects of ligand binding to GPCRs on GPCR oligomerization within family B1 is summarized in Table 2.
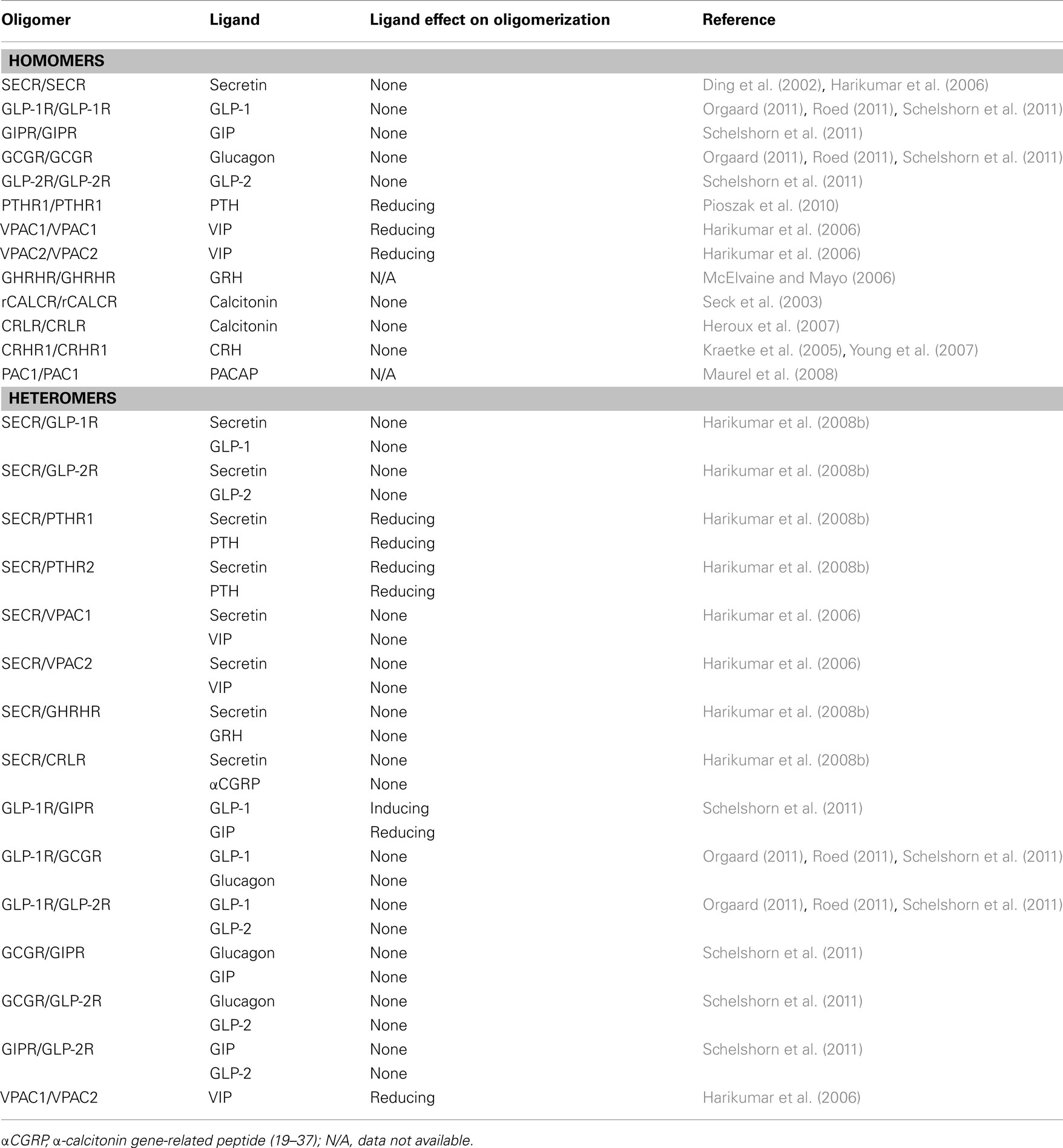
Table 2. Oligomerization of the family B1 GPCRs and the effect of ligand binding.
Another example in family B1 GPCRs where oligomerization does not influence receptor function is the VPAC1 and VPAC2 receptors. These receptors were shown to interact in both homomers and heteromers by BRET studies in 2006 (Harikumar et al., 2006). Later that same year, these interactions were further supported by Co-IP studies (Langer et al., 2006). Similar to the case of PTH binding to the PTHR1, binding of VIP to the VPACs disrupted receptor–receptor interactions (Harikumar et al., 2006). This VIP-induced VPAC oligomer disruption has not yet been followed up by structural investigations as for the PTHR1. Yet, the VPAC oligomerization was shown not to affect either ligand binding, receptor signaling, or internalization. This indicates a monomeric functional VPAC despite the presence of native constitutive receptor oligomers similar to the case of the PTHR1 (Langer et al., 2006).
The PTHR1 and VPAC receptors provide examples of the family B1 GPCRs where oligomerization does not influence receptor functionality. An example of the opposite situation where oligomerization does indeed influence the function of the GPCR can be found in the binding of GHRH to the GHRHR. Oligomerization between this receptor and a splice variant has been confirmed by Co-IP and shown to decrease ligand binding. The reduced ligand binding was not caused by a decreased receptor surface expression indicating that oligomerization has a dominant negative signaling effect on the function of GHRHR (McElvaine and Mayo, 2006). Another example of dominant negative effects of GPCR family B1 oligomerization is found in the CALCR. CALCRs form constitutive homomers which are not influenced by ligand binding as investigated by Co-IP and FRET experiments (Seck et al., 2003). As in the case of the SECR, homomerization of the CALCR was found to be important for the expression and functionality of the receptor, since heteromerization with a CALCR splice variant lead to reduced signaling. Heteromerization with the splice variant receptor thus had a dominant negative effect on the function of the WT CALCR. These studies were carried out using the rabbit CALCR (rCALCR; Seck et al., 2003). Interestingly, in a later study on the human CALCR (hCALCR), BRET experiments did not show any signals significantly different from baseline, indicating that the hCALCR does not homomerize (Harikumar et al., 2010). From alignment of the sequences of the rCALCR and the hCALCR a residue (aa 236) in the TM4 of the hCALCR (Arg) differing from rCALCR (His) as well as from all other human family B1 GPCRs was localized. Since TM4 has been assigned the interaction interface of SECR dimers, as previously described (Harikumar et al., 2007), it was speculated whether the lack of hCALCR homomerization was caused by this change in the hCALCR TM4 sequence. In support of this, mutation of the hCALCR TM4 residue to the rCALCR residue (A263H) resulted in a significant increased BRET signal indicating hCALCR homomerization. Further confirming the TM4 as the CALCR homomerization interface, the co-expression of the isolated hCALCR TM4 with rCALCR reduced the homomerization of rCALCR in a BRET competition experiment (Harikumar et al., 2010). The CALCR thus makes up an interesting example of family B1 GPCR oligomerization showing species-specific differences in the ability to engage in receptor–receptor interactions.
