 Valeria Ramundo1*
Valeria Ramundo1* Tonino Ercolino2
Tonino Ercolino2 Antongiulio Faggiano1,3
Antongiulio Faggiano1,3 Valentino Giachè2 Benedetta Ragghianti2 Elena Rapizzi2
Valentino Giachè2 Benedetta Ragghianti2 Elena Rapizzi2 Annamaria Colao1 Massimo Mannelli2
Annamaria Colao1 Massimo Mannelli2
- 1 Department of Molecular and Clinical Endocrinology and Oncology, “Federico II” University of Naples, Naples, Italy
- 2 Section of Endocrinology, Department of Clinical Pathophysiology, University of Florence, Naples, Italy
- 3 Endocrinology, National Cancer Institute, Naples, Italy
Background: Mutations in the genes encoding B, C, and D subunits of the succinate dehydrogenase (SDH) are involved in the pathogenesis of familial paraganglioma (PGL) syndrome. Many subjects with apparently sporadic extra-adrenal paragangliomas are found to be carrier for SDH mutation. Objective: Here we describe four subjects with apparently sporadic extra-adrenal paragangliomas with newly identified mutations in the SDH subunit B and the related clinical phenotype. Methods: Gene sequencing was performed to search for mutations in the SDHB (all exons), SDHC (all exons), and SDHD (all exons) genes as well as VHL (all exons) and RET (10, 11, 13, 14, 15, 16 exons) genes in all four index cases. A complete clinical, biochemical, and instrumental work-up was performed. Results: Three subjects were found to be affected with a nonsense SDHB germline mutation (Q30X, Y61X, and W201X, respectively). These mutations are predicted to encode for a truncated SDHB protein. The fourth subject presented a S195del frameshift mutation, causing a deletion of the codon AGC, encoding for a serine. Clinical presentation and course of each patient is described. Conclusions: Extra-adrenal paragangliomas, localized in the sympathetic ganglia (in the posterior thorax or in the abdomen), are very often SDHB-inherited form rather than sporadic tumor. Our data confirm the importance of genetic screening in patients affected with paragangliomas and enlarge the list of mutations responsible for the presence of these tumors.
Introduction
Extra-adrenal paragangliomas (PGLs) arising in the thorax and in the abdomen are sympathetic in origin and generally catecholamine secreting (Baysal, 2002).
Alterations of the mitochondrial complex II or succinate dehydrogenase (SDH), which catalyzes the oxidation of succinate to fumarate in the Krebs cycle and is involved in the aerobic electron transport chain, have been shown to predispose to PGL formation (King et al., 2006).
Germline mutations in genes encoding the structural (SDHD, SDHC) and catalytic (SDHB) succinate dehydrogenase subunits predispose to hereditary paraganglioma syndromes named PGL-1 (OMIM #168000), PGL-3 (OMIM #605373), and PGL-4 (OMIM #115310), respectively (Baysal et al., 2000; Astuti et al., 2001, 2003; Neumann et al., 2002).
Germline mutations in SDHD (PGL-1) are associated with non-secreting, multiple head and neck paragangliomas (HN PGLs) and less frequently with pheochromocytomas (PHEOs) and abdominal PGLs, whereas SDHC mutations (PGL-3) have been associated with benign, solitary HN PGLs (Neumann et al., 2004; Schiavi et al., 2005; Benn et al., 2006), and more recently also with abdominal PGLs (Mannelli et al., 2007). SDHB mutations (PGL-4) are mainly associated with extra-adrenal abdominal or thoracic secreting PGLs (sPGLs; Gimenez-Roqueplo et al., 2002; Neumann et al., 2004; Amar et al., 2005; Benn et al., 2006). Moreover, at variance with PGL-1 and PGL-3, which generally display a benign phenotype, PGL-4 develops into metastatic disease with higher frequency (30–70% of cases) (Gimenez-Roqueplo et al., 2003).
The SDHB gene is located at chromosome 1p35–36.1. Mutations in SDHB gene are inherited as autosomal dominant traits, and this gene behaves like an oncosuppressor.
