 Elisa Menicali1,2
Elisa Menicali1,2 Sonia Moretti1,2
Sonia Moretti1,2 Pasquale Voce1,2 Serena Romagnoli1 Nicola Avenia2,3
Pasquale Voce1,2 Serena Romagnoli1 Nicola Avenia2,3 Efisio Puxeddu1,2*
Efisio Puxeddu1,2*- 1 Dipartimento di Medicina, University of Perugia, Perugia, Italy
- 2 Centro di Proteomica e Genomica della Tiroide, University of Perugia, Perugia and Terni, Italy
- 3 Dipartimento di Chirurgia, University of Perugia, Perugia, Italy
RET gene rearrangements (RET/PTCs) represent together with BRAF point mutations the two major groups of mutations involved in papillary thyroid carcinoma (PTC) initiation and progression. In this review, we will examine the mechanisms involved in RET/PTC-induced thyroid cell transformation. In detail, we will summarize the data on the molecular mechanisms involved in RET/PTC formation and in its function as a dominant oncogene, on the activated signal transduction pathways and on the induced gene expression modifications. Moreover, we will report on the effects of RET/PTCs on the tumor microenvironment. Finally, a short review of the literature on RET/PTC prognostic significance will be presented.
Introduction
Thyroid cancer is the most common malignant tumor of the endocrine system and accounts for approximately 3% of all newly diagnosed cancer cases in the United States1. The most frequent type of thyroid cancer is papillary thyroid carcinoma (PTC), which constitutes ∼80% of all cases. PTC frequently has genetic alterations leading to the activation of the mitogen-activated protein kinase (MAPK) signaling pathway. Those include RET/PTC and NTRK1 rearrangements and BRAF and RAS point mutations (Figure 1). Mutations involving one of these genes are found in about 70% of papillary carcinomas and they rarely overlap in the same tumor (Kimura et al., 2003; Puxeddu et al., 2004; Carta et al., 2006; Kondo et al., 2006).
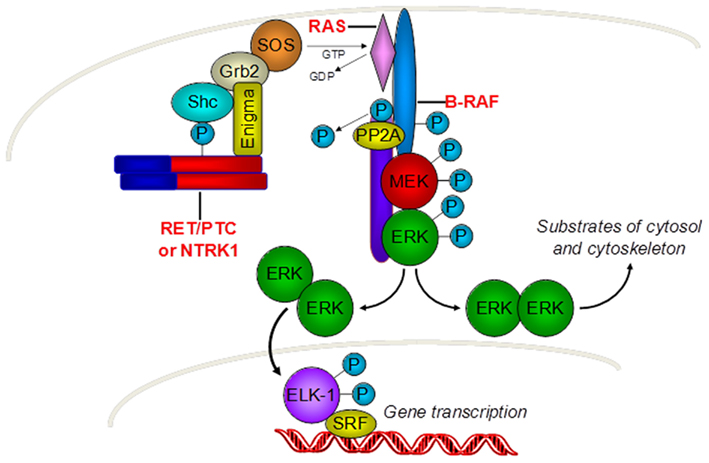
Figure 1. Gene mutations involved in PTC carcinogenesis. PTC is driven by genetic alterations leading to the activation of the mitogen-activated protein kinase (MAPK) signaling pathway. Those include RET/PTC and NTRK1 rearrangements and RAS and BRAF point mutations.
In this review, we will focus on RET/PTC rearrangements summarizing the available data on the molecular mechanisms involved in their formation and in their function as dominant oncogenes, on the activated signal transduction pathways, and on induced gene expression modifications. Moreover, we will report on their effects on tumor microenvironment.
General Features of RET/PTCs
The RET (REarranged during Transfection) proto-oncogene is located in the pericentromeric region of chromosome 10q11.2 and spans 21 exons (Takahashi et al., 1985). RET encodes for a cell membrane receptor tyrosine kinase (TK), which is expressed in neuroendocrine cells (including thyroid C cells and adrenal medullary cells), neural cells, urogenital tract cells, and testis germ cells (Wells and Santoro, 2009). RET protein displays an extracellular portion which contains four cadherin-like repeats, a calcium-binding site and a cysteine-rich region (Anders et al., 2001), a transmembrane portion and an intracellular portion, which contains the juxtamembrane domain, the TK domain, split in two subdomains by an insert of 14 amino acids, and a C-terminal tail (Knowles et al., 2006). Glial cell line-derived neurotrophic factor, neurturin, artemin, and persephin have been shown to be RET ligands (Durbec et al., 1996; Sariola and Saarma, 2003). They bind RET in conjunction with one of four glycosyl-phosphatidylinositol-anchored co-receptors, designated GDNF-family α receptors (GFRα) 1, 2, 3, and 4 (Puxeddu and Fagin, 2001).
Structurally, RET-TK adopts the typical protein kinase bi-lobate structure consisting of a small N-lobe and a large C-lobe connected by a short linker (Knowles et al., 2006). However, at variance with most kinases which are monomeric and auto-inhibited in cis by the C-lobe activation loop (A-loop) that maintains the kinase pocket closed in unstimulated conditions, RET-TK is already a dimer in the resting state. It adopts a trans-inhibited head-to-tail inactive dimer conformation in which the substrate-binding site of each monomer is occluded by the controlateral one (Knowles et al., 2006). While in other kinases ligand binding causes dimerization, rearrangement of the A-loop, and kinase activation, interaction of RET with the GDNF/GFRα complex probably causes a conformational change that relieves the trans-inhibition and favors the formation of active dimers (Knowles et al., 2006).
In thyroid follicular cells, RET is not expressed or expressed at very low levels, but RET activation can occur through chromosomal rearrangements that involve the gene. They result in the in-frame fusion of part of RET intracellular domain coding region (including that coding for the TK from residue E713 and the carboxy-terminal tail) with the 5′ end of heterologous genes (Figure 2). The resulting chimeric sequences are called RET/PTC and at least 13 variants have been reported to date, that differ according to the 5′ partner gene involved in the rearrangement (Castellone and Santoro, 2008) (Figure 3). The two most common rearrangement types are RET/PTC1 and RET/PTC3 which account alone for more than 90% of all rearrangements found in PTCs. RET/PTC1 is formed by fusion with the coiled-coil domain containing gene 6 (CCDC6, formely called H4/D10S170), and RET/PTC3 by fusion with the nuclear receptor coactivator gene-4 (NCOA4, formely called RFG/ELE1/ARA70; Grieco et al., 1990; Santoro et al., 1994). RET/PTC1 and RET/PTC3 are intra-chromosomal paracentric inversions because both genes participating in the fusion are located on chromosome 10q (Pierotti et al., 1992; Minoletti et al., 1994). At variance RET/PTC2 and the other rare types of RET/PTC are inter-chromosomal translocations (Castellone and Santoro, 2008).
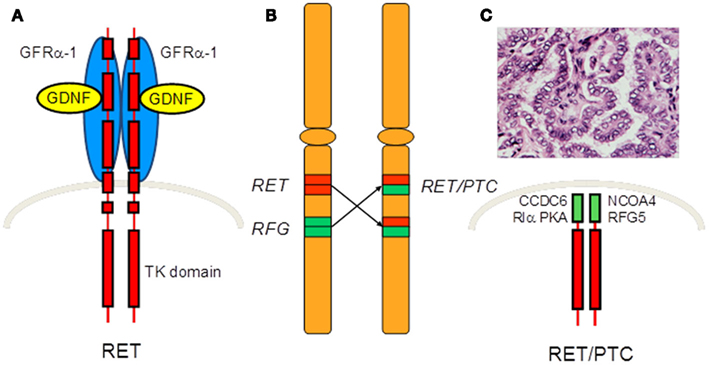
Figure 2. Formation of RET/PTC. (A) RET codes a membrane protein of the tyrosine kinase receptor family which is not expressed or expressed at very low levels in thyroid follicular cells. (B) RET activation can occur through chromosomal rearrangements that involve the gene. (C) These rearrangements result in the in-frame fusion of RET intracellular domain coding region with the 5′ end of heterologous genes. The resulting chimeric sequence codes an oncoprotein, called RET/PTC, that drives the development of PTC.
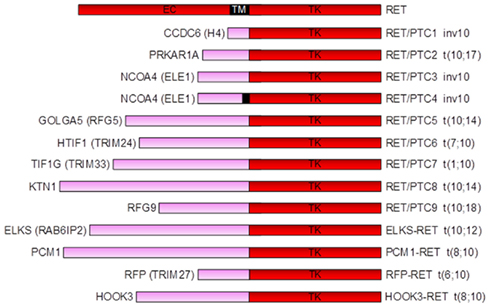
Figure 3. Schematic drawing of RET/PTC variants. At least 13 RET/PTC variants have been reported to date, that differ according to the 5′ partner gene involved in the rearrangement.
Several mechanisms sustain the conversion of RET in a dominant oncogene for thyroid follicular cells after formation of RET/PTCs (Figure 4). In first instance, the ubiquitously expressed promoter of the fusion partner warrants ectopic RET expression in the cells harboring the rearrangement. Moreover, the partner genes are predicted to provide one or more coiled-coil domains that are essential for dimerization and ligand-independent activation of the truncated RET protein (Bongarzone et al., 1993; Tong et al., 1997; Monaco et al., 2001). Furthermore, RET/PTCs lack the intracellular juxtamembrane domain which forms an integral part of the autoinhibited RET dimer interface (Knowles et al., 2006). As a consequence the TK domain is favored to adopt the active conformation. Finally, the delocalization of the rearranged protein to the cytosol is also expected to contribute to activation of the oncogene (Castellone and Santoro, 2008). In first instance, the altered subcellular localization uncouples RET/PTCs from receptor-mediated endocytosis and lysosomal degradation, leading to increased protein stability (Richardson et al., 2009). Moreover, it could prevent RET/PTC from interacting with negative regulators located at plasma membrane level, i.e., the tyrosine phosphatase Protein Tyrosine Phosphates Receptor type-J (PTPRJ; Iervolino et al., 2006).
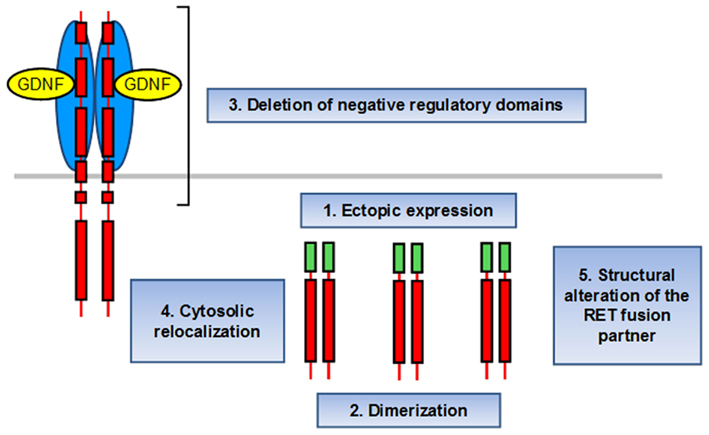
Figure 4. Mechanism of RET/PTC activation. (1) The ubiquitously expressed promoter of the fusion partner warrants ectopic RET expression in the cells harboring the rearrangement. (2) The partner genes provide one or more coiled-coil domains that are essential for dimerization and ligand-independent activation of the truncated RET protein. (3) The lack of the inhibitory intracellular juxtamembrane domain favors RET/PTC to adopt the active conformation. (4) The delocalization of the rearranged protein to the cytosol abolishes receptor-mediated endocytosis and lysosomal degradation, leading to increased protein stability, and interaction with negative regulators located at plasma membrane level. (5) RET fusion genes appear to code negative modulators of transformation. Thus, their structural alteration might contribute to the oncogenic potential of the RET/PTC rearrangements.
The possible functional interactions between RET/PTC and EGFR in PCCL3 cells was examined after the observation that the epidermal growth factor receptor (EGFR) kinase inhibitor PKI166 decreased RET/PTC kinase auto-phosphorylation and activation of downstream effectors in thyroid cells (Croyle et al., 2008). This study showed that RET/PTC induces EGFR gene expression and kinase activation in part through MAPK signaling and forms a complex with EGFR in a kinase-independent manner. In turn EGFR stimulates RET phosphorylation (Croyle et al., 2008).
It is well known that PTC in general, but those harboring RET/PTC rearrangements in particular, present relatively indolent phenotypes compared with RET-related medullary thyroid carcinomas (MTC; Elisei et al., 2008). Considering that RET/PTCs possess similar if not greater oncogenic potential than membrane-bound RET when expressed at similar levels (Richardson et al., 2009), the explanation of this phenotype difference has to be searched in divergences of the oncogene expression levels or of the cellular responsiveness to them in follicular and the C-cells in vivo. Very recently, Richardson and others demonstrated that expression of RET/PTCs via the 5′ promoter region of CCDC6 and NCOA4 could result in lower expression of these oncoproteins, relative to full-length RET, expressed off its endogenous promoter. These data indicate that also transcript level modulates the inherent oncogenicity of RET/PTC oncoproteins and that in vivo it is trumped by the substitution of relatively weak promoters upstream of the chimeric oncogene (Richardson et al., 2009).
It is plausible that also the structural alteration of the RET fusion partners might contribute to the oncogenic potential of the RET/PTC rearrangements. This is easily understandable for the RET/PTC2 fusion partner PRKAR1A which encodes a cyclic AMP-dependent protein kinase type I-regulatory subunit (Bongarzone et al., 1993). Indeed, it is known to be a bona fide tumor suppressor gene mutated in about half of Carney complex kindreds (Kirschner et al., 2000). Moreover, its ablation in the mouse induces the formation of thyroid tumors (Griffin et al., 2004). However, also CCDC6 (RET/PTC1) has been shown to possess proapoptotic activity (Celetti et al., 2004) and to repress CREB1 transcriptional activity by recruiting histone deacetylase 1 and protein phosphatase 1 proteins at the CRE site of the CREB1 target genes (Leone et al., 2010). Moreover, ERC1 (ELKS-RET) has appeared to be a NF-kB regulator (Ducut Sigala et al., 2004; Wu et al., 2006), TRIM33 (RET/PTC7) to interact with TGFβ signaling intermediates Smad (Dupont et al., 2005; He et al., 2006), and RFP (RFP-RET) to act as a transcriptional repressor disabling pRb-mediated differentiation (Shimono et al., 2000; Krützfeldt et al., 2005).
Intriguingly, some of the RET fusion partners are found rearranged in tumors other than PTC: TRIM24 (participating in RET/PTC6) rearranges with BRAF in mouse hepatocellular carcinomas (Zhong et al., 1999); PMC1 (participating in PMC1-RET) with JAK2 in chronic myeloid leukemia (Bousquet et al., 2005); CCDC6 (participating in RET/PTC1) with the kinase domain of the platelet-derived growth factor receptor-β in myeloproliferative disorders (Kulkarni et al., 2000). Moreover, in thyroid carcinoma cell lines and PTC samples CCDC6/H4 was shown to rearrange with PTEN through an intra-chromosomal inversion forming non-clonal H4/PTEN chimeric genes of unknown pathogenetic significance (Puxeddu et al., 2005b).
There are several lines of evidence pointing to RET/PTC as a key first step in thyroid cancer pathogenesis (Figure 5). In first instance, RET/PTC expression causes TSH-independent proliferation and downregulation of differentiation markers in thyroid follicular cells (Santoro et al., 1993). Moreover, thyroid-specific overexpression of either RET/PTC1 (Jhiang et al., 1996; Santoro et al., 1996) or RET/PTC3 (Powell et al., 1998) in transgenic mice leads to development of tumors with histological features consistent with PTC. Furthermore, there is a high prevalence of RET/PTC expression in occult microscopic PTC, believed to represent an early step in the development of clinically significant cancers (Viglietto et al., 1995; Sugg et al., 1998). Finally, exposure of cell lines (Ito et al., 1993) and fetal thyroid explants (Mizuno et al., 1997) to ionizing radiation results in induction of expression of RET/PTC within hours, supporting a direct role for radiation, the major known risk factor for thyroid cancer, in the illegitimate recombination of RET.
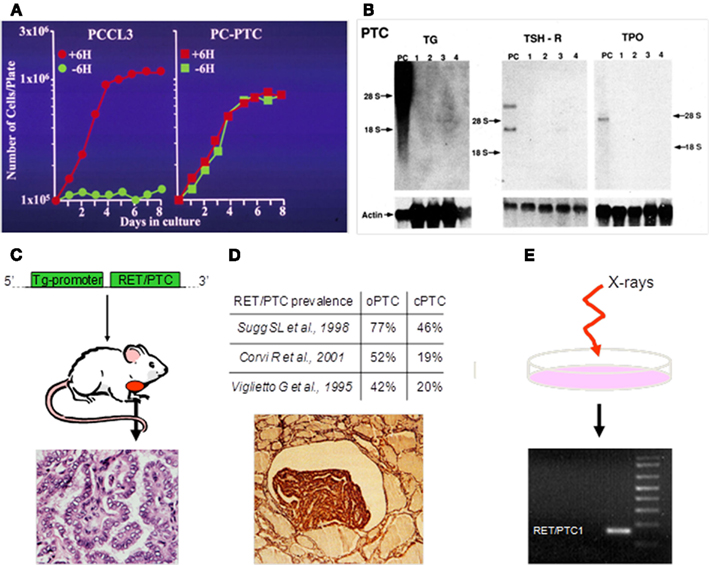
Figure 5. Lines of evidence pointing to RET/PTC as a key first step in thyroid cancer pathogenesis. (A) RET/PTC expression causes TSH-independent proliferation in thyroid follicular cells. PCCL3: Parental well differentiated rat thyroid cell line. PC-PTC: PCCL3 cells stably expressing RET/PTC1. +6H: Grown in the presence of TSH. −6H: Grown in the absence of TSH (Modified from Santoro et al. (1993) with permission of the American Association for Cancer Research). (B) RET/PTC expression causes downregulation of differentiation markers in thyroid follicular cells. PC: Control parental PCCL3 cells. 1–4: Clones of PCCL3 cells stably expressing RET/PTC1. TG: Thyroglobulin. TSH-R: TSH receptor. TPO: Thyroperoxidase. (From Santoro et al. (1993) with permission of the American Association for Cancer Research). (C) Thyroid-specific overexpression of either RET/PTC1 or RET/PTC3 in transgenic mice leads to development of tumors with histological features consistent with PTC (Jhiang et al., 1996; Santoro et al., 1996; Powell et al., 1998). (D) Occult microscopic PTCs show high prevalence of RET/PTC expression (Viglietto et al., 1995; Sugg et al., 1998). (E) Exposure of cell lines and fetal thyroid explants to ionizing radiation, the major known risk factor for PTC, results in induction of RET/PTC expression within hours (Ito et al., 1993; Mizuno et al., 1997).
