 Sylvan C. Baca1,2,3
Sylvan C. Baca1,2,3 Levi A. Garraway1,2,3,4*
Levi A. Garraway1,2,3,4*- 1 Harvard Medical School, Boston, MA, USA
- 2 Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA, USA
- 3 The Broad Institute of MIT and Harvard, Cambridge, MA, USA
- 4 Center for Cancer Genome Discovery, Dana-Farber Cancer Institute, Boston, MA, USA
Prostate cancer is a common malignancy in men, with a markedly variable clinical course. Somatic alterations in DNA drive the growth of prostate cancers and may underlie the behavior of aggressive versus indolent tumors. The accelerating application of genomic technologies over the last two decades has identified mutations that drive prostate cancer formation, progression, and therapeutic resistance. Here, we discuss exemplary somatic mutations in prostate cancer, and highlight mutated cellular pathways with biological and possible therapeutic importance. Examples include mutated genes involved in androgen signaling, cell cycle regulation, signal transduction, and development. Some genetic alterations may also predict the clinical course of disease or response to therapy, although the molecular heterogeneity of prostate tumors poses challenges to genomic biomarker identification. The widespread application of massively parallel sequencing technology to the analysis of prostate cancer genomes should continue to advance both discovery-oriented and diagnostic avenues.
Prostate cancer is the second most common cancer in men worldwide and causes over 250,000 deaths each year (Jemal et al., 2011). However, many men with prostate cancer do not develop symptomatic disease. Overtreatment of indolent tumors may result in significant morbidity. A deeper understanding of the genomic differences between lethal and indolent prostate cancer, as well as elucidation of “druggable” effectors dysregulated by genetic alterations, should improve patient stratification and speed the development of targeted therapies.
With the advance of genome characterization technologies over the last two decades, the somatic alterations that may drive prostate tumors have come into sharper focus. In this mini-review, we survey the field of prostate cancer genomics, highlight recent findings, and discuss prospects for future research.
The Mutational Spectrum of Prostate Cancer
All categories of DNA sequence alterations contribute to prostate tumorigenesis, including point mutations, small insertions or deletions, copy number changes, and chromosomal rearrangements (Figure 1). An overview of each category of alteration, and its contribution to prostate cancer biology, is presented below.
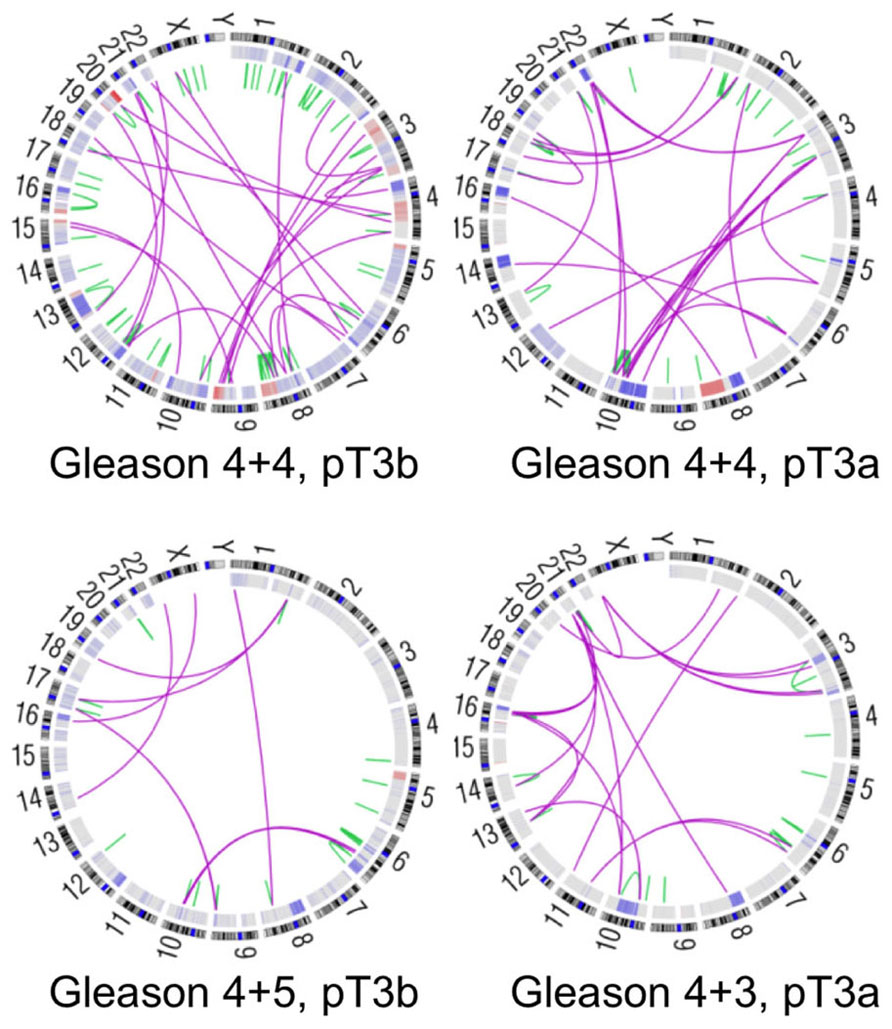
Figure 1. Genomic alterations in four high-risk prostate cancers. Circos plots depicting genomic rearrangements and copy number alterations in four prostate tumors analyzed by whole-genome sequencing (unpublished data). Green and pink lines designate intrachromosomal and interchromosomal rearrangements, respectively. Somatic copy number alterations are indicated in red (amplification) and blue (deletion) in the inner rings. Gleason scores indicate the two most prevalent histologic grades in each tumor. Pathological stage is noted as well, where pT3 indicates locally invasive disease.