Therefore SDHB mutation carriers have a greater risk to develop extra-adrenal and malignant tumors (Young et al., 2002; Gimenez-Roqueplo et al., 2003).
To date 177 SDHB gene sequence variants have been described, distributed as following: 78 missenses, 20 nonsenses, 47 frameshifts, 20 splicesites, 12 large deletions. These variations have been associated with different type of tumors, ranging from HN PGLs, PHEOs, abdominal, or thoracic PGLs, either benign or malignant.
In this paper we describe four subjects with apparently sporadic extra-adrenal paragangliomas with newly identified mutations in the SDH subunit B and the related clinical phenotype.
Patients and Methods
Patient 1
A 18-year-old man was evaluated for arterial hypertension. A clinical and morpho-functional assessment was performed: urinary catecholamines were elevated and magnetic resonance imaging (MRI) showed a 5.5 cm abdominal lesion in left paravertebral region. A scintigraphy with 123I-metaiodobenzyl-guanidine 123I-(MIBG) showed a high uptake in the corresponding site. For the control of hypertension, the patient started anti-hypertensive therapy.
The patient underwent surgical resection of the lesion, which was confirmed to be a PGL at the pathological examination. A post-surgical assessment was performed three months later: blood pressure was normal as well as urinary catecholamine levels. Anti-hypertensive drugs were discontinued and 123I-MIBG scintigraphy resulted negative.
Three years after surgery, the patient was again affected with arterial hypertension. For this reason anti-hypertensive therapy was resumed. Catecholamine levels were normal. 123I-MIBG scintigraphy and MRI were negative. At present, no other lesions, potentially associated to PGL have been detected. Genetic analysis revealed the presence of a mutation of the SDHB gene (Figure 1A). The genetic analysis was then extended to first-degree relatives and the father of the patient resulted to be carrier of the mutation without any apparent ongoing disease. Anyway, he refused to undergo biochemical and morpho-functional evaluations.
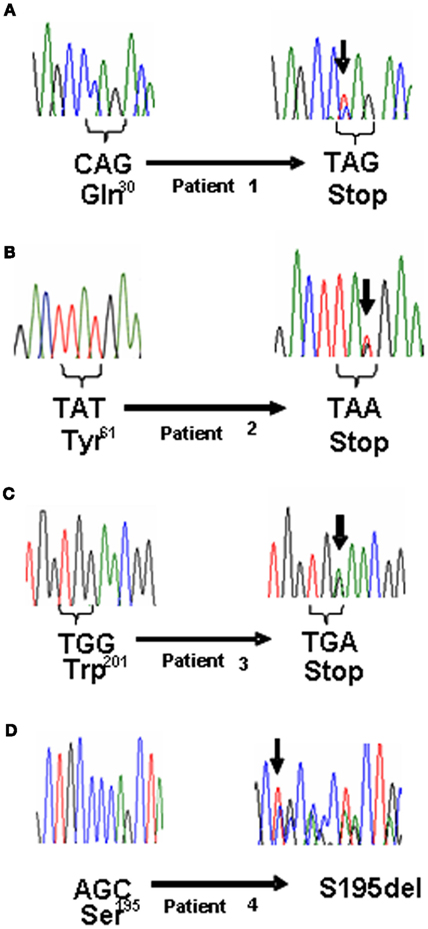
Figure 1. (A) Electropherogram showing the germline mutation denoted with arrow: wild type (left) and mutated patient 1 (right). (B) Electropherogram of wild type (left) and mutated patient 2 (right). (C) Electropherogram of wild type (left) and mutated patient 3 (right). (D) Electropherogram of wild type (left) and mutated patient 4 (right).
Interestingly, the patient’s brother developed hypertension, but the genetic analysis resulted negative (Figure 2A).