RET rearrangements have been considered exclusive of PTC. Unexpectedly, this dogma was broken very recently by three independent studies which simultaneously demonstrated the existence of a novel RET rearrangement in 1–2% of lung carcinomas. In this rearrangement, RET engages with KIF5B (the kinesin family 5B gene), located on chromosome 10p11.2, through a pericentric inversion of chromosome 10 (Kohno et al., 2011; Lipson et al., 2011; Takeuchi et al., 2011). Furthermore, one of these studies also reported the occurrence of RET/PTC1 rearrangements in two lung adenocarcinomas (Takeuchi et al., 2011).
Four variants of KIF5B-RET fusion have been described and named on the basis of the last KIF5B and first RET exon involved in the fusion: K15;R12 (variant 1), K16;R12 (variant 2), K22;R12 (variant 3), K15;R11 (variant 4). All proteins encoded by the four KIF5B-RET rearrangements appeared to retain the KIF5B coiled-coil domain necessary for homodimerization and to hold the full RET kinase domain, as other types of oncogenic RET fusion proteins observed in PTC (Kohno et al., 2011; Lipson et al., 2011; Takeuchi et al., 2011).
The mutually exclusive nature of the RET rearrangements and other oncogenic alterations suggests that the KIF5B-RET fusion is a driver mutation in lung carcinoma (Kohno et al., 2011; Lipson et al., 2011).
KIF5B-RET expression in Ba/F3 cells led to oncogenic transformation, as demonstrated by the occurrence of interleukin-3 (IL-3)-independent growth. These transformed cells resulted sensitive to sunitinib, sorafenib, and vandetanib, which are all multitarget kinase inhibitors of RET (Lipson et al., 2011), suggesting that RET kinase inhibitors should be tested in prospective clinical trials in individuals with lung carcinomas that harbor KIF5B-RET rearrangements.
RET and Medullary Thyroid Carcinoma
Medullary thyroid carcinoma (MTC) is a malignant tumor arising from thyroid C cells. It represents about 5% of all thyroid cancers and can be hereditary (about 25% of cases) or sporadic (remaining cases; Stratakis and Ball, 2000). Familial forms develop in the contest of Multiple Endocrine Neoplasia type 2 (MEN 2) syndrome in which MTC can be the sole manifestation of the disease (Familial Medullary Thyroid Carcinoma – FMTC) or be associated with pheochromocytoma (PC) and parathyroid hyperplasia/neoplasia (MEN 2A) or with PC, neuromas and body dimorphisms (MEN 2B). Germline mutations of the proto-oncogene RET confer predisposition to all forms of familial MTC. The mutations fall into two main groups (Kouvaraki et al., 2005): (a) Those affecting the extracellular domain: These primarily involve cysteine residues 609, 611, 618 and 620 (exon 10), and 634 (exon 11). (b) Those affecting the RET-TK domain: These primarily involve codons 768, 790 and 791 (exon 13), 804 (exon 14), 883 and 891 (exon 15), and 918 (exon 16).
MEN 2A is associated mainly with germline mutations in the extracellular cysteine-rich region involving codons 609, 611, 618 and 620 (exon 10), and codon 634 (exon 11; 31–32). In particular, the Cys634Arg mutation specifically correlates with a higher risk of hyperparathyroidism (Mulligan et al., 1994). Rare mutations that are more commonly associated with MEN 2A include those at codons 768, 790 and 791 (exon 13), 804 (exon 14), and 891 (exon 15; Kouvaraki et al., 2005; Evans et al., 2007). MEN 2B is caused exclusively by mutations of residues within the intracellular TK domain of the receptor. Almost 95% of cases have a Met918Thr substitution (exon 16; Eng et al., 1996), and 3–4% are associated with Ala883Phe (exon 15; Gimm et al., 1997; Smith et al., 1997). Germline mutations in FMTC kindreds are more equally distributed throughout the RET gene and show a large overlap with those detected in MEN 2A (Kouvaraki et al., 2005; Evans et al., 2007).
All point mutations of RET in MEN 2 subjects are believed to have a “gain-of-function” effect resulting in unregulated activation of the TK activity of the receptor.
Mutations affecting cysteine residues in the extracellular domain are thought to result in ligand-independent receptor dimerization (Santoro et al., 1995; Carlomagno et al., 1997; Ito et al., 1997; Chappuis-Flament et al., 1998). It is likely that the cysteine residues normally form intramolecular disulfide bridges. Substitution of one partner cysteine by another aminoacid results in an unpaired cysteine, which is thought to link with its counterpart in an adjacent molecule. This event mimics conformational changes induced by ligand binding (Santoro et al., 1995). However, signaling of MEN 2A RET mutant is not identical to ligand-mediated RET signaling, as qualitative differences were identified, i.e., in the activation of the PI3K/AKT pathway (Frêche et al., 2005).
Mutations in the TK domain of RET constitutively activate the receptor without a requirement for dimerization. This phenomenon might be linked to the fact that some of the intracellular TK mutations target positions close to the trans-inhibited dimer contact points and may activate RET by destabilizing this inactive dimer conformation (Knowles et al., 2006). Moreover, the most frequent MEN 2B mutation (Met918Thr) was found to cause a great increase in ATP binding affinity and the formation of a more stable RET-ATP complex (Gujral et al., 2006). In addition to inducing “quantitative” changes of the basal kinase activity, the Met918Thr mutation is thought to affect also the “quality” of RET-generated intracellular signals by altering the affinity of the receptor to downstream substrates (Santoro et al., 1999).
Sporadic MTCs have no detectable germline abnormalities in RET. However, a significant proportion of these cancers has acquired a mutation in RET as a somatic event during the course of tumor initiation or progression (Romei et al., 1996; Elisei et al., 2008). Here, the mutation is present only in the tumor, and most often involves codon 918 (Met918Thr), although other RET mutations have also been reported, such as some at codons 634 and 883. Estimates of prevalence of somatic RET mutations in sporadic MTC range from 23 to 70%. There are indications that tumors harboring RET mutations may carry a worse prognosis (Elisei et al., 2008).
The importance of RET mutations in the pathogenesis of either familial or sporadic MTCs has triggered the development of RET kinase inhibitors to be used in the clinical setting. At least four small molecules reached specific clinical trials in patients with unresectable, locally advanced or metastatic MTC: motesanib, vandetanib, cabozantinib, and lenvatinib (Puxeddu et al., 2011).
Motesanib was tested in a multicenter phase II study which enrolled 91 patients with advanced MTC (Schlumberger et al., 2009). The study yielded an objective response rate of only 2%, but a disease control rate (objective responses + stable disease) of 83%, with a median progression-free survival of 48 weeks and acceptable side effects. Compared to the very poor results of chemotherapy, these data appeared very promising. Unfortunately, further clinical development of motesanib was interrupted.
Vandetanib was tested in two phase II intervention studies (administering 300 and 100 mg per day, respectively) in patients with locally advanced or metastatic familial MTC (Robinson et al., 2010; Wells et al., 2010). Both studies showed objective responses in about 20% of subjects and stable disease in about another 50%, with no significant differences between the two dosages of the drug. In the 300 mg study median progression-free survival was longer than 27 months. An international, phase III, randomized, double-blinded, placebo-controlled, multicenter study to assess the efficacy of vandetanib vs. placebo in subjects with unresectable, locally advanced or metastatic MTC was recently completed (331 enrollments). The study met its primary objective of significant progression-free survival prolongation with vandetanib vs. placebo (22.6 vs 16.4 months. H.R. 0.45, 95% C.I. 0.30–0.69, P = 0.0001) (Wells et al., 2012), prompting FDA to approve the use of vandetanib to treat adult patients with late-stage (metastatic) MTC who are ineligible for surgery and who have disease that is growing or causing symptoms.
Cabozantinib was tested in a phase I dose-finding study which enrolled patients with various cancers, including an expansion cohort of 37 MTCs, showing some degree of tumor shrinkage in almost all patients of the latter group with 29% qualifying for a confirmed PR (Kurzrock et al., 2011). The good results obtained in the phase I study prompted the initiation of a registration phase III, randomized, double-blinded, placebo-controlled trial to determine the efficacy of the drug against placebo in advanced MTC patient, which is still ongoing2. Similarly, clinical efficacy of lenvantinib in advanced MTCs is tested in an ongoing phase II study (see text footnote 2).
Interestingly, all this drugs are multikinase inhibitors whose mechanism of action does not rely only on targeting RET, but also VEGF receptors and receptors for other angiogenic factors (PDGFRβ, c-KIT, c-MET) or growth factors (EGFR; Puxeddu et al., 2011). Vendetanib has been demonstrated, and the other considered TKI are expected, to inhibit also RET/PTCs and thus to represent specific targeted therapies also for PTC harboring this class of gene rearrangements.
Prevalence of RET/PTC Rearrangements
The prevalence of RET/PTC in PTCs varies significantly in different studies and geographic regions (Nikiforov, 2002). In the United States, the five largest series reported a frequency of RET/PTC ranging from 11 to 43% (Jhiang et al., 1992; Santoro et al., 1992; Lam et al., 1998; Tallini et al., 1998; Nikiforova et al., 2002), with totally 134 positive cases among 386 PTCs cumulatively studied (mean prevalence 35%). A comparable rate has been reported by other groups: Canada 40% (Sugg et al., 1999), Italy 29–35% (Santoro et al., 1992; Bongarzone et al., 1996, 1998). In other regions, a wide variation in frequency of RET/PTC has been reported, ranging from 3% in Saudi Arabia (Zou et al., 1994) to 85% in Australia (Learoyd et al., 1998). In most series, RET/PTC1 is the most common type, comprising up to 60–70% of all rearrangements, whereas RET/PTC3 accounts for 20–30% of positive cases and RET/PTC2 for less than 10% (Nikiforov, 2002).
Several studies reported a higher prevalence of RET/PTC in PTCs from children and young adults (Bongarzone et al., 1996; Nikiforov et al., 1997; Soares et al., 1998; Fenton et al., 2000), indicating that young age predisposes to the formation of these mutations. Rapid thyroid cell proliferation may account for the particular high sensitivity of children’s thyroids to chromosomal recombinations (Saad et al., 2006). In all studies RET/PTC1 was the major type of rearrangement.
Finally, the prevalence of RET/PTCs appeared significantly higher in PTC from patients with a history of radiation exposure during childhood (Nikiforov, 2002). The Chernobyl nuclear power plant accident in April 1986 resulted in the release of large amounts of iodine isotopes, mainly 131I, and, therefore, there was widespread exposure to the thyroid (Williams, 2002). A high incidence of childhood PTC was reported in contaminated areas (Figure 6). Among these PTCs, 67–87% of tumors removed 5–8 years after exposure and 49–65% of those removed 7–11 years after the accident harbored RET/PTC (Fugazzola et al., 1995; Nikiforov et al., 1997; Smida et al., 1999; Rabes et al., 2000). Interestingly, in post-Chernobyl tumors developed less than 10 years after the accident, RET/PTC3 appeared to be the most common type, whereas those removed after the longer latency had predominantly RET/PTC1 (Smida et al., 1999; Rabes et al., 2000). In patients subjected to therapeutic external irradiation during childhood the prevalence of RET/PTC was also high ranging between 54 and 84% (Bounacer et al., 1997; Elisei et al., 2001). In these cases RET/PTC1 was more represented. However, exposure to ionizing radiations promotes the fusion of RET also with unusual partners and it is in this group that the majority of rare variants of RET/PTCs were detected (Nikiforov, 2002).
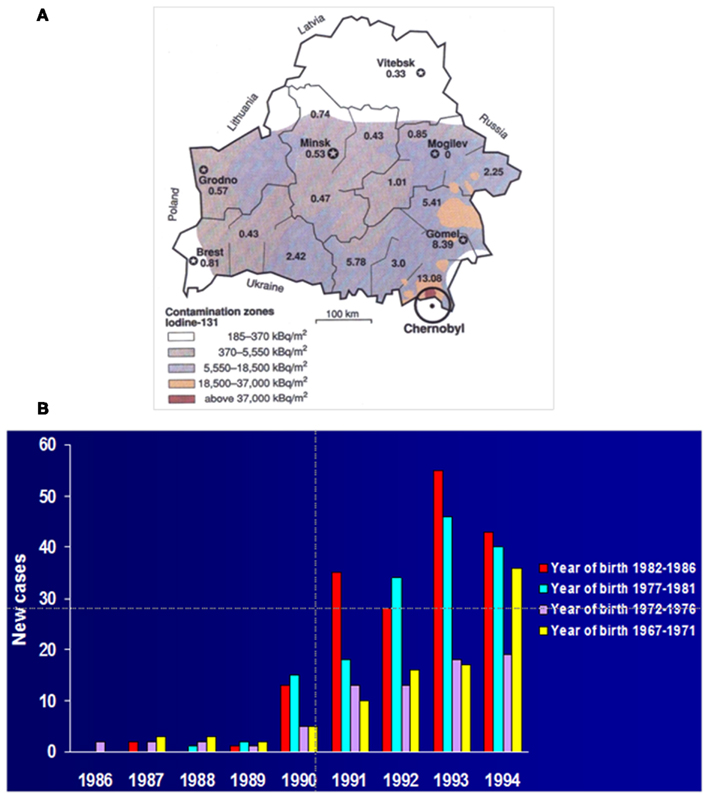
Figure 6. Effects of the Chernobyl accident. (A) The Chernobyl nuclear power plant accident on April 26th 1986 resulted in the release of large amounts of iodine isotopes, mainly 131I, that contaminated the ground of surrounding regions, specially Belarus, with widespread exposure of the thyroids of the inhabitants. A high incidence of childhood PTC was reported in the more heavily contaminated areas (in the map small numbers indicate the incidence of pediatric thyroid cancers in the different districts of Belarus, reported as number of cases per 100,000 children) [From Balter (1996) with permission of the American Association for the Advancement of Science]. (B) Unexpectedly, after the accident a rise of pediatric thyroid carcinomas was observed in Belarus with a latency as short as 4 years. The highest price was payed by the younger children at the time of the disaster [modified from Bleuer et al. (1997) with permission of Alphamed Press, INC.].
Geographic variation and differences in radiation exposure might partially explain the variability of RET/PTC frequencies in the different studies (Zhu et al., 2006). However, this cannot serve as the only explanation, because a striking variability in the frequency has been reported in the same geographical regions [8 and 85% in Australia (Learoyd et al., 1998; Chua et al., 2000), 5 and 77% in Canada (Sugg et al., 1996, 1998)]. Thus, it is conceivable that the discrepancy between the reported prevalence of RET/PTC in PTCs may also be a result of different sensitivities of the detection methods and tumor genetic heterogeneity (Zhu et al., 2006). In the first instance, it has been clearly demonstrated that efficacy of RET/PTC detection, and of consequent reported prevalence, changed using various reverse transcriptase (RT)-PCR methods, or Southern blot, or fluorescence in situ hybridization (FISH). In the second instance, it has been clearly demonstrated that PTC can harbor RET/PTC rearrangement either as a clonal mutation, affecting the majority of tumor cells, or as a “non-clonal” one, affecting a small portion of tumor cells (Rhoden et al., 2006; Zhu et al., 2006). In the first case the genetic modification is likely to be important for tumor formation, in the second one the mutation might represent only a casual alteration with no impact on the cancerogenetic process (“passenger mutation”). A counting of the second group in the RET/PTC-positive cases leads to an overestimation of the frequency of the mutation and to the inclusion of heterogeneous cases in the group of tumors driven by RET rearrangements.
The presence of RET/PTC rearrangements in non-neoplastic cells in Hashimoto’s thyroiditis is still controversial. It is possible that a heterogeneous presence of the rearrangement may account for such a controversy (Tallini and Asa, 2001; Nikiforov, 2002; Rhoden et al., 2006).
Tumor heterogeneity and multiclonality is also indicated by the presence of multiple RET/PTC variants in individual PTC samples (Sugg et al., 1998).
Among thyroid lesions, RET/PTC rearrangements have so far been identified only in PTC. They are absent in follicular adenomas and follicular thyroid carcinomas (Santoro et al., 2006).
In PTC, they are more frequent in the classic variant (Adeniran et al., 2006) and in microcarcinomas (Fusco et al., 2002), although they have been rarely described also in the cribriform variant (Cetta et al., 1998), in the Huerthle cell variant (Cheung et al., 2000; Chiappetta et al., 2002) and in hylinazing trabecular adenomas (Cheung et al., 2000; Papotti et al., 2000). In radiation- induced PTC the solid variant is associated with the RET/PTC3 oncogene, whereas the classic variant is associated with RET/PTC1 (Nikiforov et al., 1997).
There is some controversy on the occurrence of RET/PTC rearrangements in poorly differentiated (PDTC) and anaplastic thyroid carcinomas (ATC) which are believed to derive in a significant number of cases from a stepwise dedifferentiation of well-differentiated PTCs and follicular thyroid carcinomas (Jhiang et al., 1992; Bongarzone et al., 1998; Soares et al., 1998; Tallini et al., 1998; Nikiforov, 2004). A recent study analyzed the mutational profile of advanced thyroid carcinomas of follicular origin, including primary PDTC and ATC and RAI refractory-FDG-PET positive-metastatic thyroid cancer lesions (Ricarte-Filho et al., 2009). It revealed that 18% of PDTC and 9% of RAI refractory-FDG-PET positive-PDTC harbored a RET/PTC rearrangement.
Mechanisms of RET/PTC Formation
As mentioned above, thyroid cancer, and in particular PTC, frequently harbor chromosomal rearrangements, including intra-chromosomal inversions and inter-chromosomal translocations. In all rearrangements, the formation of breaks in DNA strands must occur. There are various ways in which a cell can acquire these breaks, but exposure to ionizing radiation represents the best known mode (Weterings and Chen, 2008). DNA breaks are commonly repaired by two pathways, homologous recombination or non-homologous end joining (NHEJ; Shrivastav et al., 2008), and dysfunction of these pathways can contribute to the formation of chromosomal translocations (Gasparini et al., 2007). Alternatively, a fulsome accumulation of DNA breaks could prevent these normally functioning repair mechanisms, leading to translocation events.
Chromosomal fragile sites are regions of the genome that are prone to DNA breakage and are hotspots for chromosomal translocations. They are classified as common and rare, depending on their frequency in the population. Common fragile sites can be further classified based on their mode of induction, as not all sites are induced by the same compounds, or to the same extent. Aphidicolin (APH) induces expression of the majority of common fragile sites; other known fragile site-inducing conditions are the addition of 5-bromodeoxyuridine (BrdU), 5-azacytidin, and the removal of folic acid (Sutherland, 1991). Moreover, certain dietary and environmental factors have been shown to contribute to fragile site expression, including caffeine (Yunis and Soreng, 1984), ethanol (Kuwano and Kajii, 1987), hypoxia (Coquelle et al., 1998), and pesticides (Musio and Sbrana, 1997).
Fragile sites are also known to be late replicating regions of the genome. Delayed DNA replication has been observed in all fragile sites examined to date (Handt et al., 2000; Hellman et al., 2000; Palakodeti et al., 2004; Pelliccia et al., 2008). Delayed replication at fragile sites is believed to be attributed to the high propensity of DNA to form stable secondary DNA structures (Gacy et al., 1995; Usdin and Woodford, 1995; Hewett et al., 1998; Mishmar et al., 1998; Zlotorynski et al., 2003; Zhang and Freudenreich, 2007).