Finally, the CRHR1 has been shown to form constitutive homomers which are unaffected by ligand binding (Kraetke et al., 2005; Young et al., 2007) as well as homomers of the PAC1 and the CRLR receptors have been indicated (Kraetke et al., 2005; Maurel et al., 2008). To our knowledge, the only family B1 GPCR that has not yet been investigated for oligomerization is the CRHR2.
Inter-family Oligomerization of GPCRs
As mentioned, GPCR families A, B, and C share a highly conserved general morphology, but little or no inter-family sequence homology. Hence, the search for a common sequence responsible for GPCR oligomerization has so far been fruitless (Vohra et al., 2007). Even among receptors from the same family (family A), different combinations of all seven TM domains have been identified as contact points between protomers depending on the receptor examined (GPCR-OKB, 2012). This implies that the specific residues on TM4 responsible for homodimerization of the SECR (Harikumar et al., 2007) and homomerization of CALCR (Harikumar et al., 2010) may not necessarily be the same residues that are involved in homomerization between other family B1 members, or in heteromerization between the SECR or the CALCR and other family B1 GPCRs. Interestingly, as well as intra-family heteromerization has now been demonstrated to be widespread, inter-family heteromerization has also been reported to occur.
There are a couple of reports of GPCR inter-family heteromerization involving family B1 receptors. Functional heteromers, measured by their ability to induce cAMP production upon ligand stimulation, have been reported between two family A members, β2-AR and opsin, and the family B1 member GIPR (Vrecl et al., 2006). Also, the family A vasopressin V1b receptor (V1bR) has been shown to form constitutive heteromers with the family B1 CRHR1. However, the study did not reveal any obvious effects of heteromerization on the pharmacological properties of V1bR and CRHR1 (Young et al., 2007). In line with these reports, BRET results have suggested that the family B1 GCGR is capable of forming heteromers with the family A cholecystokinin A receptor (CCKAR; Orgaard, 2011).
In addition to the ability of family B1 GPCRs to interact with other types of GPCRs, these receptors may also rely on interactions with other types of membrane proteins for proper function or in order to diversify their physiological functions. It has been shown, that the CALCR and the CRLR as well as the VPAC1 can interact with some of the receptor activity-modifying proteins, RAMP1, -2, and/or -3 thereby assembling a variety of receptor types with different specificities for endogenous peptide ligands or altered signal transduction properties (Christopoulos et al., 2003; Barwell et al., 2011).
Taken together, the accumulating evidence of various types of GPCR heteromers suggests that all GPCRs share a common propensity to heteromerize, and that tissue-specific expression patterns are probably responsible for creating a much greater diversity of GPCR signaling than would have been expected from a 1:1 GPCR:G-protein stoichiometry.
Link between Oligomerization and Binding Cooperativity in GPCRs
As described, GPCR oligomerization can have different functional effects on, e.g., receptor expression, signaling, and ligand binding. One example of the functional effects of GPCR oligomerization is the facilitation of allosterism in ligand binding to GPCRs. According to the current knowledge, most GPCRs bind their ligands in a 1:1 ligand:receptor stoichiometry (Hoare and Usdin, 2001; Kristiansen, 2004). One example of an allosteric interaction is cooperativity, in which binding of a ligand to one receptor binding site changes the binding affinity to another receptor binding site and vice versa. This is known as reciprocity and is one of the essential features of allosteric interactions (Gregory et al., 2010). For a detailed review on allostery at GPCR oligomers, Smith and Milligan (2010). The presence of cooperativity in binding of ligands to GPCRs does not correspond to the hypothesis of a 1:1 binding stoichiometry (see Figure 1), since more than one binding site is required for allosteric interactions to occur. Thus, cooperativity in GPCR ligand binding constitutes a strong indication of oligomerization (although this assumption has been challenged (Chabre et al., 2009). Cooperativity can be either positive or negative depending on whether the binding of a second ligand to a second receptor binding site increases or reduces the affinity of a pre-bound ligand to the first receptor binding site.
Over the years of GPCR investigations, several examples of negative cooperativity in the binding of ligands to these receptors have been reported. Recently, the presence of this binding phenomenon has been directly linked to GPCR oligomerization.
The classical approach for detection of negative cooperativity is to monitor the dissociation of radioactively labeled ligand in the absence or presence of unlabeled ligand using an “infinite dilution” procedure, as first proposed for the insulin receptor by De Meyts et al. (1973). In this procedure, a small fraction of the surface expressed receptors are pre-occupied by 125I-labeled ligand in an initial association step followed by dissociation in an infinite dilution of either buffer alone or buffer with an excess of unlabeled ligand. In practice, the “infinite dilution” should be sufficient to prevent rebinding of the tracer (De Meyts et al., 1973). If the presence of an excess of unlabeled ligand accelerates the dissociation of pre-bound 125I-labeled ligand, it means that binding of the unlabeled ligand to a second binding site decreases the ligand affinity at the first binding site. This is the hallmark of negative cooperativity (Koshland, 1996).