Somatic Copy Number Alteration
Most prostate cancers exhibit somatic copy number alterations (SCNAs), with genomic deletions outnumbering amplifications in early stages of disease (Visakorpi et al., 1995). Early studies relied on cytogenetics, fluorescence in situ hybridization and molecular genetic approaches to map candidate cancer genes to regions of SCNA (Brothman et al., 1999). In recent years, comparative genomic hybridization and high-density single nucleotide polymorphism arrays have allowed high-resolution genome-wide analysis of SCNAs. Statistical analyses of genome-wide copy number data have narrowed the boundaries of recurrent alterations considerably and have pinpointed novel cancer genes in these regions (Beroukhim et al., 2007; Taylor et al., 2010; Robbins et al., 2011).
The extent of SCNA is generally modest in pre-cancerous prostatic intraepithelial neoplasia (PIN), but becomes increasingly prevalent along the spectrum from localized adenocarcinoma to metastatic disease (Zitzelsberger et al., 2001). Particular recurrent SCNAs are enriched in advanced tumors. For example, tumors that fail androgen ablation therapy show frequent amplification of chromosomes 7, 8q and X (Visakorpi et al., 1995; Alers et al., 2000; Holcomb et al., 2009). Animal models of prostate cancer indicate that genes in these regions, such as the androgen receptor gene (X) and the MYC proto-oncogene (8q), contribute to cancer progression (discussed in detail below).
Point Mutations and Small Insertions–Deletions
Relative to structural alterations, recurrent point mutations are less common in primary prostate cancers (Kan et al., 2010). Primary tumors generally harbor one to two somatic variants per million base pairs – far fewer than known carcinogen-driven tumors such as lung cancer or melanoma, but comparable to breast, renal, or ovarian cancers (Greenman et al., 2007; Pleasance et al., 2010a, b; Berger et al., 2011). While most of these mutations confer no proliferative advantage, a handful of recurrent oncogenic mutations have been defined.
The reported prevalence of mutations in several known cancer genes varies widely and depends on tumor purity, stage, histological grade, and exposure to treatments. For example, RB1, TP53, and PTEN are preferentially mutated in locally advanced or metastatic tumors (Eastham et al., 1995; Tricoli et al., 1996; Cairns et al., 1997) while the androgen receptor is mutated only in metastatic or treatment-resistant disease (Linja and Visakorpi, 2004; Taylor et al., 2010). Ethnicity may influence mutation prevalence as well. Activating mutations in KRAS and BRAF occur in ~10% of Asian patients but are rare in Caucasian men, perhaps reflecting different environmental etiology or biological behavior of cancers in these populations (Watanabe et al., 1994; Konishi et al., 1997; Cho et al., 2006).
Defects in DNA mismatch repair (MMR) machinery have been reported in prostate cancers and may accelerate progression to castration-independence (Dahiya et al., 1997; Chen et al., 2001). Large-scale sequencing studies have recently identified a subset of tumors with markedly elevated rates of point mutation (Taylor et al., 2010; Kumar et al., 2011; unpublished data). It remains to be determined whether the high levels of mutation in these tumors are caused by MMR deficiency, and whether hyper-mutated cancers display more clinically aggressive behavior.
Structural Rearrangements
The discovery of ETS family gene fusions in roughly half of prostate cancers heralded a novel class of alterations in epithelial malignancies as a whole (Tomlins et al., 2005). The most common and prototypical ETS fusion places the oncogenic ERG transcription factor under control of the androgen-regulated TMPRSS2 gene, leading to high expression in the prostate epithelium. Subsequent research has identified a host of similar oncogenic fusions, where a proto-oncogene is adjoined to a highly active promoter (Tomlins et al., 2007; Kumar-Sinha et al., 2008; Palanisamy et al., 2010). Since mutation or amplification of oncogenes is less common in early-stage prostate cancer, genomic rearrangements may comprise an important means of cancer gene dysregulation in nascent tumors.