Many studies point toward the association between fragile sites and formation of cancer-specific translocation (Arlt et al., 2006). Genes participating in the two main types of RET/PTC rearrangements, RET/PTC1 and RET/PTC3, have been mapped to known fragile sites (Burrow et al., 2009). Both genes involved in the RET/PTC3 rearrangement, RET and NCOA4, are located at 10q11.2 within fragile site FRA10G, a common fragile site induced by APH. The CCDC6 gene, involved in RET/PTC1, is located at 10q21.2 within fragile site FRA10C, a common fragile site induced by BrdU. Major breakpoint cluster regions for these genes have been identified and are located within intron 11 of RET, intron 5 of NCOA4 and intron 1 of CCDC6 (Smanik et al., 1995; Nikiforov et al., 1999).
In a recent study, Gandhi and others showed the involvement of fragile sites in the formation of RET/PTC rearrangements. Using FISH and ligation-mediated PCR (LM-PCR) analysis, they provided structural and biochemical evidence that the RET, CCDC6, and NCOA4 genes are located in common fragile sites FRA10C and FRA10G and undergo DNA breakage after exposure to fragile site-inducing chemicals. Moreover, exposure of human thyroid cells to these chemicals results in the formation of cancer-specific RET/PTC rearrangements (Gandhi et al., 2010). These results provide the direct evidence for the involvement of chromosomal fragile sites in the generation of cancer-specific rearrangements in human cells. In particular, activation of FRA10C and FRA10G might contribute to spontaneous formation of RET/PTC1 and RET/PTC3 rearrangements leading to the development of spontaneous non-radiation induced PTCs harboring the rearrangements.
Exposure to ionizing radiation is a well known risk factor for thyroid cancer, especially for PTC, which represents the most common form of radiation-induced solid neoplasm (Winship and Rosvoll, 1970; Ron et al., 1995). As reported above, a high prevalence of RET/PTC is seen in PTCs of individuals with a history of radiation exposure (Bounacer et al., 1997; Nikiforov et al., 1997; Rabes et al., 2000; Elisei et al., 2001). Moreover, among PTCs of survivors of the atomic bomb in Japan, the presence of RET/PTC directly correlated with the received radiation dose (Takahashi et al., 2007; Hamatani et al., 2008). The association between RET/PTC rearrangement formation and ionizing radiation is supported by several studies demonstrating the induction of RET/PTC by irradiation of cultured human thyroid cells (Ito et al., 1993; Caudill et al., 2005) and of human fetal thyroid tissue xenografts in SCID mice (Mizuno et al., 1997, 2000).
Ionizing radiation damages DNA in a variety of ways as a result of either direct energy transfer by radiation to DNA or by secondary reactive oxygen species produced by ionization of water. Of all types of DNA damage, double-strand breaks (DSBs) are considered a crucial primary lesion for a variety of biological end points, including cell killing, chromosomal aberrations, and cell transformation (Bryant and Riches, 1989; Winegar et al., 1992). However, exactly how radiogenic DSBs lead to chromosomal rearrangements remains not fully understood. Several theories have been proposed but none of these can adequately explain the dose–effect relationship and complexity of radiation-induced aberrations (Edwards, 2002). The most widely accepted theory is the Breakage-and-Reunion theory. It postulates that chromosomal aberrations arise mainly as a result of the re-joining of two DSBs located closely in space and time (two-hit event; Savage, 1998; Hlatky et al., 2002). The initial distribution of primary breaks is thought to be random whereas the rejoining efficiency is expected to be influenced by their proximity (Yates and Morgan, 1993; Rothkamm et al., 2001).
It seems that nuclear architecture contributes to the generation of RET/PTC by placing potentially recombinogenic chromosomal loci in close proximity in the interphase nuclei of human cells. In a study that utilized FISH and three-dimensional (3D) confocal microscopy, it was demonstrated that RET and CCDC6 genes, the partners of RET/PTC1 rearrangement, were non-randomly located with respect to each other in the interphase nuclei of human thyroid cells and were much closer than expected based on their genomic separation (Nikiforova et al., 2000; Figures 7A–C). Moreover, this study showed that the proximity between potentially recombinogenic genes was cell-type specific and was present only in thyroid cells. In a more recent study, similar findings were obtained for RET and NCOA4, the partners of RET/PTC3 rearrangement (Gandhi et al., 2006;(Figures 7D–E).
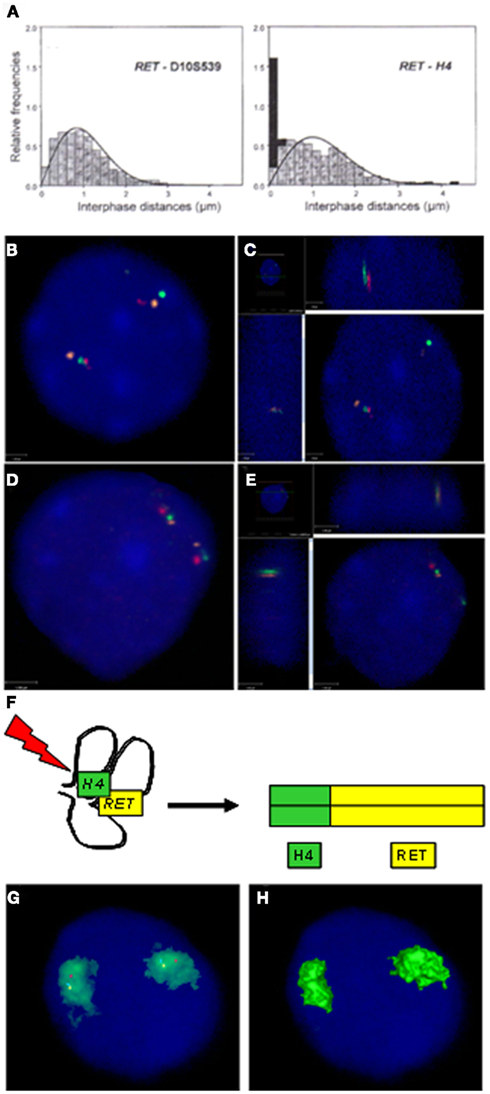
Figure 7. Proximity of chromosomal loci that participate in RET/PTC rearrangements. (A) Fluorescence in situ hybridization (FISH) analysis of the distance between RET and H4/CCDC6 loci within interphase nuclei showed that, despite a linear distance of 18 Mb, at least one pair of RET and H4/CCDC6 was juxtaposed in 35% of normal human thyroid cells (right panel). Conversely, the set of numbers obtained measuring the distance between RET and the control locus D10S539 fitted with the expected Rayleigh distribution (left panel; from Nikiforova et al. (2000) with permission of the American Association for the Advancement of Science). (B) 2D image of three-color FISH showing positioning of RET (green), NCOA4 (orange), and H4/CCDC6 (red) in interphase nuclei of thyroid cells. The panel depicts a nucleus showing two sets of RET, NCOA4, and H4/CCDC6 with one pair of RET and H4/CCDC6 positioned close to each other. (C) 3D image showing that RET and H4/CCDC6 are juxtaposed to each other in the same z plane. (D) 2D image of a nucleus showing one pair of RET and NCOA4 positioned close to each other. (E) 3D image showing that RET and NCOA4 are juxtaposed to each other in the same z plane (B–E are from Gandhi et al. (2010) with permission of Elsevier Ireland LTD). (F) Spatial contiguity of RET and the fusion partner gene may provide a structural basis for generation of RET/PTC rearrangement by allowing a single radiation track to produce a double-strand break in each gene at the same site in the nucleus (Nikiforova et al., 2000). (G) Four-color FISH showing chromosome 10 territory (green) and location of RET (blue pseudocolor), NCOA4 (yellow pseudocolor), and H4/CCDC6 (red). All three genes, RET, NCOA4, and H4/CCDC6, are positioned within the chromosome 10 territory (CT) and away from the CT edge. (H) 3D rendered image showing no signals on the surface of the CT due to the gene positioning inside the CT. (G–H are from Gandhi et al. (2010) with permission of Elsevier Ireland LTD).
In mammalian cells, DSBs are repaired by two pathways that are based on homologous or non-homologous recombination. The homology-dependent mechanism encompasses homologous recombination repair (HRR), single strand annealing (SSA), and non-allelic homologous recombination (NAHR). The non-homologous mechanism is known as non-homologous end joining (NHEJ). Another recently found repair pathway, micro-homology mediated end joining (MMEJ), combines features of the major pathways as it joins DNA ends after preliminary alignment using short homology DNA sequences located distant from the break.
Data obtained by the analysis of DNA sequences at the fusion points suggest that the formation of RET/PTC rearrangements may involve several possible DNA repair mechanisms, particularly NHEJ and MMEJ, and to lesser extent SSA (Bongarzone et al., 1997; Klugbauer et al., 2001). It remains unclear if all these pathways contribute to the generation of RET/PTC with similar frequency and if the choice is determined by specific conditions and/or individual genetic background.
Regardless of the specific DNA repair mechanism involved in recombination, spatial proximity is likely to predispose to specific rearrangements by making the neighboring regions prone to simultaneous damage by radiation or other DNA-damaging agents, and/or by facilitating mis-rejoining of free DNA ends located closely adjacent to each other (Figure 7F).
It remains unclear why specific chromosomal regions are located close to each other. For genetic loci located on the same chromosome, this is likely due to high order chromosome folding that would allow the genes to be positioned non-randomly with respect to each other. It has been found that the 18-Mb region on 10q containing RET, NCOA4, and CCDC6 has a large-scale helical folding in the interphase nuclei of human thyroid cells. This pattern of chromatin folding can offer the basis for the proximity between RET and NCOA4 and CCDC6 (Gandhi et al., 2006).
A particular feature of rearrangements found in papillary thyroid cancers is that almost all of them are intra-chromosomal inversions rather than inter-chromosomal translocations; this may be due in part to the nuclear architecture. It was found that genes involved in intra-chromosomal rearrangements were positioned at significantly greater distances away from the chromosomal territory (CT) edge and internally within their CTs as compared to genes involved in translocations that were positioned closer to the CT edge (Gandhi et al., 2009). The frequent location of RET and its recombinogenic partners within the interior of CT and the limited availability of it to interact with neighboring chromosomal territories likely predispose it to intra-chromosomal inversions, such as that seen in most cases of RET/PTC (Figures 7G–H).
Signaling Pathways Activated by RET/PTC
Several RET/PTC-activated signal transduction pathways have been identified. In detail, RET/PTC signaling, as RET signaling, depends on the auto-phosphorylation of several tyrosine residues (Figure 8). Tyrosine (Y) 905 (numbered according to integral RET amino acid sequence) is a binding site for Grb7/10 adaptors whose function has not been completely elucidated (Wells and Santoro, 2009). Y905 is also a binding site for SH2B1β, a protein that, by obstructing the SHP-1 tyrosine phosphatase, enhances RET phosphorylation (Donatello et al., 2007). Y1015 interacts with phospholipase Cγ, thereby activating protein kinase C enzymes, which in turn are key regulators of receptor TKs (Andreozzi et al., 2003). Y1062 is a highly important multi-docking binding site for several signaling mediators such as DOK1/4/5, Enigma, FRS2, IRS1/2, Shc, and ShcC. Phosphorylation of tyrosine 1062 results in the activation of major intracellular mediators, including ERK/MAPK, PI3K-AKT, NF-kB, and JNK (Wells and Santoro, 2009). Y1062-mediated MAPK activation appears to happen through the DOK4-Rap1 signaling cascade (Uchida et al., 2006; De Falco et al., 2007). Y1062 is essential for RET and RET/PTC transforming activity (Segouffin-Cariou and Billaud, 2000; Knauf et al., 2003; Melillo et al., 2005). Y1096, present only in the RET51 splicing variant, binds Grb2 and appears to have a redundant behavior with Y1062 on the activation of the AKT/MAPK pathways (Jain et al., 2006).
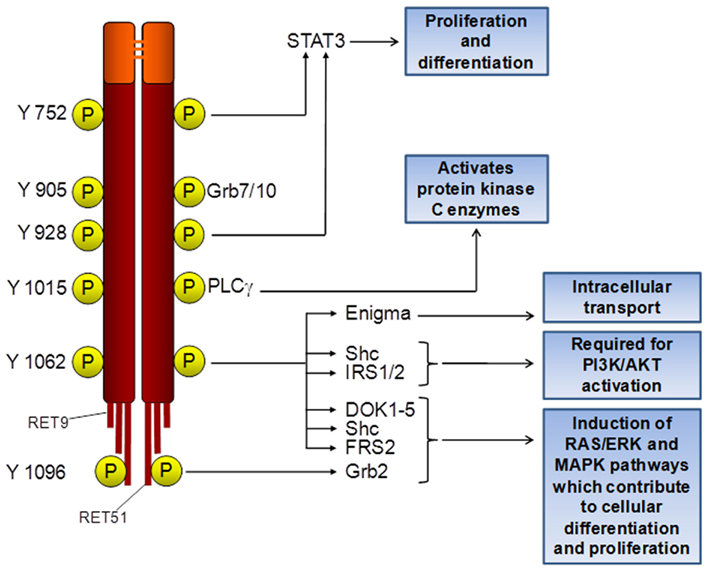
Figure 8. Signal transduction pathways regulated by RET/PTCs. Tyrosine residues (Y) are numbered according to integral RET amino acid sequence. RET9 and RET51 represent splicing variants of RET C-terminal tail.
Y752 and Y928 of MEN2A-RET appeared to be YxxV/Q Signal Transducer and Activator of Transcription 3 (STAT3) docking sites, involved in MEN2A-RET-induced phosphorylation of both Y705 and S727 residues of STAT3 (Schuringa et al., 2001). More recently, it was shown that RET/PTC1 and RET/PTC3 also interact with STAT3 and activate it through the phosphorylation of the tyrosine 705 residue (Hwang et al., 2003). However, RET/PTC1 residues Y141 and Y317, corresponding to RET residues Y752 and Y928, respectively, did not appear to be critical for the interaction with the transcription factor. Conversely, Y317 of RET/PTC1 appeared to be important for auto-phosphorylation of the kinase and for the Y705 phosphorylation of STAT3. This phosphorylation event appeared to require the intrinsic kinase activity of RET/PTC and to be independent from JAK2 and c-Src activation confirming that STAT3 is a direct substrate of RET/PTC TK in vivo. A physical interaction between RET/PTCs and the transcription factor could be demonstrated by co-immunoprecipitation experiments. Similarly, the expression of RET/PTC was found to lead to up-regulation of STAT1 expression and to the phosphorylation of the Y701 residue of this transcription factor (Hwang et al., 2004). Also in this case the dependence of the phosphorylation event from RET/PTC intrinsic kinase activity and its independence from JAKs and c-Src kinases activation pointed to the direct interaction between RET/PTCs and STAT1, although the occurrence of an association between RET/PTC and STAT1 could not be demonstrated by immune-precipitation analysis.
Among the non-phosphotyrosine mechanisms involved in RET/PTC signaling, binding of the PDZ-containing scaffold protein Shank3 to the C-tail of the RET9 splicing variant should be recalled. It contributes to sustained ERK and PI3K signaling (Schuetz et al., 2004).
Very recently, it was shown that RET/PTCs activate at least part of their pro-inflammatory programs through association with TRAF2 and TRAF6 which in turn activate NF-kB by inhibiting the constitutive proteolytic degradation of NIK kinase. This pro-inflammatory signaling pathway appeared to be independent from PI3K/AKT and RAS/BRAF/MEK/ERK pathways. Thus, the possibility was proposed that RAS/BRAF and/or PI3K/AKT pathways are required for cellular transformation and that the additional pro-inflammatory pathways of RET/PTCs shape the features of the growing tumor (Wixted et al., 2011).
Gene Expression Modifications Induced by RET/PTCs in Cellular Models
Gene expression modification induced by RET/PTCs has been studied principally and more consistently in cellular models.
A microarray analysis was conducted on well differentiated rat thyroid PCCL3 cells conditionally expressing the RET/PTC3 oncogene. Gene expression profiling 48 h after activation of RET/PTC3 identified a statistically significant modification of expression of 270 genes. Functional clustering of genes with a significant expression change revealed RET/PTC3-induced regulation of genes with key functions in apoptosis (Ripk3, Tdga), cell–cell signaling (Cdh6, Fn1), cell cycle (Il24), immune and inflammatory response (Cxcl10, Scya2, Il6, Gbp2, Oas1, Tap1, RT1Aw2, C2ta, Irf1, Lmp2, Psme2, Prkr), metabolism (Aldob, Ptges, Nd2, Gss, Gstt1), signal transduction (Socs3, Nf1, Jak2, Cpg21, Dusp6, Socs1, Stat1, Stat3, Cish), and transcription (Nr4a1, Junb, Hfh1, Runx1, Foxe1). This study clearly showed that genes coding for proteins involved in the immune response and in intracellular signal transduction pathways activated by cytokines and chemokines were strongly represented, indicating a critical role of RET/PTC3 in the activation and modulation of immune and inflammatory responses to the tumor (Figure 9) (Puxeddu et al., 2005a). The likely mediators of these expression clusters are NF-kB, STAT1, and STAT3, based on data from the microarray and the literature (Hwang et al., 2003, 2004; Wixted et al., 2011) and preliminary data from our laboratory.
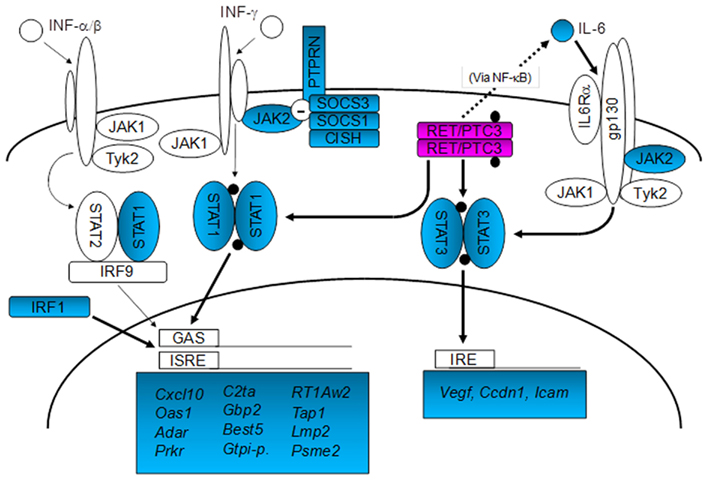
Figure 9. Schematic map of the relationships between RET/PTC3 and IFN- and cytokine-activated signal transduction pathways according to the microarray results and to the literature. Up-regulated genes are shaded, whereas black dots indicate activation by phosphorylation. RET/PTC3 induces the expression of STAT1 and STAT3, involved respectively in the regulation of IFN and cytokine (e.g., IL-6) signal transduction pathways. RET/PTC3 also activates both transcription factors through direct phosphorylation of critical tyrosine residues (Hwang et al., 2003, 2004) and through increased IL-6 expression, possibly via NF-kB (Wixted et al., 2011) (STAT3 activation). In addition RET/PTC3 induces the expression of additional components of these two pathways, such as JAK2, SOCS1 and 3, CISH and PTPRN. RET/PTC3-induced activation of STAT1 results in the up-regulation of IFN-responsive genes, whereas activation of STAT3 results in the up-regulation of VEGF, Cyclin D1, and ICAM (Hwang et al., 2003; modified from Puxeddu et al. (2005a) with permission of Bioscientifica Limited).