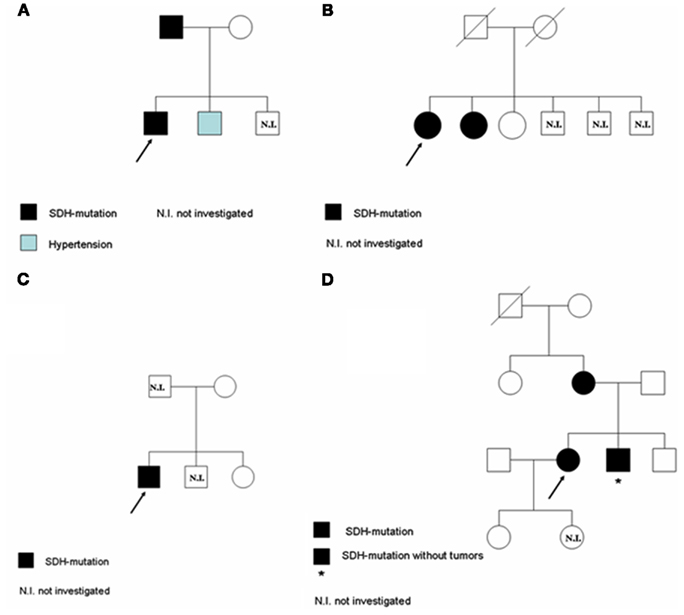
Figure 2. Pedigree of the four families under investigation: family (A) corresponds to Patient 1; family (B) corresponds to Patient 2; family (C) corresponds to Patient 3; family (D) corresponds to Patient 4.
Patient 2
A 43-year-old woman presented at the Surgical Unit for a suspicion of abdominal PGL with functional syndrome. Sixteen years before, a mass between inferior cava vein and aorta had been highlighted by a computed tomography (CT) scan. However, the patient neither decided to undergo surgery nor to refer to an endocrinologist. As soon as hypertension, tachycardia, and moderate weight loss developed, the patient was then referred to our Endocrinology Department. At the biochemical work-up, increased concentrations of urinary catecholamines and plasma chromogranin-A were found, while a 123I-MIBG scintigraphy showed a high uptake in the site of the abdominal mass. The patient underwent surgical resection of the tumor that was confirmed to be a PGL (5.5 cm of maximal diameter) at the pathological examination. The genetic analysis revealed the presence of a mutation in the SDHB gene (Figure 1B).
Six months after surgery, there was relapse of mild arterial hypertension, associated to high levels of urinary catecholamines and plasma chromogranin-A. An abdomen MRI showed loco-regional persistence of tumor mass with an extensive infiltration of the inferior cava vein. This area showed a high uptake at the somatostatin receptor scintigraphy, as well as an area of high uptake in the spinal cord, described as a bone metastasis at the MRI. For this reason, therapy with somatostatin analogs was started. Furthermore, the patient underwent trans-arterial embolization of the tumor. At the CT scan re-evaluation, the mass was unchanged. Due to the persistence of hypertension and high levels of catecholamines, therapy with sunitinib was started in combination with somatostatin analogs. A CT scan performed after three cycles of therapy with sunitinib revealed a partial response of the abdominal tumor mass and stable vertebral metastasis.
The genetic analysis was extended to two sisters: one of them was positive and her clinical history revealed she had been affected by a renal cell carcinoma, but apparently she had no signs and symptoms related to PGL (Figure 2B).
Patient 3
A 23-year-old man was referred to our Endocrinology Department for the follow-up of an abdominal PGL already operated during childhood and subsequently relapsed. At the age of 12 years, he had been operated for appendicitis. During the surgical intervention, an abdominal mass was incidentally found and an excisional biopsy was performed. The pathological examination confirmed the diagnosis of PGL. Afterward, the patient underwent two surgical interventions which were not able to completely remove the tumor mass.
At the 123I-MIBG scintigraphy, the persistent abdominal PGL was negative, while a 68GaDOTATATE-PET revealed the tumor to express somatostatin receptors. For this reason, the patient started therapy with somatostatin analogs and underwent a radiometabolic treatment with 90Y-DOTATATE.