Link between Oligomerization and Binding Cooperativity of GPCRs
In the mid 1970s, many years before the β2-AR was recognized as a GPCR, negative cooperativity was observed in the binding between this receptor and its ligand (Limbird et al., 1975; Limbird and Lefkowitz, 1976). When the β2-AR was later classified as a GPCR, a receptor type believed at that time to be a monomeric entity, the earlier finding of negative cooperativity became a matter of dispute (for review, see De Meyts, 1976, 2008). Many years later, the β2-AR was established to be a functional homomer (Hebert et al., 1996; Angers et al., 2000), an observation that offers an explanation for the observed negative binding cooperativity. However, no direct correlation between oligomerization and negative cooperativity has yet been established for the β2-AR. It has even been suggested that the monomeric β2-AR can fully couple to G-protein and exhibit cooperativity in binding (Whorton et al., 2007).
A direct correlation between negative cooperativity and receptor oligomerization was reported in 2005 for the family A GPCR thyrotrophin receptor (TSHR; Urizar et al., 2005). In this study, a combination of BRET assays and radioligand binding assays revealed that the TSHR homomerizes and displays negative cooperativity in the binding of its ligand (as shown earlier, De Meyts, 1976). In line with the accumulating evidence that most, if not all, family A GPCRs may oligomerize (Terrillon and Bouvier, 2004), this led the authors to speculate that cooperativity is a general phenomenon in family A GPCRs (Urizar et al., 2005). Indeed, several recent investigations have yielded results supporting this concept. In 2005 and 2006, negative cooperativity was found in the binding of chemokines to chemokine receptor (CCR) homo- and heteromers (El-Asmar et al., 2005; Springael et al., 2006). Furthermore, in 2008, a possible link between negative cooperativity and GPCR oligomerization was indicated for the relaxin family peptide receptor 1 (RXFP1) and RXFP2 (Svendsen et al., 2008a,b). Existence of positive cooperativity has also been reported in the binding of ligands to GPCRs. An example of this is a recent study by Gomes et al. (2011) suggesting that heteromers of μ and δ opioid receptors display strong positive cooperativity in ligand binding.
Link between Oligomerization and Binding Cooperativity in Family B1 GPCRs
In family B1 GPCRs, the extensive studies of the SECR by Miller and his group have also revealed a role for negative cooperativity in binding of secretin to the SECR. After localizing the specific residues responsible for SECR homodimer formation to TM4 (Harikumar et al., 2007), these residues were mutated producing a monomeric SECR. The dimeric WT SECR was then shown to display negative cooperativity in the binding of secretin, whereas the mutated monomeric SECR had lost this ability (Gao et al., 2009). These results provide the ultimate link between negative cooperativity and GPCR oligomerization. Another example of a link between negative cooperativity and receptor oligomerization of the family B1 GPCRs is indicated for the GLP-1R and GCGRs. As described previously and indicated by BRET saturation studies in Figure 2, these receptors might form constitutive oligomers. In radioligand binding assays, an accelerated dissociation of pre-bound 125I-labeled ligand in the presence of excess of unlabeled ligand suggested the existence of negative cooperativity in the binding between GLP-1 and the GLP-1R as well as in the binding between glucagon and the GCGR (Figure 3; Orgaard, 2011; Roed, 2011). Negative cooperativity in ligand binding to the GLP-1R and GCGR suggests the presence of at least two binding sites for each of the receptors. Since only one ligand bind per family B1 GPCR, as suggested by Hoare and Usdin, 2001; Figure 1), the presence of negative cooperativity supports the otherwise invalidated BRET saturation studies (Figures 2A,B; Orgaard, 2011; Roed, 2011) indicating homomerization of GLP-1R and GCGR. Thus, negative cooperativity in ligand binding to GPCRs can provide a functional support for GPCR oligomerization. Since the link between negative cooperativity and receptor oligomerization has been shown for the prototypical family B1 receptor, the SECR, as well as indicated for the GLP-1R and the GCGR, it can be speculated that this is a general phenomenon within family B1. However, this will have to be further investigated.
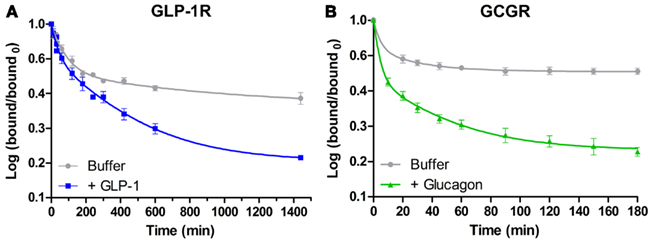
Figure 3. Dissociation experiment investigating negative cooperativity in the binding of GLP-1 and glucagon to the GLP-1R and the GCGR, respectively. These dissociation assays were carried out as described by De Meyts et al. (1973) for the insulin receptor. In this procedure, a small fraction of the surface expressed receptors are pre-occupied by 125I-labeled ligand in an initial association step followed by dissociation in an “infinite dilution” of either buffer of buffer containing an excess of unlabeled ligand. Accelerated dissociation of 125I-labeled ligand in the presence of unlabeled ligand indicates the presence of negative binding cooperativity. The data are plotted as the logarithm of bound/bound0 as a function of time in minutes and fitted to a two-site exponential decay model in GraphPad Prism. All data represent mean ± SD of three independent experiments carried out in duplicates. (A) Dissociation of GLP-1 from the GLP-1R. A concentration of 5 × 106 cells/ml BHK cells stably transfected with the GLP-1R were incubated with 150,000 cpm 125I-labeled GLP-1 for 3 h at 15°C. Subsequently, the unbound 125I-GLP-1 was aspired and the cells were diluted 1:40 in either HEPES binding buffer (gray) or HEPES binding buffer with 167 nM unlabeled GLP-1 (blue) and incubated at 25°C allowing ligand dissociation for up to 1440 min (24 h) (Roed, 2011). (B) Dissociation of glucagon from the GCGR. A concentration of 5 × 106 cells/ml BHK cells stably transfected with the GCGR were incubated with 150,000 cpm 125I-labeled glucagon for 1 h at 15°C. Subsequently, the unbound 125I-glucagon was removed and the cells were diluted 1:40 in either HEPES binding buffer (gray) or HEPES binding buffer with 167 nM unlabeled glucagon (green) and incubated at 25°C allowing ligand dissociation for up to 180 min (3 h) (Orgaard, 2011).