Similarly, exogenous expression of RET/PTC1 in primary normal human thyrocytes induced the expression of a set of genes involved in inflammatory/immune response including those encoding chemokines (CCL2, CCL20, CXCL8, and CXCL12), chemokine receptors (CXCR4) and cytokines (IL1B, CSF-1, GM-CSF, and G-CSF). Moreover, the modulation of genes involved in tumor invasion could be demonstrated, including matrix-degrading enzymes (metalloproteases and urokinase-type plasminogen activator and its receptor), and adhesion molecules (L-selectin). All these effects were strictly dependent on the presence of the RET/PTC1 Y451 residue (corresponding to RET Y1062 multidocking site). Selected relevant genes (CCL20, CCL2, CXCL8, CXCR4, L-selectin, GM-CSF, IL1B, MMP9, UPA, and SPP1/OPN) were found up-regulated also in clinical samples of PTC, particularly those characterized by RET/PTC activation, local extrathyroid spread, and lymph node metastases (Borrello et al., 2005).
Another study analyzed the gene expression profile of the TPC1 cell line, derived from a papillary thyroid cancer lymph node metastasis and naturally harboring a RET/PTC1 rearrangement, and of the thyroid follicular cell line Nthy-ori 3-1 stably transduced with the oncogene. Among the genes found to be overexpressed, those involved in biological processes such as cell differentiation, proliferation, and cell signaling were over-represented. Such genes included CEBPB, CCNG1, IFITM3, HTRA1, SEMA3F, and P300/CBP-associated factor (PCAF). Among the genes shown to be down-regulated in RET/PTC1-expressing cell lines when compared to normal control, those involved in cell structure and motility, cytoskeletal remodeling, and cell signaling were over-represented. Examples of such genes included DCTN5, TPM1, TPM3, CRP1, Keratin type 1, Tropomyosin, PSMD2 (TRAP2), and RAB32. Of particular interest in this study was the discovery of the under-expression of DROSHA in PTC cell lines. This is the core nuclease that executes the initiation step of miRNA processing in the nucleus. DROSHA collaborates with Dicer in stepwise processing of miRNAs and has a key role in miRNA mediated gene regulation processes such as development and differentiation. Interestingly, evaluation of the miRNA expression profile in the same cell lines revealed 21 overexpressed miRNAs and 14 underexpressed miRNAs in both cell lines when compared to Nthy-ori 3-1. Thus, miRNAs expression also appeared to be significantly affected by RET/PTC1 expression (Cahill et al., 2006).
Another study compared the transformed phenotype and the gene expression profile of rat thyroid PCCL3 cells stably expressing RET/PTC3 and its mutant RET/PTC3(Y1062F), BRAF(V600E), and H-RAS(V12). In the first instance, the authors could demonstrate that the oncogenic proteins involved in the initiation of PTC work along the same signaling cascade. This pathway starts at the level of RET tyrosine 1062 and sequentially triggers RAS, BRAF, and ERK stimulation. However, the analysis of transcriptional profiles indicated that the three oncoproteins are not completely equivalent. Indeed, in addition to targets common to RET/PTC3, HRAS, and BRAF, there were relatively large sets of genes specifically modulated by only one or two of the three oncogenes. Many of the commonly modulated genes were involved in hallmarks of neoplastic transformation, e.g., altered cell morphology, uncontrolled growth, loss of differentiation, and apoptosis. RET/PTC3-induced genes included metalloproteinases (Mmp3, -10, -12, -13), those encoding adhesion/structure-associated proteins, such as collagen 1 (Col1α1), thymosinβ4, and galectin-3, and the dual-specificity phosphatase Dusp6. Moreover, the RET/PTC-RAS-BRAF signaling cascade appeared to stimulate the overexpression of some chemokines/cytokines (Cxcl1, Cxcl10, and Ccl2). Thus, this study too confirmed a strict relationship between thyroid cancer and inflammation in which tumors appear to use molecules of the innate immune system not only to recruit leukocytes, but also for growth, survival, and metastasis (Melillo et al., 2005).
A final study which evaluated RET/PTC3-induced gene expression profile in a cellular model was a follow-up study of the first one described in this paragraph (Puxeddu et al., 2005a). It compared PCCL3 cells with conditional expression of RET/PTC3 to PCCL3 cells with conditional expression of BRAF(V600E) or RET/PTC3 in the presence of small interfering RNA-mediated knockdown of BRAF. Among the RET/PTC3-induced genes, 2,552 did not require BRAF as they were similarly regulated by RET/PTC3 with or without BRAF knockdown and not by expression of BRAF(V600E). Acquired immune response and IFN-related genes were highly represented in this group. Conversely, about 24% of RET/PTC3-regulated genes were BRAF dependent, as they were similarly modified by RET/PTC3 and BRAF(V600E) but not in cells expressing RET/PTC3 with knockdown of BRAF. A significant subset of genes involved in innate immune responses belong to this group. Among them were the monocyte-macrophage chemo-attractants Mcp1 (Ccl2), Mcp3, Gm-Csf, and Ccl15. Moreover, a gene cluster coding for components of the mitochondrial electron transport chain pathway was down-regulated in this group, potentially altering regulation of cell viability. Genes coding for metalloproteinases were also preferentially induced by BRAF, particularly Mmp3, Mmp9, and Mmp13. This study recapitulated most of the data reported by the previous ones indicating a fairly clear demarcation between some of the functional gene programs activated by RET/PTC3 and BRAF in thyroid cells. RET/PTC3 appeared to induce expression of a remarkably rich array of genes involved in acquired immunity, whereas BRAF appeared to evoke expression of genes that promote activation of an innate immune reaction. Moreover, although both genes induced expression of genes involved in matrix remodeling, RET/PTC did so less robustly, which could account in part for the lower predisposition of PTC harboring RET/PTC rearrangements to invade surrounding tissues compared to those harboring BRAF mutations (Mesa et al., 2006).
Insights on RET/PTC Influences on Tumor Microenvironment
RET/PTC oncoproteins are believed to take part in several mechanisms that allow tumor growth and spread, including angiogenesis, invasion, metastasis, and immune escape. RET/PTC induces these significant phenotypic changes oriented toward neoplastic transformation affecting the tumor microenvironment.
Eicosanoids are thought to play an important role in survival, growth and metabolic support of tumor cells (Koki et al., 2002). Significant among them are prostaglandins, of which prostaglandin E2 (PGE2) is the most abundant in nature. The synthesis of PGE2 from arachidonic acid requires two enzymes acting sequentially. The first step is catalyzed by cyclooxygenase (COX), which transforms arachidonic acid in the endoperoxide prostaglandin H2 (PGH2). The second step is catalyzed by prostaglandin E synthase (PGES), which converts PGH2 in PGE2. There are two COX isoforms: COX-1 and COX-2. COX-1 is constitutively expressed in most tissues whereas COX-2 is normally not expressed but is induced by growth factors, cytokines and certain oncogenes (Smith and Langenbach, 2001). A variety of epithelial cancers have increased expression of COX-2, including colorectal (Eberhart et al., 1994; Yoshimatsu et al., 2001) and thyroid carcinomas (Nose et al., 2002; Specht et al., 2002). There are three PGES isoforms: cytosolic PGES (cPGES), microsomal PGES-1 (mPGES-1) and mPGES-2. cPGES is expressed constitutively and has been proposed to exhibit preferential functional coupling with COX-1 (Tanioka et al., 2000). mPGES-1 is induced by pro-inflammatory stimuli and increases during the period when COX-2-dependent PGE2 generation is ongoing (Murakami et al., 2000; Mancini et al., 2001). mPGES-2 does not show homology with mPGES-1 (Tanikawa et al., 2002) and has been proposed to couple functionally to both COX-1 and COX-2 (Murakami et al., 2003). mPGES-1 is overexpressed in most colorectal adenomas and carcinomas, suggesting that this may contribute to the increased amounts of PGE2 in these tumors.
Conditional expression of RET/PTC1 or RET/PTC3 in PCCL3 thyroid cells markedly induced both COX-2 and mPGES-1 mRNA and protein. Accordingly, conditioned media of these cells, analyzed by HPLC or ELISA, showed a significant increase of PGE2 concentration (Figure 10). Based on signal transduction dissection experiments, RET/PTC was found to regulate mPGES-1 expression through the MAPK pathway (Puxeddu et al., 2003a). These data indicate a direct relationship between RET/PTC activation and regulation of PGE2 synthesis. It can be speculated that paracrine action of PGE2 might influence tumor development and progression regulating angiogenesis and the anti-tumor immune response. However, further investigation is needed to fully understand these phenomena.
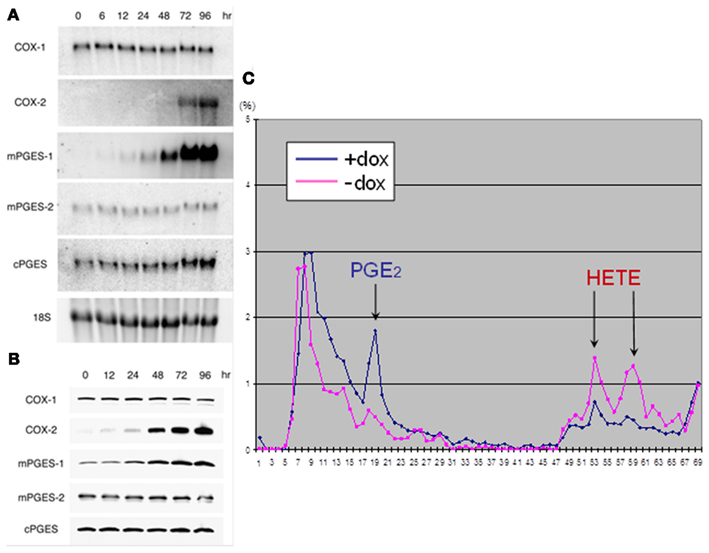
Figure 10. RET/PTC3 regulates PGE2 biosynthesis in PCCL3 cells characterized by the doxycycline (DOX)-inducible expression of the oncogene (PTC3-5 cells). (A) Northern blots showing that treatment with DOX for up to 96 h induces COX-2 and mPGES-1 mRNA but not COX-1, mPGES-2, or cPGES. (B) Western blots confirming that activation of RET/PTC3 expression for up to 96 h induces COX-2 and mPGES-1 protein but not COX-1, mPGES-2, or cPGES. (C) HPLC analysis of conditioned medium of PTC3-5 cells metabolically labeled with [3H]arachidonic acid showing that doxycycline (DOX) treatment induces PGE2 synthesis and secretion in the medium [modified from Puxeddu et al. (2003a)].
It has been hypothesized that the RET/PTC oncogene contributes to tumor invasion. Castellone and others, explored the transcriptional response of PCCL3 cells to RET/PTC oncogene expression looking for the overexpression of genes involved in tumor cell diffusion. They found that among several genes induced by RET/PTC, CXCR4 showed approximately a threefold increase. Western blot with anti-CXCR4 polyclonal antibody confirmed the overexpression of CXCR4 in rat transformed thyroid cells (Castellone et al., 2004). CXCR4 is the receptor for the chemokine CXCL12/SDF-1α/β. Chemokines are chemoattractant cytokines that play a major role in the recruitment of leukocytes to sites of inflammation. They are secreted in the tumor microenvironment by infiltrating inflammatory cells and by tumor cells (Balkwill and Mantovani, 2001; Coussens and Werb, 2002). Chemokines bind to seven-transmembrane receptors present on the cell surface that are coupled to G proteins, such as CXCR4, and activate a cascade of cellular events that result in cell polarization, adhesion, and migration (Melillo et al., 2005). Castellone and others observed that CXCR4 expression correlated with the integrity of RET/PTC catalytic domain and depended on the activation of the RET/PTC-RAS-ERK signaling pathway. They also found that CXCR4 was expressed in RET/PTC-positive human thyroid cancer cells but not in normal thyroid cells. Treatment with SDF-1α caused an increase of the proliferation and survival of PCCL3 cells expressing RET/PTC1 (PC-PTC1 cells) and of Matrigel invasion of PC-PTC1 and human FB2 cells (PTC-derived cell line expressing RET/PTC1; Castellone et al., 2004). Taken together, these results suggest that human thyroid cancers harboring RET/PTC rearrangements may use the CXCR4/SDF-1α receptor-ligand pathway to proliferate, survive, and migrate.
Moreover, there are other reasons why the expression of CXCR4 in human PTCs could be important. In the first instance, it might contribute to the preferential localization of PTC metastases to lymph nodes. Indeed, lymph nodes have been indicated as sites of high production of SDF-1α (Müller et al., 2001). Moreover, CXCR4 expression might enhance the tumor inflammatory infiltrate through the activation of signal transduction pathways that lead to the secretion of cytokines. Indeed, it has been shown that stimulation of CXCR4 in ovarian cancer leads to the production of TNF-α, which, in turn, can behave as a growth factor for cancer cells or mediate other events such as recruitment of leukocytes to the tumor site (Scotton et al., 2002).
As already reported above, Melillo showed that RET/PTC3, HRAS(V12), and BRAF(V600E) oncogenes activate a common transcriptional program in thyroid cells that includes CXCL1/growth-related oncogene-α (CXCL1/GRO-α) and CXCL10/interferon-γ-inducible protein 10 (CXCL10/IP-10) chemokines. According to the microarray results, QPCR data demonstrated that CXCL1 and CXCL10 were up-regulated also in human PTCs. CXCR2 (the CXCL1 receptor) and CXCR3 (the CXCL10 receptor) were expressed in parental and transformed PCCL3 cells and up-regulated in all the PTC cell lines examined in comparison to normal thyroid follicular cells. Treatment of TPC1 cells with recombinant CXCL1 and CXCL10 stimulated DNA synthesis and cell invasiveness through Matrigel (Melillo et al., 2005).
These data are very interesting in light of the fact that at least 30% of PTC present a chronic inflammatory reaction (Rosai et al., 1992; Scarpino et al., 2000). A molecular explanation of this phenomenon could be the fact that the RET/PTC-RAS-BRAF signaling cascade stimulates the overexpression of several chemokines. In turn, chemokines secreted by cancer cells can recruit leukocytes to the tumor site (Luster, 1998; Mellado et al., 2001). Moreover, it can be envisaged that these chemokines secreted by the cancer cells in the tumor microenvironment can act in an autocrine/paracrine fashion to support proliferation and cell motility of cancer cells themselves.
By using PCCL3 cells, osteopontin (OPN) has been identified as a major transcriptional target of RET/PTC in thyroid cells. OPN, also known as SPP1 (secreted phosphoprotein 1) is a highly acidic calcium-binding glycosylated phosphoprotein that binds to the cell surface receptors αv- or β1-containing integrins and CD44 (Weber, 2001). It is also regarded as a cytokine (ETA-1, Early T-lymphocyte antigen, or IL-28) regulating T helper cell-1 function (Ashkar et al., 2000; Chabas et al., 2001). OPN supports several functions including chemotaxis, cell attachment, and cell migration; involvement of OPN in tumorigenesis and metastasis formation has elicited much interest (Weber, 2001).
CD44 is a cell surface glycoprotein that can be expressed as a standard receptor (CD44s) or as multiple splice isoforms (CD44v) whose expression is altered during tumor growth and progression. Expression of the v6 exon variant is necessary for efficient OPN binding (Weber, 2001; Ponta et al., 2003). Normally, only CD44s is expressed on the cell surface of non-proliferating thyrocytes, whereas CD44v6 is invariably overexpressed in PTC samples (Ermak et al., 1996; Bartolazzi et al., 2001). CD44 has been implicated in cell–cell and cell–matrix interactions and homing of tumor cell metastasis (Ponta et al., 2003).
OPN is expressed in several human tumors, such as colon, breast, prostate, ovarian, gastric, and lung carcinomas. Moreover, OPN expression often correlates with a poor prognosis (Rittling and Chambers, 2004). It has been shown that OPN produced by RET/PTC-transformed thyrocytes cooperates with RET/PTC to increase DNA synthesis and invasiveness of PCCL3 cells (Castellone et al., 2004).
In a more recent work, OPN and CD44 expression were evaluated by immunohistochemistry with specific monoclonal antibodies. It has been found that the prevalence and intensity of OPN staining significantly correlated with the presence of lymph node metastases and tumor size. It has also been shown that treatment of human PTC cells with recombinant exogenous OPN stimulated Matrigel invasion and activated the ERK and AKT/PKB signaling pathways (Guarino et al., 2005).
Because OPN and CD44v6 overexpression is a common feature not only of PTC cells that harbor RET/PTC but also of those expressing BRAF(V600E), the activation of the OPN-CD44v6 axis appears as one of the end points of the RET-RAS-BRAF-MAPK oncogenic cascade. This model is consistent with the idea that OPN expression is also induced by other oncogenes such as RAS and is dependent on the MAPK pathway and that CD44 splicing is under control of the RAS-MAPK pathway as well (Geissinger et al., 2002; Matter et al., 2002).
The association between lymphocytic infiltrates and thyroid cancer, particularly PTC, in humans is also well documented (Dailey et al., 1955; Matsubayashi et al., 1995).
The observation that lymphocytic infiltration of the thyroid coexists with cancer suggests that antigens expressed by tumor cells are responsible for inducing an acquired anti-tumor immune response (Segal et al., 1985). The study of immune responses to tumors has primarily focused on the identification of tumor-associated antigens capable of serving as antigenic targets of anti-tumor immune responses (Van den Eynde et al., 1991; Rosenberg, 1997). Despite the presence of a unique tumor antigen and demonstration of an anti-tumor T cell response, cancer often persists in mouse tumor models as well as in cancer patients (Lee et al., 1999; Yu et al., 2005). These observations led to a new vision of inflammatory cells as a means of tumor support and a necessity for cancer progression. Inflammatory cells provide cytokines, growth factors, proangiogenetic mediators, and activating anti-apoptotic pathways that supply a supportive environment for cancer cell survival and tumor development (Balkwill et al., 2005; de Visser et al., 2006). These cytokines and growth factors also promote recruitment and establishment of non-transformed host-derived fibroblast and endothelial cells, creating a supportive framework for cancer cells (Moore et al., 1999; Olumi et al., 1999). Moreover, chronically activated inflammatory cells within tumors can play a protective role in suppressing adaptive anti-tumor responses (Zou, 2005).