When the patient was first referred to our Endocrinology Department, he had local relapse of the disease, associated with multiple bone metastases. At the biochemical evaluation, chromogranin-A levels were high, as well as urinary catecholamines and urinary vanilmandelic acid. At the radiological assessment there was evidence of progressive abdominal disease. Therapy with somatostatin analogs was continued and therapy with sunitinib was started. The genetic analysis revealed the presence of a mutation in the SDHB gene (Figure 1C). Patient’s mother and sister underwent the genetic analysis and they both resulted negative. Patient’s father has been never investigated because he was unavailable (Figure 2C).
Patient 4
A 25-year-old pregnant woman was evaluated for hypertension. Due to the unsatisfactory blood pressure control she underwent cesarean surgery during the seventh month of pregnancy. After delivery, a CT scan of the abdomen revealed the presence of two abdominal lesions: the first, on the right side, 5.5 cm of maximal diameter, apparently was not clearly separable from the underlying tissues, the inferior cava vein and the aorta; the second, on the left side, 2 cm of maximal diameter, was localized at para-aortic level. A strong uptake at the 123I-MIBG scintigraphy and elevated urinary catecholamine concentrations were consistent with the diagnosis of PGLs. On this basis, the patient underwent surgical resection of the abdominal lesions. The pathological examination confirmed the diagnosis of PGL. The genetic analysis revealed the presence of a mutation in the SDHB gene (Figure 1D). Interestingly, an accurate clinical history revealed that the patient’s mother had been already operated for a PGL. Therefore, mother, brother and daughter of the patient underwent genetic analysis. The first two were positive, while the third one was negative. Anyway, the brother had no apparent disease (Figure 2D).
After surgery, blood pressure levels progressively improved until a complete normalization. At the last control, 18 months after surgery, urinary metanephrines, and catecholamines as well as blood pressure levels were normal. Later on she moved into another city and was unavailable for follow-up.
Methods
Written informed consent was obtained from all the four patients undergoing genetic test in agreement with the guidelines of our Ethical Committees.
DNA was extracted from peripheral blood leukocytes using the commercial kit NucleoSpin Blood L (Macherey-Nagel, Düren, Germany) following the manufacturer’s instructions.
Searching for germline mutations we analyzed SDHB (all exons), SDHC (all exons), and SDHD (all exons) genes as well as VHL (all exons) and RET (10, 11, 13, 14, 15, 16 exons) genes. All the gene coding regions and exon-intron boundaries were amplified by PCR using the appropriate primers as previously described (Simi et al., 2005).
PCR products, purified using a PCR purification kit (Qiagen, Milan, Italy) following the protocol instructions and semi-quantified in a 2% agarose ethidium bromide gel using DNA molecular weight (Roche, IN, USA), were sequenced by ABI PRISM 310 Genetic Analyzer (Applied Biosystems Milan, Italy).
Genetic analysis was extended to the family members who consented to be studied. After informed consent, genomic DNA was screened for directed research of described SDHB mutations using PCR and sequencing. Family members who were found to be carriers of the mutation underwent extensive clinical and laboratory investigations, aimed at diagnosing chromaffin tumors.
Results and Discussion
Pheochromocytomas and PGLs can be sporadic or associated to hereditary syndromes. In our institution, all patients with apparently sporadic PHEO and PGL are offered to perform genetic screening to exclude a hereditary disease.
Extra-adrenal PGLs are the most frequent lesions found in SDHB mutation carriers. Therefore, in these patients, genetic analysis should start with SDHx genes sequencing. The sequencing might be preceded by a less expensive immunohistochemistry on tumor tissue using an anti-SDHB antibody. In fact, a positive immunostaining excludes the involvement of SDHx genes while a negative one indicates the degradation of the complex due to a mutation in the SDHB, SDHC, or SDHD genes (van Nederveen et al., 2009).