Summary and Conclusion
Here, we have reviewed the currently available data on oligomerization of the family B1 GPCRs. Studies on this family have revealed indications on homomerization of almost all members belonging to this subfamily of GPCRs. Even though not all of these studies have been as extensive as the studies on the prototypical family B1 GPCR, the SECR, the accumulating data and evidence points in the direction of homomerization as a general phenomenon for family B1 GPCRs. This is in line with the many publications on oligomerization of other classes of GPCRs (Milligan, 2001; George et al., 2002).
In addition to the increasing evidence of homomerization within family B1 GPCRs, several studies have also indicated heteromerization between different receptors of this family. The extensive study on the SECR indicating heteromerization between this prototypic receptor and almost all other family B1 GPCRs is a good example (Harikumar et al., 2008b). However, the study by Harikumar et al. also revealed a single family B1 GPCR that did not interact with the SECR, namely the CALCR with the lack of interaction likely caused by differences in key residues in TM4. Thus, there seems to be exceptions from the rule of a general heteromeric communication network in the family B1 GPCRs. Also, it is important to remember, that the physiological importance of such an intra- and possibly also inter-family cross-talk network for B1 GPCRs is dependent on the co-expression of the involved receptors in native tissue.
Interestingly, despite the many studies on family B1 GPCR oligomerization, no general effect of ligand binding to family B1 oligomers has been found. Indeed, most of these receptors seem to form constitutive oligomers with no effect of ligand binding (Table 2). Yet, examples on both the induction (GLP-1R and GIPR; Schelshorn et al., 2011) and disruption of receptor oligomerization (PTHR1; Pioszak et al., 2010 and VPAC1/2; Harikumar et al., 2006) in response to ligand binding have been indicated. This is interesting, since a common effect of ligand binding on GPCR oligomerization would be expected from the common model for the binding mechanism of ligands to family B1 GPCRs, as suggested by Hoare and Usdin(2001; see Figure 1).
Even though the many evidences on GPCR oligomerization cannot be neglected, the functionality of these receptor–receptor interactions is controversial and extensively debated. In family B1 GPCRs, only a few examples on GPCR oligomer functionality are available, e.g., the importance of SECR homodimerization for receptor expression and full intracellular G-protein coupling (Harikumar et al., 2007). However, family B1 GPCRs also provide examples on receptors which are fully functional in their monomeric form, such as the PTHR1 (Pioszak et al., 2010).
An important functional aspect of GPCR oligomerization is the possibility of cooperativity in ligand binding to these receptors. A strong link between negative cooperativity and GPCR oligomerization has been found for a family A GPCR, the THSR, as described previously (Urizar et al., 2005). Yet, to our knowledge, the only published data on such a link for the family B1 GPCRs is the SECR (Gao et al., 2009). In addition, we have here presented data indicating a similar link between oligomerization, as indicated by BRET experiments, and negative cooperativity, as indicated by dissociation binding experiments, for the GLP-1R and GCGR (Figures 2A,B and 3; Orgaard, 2011; Roed, 2011). However, to obtain a strong direct link between these two phenomena, further studies like mutational studies and functional complementation studies are required.
In conclusion, homomerization and to some extent also heteromerization of family B1 GPCRs seem to be a general phenomenon. However, only a few studies on the functionality of these receptor–receptor interactions are currently available. The family B1 GPCRs are very important receptors since they are almost all involved in diseases affecting millions of people worldwide, such as diabetes, osteoporosis and chronic inflammation. Thus, further studies are needed to understand the importance of family B1 GPCR oligomerization and its possible role in pathophysiological conditions, in order to improve the development of treatments for these diseases.
Conflict of Interest Statement
Sarah Norklit Roed, Rasmus Jorgensen, and Pierre De Meyts own Novo Nordisk A/S company stocks.
Acknowledgments
We kindly thank Jane Nohr Larsen for cloning of the tagged receptor constructs for the BRET studies as well as for great supervision. Also, we are thankful to Milka Vrecl and Nikolaj Kulahin for fruitful discussions and help on BRET result interpretations.
Abbreviations
αCGRP, α-calcitonin gene-related peptide (19–37); β-AR, β-adrenergic receptor; BiFC, bimolecular fluorescence complementation; BiLC, bimolecular luminescence complementation; BRET, bioluminescence resonance energy transfer; CALCR, calcitonin receptor; CaSR, Ca2+ sensing receptor; CCKAR, cholecystokinin A receptor; CCR, chemokine receptor; CFP, cyan fluorescent protein; co-IP, co-immunoprecipitation; CRH, corticotrophin-releasing hormone; CRHR1 and CRHR2, corticotrophin-releasing hormone receptor-1 and -2; CRLR, calcitonin receptor-like receptor; ECD, extracellular domain; ECL, extracellular loop; FRET, fluorescence resonance energy transfer; GABABR, γ-aminobutyric acid receptor; GCGR, glucagon receptor; GFP, green fluorescent protein; GHRHR, growth hormone-releasing hormone receptor; GIP, glucose-dependent insulinotropic polypeptide; GIPR, glucose-dependent insulinotropic polypeptide receptor; GLP-1 and GLP-2, glucagon-like peptide-1 and -2; GLP-1R and GLP-2R, glucagon-like peptide-1 and -2 receptors; GPCR, G-protein-coupled receptor; GRH, growth hormone-releasing hormone; mGluR, metabotropic glutamate receptor; PACAP, pituitary adenylate cyclase-activating peptide; PAC1, pituitary adenylate cyclase-activating peptide receptor; PTH, parathyroid hormone; PTHR1 and PTHR2, parathyroid hormone receptor-1 and -2; RAMP, receptor activity-modifying protein; Rluc, Renilla luciferase; RXFP1 and RXFP2, relaxin family peptide receptor-1 and -2; SECR, secretin receptor; TR-FRET, time-resolved FRET; TSHR, thyrotrophin receptor; VIP, vasoactive intestinal peptide; VPAC1 and VPAC2, vasoactive intestinal peptide receptor-1 and -2; V1bR, vasopressin V1b receptor; YFP, yellow fluorescent protein; 7TM, seven transmembrane.