The study of the properties of RET/PTC3 has revealed dual immunological features. First, its constitutive activation can induce the nuclear transactivation of the NF-κB protein complex (Visconti et al., 1997; Hayashi et al., 2000; Ludwig et al., 2001), driving the expression of several pro-inflammatory mediators (Ghosh and Karin, 2002; Puxeddu et al., 2005a). Second, RET/PTC3 encodes a chimeric protein that includes peptides that may be targets of anti-tumor immune responses (Viglietto et al., 1995; Fusco et al., 2002).
Pufnock and Rothstein hypothesized that combined immune and neoplastic properties of RET/PTC3 play a role in tumor progression and that these properties are dependent on the immune system. They studied the immunological functions of RET/PTC3 in an in vivo cancer model by measuring the recruitment of inflammatory cells in the tumor microenvironment and the effects on tumor growth. They found that tumor take in immunocompetent mice was more frequent using RET/PTC3 tumor tissue rather than the control RET/PTCY588F one (mutation of tyrosine 588 eliminates the transforming ability of RET/PTC3). RET/PTC3 expression correlated with increased CD8+ T cell recruitment at an early stage of tumor progression and induced recruitment of myeloid-derived CD11b+ Gr1+ cells into tumors. Thus, tumors expressing the functional form of RET/PTC3 showed enhanced infiltration of CD8+ lymphocytes, myeloid-derived CD11b+ Gr1+ cells and enhanced growth in immunocompetent mice. In contrast, RET/PTC3 signaling mutant-expressing tumors maintained enhanced infiltration of CD8+ lymphocytes but showed a lower recruitment of CD11b+ Gr1+ cells and a decreased tumor incidence. Pufnock and Rothstein (2009) proposed that enhanced transactivation of NF-κB by RET/PTC3 increases the quantity of secreted cytokines, thereby increasing the number of myeloid-derived CD11b+ Gr1+ cells recruited into tumor tissue.
Other studies showed that CD11b+ Gr1+ cells had strong suppressive activity on CD8+ tumor-specific responses (Serafini et al., 2004). An increase in CD11b+ Gr1+ cells in RET/PTC3-expressing tumors at an early stage of tumor progression may account for both the enhanced tumor incidence and larger tumor size observed despite the high number of CD8+ cells in the tumors.
Recently, it was demonstrated that IFN-γ, derived from intratumoral-activated T cells, was necessary to activate the suppressive function of CD11b+ Gr1+ cells (Gallina et al., 2006). These data confirm the notion that the increase of T cell activation at the tumor site, when combined with localization of myeloid-derived CD11b+ Gr1+ cells, may promote an environment supporting cancer progression rather than regression, in the contest of a potential anti-tumor response. Although Pufnock and Rothstein suggested that immature CD11b+ Gr1+ cells act in a suppressive manner on CD8+ cells, further evaluations are necessary to determine whether this interaction provides direct help to growing tumors through the secretion of growth factors rather than, or in addition to, suppression of directed anti-tumor CD8+ cytolytic responses. At any rate, these data suggest that RET/PTC3 enzymatic functions might independently regulate cellular transformation and anti-tumor lymphocytic responses promoting tumor progression from an early stage. The immunological functions seem to influence both innate and adaptive immune cells present at the site of the growing tumor. Furthermore, RET/PTC3-induced NF-κB activation may be the key pathway governing the immunomodulatory functions of the oncoprotein.
CD8+ T cell infiltration of the tumors has been hypothesized to represent an oncoprotein-reactive lymphocytic accumulation. The immunogenic capacity of RET/PTC3 fusion protein has been explored. It was demonstrated that the fusion oncoprotein behaves like an antigenic non-self protein and a thyroid-specific tumor antigen. Although RET/PTC3 is the fusion of two self proteins to which immunological tolerance is most likely induced, it was found that the fusion modifies the immunological properties of the molecules. Interestingly, it was detected that the immunogenic response to RET/PTC3 was not directed against the peptide comprising the unique fusion region but rather against the carboxyl-terminal portion of RET/PTC3 that derives from the self protein c-RET. Furthermore, transplantation of RET/PTC3-expressing thyroid cancers into naïve mice resulted in leukocytic infiltration, tumor rejection and induction of RET/PTC3-specific T cells (Powell et al., 2003). Thus, the somatic fusion of two unrelated self proteins appears able to trigger a uniquely immunogenic response directed against self epitopes within RET/PTC3.
In summary, RET/PTC appears to activate innate and acquired immunity in several ways. In a first phase, these processes might be instrumental to tumor elimination or to instauration of equilibrium between the tumor and the immune system. However, with time the cancer cell develops progressive immune resistance and gains the ability to tilt the immune response from restraining to advantageous for the tumor growth. The iteration of evasion mechanisms, associated with the suppression of different components of the immune system, results finally in immune escape. Moreover, several humoral and cellular components of the immune system itself may start to contribute to cancer maintenance and progression.
Does RET/PTC Impact on the Prognostic Stratification of Thyroid Carcinoma?
Considering the above-described features of RET/PTCs and their transforming mechanisms, the question of whether RET/PTC has a role as a thyroid carcinoma tumor marker arises. Regarding thyroid cancer diagnostics the answer is yes, and because this issue goes beyond the aims of this review, we refer to a very recent work on this topic (Nikiforov, 2011). Regarding prognostic stratification, the problem appears to still not be solved. Because of the lack of extensive reviews on this problem, we dedicate this paragraph to review the impact of RET/PTC on the prognostic stratification of thyroid carcinoma.
Several studies have tried to associate the presence of a rearranged RET with clinical parameters. Unfortunately, the results obtained are controversial. Some studies indicated an association of RET/PTC with a poor prognosis. Conversely, others indicated an association with a good prognosis. Finally, a last group could not find any association with prognosis at all.
In post-Chernobyl cases, rearrangement-positive PTCs appeared in a more advanced pT category and more frequently in the pN1 category at presentation than rearrangement-negative PTCs (Rabes et al., 2000). Moreover, RET/PTC3, the dominant rearrangement in the post-Chernobyl PTCs presenting with short latency, was related to the solid variant of PTC, considered by some researchers as a more malignant histotype (Thomas et al., 1999; Rabes et al., 2000). Accordingly, also in sporadic PTCs it was suggested that the rearrangement of the RET proto-oncogene may be involved in the development of local invasion (pT4) and distant metastases (Jhiang et al., 1992; Mayr et al., 1997; Bongarzone et al., 1998).
Other studies indicated the exact opposite. Viglietto and co-workers showed a higher frequency of RET/PTC rearrangements in papillary thyroid microcarcinomas compared to clinically significant PTCs (42 vs 20%; Viglietto et al., 1995). Soares et al. (1998) showed the occurrence of the rearrangement in well differentiated slow growing neoplasms characterized in their view by a “Bonsai” phenotype. Finally, Tallini et al. (1998) confirmed the association of RET/PTC rearrangements with well differentiated or under-centimetric PTCs and showed that in their hands the subset of RET/PTC-positive PTCs do not progress to more aggressive, less differentiated tumor phenotypes.
Finally, at least three studies did not show any correlation between RET/PTC activation and age, sex, tumor size, TNM staging, number of neoplastic foci, and histological subtype (Learoyd et al., 1998; Elisei et al., 2001; Puxeddu et al., 2003b).
It is likely that these studies have all been impaired by the lack of a unique and validated technique to detect RET/PTC rearrangements (see “Prevalence of RET/PTC Rearrangements”). Moreover, most of them are characterized by a far too small size to arrive to any consistent conclusion. However, probably with the exception of radiation-induced PTCs in which RET/PTC might define a subset of more aggressive neoplasms, some evidence points to a good prognostic impact of the rearrangements compared to other genetic alterations. In the first instance, it is clear that PTC harboring BRAF mutations have the worst outcome (Xing, 2010). Second, transgenic mice models confirmed the weaker carcinogenetic potential on the thyroid gland of RET/PTC rearrangements (Jhiang et al., 1996; Santoro et al., 1996; Powell et al., 1998) compared to other alterations such as BRAF (Knauf et al., 2005) and RAS (Vitagliano et al., 2006) mutations. Third, as mentioned above, the low expression levels of the chimeric oncogenes in vivo, driven by weak promoters of the RET fusion partner genes, hamper their transforming potentials (Richardson et al., 2009).
Conclusion
In this review, we have tried to summarize 25 years of research on RET/PTC rearrangement started with its discovery in 1987 (Fusco et al., 1987). Many points regarding its properties of dominant oncogene, its epidemiology, the molecular mechanisms of its formation, and its transforming features have been clarified. Doubts remain as to the best laboratory method for its detection, on its real prevalence and on its impact on clinical behavior of PTC. Moreover, more complete information on activated signal transduction pathways, induced gene expression modification and influences on tumor microenvironment still need to be collected. However, what we have learned up to now is sufficient to target RET/PTC with potentially specific small molecules that inhibit its tyrosine kinase function (TKIs) in RAI-refractory advanced PTCs or to develop new compounds with these features. In the near future we expect that a deeper knowledge of the key biological events that drive RET/PTC induced carcinogenesis will allow us to obtain additional negative modulators of this process to be placed beside RET/PTC specific TKIs. In addition, standardization of the detection method and development of multicenter studies might allow clarification of its real prevalence and impact on PTC prognosis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We wish to thank Mrs. Katherine Brandt Tonato for her editorial assistance. The work of our laboratory was supported by grants from the “Fondazione Cassa di Risparmio di Perugia” and “Associazione Italiana per la Ricerca sul Cancro (IG 9338)” and by a liberal contribution from the Beadle Family Foundation (San Antonio, TX, USA) to Efisio Puxeddu.
Footnotes
References
Anders, J., Kjar, S., and Ibáñez, C. F. (2001). Molecular modeling of the extracellular domain of the RET receptor tyrosine kinase reveals multiple cadherin-like domains and a calcium-binding site. J. Biol. Chem. 276, 35808–35817.
Andreozzi, F., Melillo, R. M., Carlomagno, F., Oriente, F., Miele, C., Fiory, F., Santopietro, S., Castellone, M. D., Beguinot, F., Santoro, M., and Formisano, P. (2003). Protein kinase Calpha activation by RET: evidence for a negative feedback mechanism controlling RET tyrosine kinase. Oncogene 22, 2942–2949.
Arlt, M. F., Durkin, S. G., Ragland, R. L., and Glover, T. W. (2006). Common fragile sites as targets for chromosome rearrangements. DNA Repair (Amst.) 5, 1126–1135.
Ashkar, S., Weber, G. F., Panoutsakopoulou, V., Sanchirico, M. E., Jansson, M., Zawaideh, S., Rittling, S. R., Denhardt, D. T., Glimcher, M. J., and Cantor, H. (2000). Eta-1 (osteopontin): an early component of type-1 (cell-mediated) immunity. Science 287, 860–864.
Adeniran, A. J., Zhu, Z., Gandhi, M., Steward, D. L., Fidler, J. P., Giordano, T. J., Biddinger, P. W., and Nikiforov, Y. E. (2006). Correlation between genetic alterations and microscopic features, clinical manifestations, and prognostic characteristics of thyroid papillary carcinomas. Am. J. Surg. Pathol. 30, 216–222.
Balkwill, F., Charles, K. A., and Mantovani, A. (2005). Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell 7, 211–217.
Balkwill, F., and Mantovani, A. (2001). Inflammation and cancer: back to Virchow? Lancet 357, 539–545.
Bartolazzi, A., Gasbarri, A., Papotti, M., Bussolati, G., Lucante, T., Khan, A., Inohara, H., Marandino, F., Orlandi, F., Nardi, F., Vecchione, A., Tecce, R., Larsson, O., and Thyroid Cancer Study Group. (2001) Application of an immunodiagnostic method for improving preoperative diagnosis of nodular thyroid lesions. Lancet 357, 1644–1650.
Bleuer, J. P., Averkin, Y. I., and Abelin, T. (1997). Chernobyl-related thyroid cancer: what evidence for role of short-lived iodines? Environ. Health Perspect. 105, 1483–1486.
Bongarzone, I., Butti, M. G., Fugazzola, L., Pacini, F., Pinchera, A., Vorontsova, T. V., Demidchik, E. P., and Pierotti, M. A. (1997). Comparison of the breakpoint regions of ELE1 and RET genes involved in the generation of RET/PTC3 oncogene in sporadic and in radiation-associated papillary thyroid carcinomas. Genomics 42, 252–259.
Bongarzone, I., Fugazzola, L., Vigneri, P., Mariani, L., Mondellini, P., Pacini, F., Basolo, F., Pinchera, A., Pilotti, S., and Pierotti, M. A. (1996). Age-related activation of the tyrosine kinase receptor protooncogenes RET and NTRK1 in papillary thyroid carcinoma. J. Clin. Endocrinol. Metab. 81, 2006–2009.
Bongarzone, I., Monzini, N., Borrello, M. G., Carcano, C., Ferraresi, G., Arighi, E., Mondellini, P., Della Porta, G., and Pierotti, M. A. (1993). Molecular characterization of a thyroid tumor-specific transforming sequence formed by the fusion of ret tyrosine kinase and the regulatory subunit RI alpha of cyclic AMP-dependent protein kinase A. Mol. Cell Biol. 13, 358–366.
Bongarzone, I., Vigneri, P., Mariani, L., Collini, P., Pilotti, S., and Pierotti, M. A. (1998). RET/NTRK1 rearrangements in thyroid gland tumors of the papillary carcinoma family: correlation with clinicopathological features. Clin. Cancer Res. 4, 223–228.
Borrello, M. G., Alberti, L., Fischer, A., Degl’innocenti, D., Ferrario, C., Gariboldi, M., Marchesi, F., Allavena, P., Greco, A., Collini, P., Pilotti, S., Cassinelli, G., Bressan, P., Fugazzola, L., Mantovani, A., and Pierotti, M. A. (2005). Induction of a proinflammatory program in normal human thyrocytes by the RET/PTC1 oncogene. Proc. Natl. Acad. Sci. U.S.A. 102, 14825–14830.
Bounacer, A., Wicker, R., Caillou, B., Cailleux, A. F., Sarasin, A., Schlumberger, M., and Suárez, H. G. (1997). High prevalence of activating ret proto-oncogene rearrangements, in thyroid tumors from patients who had received external radiation. Oncogene 15, 1263–1273.
Bousquet, M., Quelen, C., De Mas, V., Duchayne, E., Roquefeuil, B., Delsol, G., Laurent, G., Dastugue, N., and Brousset, P. (2005). The t(8;9)(p22;p24) translocation in atypical chronic myeloid leukaemia yields a new PCM1-JAK2 fusion gene. Oncogene 24, 7248–7252.
Bryant, P. E., and Riches, A. C. (1989). Oncogenic transformation of murine C3H 10T1/2 cells resulting from DNA double-strand breaks induced by a restriction endonuclease. Br. J. Cancer 60, 852–854.
Burrow, A. A., Williams, L. E., Pierce, L. C., and Wang, Y. H. (2009). Over half of breakpoints in gene pairs involved in cancer-specific recurrent translocations are mapped to human chromosomal fragile sites. BMC Genomics 10, 59. doi:10.1186/1471-2164-10-59
Cahill, S., Smyth, P., Finn, S. P., Denning, K., Flavin, R., O’Regan, E. M., Li, J., Potratz, A., Guenther, S. M., Henfrey, R., O’Leary, J. J., and Sheils, O. (2006). Effect of ret/PTC 1 rearrangement on transcription and post-transcriptional regulation in a papillary thyroid carcinoma model. Mol. Cancer 5, 70.
Carlomagno, F., Salvatore, G., Cirafici, A. M., De Vita, G., Melillo, R. M., de Franciscis, V., Billaud, M., Fusco, A., and Santoro, M. (1997). The different ret-activating capability of mutations of cysteine 620 or cysteine 634 correlates with the multiple endocrine neoplasia type 2 disease phenotype. Cancer Res. 57, 391–395.
Carta, C., Moretti, S., Passeri, L., Barbi, F., Avenia, N., Cavaliere, A., Monacelli, M., Macchiarulo, A., Santeusanio, F., Tartaglia, M., and Puxeddu, E. (2006). Genotyping of an Italian papillary thyroid carcinoma cohort revealed high prevalence of BRAF mutations, absence of RAS mutations and allowed the detection of a new mutation of BRAF oncoprotein (BRAF(V599Ins)). Clin. Endocrinol. (Oxf.) 64, 105–109.
Castellone, M. D., Guarino, V., De Falco, V., Carlomagno, F., Basolo, F., Faviana, P., Kruhoffer, M., Orntoft, T., Russell, J. P., Rothstein, J. L., Fusco, A., Santoro, M., and Melillo, R. M. (2004). Functional expression of the CXCR4 chemokine receptor is induced by RET/PTC oncogenes and is a common event in human papillary thyroid carcinomas. Oncogene 23, 5958–5967.
Castellone, M. D., and Santoro, M. (2008). Dysregulated RET signaling in thyroid cancer. Endocrinol. Metab. Clin. North Am. 37, 363–374.
Caudill, C. M., Zhu, Z., Ciampi, R., Stringer, J. R., and Nikiforov, Y. E. (2005). Dose-dependent generation of RET/PTC in human thyroid cells after in vitro exposure to gamma-radiation: a model of carcinogenic chromosomal rearrangement induced by ionizing radiation. J. Clin. Endocrinol. Metab. 90, 2364–2369.
Celetti, A., Cerrato, A., Merolla, F., Vitagliano, D., Vecchio, G., and Grieco, M. (2004). H4(D10S170), a gene frequently rearranged with RET in papillary thyroid carcinomas: functional characterization. Oncogene 23, 109–121.
Cetta, F., Chiappetta, G., Melillo, R. M., Petracci, M., Montalto, G., Santoro, M., and Fusco, A. (1998). The ret/ptc1 oncogene is activated in familial adenomatous polyposis-associated thyroid papillary carcinomas. J. Clin. Endocrinol. Metab. 83, 1003–1006.
Chabas, D., Baranzini, S. E., Mitchell, D., Bernard, C. C., Rittling, S. R., Denhardt, D. T., Sobel, R. A., Lock, C., Karpuj, M., Pedotti, R., Heller, R., Oksenberg, J. R., and Steinman, L. (2001). The influence of the proinflammatory cytokine, osteopontin, on autoimmune demyelinating disease. Science 294, 1731–1735.
Chappuis-Flament, S., Pasini, A., and De Vita, G. (1998). MEN 2 mutations affecting specific extracytoplasmic cysteines exert a dual effect on the ret receptor. Oncogene 17, 2851–2861.
Cheung, C. C., Ezzat, S., Ramyar, L., Freeman, J. L., and Asa, S. L. (2000). Molecular basis off hurthle cell papillary thyroid carcinoma. J. Clin. Endocrinol. Metab. 85, 878–882.
Chiappetta, G., Toti, P., Cetta, F., Giuliano, A., Pentimalli, F., Amendola, I., Lazzi, S., Monaco, M., Mazzuchelli, L., Tosi, P., Santoro, M., and Fusco, A. (2002). The RET/PTC oncogene is frequently activated in oncocytic thyroid tumors (Hurthle cell adenomas and carcinomas), but not in oncocytic hyperplastic lesions. J. Clin. Endocrinol. Metab. 87, 364–369.