In this report we described four novel mutations in the SDHB gene in patients affected by extra-adrenal sPGLs (Table 1). Patient 1 was heterozygous for the p.Q30X, which consists in a C to T substitution in a glutamine coding triplet CAG that is then converted in a stop codon. Patient 2 was heterozygous for the p.Y61X, which consists in a T to A substitution in a tyrosine coding triplet TAT that is then converted in a stop codon. Patient 3 was heterozygous for the p.W201X, which consists in a G to A substitution in a tryptophan coding triplet TGG that is then converted in a stop codon. These mutations are predicted to encode for a truncated SDHB protein. The fourth mutation is a S195del frameshift mutation, which consists in a deletion of the codon AGC encoding for a serine. These mutations were not detected in 124 normal alleles. To our knowledge the mutations above described has not been previously reported.
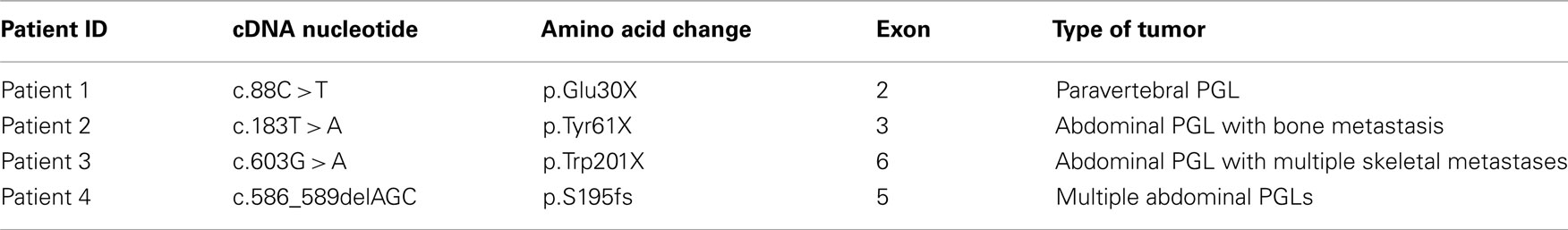
Table 1. Germline mutations affecting SDHB and related phenotype.
All four patients presented one or more symptoms related to PHEO/PGL syndrome and in all arterial hypertension was found. The biochemical examination revealed in all patients elevated concentrations of urinary catecholamines and plasma chromogranin-A. Size and location of tumors were detailed by MRI, CT, and 123I-MIBG. The diagnosis of extra-adrenal PGLs was then confirmed at the pathological examination.
Extra-adrenal PHEOs, also known as sPGLs, account for 10–20% of catecholamine-producing tumors.
So far clinical and research studies have shown that SDHB mutation carriers develop extra-adrenal PGLs in the most of cases and metastatic disease in 30–50% of cases (Gimenez-Roqueplo et al., 2003; Neumann et al., 2004; Timmers et al., 2007).
The highlights of this study are that all these patients with apparently sporadic sPGL undergone SDH gene analysis were SDHB mutation carriers and two of them developed malignant traits with bone metastases. These findings strongly suggest to perform SDH gene analysis in all patients with apparently sporadic sPGL and to perform a careful and periodical clinical follow-up in those who result to be SDHB mutation carriers because of tumor aggressive behavior.
The mechanisms by which SDHB mutations are more strongly associated with extra-adrenal location, hormone activity, malignancy and onset at an early age remain to be clarified.
Among the three SDHB mutated family members diagnosed by the genetic screening, none presented clinical manifestations of PGL, confirming the low penetrance of PGL-4 syndrome, as reported in the literature.
In conclusion, these findings underline the central role of genetic screening in patients affected by PHEOs or PGLs and enlarge the list of SDHB mutations responsible for the presence of these tumors. Apparently sporadic extra-adrenal sPGL have to be preferentially tested for SDHB gene and SDHB mutated patients have to be investigated for malignant disease.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the patients participating in this study and the clinicians who provided to collect blood samples and clinical information. This work was partially supported by a grant from the Italian Minister of Research and University in Rome (no. 2008LFK7J5).