References
AbdAlla, S., Lother, H., el, M. A., and Quitterer, U. (2001). Increased AT(1) receptor heterodimers in preeclampsia mediate enhanced angiotensin II responsiveness. Nat. Med. 7, 1003–1009.
Albizu, L., Cottet, M., Kralikova, M., Stoev, S., Seyer, R., Brabet, I., Roux, T., Bazin, H., Bourrier, E., Lamarque, L., Breton, C., Rives, M. L., Newman, A., Javitch, J., Trinquet, E., Manning, M., Pin, J. P., Mouillac, B., and Durroux, T. (2010). Time-resolved FRET between GPCR ligands reveals oligomers in native tissues. Nat. Chem. Biol. 6, 587–594.
Ali, S., and Drucker, D. J. (2009). Benefits and limitations of reducing glucagon action for the treatment of type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 296, E415–E421.
Angers, S., Salahpour, A., Joly, E., Hilairet, S., Chelsky, D., Dennis, M., and Bouvier, M. (2000). Detection of beta 2-adrenergic receptor dimerization in living cells using bioluminescence resonance energy transfer (BRET). Proc. Natl. Acad. Sci. U.S.A. 97, 3684–3689.
Ayoub, M. A., and Pfleger, K. D. (2010). Recent advances in bioluminescence resonance energy transfer technologies to study GPCR heteromerization. Curr. Opin. Pharmacol. 10, 44–52.
Bagger, J. I., Knop, F. K., Holst, J. J., and Vilsboll, T. (2011). Glucagon antagonism as a potential therapeutic target in type 2 diabetes. Diabetes Obes. Metab. 13, 965–971.
Barwell, J., Gingell, J. J., Watkins, H. A., Archbold, J. K., Poyner, D. R., and Hay, D. L. (2011). Calcitonin and calcitonin receptor-like receptors: common themes with family B GPCRs? Br. J. Pharmacol. 166, 51–65.
Bockaert, J., and Pin, J. P. (1999). Molecular tinkering of G protein-coupled receptors: an evolutionary success. EMBO J. 18, 1723–1729.
Campbell, R. M., Bongers, J., and Felix, A. M. (1995). Rational design, synthesis, and biological evaluation of novel growth hormone releasing factor analogues. Biopolymers 37, 67–88.
Chabre, M., Deterre, P., and Antonny, B. (2009). The apparent cooperativity of some GPCRs does not necessarily imply dimerization. Trends Pharmacol. Sci. 30, 182–187.
Christopoulos, A., Christopoulos, G., Morfis, M., Udawela, M., Laburthe, M., Couvineau, A., Kuwasako, K., Tilakaratne, N., and Sexton, P. M. (2003). Novel receptor partners and function of receptor activity-modifying proteins. J. Biol. Chem. 278, 3293–3297.
Comps-Agrar, L., Kniazeff, J., Norskov-Lauritsen, L., Maurel, D., Gassmann, M., Gregor, N., Prezeau, L., Bettler, B., Durroux, T., Trinquet, E., and Pin, J. P. (2011). The oligomeric state sets GABA(B) receptor signalling efficacy. EMBO J. 30, 2336–2349.
D’Alessio, D. (2011). The role of dysregulated glucagon secretion in type 2 diabetes. Diabetes Obes. Metab. 13(Suppl. 1), 126–132.
De, A., Loening, A. M., and Gambhir, S. S. (2007). An improved bioluminescence resonance energy transfer strategy for imaging intracellular events in single cells and living subjects. Cancer Res. 67, 7175–7183.
De Meyts, P. (1976). Cooperative properties of hormone receptors in cell membranes. J. Supramol. Struct. 4, 241–258.
De Meyts, P. (2008). The insulin receptor: a prototype for dimeric, allosteric membrane receptors? Trends Biochem. Sci. 33, 376–384.
De Meyts, P., Roth, J., Neville, D. M. Jr., Gavin, J. R. III, and Lesniak, M. A. (1973). Insulin interactions with its receptors: experimental evidence for negative cooperativity. Biochem. Biophys. Res. Commun. 55, 154–161.
de Paula, F. J., and Rosen, C. J. (2010). Back to the future: revisiting parathyroid hormone and calcitonin control of bone remodeling. Horm. Metab. Res. 42, 299–306.
Deacon, C. F. (2007). Incretin-based treatment of type 2 diabetes: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors. Diabetes Obes. Metab. 9(Suppl. 1), 23–31.
Ding, W. Q., Cheng, Z. J., McElhiney, J., Kuntz, S. M., and Miller, L. J. (2002). Silencing of secretin receptor function by dimerization with a misspliced variant secretin receptor in ductal pancreatic adenocarcinoma. Cancer Res. 62, 5223–5229.
Dong, M., Gao, F., Pinon, D. I., and Miller, L. J. (2008a). Insights into the structural basis of endogenous agonist activation of family B G protein-coupled receptors. Mol. Endocrinol. 22, 1489–1499.
Dong, M., Pinon, D. I., and Miller, L. J. (2008b). Exploration of the endogenous agonist mechanism for activation of secretin and VPAC1 receptors using synthetic glycosylated peptides. J. Mol. Neurosci. 36, 254–259.
Dong, M., Pinon, D. I., Asmann, Y. W., and Miller, L. J. (2006). Possible endogenous agonist mechanism for the activation of secretin family G protein-coupled receptors. Mol. Pharmacol. 70, 206–213.
El-Asmar, L., Springael, J. Y., Ballet, S., Andrieu, E. U., Vassart, G., and Parmentier, M. (2005). Evidence for negative binding cooperativity within CCR5-CCR2b heterodimers. Mol. Pharmacol. 67, 460–469.