Chua, E. L., Wu, W. M., Tran, K. T., McCarthy, S. W., Lauer, C. S., Dubourdieu, D., Packham, N., O’Brien, C. J., Turtle, J. R., and Dong, Q. (2000). Prevalence and distribution of ret/ptc 1, 2, and 3 in papillary thyroid carcinoma in New Caledonia and Australia. J. Clin. Endocrinol. Metab. 85, 2733–2739.
Coquelle, A., Toledo, F., Stern, S., Bieth, A., and Debatisse, M. (1998). A new role for hypoxia in tumor progression: induction of fragile site triggering genomic rearrangements and formation of complex DMs and HSRs. Mol. Cell 2, 259–265.
Croyle, M., Akeno, N., Knauf, J. A., Fabbro, D., Chen, X., Baumgartner, J. E., Lane, H. A., and Fagin, J. A. (2008). RET/PTC-induced cell growth is mediated in part by epidermal growth factor receptor (EGFR) activation: evidence for molecular and functional interactions between RET and EGFR. Cancer Res. 68, 4183–4191.
Dailey, M. E., Lindsay, S., and Skahen, R. (1955). Relation of thyroid neoplasms to Hashimoto disease of the thyroid gland. Arch. Surg. 70, 291–297.
De Falco, V., Castellone, M. D., De Vita, G., Cirafici, A. M., Hershman, J. M., Guerrero, C., Fusco, A., Melillo, R. M., and Santoro, M. (2007). RET/papillary thyroid carcinoma oncogenic signaling through the Rap1 small GTPase. Cancer Res. 67, 381–390.
de Visser, K. E., Eichten, A., and Coussens, L. M. (2006). Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 6, 24–37.
Donatello, S., Fiorino, A., Degl’Innocenti, D., Alberti, L., Miranda, C., Gorla, L., Bongarzone, I., Rizzetti, M. G., Pierotti, M. A., and Borrello, M. G. (2007). SH2B1beta adaptor is a key enhancer of RET tyrosine kinase signaling. Oncogene 26, 6546–6559.
Ducut Sigala, J. L., Bottero, V., Young, D. B., Shevchenko, A., Mercurio, F., and Verma, I. M. (2004). Activation of transcription factor NF-kappaB requires ELKS, an IkappaB kinase regulatory subunit. Science 304, 1963–1967.
Dupont, S., Zacchigna, L., Cordenonsi, M., Soligo, S., Adorno, M., Rugge, M., and Piccolo, S. (2005). Germ-layer specification and control of cell growth by Ectodermin, a Smad4 ubiquitin ligase. Cell 121, 87–99.
Durbec, P., Marcos-Gutierrez, C. V., Kilkenny, C., Grigoriou, M., Wartiowaara, K., Suvanto, P., Smith, D., Ponder, B., Costantini, F., and Saarma, M. (1996). GDNF signalling through the Ret receptor tyrosine kinase. Nature 381, 789–793.
Eberhart, C. E., Coffey, R. J., Radhika, A., Giardiello, F. M., Ferrenbach, S., and DuBois, R. N. (1994). Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology 107, 1183–1188.
Edwards, A. A. (2002). Modelling radiation-induced chromosome aberrations. Int. J. Radiat. Biol. 78, 551–558.
Elisei, R., Cosci, B., Romei, C., Bottici, V., Renzini, G., Molinaro, E., Agate, L., Vivaldi, A., Faviana, P., Basolo, F., Miccoli, P., Berti, P., Pacini, F., and Pinchera, A. (2008). Prognostic significance of somatic RET oncogene mutations in sporadic medullary thyroid cancer: a 10-year follow-up study. J. Clin. Endocrinol. Metab. 93, 682–687.
Elisei, R., Romei, C., Vorontsova, T., Cosci, B., Veremeychik, V., Kuchinskaya, E., Basolo, F., Demidchik, E. P., Miccoli, P., Pinchera, A., and Pacini, F. (2001). RET/PTC rearrangements in thyroid nodules: studies in irradiated and not irradiated, malignant and benign thyroid lesions in children and adults. J. Clin. Endocrinol. Metab. 86, 3211–3216.
Eng, C., Clayton, D., Schuffenecker, I., Lenoir, G., Cote, G., Gagel, R. F., van Amstel, H. K., Lips, C. J., Nishisho, I., Takai, S. I., Marsh, D. J., Robinson, B. G., Frank-Raue, K., Raue, F., Xue, F., Noll, W. W., Romei, C., Pacini, F., Fink, M., Niederle, B., Zedenius, J., Nordenskjöld, M., Komminoth, P., Hendy, G. N., Mulligan, L. M., Gharib, H., Thibodeau, S. N., Lacroix, A., Frilling, A., and Ponder, B.A. J. (1996). The relationship between specific ret proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2: International Ret Mutation Consortium. JAMA 276, 1575–1579.
Ermak, G., Jennings, T., Robinson, L., Ross, J. S., and Figge, J. (1996). Restricted patterns of CD44 variant exon expression in human papillary thyroid carcinoma. Cancer Res. 56, 1037–1042.
Evans, D. B., Shapiro, S. E., and Cote, G. J. (2007). Invited commentary: medullary thyroid cancer: the importance of RET testing. Surgery 141, 96–99.
Fenton, C. L., Lukes, Y., Nicholson, D., Dinauer, C. A., Francis, G. L., and Tuttle, R. M. (2000). The ret/PTC mutations are common in sporadic papillary thyroid carcinoma of children and young adults. J. Clin. Endocrinol. Metab. 85, 1170–1175.
Frêche, B., Guillaumot, P., Charmetant, J., Pelletier, L., Luquain, C., Christiansen, D., Billaud, M., and Manié, S. N. (2005). Inducible dimerization of RET reveals a specific AKT deregulation in oncogenic signaling. J. Biol. Chem. 280, 36584–36591.
Fugazzola, L., Pilotti, S., Pinchera, A., Vorontsova, T. V., Mondellini, P., Bongarzone, I., Greco, A., Astakhova, L., Butti, M. G., Demidchik, E. P., Pacini, F., and Pierotti, M. A. (1995). Oncogenic rearrangements of the RET proto-oncogene in papillary thyroid carcinomas from children exposed to the Chernobyl nuclear accident. Cancer Res. 55, 5617–5620.
Fusco, A., Chiappetta, G., Hui, P., Garcia-Rostan, G., Golden, L., Kinder, B. K., Dillon, D. A., Giuliano, A., Cirafici, A. M., Santoro, M., Rosai, J., and Tallini, G. (2002). Assessment of RET/PTC oncogene activation and clonality in thyroid nodules with incomplete morphological evidence of papillary carcinoma: a search for the early precursors of papillary cancer. Am. J. Pathol. 160, 2157–2167.
Fusco, A., Grieco, M., Santoro, M., Berlingieri, M. T., Pilotti, S., Pierotti, M. A., Della Porta, G., and Vecchio, G. (1987). A new oncogene in human thyroid papillary carcinomas and their lymph-nodal metastases. Nature 328, 170–172.
Gacy, A. M., Goellner, G., Juranic, N., Macura, S., and McMurray, C. T. (1995). Trinucleotide repeats that expand in human disease form hairpin structures in vitro. Cell 81, 533–540.
Gallina, G., Dolcetti, L., Serafini, P., De Santo, C., Marigo, I., Colombo, M. P., Basso, G., Brombacher, F., Borrello, I., Zanovello, P., Bicciato, S., and Bronte, V. (2006). Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J. Clin. Invest. 116, 2777–2790.
Gandhi, M., Dillon, L. W., Pramanik, S., Nikiforov, Y. E., and Wang, Y. H. (2010). DNA breaks at fragile sites generate oncogenic RET/PTC rearrangements in human thyroid cells. Oncogene 29, 2272–2280.
Gandhi, M., Medvedovic, M., Stringer, J. R., and Nikiforov, Y. E. (2006). Interphase chromosome folding determines spatial proximity of genes participating in carcinogenic RET/PTC rearrangements. Oncogene 25, 2360–2366.
Gandhi, M. S., Stringer, J. R., Nikiforova, M. N., Medvedovic, M., and Nikiforov, Y. E. (2009). Gene position within chromosome territories correlates with their involvement in distinct rearrangement types in thyroid cancer cells. Genes Chromosomes Cancer 48, 222–228.
Gasparini, P., Sozzi, G., and Pierotti, M. A. (2007). The role of chromosomal alterations in human cancer development. J. Cell. Biochem. 102, 320–331.
Geissinger, E., Weisser, C., Fischer, P., Schartl, M., and Wellbrock, C. (2002). Autocrine stimulation by osteopontin contributes to antiapoptotic signalling of melanocytes in dermal collagen. Cancer Res. 62, 4820–4828.
Gimm, O., Marsh, D. J., Andrew, S. D., Frilling, A., Dahia, P. L., Mulligan, L. M., Zajac, J. D., Robinson, B. G., and Eng, C. (1997). Germline dinucleotide mutation in codon 883 of the ret proto-oncogene in multiple endocrine neoplasia type 2B without codon 918 mutation. J. Clin. Endocrinol. Metab. 82, 3902–3904.
Grieco, M., Santoro, M., Berlingieri, M. T., Melillo, R. M., Donghi, R., Bongarzone, I., Pierotti, M. A., Della Porta, G., Fusco, A., and Vecchio, G. (1990). PTC is a novel rearranged form of the ret proto-oncogene and is frequently detected in vivo in human thyroid papillary carcinomas. Cell 60, 557–563.
Griffin, K. J., Kirschner, L. S., Matyakhina, L., Stergiopoulos, S. G., Robinson-White, A., Lenherr, S. M., Weinberg, F. D., Claflin, E. S., Batista, D., Bourdeau, I., Voutetakis, A., Sandrini, F., Meoli, E. M., Bauer, A. J., Cho-Chung, Y. S., Bornstein, S. R., Carney, J. A., and Stratakis, C. A. (2004). A transgenic mouse bearing an antisense construct of regulatory subunit type 1A of protein kinase A develops endocrine and other tumours: comparison with Carney complex and other PRKAR1A induced lesions. J. Med. Genet. 41, 923–931.
Guarino, V., Faviana, P., Salvatore, G., Castellone, M. D., Cirafici, A. M., De Falco, V., Celetti, A., Giannini, R., Basolo, F., Melillo, R. M., and Santoro, M. (2005). Osteopontin is overexpressed in human papillary thyroid carcinomas and enhances thyroid carcinoma cell invasiveness. J. Clin. Endocrinol. Metab. 90, 5270–5278.
Gujral, T. S., Singh, V. K., Jia, Z., and Mulligan, L. M. (2006). Molecular mechanisms of RET receptor-mediated oncogenesis in multiple endocrine neoplasia 2B. Cancer Res. 66, 10741–10749.
Hamatani, K., Eguchi, H., Ito, R., Mukai, M., Takahashi, K., Taga, M., Imai, K., Cologne, J., Soda, M., Arihiro, K., Fujihara, M., Abe, K., Hayashi, T., Nakashima, M., Sekine, I., Yasui, W., Hayashi, Y., and Nakachi, K. (2008). RET/PTC rearrangements preferentially occurred in papillary thyroid cancer among atomic bomb survivors exposed to high radiation dose. Cancer Res. 68, 7176–7182.
Handt, O., Sutherland, G. R., and Richards, R. I. (2000). Fragile sites and minisatellite repeat instability. Mol. Genet. Metab. 70, 99–105.
Hayashi, H., Ichihara, M., Iwashita, T., Murakami, H., Shimono, Y., Kawai, K., Kurokawa, K., Murakumo, Y., Imai, T., Funahashi, H., Nakao, A., and Takahashi, M. (2000). Characterization of intracellular signals via tyrosine 1062 in RET activated by glial cell line-derived neurotrophic factor. Oncogene 19, 4469–4475.
He, W., Dorn, D. C., Erdjument-Bromage, H., Tempst, P., Moore, M. A., and Massagué, J. (2006). Hematopoiesis controlled by distinct TIF1gamma and Smad4 branches of the TGFbeta pathway. Cell 125, 929–941.
Hellman, A., Rahat, A., Scherer, S. W., Darvasi, A., Tsui, L. C., and Kerem, B. (2000). Replication delay along FRA7H, a common fragile site on human chromosome 7, leads to chromosomal instability. Mol. Cell Biol. 20, 4420–4427.
Hewett, D. R., Handt, O., Hobson, L., Mangelsdorf, M., Eyre, H. J., Baker, E., Sutherland, G. R., Schuffenhauer, S., Mao, J. I., and Richards, R. I. (1998). FRA10B structure reveals common elements in repeat expansion and chromosomal fragile site genesis. Mol. Cell 1, 773–781.
Hlatky, L., Sachs, R. K., Vazquez, M., and Cornforth, M. N. (2002). Radiation-induced chromosome aberrations: insights gained from biophysical modeling. Bioessays 24, 714–723.
Hwang, E. S., Kim, D. W., Hwang, J. H., Jung, H. S., Suh, J. M., Park, Y. J., Chung, H. K., Song, J. H., Park, K. C., Park, S. H., Yun, H. J., Kim, J. M., and Shong, M. (2004). Regulation of signal transducer and activator of transcription 1 (STAT1) and STAT1-dependent genes by RET/PTC (rearranged in transformation/papillary thyroid carcinoma) oncogenic tyrosine kinases. Mol. Endocrinol. 18, 2672–2684.
Hwang, J. H., Kim, D. W., Suh, J. M., Kim, H., Song, J. H., Hwang, E. S., Park, K. C., Chung, H. K., Kim, J. M., Lee, T. H., Yu, D. Y., and Shong, M. (2003). Activation of signal transducer and activator of transcription 3 by oncogenic RET/PTC (rearranged in transformation/papillary thyroid carcinoma) tyrosine kinase: roles in specific gene regulation and cellular transformation. Mol. Endocrinol. 17, 1155–1166.
Iervolino, A., Iuliano, R., Trapasso, F., Viglietto, G., Melillo, R. M., Carlomagno, F., Santoro, M., and Fusco, A. (2006). The receptor-type protein tyrosine phosphatase J antagonizes the biochemical and biological effects of RET-derived oncoproteins. Cancer Res. 66, 6280–6287.
Ito, S., Iwashita, T., Asai, N., Murakami, H., Iwata, Y., Sobue, G., and Takahashi, M. (1997). Biological properties of ret with cysteine mutations correlate with multiple endocrine neoplasia type 2A, familial medullary thyroid carcinoma, and Hirschprung’s disease phenotype. Cancer Res. 57, 2870–2872.
Ito, T., Seyama, T., Iwamoto, K. S., Hayashi, T., Mizuno, T., Tsuyama, N., Dohi, K., Nakamura, N., and Akiyama, M. (1993) In vitro irradiation is able to cause RET oncogene rearrangement. Cancer Res. 53, 2940–2943.
Jain, S., Encinas, M., Johnson, E. M. Jr., and Milbrandt, J. (2006). Critical and distinct roles for key RET tyrosine docking sites in renal development. Genes Dev. 20, 321–333.
Jhiang, S. M., Caruso, D. R., Gilmore, E., Ishizaka, Y., Tahira, T., Nagao, M., Chiu, I. M., and Mazzaferri, E. L. (1992). Detection of the PTC/retTPC oncogene in human thyroid cancers. Oncogene 7, 1331–1337.
Jhiang, S. M., Sagartz, J. E., Tong, Q., Parker-Thornburg, J., Capen, C. C., Cho, J. Y., Xing, S., and Ledent, C. (1996). Targeted expression of the ret/PTC1 oncogene induces papillary thyroid carcinomas. Endocrinology 137, 375–378.
Kimura, E. T., Nikiforova, M. N., Zhu, Z., Knauf, J. A., Nikiforov, Y. E., and Fagin, J. A. (2003). High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. 63, 1454–1457.
Kirschner, L. S., Sandrini, F., Monbo, J., Lin, J. P., Carney, J. A., and Stratakis, C. A. (2000). Genetic heterogeneity and spectrum of mutations of the PRKAR1A gene in patients with the carney complex. Hum. Mol. Genet. 9, 3037–3046.
Klugbauer, S., Pfeiffer, P., Gassenhuber, H., Beimfohr, C., and Rabes, H. M. (2001). RET rearrangements in radiation-induced papillary thyroid carcinomas: high prevalence of topoisomerase I sites at breakpoints and microhomology-mediated end joining in ELE1 and RET chimeric genes. Genomics 73, 149–160.
Knauf, J. A., Kuroda, H., Basu, S., and Fagin, J. A. (2003). RET/PTC-induced dedifferentiation of thyroid cells is mediated through Y1062 signaling through SHC-RAS-MAP kinase. Oncogene 22, 4406–4412.
Knauf, J. A., Ma, X., Smith, E. P., Zhang, L., Mitsutake, N., Liao, X. H., Refetoff, S., Nikiforov, Y. E., and Fagin, J. A. (2005). Targeted expression of BRAF(V600E) in thyroid cells of transgenic mice results in papillary thyroid cancers that undergo dedifferentiation. Cancer Res. 65, 4238–4245.
Knowles, P. P., Murray-Rust, J., Kjaer, S., Scott, R. P., Hanrahan, S., Santoro, M., Ibáñez, C. F., and McDonald, N. Q. (2006). Structure and chemical inhibition of the RET tyrosine kinase domain. J. Biol. Chem. 281, 33577–33587.
Kohno, T., Ichikawa, H., Totoki, Y., Yasuda, K., Hiramoto, M., Nammo, T., Sakamoto, H., Tsuta, K., Furuta, K., Shimada, Y., Iwakawa, R., Ogiwara, H., Oike, T., Enari, M., Schetter, A. J., Okayama, H., Haugen, A., Skaug, V., Chiku, S., Yamanaka, I., Arai, Y., Watanabe, S., Sekine, I., Ogawa, S., Harris, C. C., Tsuda, H., Yoshida, T., Yokota, J., and Shibata, T. (2011). KIF5B-RET fusions in lung adenocarcinoma. Nat. Med. 18, 375–377.
Koki, A., Khan, N. K., Woerner, B. M., Dannenberg, A. J., Olson, L., Seibert, K., Edwards, D., Hardy, M., Isakson, P., and Masferrer, J. L. (2002). Cyclooxygenase-2 in human pathological disease. Adv. Exp. Med. Biol. 507, 177–184.
Kondo, T., Ezzat, S., and Asa, S. L. (2006). Pathogenetic mechanisms in thyroid follicular-cell neoplasia. Nat. Rev. Cancer 6, 292–306.
Kouvaraki, M. A., Shapiro, S. E., Perrier, N. D., Cote, G. J., Gagel, R. F., Hoff, A. O., Sherman, S. I., Lee, J. E., and Evans, D. B. (2005). RET proto-oncogene: a review and update of genotype-phenotype correlations in hereditary medullary thyroid cancer and associated endocrine tumors. Thyroid 15, 531–544.