References
Amar, L., Bertherat, J., Baudin, E., Ajzenberg, C., Bressac-de Paillerets, B., Chabre, O., Chamontin, B., Delemer, B., Giraud, S., Murat, A., Niccoli-Sire, P., Richard, S., Rohmer, V., Sadoul, J. L., Strompf, L., Schlumberger, M., Bertagna, X., Plouin, P. F., Jeunemaitre, X., and Gimenez-Roqueplo, A. P. (2005). Genetic testing in pheochromocytoma or functional paraganglioma. J. Clin. Oncol. 23, 8812–8818.
Astuti, D., Hart-Holden, N., Latif, F., Lalloo, F., Black, G. C., Lim, C., Moran, A., Grossman, A. B., Hodgson, S. V., Freemont, A., Ramsden, R., Eng, C., Evans, D. G., and Maher, E. R. (2003). Genetic analysis of mitochondrial complex II subunits SDHD, SDHB and SDHC in paraganglioma and pheochromocytoma susceptibility. Clin. Endocrinol. (Oxf.) 59, 728–733.
Astuti, D., Latif, F., Dallol, A., Dahia, P. L., Douglas, F., George, E., Skoldberg, F., Husebye, E. S., Eng, C., and Maher, E. R. (2001). Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am. J. Hum. Genet. 69, 49–54.
Baysal, B. E. (2002). Hereditary paraganglioma targets diverse paraganglia. J. Med. Genet. 39, 617–622.
Baysal, B. E., Ferrell, R. E., Willett-Brozick, J. E., Lawrence, E. C., Myssiorek, D., Bosch, A., van der Mey, A., Taschner, P. E., Rubinstein, W. S., Myers, E. N., Richard, C. W. III, Cornelisse, C. J., Devilee, P., and Devlin, B. (2000). Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 287, 848–851.
Benn, D. E., Gimenez-Roqueplo, A. P., Reilly, J. R., Bertherat, J., Burgess, J., Byth, K., Croxson, M., Dahia, P. L., Elston, M., Gimm, O., Henley, D., Herman, P., Murday, V., Niccoli-Sire, P., Pasieka, J. L., Rohmer, V., Tucker, K., Jeunemaitre, X., Marsh, D. J., Plouin, P. F., and Robinson, B. G. (2006). Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J. Clin. Endocrinol. Metab. 91, 827–836.
Gimenez-Roqueplo, A. P., Favier, J., Rustin, P., Rieubland, C., Crespin, M., Nau, V., Khau Van Kien, P., Corvol, P., Plouin, P. F., and Jeunemaitre, X. (2003). Mutations in the SDHB gene are associated with extra-adrenal and/or malignant pheochromocytomas. Cancer Res. 63, 5615–5621.
Gimenez-Roqueplo, A. P., Favier, J., Rustin, P., Rieubland, C., Keplan, V., Plouin, P. F., Rötig, A., and Jeunemaitre, X. (2002). Functional consequences of a SDHB gene mutation in apparently sporadic pheochromocytoma. J. Clin. Endocrinol. Metab. 87, 4771–4774.
King, A., Selak, M. A., and Gottlieb, E. (2006). Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene 25, 4675–4682.
Mannelli, M., Ercolino, T., Giachè, V., Simi, L., Cirami, C., and Parenti, G. (2007). Genetic screening for pheochromocytoma: should SDHC gene analysis be included? J. Med. Genet. 44, 586–587.
Neumann, H. P., Bausch, B., McWhinney, S. R., Bender, B. U., Gimm, O., Franke, G., Schipper, J., Klisch, J., Altehoefer, C., Zerres, K., Januszewicz, A., Eng, C., Smith, W. M., Munk, R., Manz, T., Glaesker, S., Apel, T. W., Treier, M., Reineke, M., Walz, M. K., Hoang-Vu, C., Brauckhoff, M., Klein-Franke, A., Klose, P., Schmidt, H., Maier-Woelfle, M., Peczkowska, M., Szmigielski, C., and Eng, C. (2002). Germ-line mutations in nonsyndromic pheochromocytoma. N. Engl. J. Med. 346, 1459–1466.