Fredriksson, R., Lagerstrom, M. C., Lundin, L. G., and Schioth, H. B. (2003). The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 63, 1256–1272.
Galvez, T., Duthey, B., Kniazeff, J., Blahos, J., Rovelli, G., Bettler, B., Prezeau, L., and Pin, J. P. (2001). Allosteric interactions between GB1 and GB2 subunits are required for optimal GABA(B) receptor function. EMBO J. 20, 2152–2159.
Gao, F., Harikumar, K. G., Dong, M., Lam, P. C. H., Sexton, P. M., Christopoulos, A., Bordner, A., Abagyan, R., and Miller, L. J. (2009). Functional importance of a structurally distinct homodimeric complex of the family B G protein-coupled secretin receptor. Mol. Pharmacol. 76, 264–274.
George, S. R., O’Dowd, B. F., and Lee, S. P. (2002). G-protein-coupled receptor oligomerization and its potential for drug discovery. Nat. Rev. Drug Discov. 1, 808–820.
Gomes, I., Ijzerman, A. P., Ye, K., Maillet, E. L., and Devi, L. A. (2011). G protein-coupled receptor heteromerization: a role in allosteric modulation of ligand binding. Mol. Pharmacol. 79, 1044–1052.
Gonzalez-Rey, E., Chorny, A., Fernandez-Martin, A., Varela, N., and Delgado, M. (2005). Vasoactive intestinal peptide family as a therapeutic target for Parkinson’s disease. Expert Opin. Ther. Targets 9, 923–929.
GPCR-OKB. (2012). The G-protein Coupled Receptor-Oligomerization Knowledge Base Project. Available at: http://data.gpcr-okb.org/gpcr-okb/(2012/3/4)
Grace, C. R., Perrin, M. H., DiGruccio, M. R., Miller, C. L., Rivier, J. E., Vale, W. W., and Riek, R. (2004). NMR structure and peptide hormone binding site of the first extracellular domain of a type B1 G protein-coupled receptor. Proc. Natl. Acad. Sci. U.S.A. 101, 12836–12841.
Gregory, K. J., Sexton, P. M., and Christopoulos, A. (2010). Overview of receptor allosterism. Curr. Protoc. Pharmacol. Chapter 1, Unit 1.21.
Hamdan, F. F., Percherancier, Y., Breton, B., and Bouvier, M. (2006). Monitoring protein-protein interactions in living cells by bioluminescence resonance energy transfer (BRET). Curr. Protoc. Neurosci. Chapter 5, Unit.
Hansen, J. L., Hansen, J. T., Speerschneider, T., Lyngs, C., Erikstrup, N., Burstein, E. S., Weiner, D. M., Walther, T., Makita, N., Iiri, T., Merten, N., Kostenis, E., and Sheikh, S. P. (2009). Lack of evidence for AT1R/B2R heterodimerization in COS-7, HEK293, and NIH3T3 cells: how common is the AT1R/B2R heterodimer? J. Biol. Chem. 284, 1831–1839.
Harikumar, K. G., Ball, A. M., Sexton, P. M., and Miller, L. J. (2010). Importance of lipid-exposed residues in transmembrane segment four for family B calcitonin receptor homo-dimerization. Regul. Pept. 164, 113–119.
Harikumar, K. G., Happs, R. M., and Miller, L. J. (2008a). Dimerization in the absence of higher-order oligomerization of the G protein-coupled secretin receptor. Biochim. Biophys. Acta 1778, 2555–2563.
Harikumar, K. G., Morfis, M. M., Sexton, P. M., and Miller, L. J. (2008b). Pattern of intra-family hetero-oligomerization involving the G-protein-coupled secretin receptor. J. Mol. Neurosci. 36, 279–285.
Harikumar, K. G., Morfis, M. M., Lisenbee, C. S., Sexton, P. M., and Miller, L. J. (2006). Constitutive formation of oligomeric complexes between family B G protein-coupled vasoactive intestinal polypeptide and secretin receptors. Mol. Pharmacol. 69, 363–373.
Harikumar, K. G., Pinon, D. I., and Miller, L. J. (2007). Transmembrane segment IV contributes a functionally important interface for oligomerization of the class II G protein-coupled secretin receptor. J. Biol. Chem. 282, 30363–30372.
Hebert, T. E., Moffett, S., Morello, J. P., Loisel, T. P., Bichet, D. G., Barret, C., and Bouvier, M. (1996). A peptide derived from a beta2-adrenergic receptor transmembrane domain inhibits both receptor dimerization and activation. J. Boil. Chem. 271, 16384–16392.
Heroux, M., Breton, B., Hogue, M., and Bouvier, M. (2007). Assembly and Signaling of CRLR and RAMP1 complexes assessed by BRET. Biochemistry 46, 7022–7033.
Hoare, S. R. (2005). Mechanisms of peptide and nonpeptide ligand binding to Class B G-protein-coupled receptors. Drug Discov. Today 10, 417–427.
Hoare, S. R., and Usdin, T. B. (2001). Molecular mechanisms of ligand recognition by parathyroid hormone 1 (PTH1) and PTH2 receptors. Curr. Pharm. Des. 7, 689–713.
Holst, J. J., Christensen, M., Lund, A., de, H. J., Svendsen, B., Kielgast, U., and Knop, F. K. (2011). Regulation of glucagon secretion by incretins. Diabetes Obes. Metab. 13(Suppl. 1), 89–94.
Holst, J. J., Vilsboll, T., and Deacon, C. F. (2009). The incretin system and its role in type 2 diabetes mellitus. Mol. Cell. Endocrinol. 297, 127–136.
Hornby, P. J., and Moore, B. A. (2011). The therapeutic potential of targeting the glucagon-like peptide-2 receptor in gastrointestinal disease. Expert Opin. Ther. Targets 15, 637–646.
Ishihara, T., Nakamura, S., Kaziro, Y., Takahashi, T., Takahashi, K., and Nagata, S. (1991). Molecular cloning and expression of a cDNA encoding the secretin receptor. EMBO J. 10, 1635–1641.