Krützfeldt, M., Ellis, M., Weekes, D. B., Bull, J. J., Eilers, M., Vivanco, M. D., Sellers, W. R., and Mittnacht, S. (2005). Selective ablation of retinoblastoma protein function by the RET finger protein. Mol. Cell 18, 213–224.
Kulkarni, S., Heath, C., Parker, S., Chase, A., Iqbal, S., Pocock, C. F., Kaeda, J., Cwynarski, K., Goldman, J. M., and Cross, N. C. (2000). Fusion of H4/D10S170 to the platelet-derived growth factor receptor beta in BCR-ABL-negative myeloproliferative disorders with a t(5;10)(q33;q21). Cancer Res. 60, 3592–3598.
Kurzrock, R., Sherman, S. I., Ball, D. W., Forastiere, A. A., Cohen, R. B., Mehra, R., Pfister, D. G., Cohen, E. E., Janisch, L., Nauling, F., Hong, D. S., Ng, C. S., Ye, L., Gagel, R. F., Frye, J., Müller, T., Ratain, M. J., and Salgia, R. (2011). Activity of XL184 (Cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J. Clin. Oncol. 29, 2660–2666.
Kuwano, A., and Kajii, T. (1987). Synergistic effect of aphidicolin and ethanol on the induction of common fragile sites. Hum. Genet. 75, 75–78.
Lam, A. K., Montone, K. T., Nolan, K. A., and Livolsi, V. A. (1998). Ret oncogene activation in papillary thyroid carcinoma: prevalence and implication on the histological parameters. Hum. Pathol. 29, 565–568.
Learoyd, D. L., Messina, M., Zedenius, J., Guinea, A. I., Delbridge, L. W., and Robinson, B. G. (1998). RET/PTC and RET tyrosine kinase expression in adult papillary thyroid carcinomas. J. Clin. Endocrinol. Metab. 83, 3631–3635.
Lee, P. P., Yee, C., Savage, P. A., Fong, L., Brockstedt, D., Weber, J. S., Johnson, D., Swetter, S., Thompson, J., Greenberg, P. D., Roederer, M., and Davis, M. M. (1999). Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat. Med. 5, 677–685.
Leone, V., Mansueto, G., Pierantoni, G. M., Tornincasa, M., Merolla, F., Cerrato, A., Santoro, M., Grieco, M., Scaloni, A., Celetti, A., and Fusco, A. (2010). CCDC6 represses CREB1 activity by recruiting histone deacetylase 1 and protein phosphatase 1. Oncogene 29, 4341–4351.
Lipson, D., Capelletti, M., Yelensky, R., Otto, G., Parker, A., Jarosz, M., Curran, J. A., Balasubramanian, S., Bloom, T., Brennan, K. W., Donahue, A., Downing, S. R., Frampton, G. M., Garcia, L., Juhn, F., Mitchell, K. C., White, E., White, J., Zwirko, Z., Peretz, T., Nechushtan, H., Soussan-Gutman, L., Kim, J., Sasaki, H., Kim, H. R., Park, S. I., Ercan, D., Sheehan, C. E., Ross, J. S., Cronin, M. T., Jänne, P. A., and Stephens, P. J. (2011). Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat. Med. 18, 382–384.
Ludwig, L., Kessler, H., Wagner, M., Hoang-Vu, C., Dralle, H., Adler, G., Böhm, B. O., and Schmid, R. M. (2001). Nuclear factor-kappaB is constitutively active in C-cell carcinoma and required for RET-induced transformation. Cancer Res. 61, 4526–4535.
Luster, A. D. (1998). Chemokines – chemotactic cytokines that mediate inflammation. N. Engl. J. Med. 338, 436–445.
Mancini, J. A., Blood, K., Guay, J., Gordon, R., Claveau, D., Chan, C. C., and Riendeau, D. (2001). Cloning, expression, and up-regulation of inducible rat prostaglandin e synthase during lipopolysaccharide-induced pyresis and adjuvant-induced arthritis. J. Biol. Chem. 276, 4469–4475.
Matsubayashi, S., Kawai, K., Matsumoto, Y., Mukuta, T., Morita, T., Hirai, K., Matsuzuka, F., Kakudoh, K., Kuma, K., and Tamai, H. (1995). The correlation between papillary thyroid carcinoma and lymphocytic infiltration in the thyroid gland. J. Clin. Endocrinol. Metab. 80, 3421–3424.
Matter, N., Herrlich, P., and König, H. (2002). Signal-dependent regulation of splicing via phosphorylation of Sam68. Nature 420, 691–695.
Mayr, B., Brabant, G., Goretzki, P., Rüschoff, J., Dietmaier, W., and Dralle, H. (1997). Ret/PTC-1, -2, and -3 oncogene rearrangements in human thyroid carcinomas: implications for metastatic potential? J. Clin. Endocrinol. Metab. 82, 1306–1307.
Melillo, R. M., Castellone, M. D., Guarino, V., De Falco, V., Cirafici, A. M., Salvatore, G., Caiazzo, F., Basolo, F., Giannini, R., Kruhoffer, M., Orntoft, T., Fusco, A., and Santoro, M. (2005). The RET/PTC-RAS-BRAF linear signaling cascade mediates the motile and mitogenic phenotype of thyroid cancer cells. J. Clin. Invest. 115, 1068–1081.
Mellado, M., Rodríguez-Frade, J. M., Mañes, S., and Martínez-A, C. (2001). Chemokine signaling and functional responses: the role of receptor dimerization and TK pathway activation. Annu. Rev. Immunol. 19, 397–421.
Mesa, C. Jr., Mirza, M., Mitsutake, N., Sartor, M., Medvedovic, M., Tomlinson, C., Knauf, J. A., Weber, G. F., and Fagin, J. A. (2006). Conditional activation of RET/PTC3 and BRAF(V600E) in thyroid cells is associated with gene expression profiles that predict a preferential role of BRAF in extracellular matrix remodeling. Cancer Res. 66, 6521–6529.
Minoletti, F., Butti, M. G., Coronelli, S., Miozzo, M., Sozzi, G., Pilotti, S., Tunnacliffe, A., Pierotti, M. A., and Bongarzone, I. (1994). The two genes generating RET/PTC3 are localized in chromosomal band 10q11.2. Genes Chromosomes Cancer 11, 51–57.
Mishmar, D., Rahat, A., Scherer, S. W., Nyakatura, G., Hinzmann, B., Kohwi, Y., Mandel-Gutfroind, Y., Lee, J. R., Drescher, B., Sas, D. E., Margalit, H., Platzer, M., Weiss, A., Tsui, L. C., Rosenthal, A., and Kerem, B. (1998). Molecular characterization of a common fragile site (FRA7H) on human chromosome 7 by the cloning of a simian virus 40 integration site. Proc. Natl. Acad. Sci. U.S.A. 95, 8141–8146.
Mizuno, T., Iwamoto, K. S., Kyoizumi, S., Nagamura, H., Shinohara, T., Koyama, K., Seyama, T., and Hamatani, K. (2000). Preferential induction of RET/PTC1 rearrangement by X-ray irradiation. Oncogene 19, 438–443.
Mizuno, T., Kyoizumi, S., Suzuki, T., Iwamoto, K. S., and Seyama, T. (1997). Continued expression of a tissue specific activated oncogene in the early steps of radiation-induced human thyroid carcinogenesis. Oncogene 15, 1455–1460.
Monaco, C., Visconti, R., Barone, M. V., Pierantoni, G. M., Berlingieri, M. T., De Lorenzo, C., Mineo, A., Vecchio, G., Fusco, A., and Santoro, M. (2001). The RFG oligomerization domain mediates kinase activation and re-localization of the RET/PTC3 oncoprotein to the plasma membrane. Oncogene 20, 599–608.
Moore, R. J., Owens, D. M., Stamp, G., Arnott, C., Burke, F., East, N., Holdsworth, H., Turner, L., Rollins, B., Pasparakis, M., Kollias, G., and Balkwill, F. (1999). Mice deficient in tumor necrosis factor-alpha are resistant to skin carcinogenesis. Nat. Med. 5, 828–831.
Müller, A., Homey, B., Soto, H., Ge, N., Catron, D., Buchanan, M. E., McClanahan, T., Murphy, E., Yuan, W., Wagner, S. N., Barrera, J. L., Mohar, A., Verástegui, E., and Zlotnik, A. (2001). Involvement of chemokine receptors in breast cancer metastasis. Nature 410, 50–56.
Mulligan, L. M., Eng, C., Healey, C. S., Clayton, D., Kwok, J. B., Gardner, E., Ponder, M. A., Frilling, A., Jackson, C. E., Lehnert, H., Neumann, H. P. H., Thibodeau, S. N., and Ponder, B. A. J. (1994). Specific mutations of the ret proto-oncogene are related to disease phenotype in MEN 2A and FMTC. Nat. Genet. 6, 70–74.
Murakami, M., Nakashima, K., Kamei, D., Masuda, S., Ishikawa, Y., Ishii, T., Ohmiya, Y., Watanabe, K., and Kudo, I. (2003). Cellular prostaglandin E2 production by membrane-bound prostaglandin E synthase-2 via both cyclooxygenases-1 and -2. J. Biol. Chem. 278, 37937–37947.
Murakami, M., Naraba, H., Tanioka, T., Semmyo, N., Nakatani, Y., Kojima, F., Ikeda, T., Fueki, M., Ueno, A., Oh, S., and Kudo, I. (2000). Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J. Biol. Chem. 275, 32783–32792.
Musio, A., and Sbrana, I. (1997). Aphidicolin-sensitive specific common fragile sites: a biomarker of exposure to pesticides. Environ. Mol. Mutagen. 29, 250–255.
Nikiforov, Y. E. (2004). Genetic alterations involved in the transition from well-differentiated to poorly differentiated and anaplastic thyroid carcinomas. Endocr. Pathol. 15, 319–327.
Nikiforov, Y. E. (2011). Molecular diagnostics of thyroid tumors. Arch. Pathol. Lab. Med. 135, 569–577.
Nikiforov, Y. E., Koshoffer, A., Nikiforova, M., Stringer, J., and Fagin, J. A. (1999). Chromosomal breakpoint positions suggest a direct role for radiation in inducing illegitimate recombination between the ELE1 and RET genes in radiation-induced thyroid carcinomas. Oncogene 18, 6330–6334.
Nikiforov, Y. E., Rowland, J. M., Bove, K. E., Monforte-Munoz, H., and Fagin, J. A. (1997). Distinct pattern of ret oncogene rearrangements in morphological variants of radiation-induced and sporadic thyroid papillary carcinomas in children. Cancer Res. 57, 1690–1694.
Nikiforova, M. N., Caudill, C. M., Biddinger, P., and Nikiforov, Y. E. (2002). Prevalence of RET/PTC rearrangements in Hashimoto’s thyroiditis and papillary thyroid carcinomas. Int. J. Surg. Pathol. 10, 15–22.
Nikiforova, M. N., Stringer, J. R., Blough, R., Medvedovic, M., Fagin, J. A., and Nikiforov, Y. E. (2000). Proximity of chromosomal loci that participate in radiation-induced rearrangements in human cells. Science 290, 138–141.
Nose, F., Ichikawa, T., Fujiwara, M., and Okayasu, I. (2002). Up-regulation of cyclooxygenase-2 expression in lymphocytic thyroiditis and thyroid tumors: significant correlation with inducible nitric oxide synthase. Am. J. Clin. Pathol. 117, 546–551.
Olumi, A. F., Grossfeld, G. D., Hayward, S. W., Carroll, P. R., Tlsty, T. D., and Cunha, G. R. (1999). Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 59, 5002–5011.
Palakodeti, A., Han, Y., Jiang, Y., and Le Beau, M. M. (2004). The role of late/slow replication of the FRA16D in common fragile site induction. Genes Chromosomes Cancer 39, 71–76.
Papotti, M., Volante, M., Giuliano, A., Fassina, A., Fusco, A., Bussolati, G., Santoro, M., and Chiappetta, G. (2000). RET/PTC activation in hyalinizing trabecular tumors of the thyroid. Am. J. Surg. Pathol. 24, 1615–1621.
Pelliccia, F., Bosco, N., Curatolo, A., and Rocchi, A. (2008). Replication timing of two human common fragile sites: FRA1H and FRA2G. Cytogenet. Genome Res. 121, 196–200.
Pierotti, M. A., Santoro, M., Jenkins, R. B., Sozzi, G., Bongarzone, I., Grieco, M., Monzini, N., Miozzo, M., Herrmann, M. A., and Fusco, A. (1992). Characterization of an inversion on the long arm of chromosome 10 juxtaposing D10S170 and RET and creating the oncogenic sequence RET/PTC. Proc. Natl. Acad. Sci. U.S.A. 89, 1616–1620.
Ponta, H., Sherman, L., and Herrlich, P. A. (2003). CD44: from adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell Biol. 4, 33–45.
Powell, D. J. Jr., Eisenlohr, L. C., and Rothstein, J. L. (2003). A thyroid tumor-specific antigen formed by the fusion of two self proteins. J. Immunol. 170, 861–869.
Powell, D. J. Jr., Russell, J., Nibu, K., Li, G., Rhee, E., Liao, M., Goldstein, M., Keane, W. M., Santoro, M., Fusco, A., and Rothstein, J. L. (1998). The RET/PTC3 oncogene: metastatic solid-type papillary carcinomas in murine thyroids. Cancer Res. 58, 5523–5528.
Pufnock, J. S., and Rothstein, J. L. (2009). Oncoprotein signaling mediates tumor-specific inflammation and enhances tumor progression. J. Immunol. 182, 5498–5506.
Puxeddu, E., and Fagin, J. A. (2001). Genetic markers in thyroid neoplasia. Endocrinol. Metab. Clin. North Am. 30, 493–513.
Puxeddu, E., Knauf, J. A., Sartor, M. A., Mitsutake, N., Smith, E. P., Medvedovic, M., Tomlinson, C. R., Moretti, S., and Fagin, J. A. (2005a). RET/PTC-induced gene expression in thyroid PCCL3 cells reveals early activation of genes involved in regulation of the immune response. Endocr. Relat. Cancer 12, 319–334.
Puxeddu, E., Zhao, G., Stringer, J. R., Medvedovic, M., Moretti, S., and Fagin, J. A. (2005b). Characterization of novel non-clonal intrachromosomal rearrangements between the H4 and PTEN genes (H4/PTEN) in human thyroid cell lines and papillary thyroid cancer specimens. Mutat. Res. 570, 17–32.
Puxeddu, E., Mitsutake, N., Knauf, J. A., Moretti, S., Kim, H. W., Seta, K. A., Brockman, D., Myatt, L., Millhorn, D. E., and Fagin, J. A. (2003a). Microsomal prostaglandin E2 synthase-1 is induced by conditional expression of RET/PTC in thyroid PCCL3 cells through the activation of the MEK-ERK pathway. J. Biol. Chem. 278, 52131–52138.
Puxeddu, E., Moretti, S., Giannico, A., Martinelli, M., Marino, C., Avenia, N., Cristofani, R., Farabi, R., Reboldi, G., Ribacchi, R., Pontecorvi, A., and Santeusanio, F. (2003b). Ret/PTC activation does not influence clinical and pathological features of adult papillary thyroid carcinomas. Eur. J. Endocrinol. 148, 505–513.
Puxeddu, E., Moretti, S., Elisei, R., Romei, C., Pascucci, R., Martinelli, M., Marino, C., Avenia, N., Rossi, E. D., Fadda, G., Cavaliere, A., Ribacchi, R., Falorni, A., Pontecorvi, A., Pacini, F., Pinchera, A., and Santeusanio, F. (2004). BRAF(V599E) mutation is the leading genetic event in adult sporadic papillary thyroid carcinomas. J. Clin. Endocrinol. Metab. 89, 2414–2420.
Puxeddu, E., Romagnoli, S., and Dottorini, M. E. (2011). Targeted therapies for advanced thyroid cancer. Curr. Opin. Oncol. 23, 13–21.
Rabes, H. M., Demidchik, E. P., Sidorow, J. D., Lengfelder, E., Beimfohr, C., Hoelzel, D., and Klugbauer, S. (2000). Pattern of radiation-induced RET and NTRK1 rearrangements in 191 post-chernobyl papillary thyroid carcinomas: biological, phenotypic, and clinical implications. Clin. Cancer Res. 6, 1093–1103.
Rhoden, K. J., Unger, K., Salvatore, G., Yilmaz, Y., Vovk, V., Chiappetta, G., Qumsiyeh, M. B., Rothstein, J. L., Fusco, A., Santoro, M., Zitzelsberger, H., and Tallini, G. (2006). RET/papillary thyroid cancer rearrangement in nonneoplastic thyrocytes: follicular cells of Hashimoto’s thyroiditis share low-level recombination events with a subset of papillary carcinoma. J. Clin. Endocrinol. Metab. 91, 2414–2423.
Ricarte-Filho, J. C., Ryder, M., Chitale, D. A., Rivera, M., Heguy, A., Ladanyi, M., Janakiraman, M., Solit, D., Knauf, J. A., Tuttle, R. M., Ghossein, R. A., and Fagin, J. A. (2009). Mutational profile of advanced primary and metastatic radioactive iodine-refractory thyroid cancers reveals distinct pathogenetic roles for BRAF, PIK3CA, and AKT1. Cancer Res. 69, 4885–4893.
Richardson, D. S., Gujral, T. S., Peng, S., Asa, S. L., and Mulligan, L. M. (2009). Transcript level modulates the inherent oncogenicity of RET/PTC oncoproteins. Cancer Res. 69, 4861–4869.
Rittling, S. R., and Chambers, A. F. (2004). Role of osteopontin in tumour progression. Br. J. Cancer 90, 1877–1881.
Robinson, B. G., Paz-Ares, L., Krebs, A., Vasselli, J., and Haddad, R. (2010). Vandetanib (100 mg) in patients with locally advanced or metastatic hereditary medullary thyroid cancer. J. Clin. Endocrinol. Metab. 95, 2664–2671.
Romei, C., Elisei, R., Pinchera, A., Ceccherini, I., Molinaro, E., Mancusi, F., Martino, E., Romeo, G., and Pacini, F. (1996). Somatic mutations of the ret proto-oncogene in sporadic medullary thyroid carcinoma are not restricted to exon 16 and are associated with tumor recurrence. J. Clin. Endocrinol. Metab. 81, 1619–1622.
Ron, E., Lubin, J. H., Shore, R. E., Mabuchi, K., Modan, B., Pottern, L. M., Schneider, A. B., Tucker, M. A., and Boice, J. D. Jr. (1995). Thyroid cancer after exposure to external radiation: a pooled analysis of seven studies. Radiat. Res. 141, 259–277.
Rosai, J., Carcangiu, M. L., and DeLellis, R. A. (1992). Atlas of Tumor Pathology: Tumors of the Thyroid Gland, 3rd Series. Washington, DC: Armed Forces Institute of Pathology.
Rosenberg, S. A. (1997). Cancer vaccines based on the identification of genes encoding cancer regression antigens. Immunol. Today 18, 175–182.