Neumann, H. P., Pawlu, C., Peczkowska, M., Bausch, B., McWhinney, S. R., Muresan, M., Buchta, M., Franke, G., Klisch, J., Bley, T. A., Hoegerle, S., Boedeker, C. C., Opocher, G., Schipper, J., Januszewicz, A., and Eng, C. (2004). Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA 292, 943–951.
Schiavi, F., Boedeker, C. C., Bausch, B., Peçzkowska, M., Gomez, C. F., Strassburg, T., Pawlu, C., Buchta, M., Salzmann, M., Hoffmann, M. M., Berlis, A., Brink, I., Cybulla, M., Muresan, M., Walter, M. A., Forrer, F., Välimäki, M., Kawecki, A., Szutkowski, Z., Schipper, J., Walz, M. K., Pigny, P., Bauters, C., Willet-Brozick, J. E., Baysal, B. E., Januszewicz, A., Eng, C., Opocher, G., and Neumann, H. P. (2005). Predictors and prevalence of paraganglioma syndrome associated with mutations of the SDHC gene. JAMA 29, 2057–2063.
Simi, L., Sestini, R., Ferruzzi, P., Gaglianò, M. S., Gensini, F., Mascalchi, M., Guerrini, L., Pratesi, C., Pinzani, P., Nesi, G., Ercolino, T., Genuardi, M., and Mannelli, M. (2005). Phenotype variability of neural crest derived tumours in six Italian families segregating the same founder SDHD mutation Q109X. J. Med. Genet. 42, e52.
Timmers, H. J., Kozupa, A., Eisenhofer, G., Raygada, M., Adams, K. T., Solis, D., Lenders, J. W., and Pacak, K. (2007). Clinical presentations, biochemical phenotypes, and genotype-phenotype correlations in patients with succinate dehydrogenase subunit B-associated pheochromocytomas and paragangliomas. J. Clin. Endocrinol. Metab. 92, 779–786.
van Nederveen, F. H., Gaal, J., Favier, J., Korpershoek, E., Oldenburg, R. A., de Bruyn, E. M., Sleddens, H. F., Derkx, P., Rivière, J., Dannenberg, H., Petri, B. J., Komminoth, P., Pacak, K., Hop, W. C., Pollard, P. J., Mannelli, M., Bayley, J. P., Perren, A., Niemann, S., Verhofstad, A. A., de Bruïne, A. P., Maher, E. R., Tissier, F., Méatchi, T., Badoual, C., Bertherat, J., Amar, L., Alataki, D., Van Marck, E., Ferrau, F., François, J., de Herder, W. W., Peeters, M. P., van Linge, A., Lenders, J. W., Gimenez-Roqueplo, A. P., de Krijger, R. R., and Dinjens, W. N. (2009). An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis. Lancet Oncol. 10, 764–771.
Keywords: extra-adrenal paragangliomas, germ-line mutations, succinate dehydrogenase subunit B, genetic screening
Citation: Ramundo V, Ercolino T, Faggiano A, Giachè V, Ragghianti B, Rapizzi E, Colao A and Mannelli M (2012) Genetic-clinical profile of subjects with apparently sporadic extra-adrenal paragangliomas. Front. Endocrin. 3:65. doi: 10.3389/fendo.2012.00065
Received: 27 January 2012; Accepted: 28 April 2012;
Published online: 18 May 2012.
Edited by:
Antonino Belfiore, University Magna Graecia of Catanzaro, ItalyReviewed by:
Jean-Yves Scoazec, Université Lyon 1, FranceSalvatore Cannavo, University of Messina, Italy
Copyright: © 2012 Ramundo, Ercolino, Faggiano, Giachè, Ragghianti, Rapizzi, Colao and Mannelli. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Valeria Ramundo, Department of Molecular and Clinical Endocrinology and Oncology, “Federico II” University of Naples, Via S. Pansini, 5 – 80131 Naples, Italy. e-mail:dmFsZXJpYS5yYW11bmRvODNAbGliZXJvLml0