Jordan, B. A., and Devi, L. A. (1999). G-protein-coupled receptor heterodimerization modulates receptor function. Nature 399, 697–700.
Kaczor, A. A., and Selent, J. (2011). Oligomerization of G protein-coupled receptors: biochemical and biophysical methods. Curr. Med. Chem. 18, 4606–4634.
Kenakin, T., and Miller, L. J. (2010). Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol. Rev. 62, 265–304.
Knop, F. K., Vilsboll, T., and Holst, J. J. (2009). Incretin-based therapy of type 2 diabetes mellitus. Curr. Protein Pept. Sci. 10, 46–55.
Kolakowski, L. F. Jr. (1994). GCRDb: a G-protein-coupled receptor database. Recept. Channels 2, 1–7.
Koshland, J. (1996). The structural basis of negative cooperativity: receptors and enzymes. Curr. Opin. Struct. Biol. 6, 757–761.
Kraetke, O., Wiesner, B., Eichhorst, J., Furkert, J., Bienert, M., and Beyermann, M. (2005). Dimerization of corticotropin-releasing factor receptor type 1 is not coupled to ligand binding. J. Recept. Signal Transduct. Res. 25, 251–276.
Kristiansen, K. (2004). Molecular mechanisms of ligand binding, signaling, and regulation within the superfamily of G-protein-coupled receptors: molecular modeling and mutagenesis approaches to receptor structure and function. Pharmacol. Ther. 103, 21–80.
Kunishima, N., Shimada, Y., Tsuji, Y., Sato, T., Yamamoto, M., Kumasaka, T., Nakanishi, S., Jingami, H., and Morikawa, K. (2000). Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature 407, 971–977.
Langer, I., Gaspard, N., and Robberecht, P. (2006). Pharmacological properties of Chinese hamster ovary cells coexpressing two vasoactive intestinal peptide receptors (hVPAC1 and hVPAC2). Br. J. Pharmacol. 148, 1051–1059.
Limbird, L. E., and Lefkowitz, R. J. (1976). Negative cooperativity among beta-adrenergic receptors in frog erythrocyte membranes. J. Biol. Chem. 251, 5007–5014.
Limbird, L. E., Meyts, P. D., and Lefkowitz, R. J. (1975). Beta-adrenergic receptors: evidence for negative cooperativity. Biochem. Biophys. Res. Commun. 64, 1160–1168.
Lisenbee, C. S., and Miller, L. J. (2006). Secretin receptor oligomers form intracellularly during maturation through receptor core domains. Biochemistry 45, 8216–8226.
Maurel, D., Comps-Agrar, L., Brock, C., Rives, M. L., Bourrier, E., Ayoub, M. A., Bazin, H., Tinel, N., Durroux, T., Prezeau, L., Trinquet, E., and Pin, J. P. (2008). Cell-surface protein-protein interaction analysis with time-resolved FRET and snap-tag technologies: application to GPCR oligomerization. Nat. Methods 5, 561–567.
McElvaine, A. T., and Mayo, K. E. (2006). A dominant-negative human growth hormone-releasing hormone (GHRH) receptor splice variant inhibits GHRH binding. Endocrinology 147, 1884–1894.
Milligan, G., and Bouvier, M. (2005). Methods to monitor the quaternary structure of G protein-coupled receptors. FEBS J. 272, 2914–2925.
Orgaard, A. (2011). Binding and Dimerization Studies on the Glucagon Receptor. University of Copenhagen, Copenhagen.
Pal, K., Swaminathan, K., Xu, H. E., and Pioszak, A. A. (2010). Structural basis for hormone recognition by the Human CRFR2{alpha} G protein-coupled receptor. J. Biol. Chem. 285, 40351–40361.
Parthier, C., Kleinschmidt, M., Neumann, P., Rudolph, R., Manhart, S., Schlenzig, D., Fanghanel, J., Rahfeld, J. U., Demuth, H. U., and Stubbs, M. T. (2007). Crystal structure of the incretin-bound extracellular domain of a G protein-coupled receptor. Proc. Natl. Acad. Sci. U.S.A. 104, 13942–13947.
Pfleger, K. D. G., and Eidne, K. A. (2006). Illuminating insights into protein-protein interactions using bioluminescence resonance energy transfer (BRET). Nat. Methods 3, 165–169.
Pioszak, A. A., Harikumar, K. G., Parker, N. R., Miller, L. J., and Xu, H. E. (2010). Dimeric arrangement of the parathyroid hormone receptor and a structural mechanism for ligand-induced dissociation. J. Biol. Chem. 285, 12435–12444.
Pioszak, A. A., Parker, N. R., Gardella, T. J., and Xu, H. E. (2009). Structural basis for parathyroid hormone-related protein binding to the parathyroid hormone receptor and design of conformation-selective peptides. J. Biol. Chem. 284, 28382–28391.
Pioszak, A. A., Parker, N. R., Suino-Powell, K., and Xu, H. E. (2008). Molecular recognition of corticotropin-releasing factor by its G-protein-coupled receptor CRFR1. J. Biol. Chem. 283, 32900–32912.
Pioszak, A. A., and Xu, H. E. (2008). Molecular recognition of parathyroid hormone by its G protein-coupled receptor. Proc. Natl. Acad. Sci. U.S.A. 105, 5034–5039.
Roed, S. N. (2011). Receptor Dimerization and Binding Profile of the Incretin Glucagon-Like Peptide-1. University of Copenhagen, Copenhagen.
Runge, S., Thogersen, H., Madsen, K., Lau, J., and Rudolph, R. (2008). Crystal structure of the ligand-bound glucagon-like peptide-1 receptor extracellular domain. J. Biol. Chem. 283, 11340–11347.
Schelshorn, D. W., Joly, F., Mutel, S., Hampe, C., Breton, B., Mutel, V., and Lutjens, R. (2011). Lateral allosterism in the glucagon receptor family: GLP-1 induces GPCR heteromer formation. Mol. Pharmacol. 81, 309–318.