Rothkamm, K., Kühne, M., Jeggo, P. A., and Löbrich, M. (2001). Radiation-induced genomic rearrangements formed by nonhomologous end-joining of DNA double-strand breaks. Cancer Res. 61, 3886–3893.
Saad, A. G., Kumar, S., Ron, E., Lubin, J. H., Stanek, J., Bove, K. E., and Nikiforov, Y. E. (2006). Proliferative activity of human thyroid cells in various age groups and its correlation with the risk of thyroid cancer after radiation exposure. J. Clin. Endocrinol. Metab. 91, 2672–2677.
Santoro, M., Carlomagno, F., Hay, I. D., Herrmann, M. A., Grieco, M., Melillo, R., Pierotti, M. A., Bongarzone, I., Della Porta, G., and Berger, N. (1992). Ret oncogene activation in human thyroid neoplasms is restricted to the papillary cancer subtype. J. Clin. Invest. 89, 1517–1522.
Santoro, M., Carlomagno, F., Melillo, R. M., Billaud, M., Vecchio, G., and Fusco, A. (1999). Molecular mechanisms of ret activation in human neoplasia. J. Endocrinol. Invest. 22, 811–819.
Santoro, M., Carlomagno, F., Romano, A., Bottaro, D. P., Dathan, N. A., Grieco, M., Fusco, A., Vecchio, G., Matoskova, B., Kraus, M. H., and Di Fiore, P. P. (1995). Activation of RET as a dominant transforming gene by germline mutations of MEN2A and MEN2B. Science 267, 381–383.
Santoro, M., Chiappetta, G., Cerrato, A., Salvatore, D., Zhang, L., Manzo, G., Picone, A., Portella, G., Santelli, G., Vecchio, G., and Fusco, A. (1996). Development of thyroid papillary carcinomas secondary to tissue-specific expression of the RET/PTC1 oncogene in transgenic mice. Oncogene 12, 1821–1826.
Santoro, M., Dathan, N. A., Berlingieri, M. T., Bongarzone, I., Paulin, C., Grieco, M., Pierotti, M. A., Vecchio, G., and Fusco, A. (1994). Molecular characterization of RET/PTC3; a novel rearranged version of the RET proto-oncogene in a human thyroid papillary carcinoma. Oncogene 9, 509–516.
Santoro, M., Melillo, R. M., and Fusco, A. (2006). RET/PTC activation in papillary thyroid carcinoma: European journal of endocrinology prize lecture. Eur. J. Endocrinol. 155, 645–653.
Santoro, M., Melillo, R. M., Grieco, M., Berlingieri, M. T., Vecchio, G., and Fusco, A. (1993). The TRK and RET tyrosine kinase oncogenes cooperate with ras in the neoplastic transformation of a rat thyroid epithelial cell line. Cell Growth Differ. 4, 77–84.
Sariola, H., and Saarma, M. (2003). Novel functions and signalling pathways for GDNF. J. Cell. Sci. 116, 3855–3862.
Scarpino, S., Stoppacciaro, A., Ballerini, F., Marchesi, M., Prat, M., Stella, M. C., Sozzani, S., Allavena, P., Mantovani, A., and Ruco, L. P. (2000). Papillary carcinoma of the thyroid: hepatocyte growth factor (HGF) stimulates tumor cells to release chemokines active in recruiting dendritic cells. Am. J. Pathol. 156, 831–837.
Schlumberger, M., Elisei, R., Bastholt, L., Wirth, L. J., Martins, R. G., Locati, L. D., Jarzab, B., Pacini, F., Daumerie, C., Droz, J. P., Eschenberg, M. J., Sun, Y. N., Juan, T., Stepan, D. E., and Sherman, S. I. (2009). Phase II study of safety and efficacy of motesanib in patients with progressive or symptomatic, advanced or metastatic medullary thyroid cancer. J. Clin. Oncol. 27, 3794–3801.
Schuetz, G., Rosário, M., Grimm, J., Boeckers, T. M., Gundelfinger, E. D., and Birchmeier, W. (2004). The neuronal scaffold protein Shank3 mediates signaling and biological function of the receptor tyrosine kinase Ret in epithelial cells. J. Cell Biol. 167, 945–952.
Schuringa, J. J., Wojtachnio, K., Hagens, W., Vellenga, E., Buys, C. H., Hofstra, R., and Kruijer, W. (2001). MEN2A-RET-induced cellular transformation by activation of STAT3. Oncogene 20, 5350–5358.
Scotton, C. J., Wilson, J. L., Scott, K., Stamp, G., Wilbanks, G. D., Fricker, S., Bridger, G., and Balkwill, F. R. (2002). Multiple actions of the chemokine CXCL12 on epithelial tumor cells in human ovarian cancer. Cancer Res. 62, 5930–5938.
Segal, K., Ben-Bassat, M., Avraham, A., Har-El, G., and Sidi, J. (1985). Hashimoto’s thyroiditis and carcinoma of the thyroid gland. Int. Surg. 70, 205–209.
Segouffin-Cariou, C., and Billaud, M. (2000). Transforming ability of MEN2A-RET requires activation of the phosphatidylinositol 3-kinase/AKT signaling pathway. J. Biol. Chem. 275, 3568–3576.
Serafini, P., De Santo, C., Marigo, I., Cingarlini, S., Dolcetti, L., Gallina, G., Zanovello, P., and Bronte, V. (2004). Derangement of immune responses by myeloid suppressor cells. Cancer Immunol. Immunother. 53, 64–72.
Shimono, Y., Murakami, H., Hasegawa, Y., and Takahashi, M. (2000). RET finger protein is a transcriptional repressor and interacts with enhancer of polycomb that has dual transcriptional functions. J. Biol. Chem. 275, 39411–39419.
Shrivastav, M., De Haro, L. P., and Nickoloff, J. A. (2008). Regulation of DNA double-strand break repair pathway choice. Cell Res. 18, 134–147.
Smanik, P. A., Furminger, T. L., Mazzaferri, E. L., and Jhiang, S. M. (1995). Breakpoint characterization of the ret/PTC oncogene in human papillary thyroid carcinoma. Hum. Mol. Genet. 4, 2313–2318.
Smida, J., Salassidis, K., Hieber, L., Zitzelsberger, H., Kellerer, A. M., Demidchik, E. P., Negele, T., Spelsberg, F., Lengfelder, E., Werner, M., and Bauchinger, M. (1999). Distinct frequency of ret rearrangements in papillary thyroid carcinomas of children and adults from Belarus. Int. J. Cancer 80, 32–38.
Smith, D. P., Houghton, C., and Ponder, B. A. J. (1997). Germline mutation of ret codon 883 in two cases of de novo MEN 2B. Oncogene 15, 1213–1217.
Smith, W. L., and Langenbach, R. (2001). Why there are two cyclooxygenase isozymes. J. Clin. Invest. 107, 1491–1495.
Soares, P., Fonseca, E., Wynford-Thomas, D., and Sobrinho-Simões, M. (1998). Sporadic ret-rearranged papillary carcinoma of the thyroid: a subset of slow growing, less aggressive thyroid neoplasms? J. Pathol. 185, 71–78.
Specht, M. C., Tucker, O. N., Hocever, M., Gonzalez, D., Teng, L., and Fahey TJ, 3. r. d. (2002). Cyclooxygenase-2 expression in thyroid nodules. J. Clin. Endocrinol. Metab. 87, 358–363.
Stratakis, C. A., and Ball, D. W. (2000). A concise genetic and clinical guide to multiple endocrine neoplasias and related syndromes. J. Pediatr. Endocrinol. Metab. 13, 457–465.
Sugg, S. L., Ezzat, S., Rosen, I. B., Freeman, J. L., and Asa, S. L. (1998). Distinct multiple RET/PTC gene rearrangements in multifocal papillary thyroid neoplasia. J. Clin. Endocrinol. Metab. 83, 4116–4122.
Sugg, S. L., Ezzat, S., Zheng, L., Freeman, J. L., Rosen, I. B., and Asa, S. L. (1999). Oncogene profile of papillary thyroid carcinoma. Surgery 125, 46–52.
Sugg, S. L., Zheng, L., Rosen, I. B., Freeman, J. L., Ezzat, S., and Asa, S. L. (1996). ret/PTC-1, -2, and -3 oncogene rearrangements in human thyroid carcinomas: implications for metastatic potential? J. Clin. Endocrinol. Metab. 81, 3360–3365.
Takahashi, K., Eguchi, H., Arihiro, K., Ito, R., Koyama, K., Soda, M., Cologne, J., Hayashi, Y., Nakata, Y., Nakachi, K., and Hamatani, K. (2007). The presence of BRAF point mutation in adult papillary thyroid carcinomas from atomic bomb survivors correlates with radiation dose. Mol. Carcinog. 46, 242–248.
Takahashi, M., Ritz, J., and Cooper, G. M. (1985). Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell 42, 581–588.
Takeuchi, K., Soda, M., Togashi, Y., Suzuki, R., Sakata, S., Hatano, S., Asaka, R., Hamanaka, W., Ninomiya, H., Uehara, H., Lim Choi, Y., Satoh, Y., Okumura, S., Nakagawa, K., Mano, H., and Ishikawa, Y. (2011). RET, ROS1 and ALK fusions in lung cancer. Nat. Med. 18, 378–381.
Tallini, G., and Asa, S. L. (2001). RET oncogene activation in papillary thyroid carcinoma. Adv. Anat. Pathol. 8, 345–354.
Tallini, G., Santoro, M., Helie, M., Carlomagno, F., Salvatore, G., Chiappetta, G., Carcangiu, M. L., and Fusco, A. (1998). RET/PTC oncogene activation defines a subset of papillary thyroid carcinomas lacking evidence of progression to poorly differentiated or undifferentiated tumor phenotypes. Clin. Cancer Res. 4, 287–294.
Tanikawa, N., Ohmiya, Y., Ohkubo, H., Hashimoto, K., Kangawa, K., Kojima, M., Ito, S., and Watanabe, K. (2002). Identification and characterization of a novel type of membrane-associated prostaglandin E synthase. Biochem. Biophys. Res. Commun. 291, 884–889.
Tanioka, T., Nakatani, Y., Semmyo, N., Murakami, M., and Kudo, I. (2000). Molecular identification of cytosolic prostaglandin E2 synthase that is functionally coupled with cyclooxygenase-1 in immediate prostaglandin E2 biosynthesis. J. Biol. Chem. 275, 32775–32782.
Thomas, G. A., Bunnell, H., Cook, H. A., Williams, E. D., Nerovnya, A., Cherstvoy, E. D., Tronko, N. D., Bogdanova, T. I., Chiappetta, G., Viglietto, G., Pentimalli, F., Salvatore, G., Fusco, A., Santoro, M., and Vecchio, G. (1999). High prevalence of RET/PTC rearrangements in Ukrainian and Belarussian post-Chernobyl thyroid papillary carcinomas: a strong correlation between RET/PTC3 and the solid-follicular variant. J. Clin. Endocrinol. Metab. 84, 4232–4238.
Tong, Q., Xing, S., and Jhiang, S. M. (1997). Leucine zipper-mediated dimerization is essential for the PTC1 oncogenic activity. J. Biol. Chem. 272, 9043–9047.
Uchida, M., Enomoto, A., Fukuda, T., Kurokawa, K., Maeda, K., Kodama, Y., Asai, N., Hasegawa, T., Shimono, Y., Jijiwa, M., Ichihara, M., Murakumo, Y., and Takahashi, M. (2006). Dok-4 regulates GDNF-dependent neurite outgrowth through downstream activation of Rap1 and mitogen-activated protein kinase. J. Cell. Sci. 119, 3067–3077.
Usdin, K., and Woodford, K. J. (1995). CGG repeats associated with DNA instability and chromosome fragility form structures that block DNA synthesis in vitro. Nucleic Acids Res. 23, 4202–4209.
Van den Eynde, B., Lethé, B., Van Pel, A., De Plaen, E., and Boon, T. (1991). The gene coding for a major tumor rejection antigen of tumor P815 is identical to the normal gene of syngeneic DBA/2 mice. J. Exp. Med. 173, 1373–1384.
Viglietto, G., Chiappetta, G., Martinez-Tello, F. J., Fukunaga, F. H., Tallini, G., Rigopoulou, D., Visconti, R., Mastro, A., Santoro, M., and Fusco, A. (1995). RET/PTC oncogene activation is an early event in thyroid carcinogenesis. Oncogene 11, 1207–1210.
Visconti, R., Cerutti, J., Battista, S., Fedele, M., Trapasso, F., Zeki, K., Miano, M. P., de Nigris, F., Casalino, L., Curcio, F., Santoro, M., and Fusco, A. (1997). Expression of the neoplastic phenotype by human thyroid carcinoma cell lines requires NFkappaB p65 protein expression. Oncogene 15, 1987–1994.
Vitagliano, D., Portella, G., Troncone, G., Francione, A., Rossi, C., Bruno, A., Giorgini, A., Coluzzi, S., Nappi, T. C., Rothstein, J. L., Pasquinelli, R., Chiappetta, G., Terracciano, D., Macchia, V., Melillo, R. M., Fusco, A., and Santoro, M. (2006). Thyroid targeting of the N-ras(Gln61Lys) oncogene in transgenic mice results in follicular tumors that progress to poorly differentiated carcinomas. Oncogene 25, 5467–5474.
Weber, G. F. (2001). The metastasis gene osteopontin: a candidate target for cancer therapy. Biochim. Biophys. Acta 1552, 61–85.
Wells, S. A. Jr., Gosnell, J. E., Gagel, R. F., Moley, J., Pfister, D., Sosa, J. A., Skinner, M., Krebs, A., Vasselli, J., and Schlumberger, M. (2010). Vandetanib for the treatment of patients with locally advanced or metastatic hereditary medullary thyroid cancer. J. Clin. Oncol. 28, 767–772.
Wells, S. A. Jr., Robinson, B. G., Gagel, R. F., Dralle, H., Fagin, J. A., Santoro, M., Baudin, E., Elisei, R., Jarzab, B., Vasselli, J. R., Read, J., Langmuir, P., Ryan, A. J., and Schlumberger, M. J. (2012). Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J. Clin. Oncol. 30, 134–141.
Wells, S. A. Jr., and Santoro, M. (2009). Targeting the RET pathway in thyroid cancer. Clin. Cancer Res. 15, 7119–7123.
Weterings, E., and Chen, D. J. (2008). The endless tale of non-homologous end-joining. Cell Res. 18, 114–124.
Williams, D. (2002). Cancer after nuclear fallout: lessons from the Chernobyl accident. Nat. Rev. Cancer 2, 543–549.
Winegar, R. A., Lutze, L. H., Rufer, J. T., and Morgan, W. F. (1992). Spectrum of mutations produced by specific types of restriction enzyme-induced double-strand breaks. Mutagenesis 7, 439–445.
Winship, T., and Rosvoll, R. V. (1970). Cancer of the thyroid in children. Proc. Natl. Cancer Conf. 6, 677–681.
Wixted, J. H., Rothstein, J. L., and Eisenlohr, L. C. (2011). Identification of functionally distinct TRAF proinflammatory and PI3K/MEK transforming activities emanating from the RET/PTC fusion oncoprotein. J. Biol. Chem. 287, 3691–3703.
Wu, Z. H., Shi, Y., Tibbetts, R. S., and Miyamoto, S. (2006). Molecular linkage between the kinase ATM and NF-kappaB signaling in response to genotoxic stimuli. Science 311, 1141–1146.
Xing, M. (2010). Prognostic utility of BRAF mutation in papillary thyroid cancer. Mol. Cell. Endocrinol. 321, 86–93.
Yates, B. L., and Morgan, W. F. (1993). Nonhomologous DNA end rejoining in chromosomal aberration formation. Mutat. Res. 285, 53–60.
Yoshimatsu, K., Golijanin, D., Paty, P. B., Soslow, R. A., Jakobsson, P. J., DeLellis, R. A., Subbaramaiah, K., and Dannenberg, A. J. (2001). Inducible microsomal prostaglandin E synthase is overexpressed in colorectal adenomas and cancer. Clin. Cancer Res. 7, 3971–3976.
Yu, P., Lee, Y., Liu, W., Krausz, T., Chong, A., Schreiber, H., and Fu, Y. X. (2005). Intratumor depletion of CD4+ cells unmasks tumor immunogenicity leading to the rejection of late-stage tumors. J. Exp. Med. 201, 779–791.
Yunis, J. J., and Soreng, A. L. (1984). Constitutive fragile sites and cancer. Science 226, 1199–1204.
Zhang, H., and Freudenreich, C. H. (2007). An AT-rich sequence in human common fragile site FRA16D causes fork stalling and chromosome breakage in S. cerevisiae. Mol. Cell 27, 367–379.
Zhong, S., Delva, L., Rachez, C., Cenciarelli, C., Gandini, D., Zhang, H., Kalantry, S., Freedman, L. P., and Pandolfi, P. P. (1999). A RA-dependent, tumour-growth suppressive transcription complex is the target of the PML-RARalpha and T18 oncoproteins. Nat. Genet. 23, 287–295.
Zhu, Z., Ciampi, R., Nikiforova, M. N., Gandhi, M., and Nikiforov, Y. E. (2006). Prevalence of RET/PTC rearrangements in thyroid papillary carcinomas: effects of the detection methods and genetic heterogeneity. J. Clin. Endocrinol. Metab. 91, 3603–3610.
Zlotorynski, E., Rahat, A., Skaug, J., Ben-Porat, N., Ozeri, E., Hershberg, R., Levi, A., Scherer, S. W., Margalit, H., and Kerem, B. (2003). Molecular basis for expression of common and rare fragile sites. Mol. Cell Biol. 23, 7143–7151.
Zou, M., Shi, Y., and Farid, N. R. (1994). Low rate of ret proto-oncogene activation (PTC/retTPC) in papillary thyroid carcinomas from Saudi Arabia. Cancer 73, 176–180.
Keywords: thyroid carcinoma, papillary thyroid carcinoma, RET/PTC
Citation: Menicali E, Moretti S, Voce P, Romagnoli S, Avenia N and Puxeddu E (2012) Intracellular signal transduction and modification of the tumor microenvironment induced by RET/PTCs in papillary thyroid carcinoma. Front. Endocrin. 3:67. doi: 10.3389/fendo.2012.00067
Received: 05 February 2012; Paper pending published: 25 February 2012;
Accepted: 30 April 2012; Published online: 22 May 2012.
Edited by:
Carmelo Nucera, Beth Israel Deaconess Medical Center and Harvard Medical School, USAReviewed by:
Marco Alessandro Pierotti, Fondazione IRCCS IStituto Nazionale dei Tumori – Milano, ItalyPaula Soares, University of Porto, Portugal
Copyright: © 2012 Menicali, Moretti, Voce, Romagnoli, Avenia and Puxeddu. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Efisio Puxeddu, Dipartimento di Medicina, Sezione MIENDO, Via Enrico dal Pozzo – Padiglione X, 06126 Perugia, Italy. e-mail:ZWZpc2lvcHV4ZWRkdUBob3RtYWlsLmNvbQ==