Seck, T., Baron, R., and Horne, W. C. (2003). The alternatively spliced deltae13 transcript of the rabbit calcitonin receptor dimerizes with the C1a isoform and inhibits its surface expression. J. Biol. Chem. 278, 23085–23093.
See, H. B., Seeber, R. M., Kocan, M., Eidne, K. A., and Pfleger, K. D. (2011). Application of G protein-coupled receptor-heteromer identification technology to monitor & beta;-arrestin recruitment to G protein-coupled receptor heteromers. Assay Drug Dev. Technol. 9, 21–30.
Smith, N. J., and Milligan, G. (2010). Allostery at G protein-coupled receptor homo- and heteromers: uncharted pharmacological landscapes. Pharmacol. Rev. 62, 701–725.
Springael, J. Y., Le Minh, P. N., Urizar, E., Costagliola, S., Vassart, G., and Parmentier, M. (2006). Allosteric modulation of binding properties between units of chemokine receptor homo- and hetero-oligomers. Mol. Pharmacol. 69, 1652–1661.
Stengel, A., and Tache, Y. (2010). Corticotropin-releasing factor signaling and visceral response to stress. Exp. Biol. Med. (Maywood) 235, 1168–1178.
Svendsen, A. M., Vrecl, M., Ellis, T. M., Heding, A., Kristensen, J. B., Wade, J. D., Bathgate, R. A., De Meyts, P., and Nohr, J. (2008a). Cooperative binding of insulin-like Peptide 3 to a dimeric relaxin family peptide receptor 2. Endocrinology 149, 1113–1120.
Svendsen, A. M., Zalesko, A., Konig, J., Vrecl, M., Heding, A., Kristensen, J. B., Wade, J. D., Bathgate, R. A. D., De Meyts, P., and Nohr, J. (2008b). Negative cooperativity in H2 relaxin binding to a dimeric relaxin family peptide receptor 1. Mol. Cell. Endocrinol. 296, 10–17.
Tan, Y. V., Couvineau, A., Murail, S., Ceraudo, E., Neumann, J. M., Lacapere, J. J., and Laburthe, M. (2006). Peptide agonist docking in the N-terminal ectodomain of a class II G protein-coupled receptor, the VPAC1 receptor. Photoaffinity, NMR, and molecular modeling. J. Biol. Chem. 281, 12792–12798.
ter Haar, E., Koth, C. M., bdul-Manan, N., Swenson, L., Coll, J. T., Lippke, J. A., Lepre, C. A., Garcia-Guzman, M., and Moore, J. M. (2010). Crystal structure of the ectodomain complex of the CGRP receptor, a class-B GPCR, reveals the site of drug antagonism. Structure 18, 1083–1093.
Terrillon, S., and Bouvier, M. (2004). Roles of G-protein-coupled receptor dimerization. EMBO Rep. 5, 30–34.
Ulrich, C. D., Holtmann, M., and Miller, L. J. (1998). Secretin and vasoactive intestinal peptide receptors: members of a unique family of G protein-coupled receptors. Gastroenterology 114, 382–397.
Urizar, E., Montanelli, L., Loy, T., Bonomi, M., Swillens, S., Gales, C., Bouvier, M., Smits, G., Vassart, G., and Costagliola, S. (2005). Glycoprotein hormone receptors: link between receptor homodimerization and negative cooperativity. EMBO J. 24, 1954–1964.
Valdez, G. R. (2009). CRF receptors as a potential target in the development of novel pharmacotherapies for depression. Curr. Pharm. Des. 15, 1587–1594.
Verhaar, H. J., and Lems, W. F. (2010). PTH analogues and osteoporotic fractures. Expert Opin. Biol. Ther. 10, 1387–1394.
Vidi, P. A., Ejendal, K. F., Przybyla, J. A., and Watts, V. J. (2011). Fluorescent protein complementation assays: new tools to study G protein-coupled receptor oligomerization and GPCR-mediated signaling. Mol. Cell. Endocrinol. 331, 185–193.
Vohra, S., Chintapalli, S. V., Illingworth, C. J., Reeves, P. J., Mullineaux, P. M., Clark, H. S., Dean, M. K., Upton, G. J., and Reynolds, C. A. (2007). Computational studies of family A and family B GPCRs. Biochem. Soc. Trans. 35, 749–754.
Vrecl, M., Drinovec, L., Elling, C., and Heding, A. (2006). Opsin oligomerization in a heterologous cell system. J. Recept. Signal Transduct. Res. 26, 505–526.
WHO. (2011). World Healthcare Organization. Available at: http://www.who.int/en/(2012/3/4)
Whorton, M. R., Bokoch, M. P., Rasmussen, S. G., Huang, B., Zare, R. N., Kobilka, B., and Sunahara, R. K. (2007). A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc. Natl. Acad. Sci. U.S.A. 104, 7682–7687.
Keywords: GPCRs, family B1, oligomerization, BRET, binding cooperativity
Citation: Roed SN, Orgaard A, Jorgensen R and Meyts PD (2012) Receptor oligomerization in family B1 of G-protein-coupled receptors: focus on BRET investigations and the link between GPCR oligomerization and binding cooperativity. Front. Endocrin. 3:62. doi: 10.3389/fendo.2012.00062
Received: 03 April 2012; Paper pending published: 12 April 2012;
Accepted: 20 April 2012; Published online: 07 May 2012.
Edited by:
Milka Vrecl, Veterinary faculty of the University in Ljubljana, SloveniaReviewed by:
Laurence J. Miller, Mayo Clinic, USANicola J. Smith, Victor Chang Cardiac Research Institute, Australia
Copyright: © 2012 Roed, Orgaard, Jorgensen and Meyts. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Sarah Norklit Roed, Department of Incretin Biology, Diabetes Biology and Hagedorn Research Institute, Novo Nordisk A/S, Niels Steensensvej 6, 2820 Gentofte, Denmark. e-mail:c25yb0Bub3Zvbm9yZGlzay5jb20=
†Sarah Norklit Roed and Anne Orgaard have contributed equally to this work.