Complete sequencing of prostate cancer genomes has provided further insight into chromosomal rearrangements in prostate cancer. Primary tumors may harbor an average of approximately 100 rearrangements, including translocations, deletions, insertions, and inversions (Figure 1; Berger et al., 2011). Some tumors display “closed chains” of balanced rearrangements, in which multiple DNA breaks occur throughout the genome and the resulting fragments are shuffled and rejoined to one another. These rearrangements may arise when the affected genetic loci are physically proximal to each other, possibly due to co-regulation by transcriptional machinery or nuclear co-localization in open- or closed-chromatin compartments (Osborne et al., 2004; Berger et al., 2011). Consistent with this hypothesis, androgen stimulation can induce physical co-localization of TMPRSS2 and ERG and permit fusion of these genes de novo via a topoisomerase 2B-mediated mechanism (Haffner et al., 2010).
The diverse categories of genomic aberrations underscore the need for comprehensive genomic analyses both to understand tumor biology and to direct targeted therapies on a genotype-specific basis (Roychowdhury et al., 2011).
Cellular Pathways Dysregulated by Recurrent Prostate Cancer Genomic Alterations
Genomic alterations in prostate cancer can increasingly be conceptualized in terms of the molecular processes and pathways on which they impinge (Taylor et al., 2010). Mutations in prostate cancer may affect signal transduction pathways that regulate growth and proliferation, as well as genes involved in the normal development of the prostate. Below, we highlight several themes and pathways that provide a framework for understanding genomic alterations in prostate cancer.
PI3K and MAPK Signaling
The phosphoinositide 3-kinase (PI3K) pathway is a central mediator of cellular proliferation and growth that is aberrantly activated in prostate cancer. In response to pro-proliferative signals, PI3K catalyzes the formation of phosphatidylinositol (3,4,5)-triphosphate (PIP3), which recruits Akt to the plasma membrane. Upon phospho-activation at the plasma membrane, Akt phosphorylates a wide array of substrates that promote proliferation and cell survival.
Prostate tumors achieve activation of PI3K signaling most frequently via inactivation of the tumor-suppressor gene PTEN (Figure 2). PTEN encodes a lipid–protein phosphatase that counteracts signaling by PI3K via dephosphorylation of PIP3. Loss of heterozygosity at the PTEN locus is found in up to 70% of primary prostate cancers and inactivating mutations occur in 5–10% (Cairns et al., 1997; Gray et al., 1998; Barbieri et al., 2012). Inactivation of PTEN is enriched in advanced tumors and correlates with decreased cancer-specific survival (McMenamin et al., 1999; Sircar et al., 2009). PTEN loss in the mouse prostate collaborates with other tumor-promoting events such as loss of TP53 and overexpression of c-Myc or ERG (Chen et al., 2005; King et al., 2009; Kim et al., 2012).
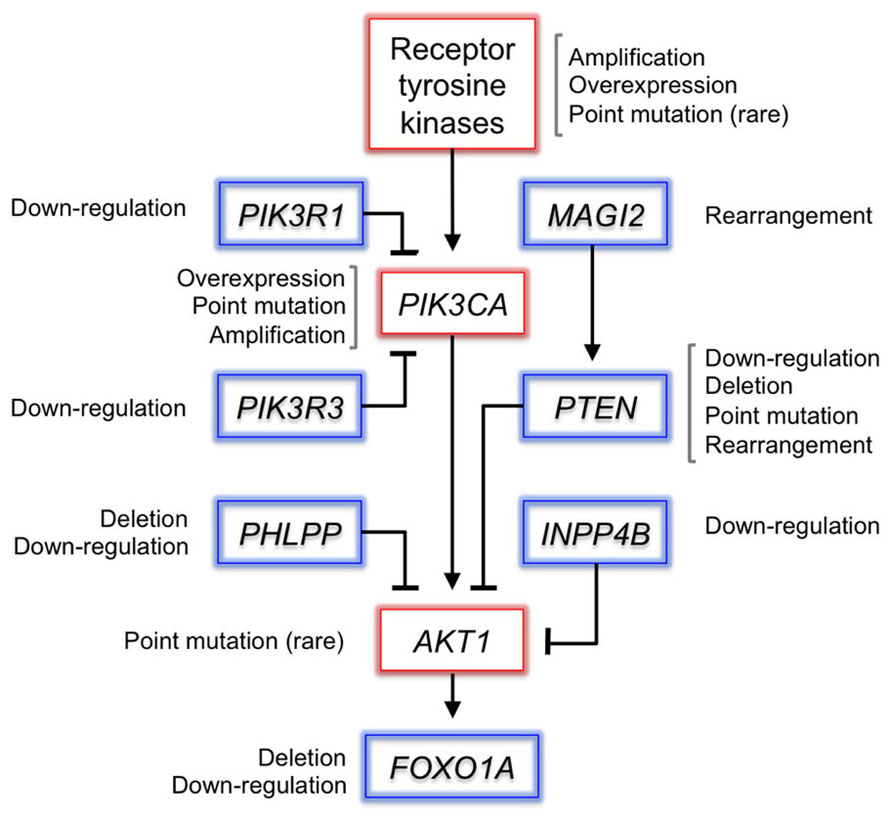
Figure 2. Somatic alterations in the PI3K pathway in prostate cancer. Selected genes in the PI3K pathway are depicted, alongside the mechanisms by which they are altered in prostate cancer. Putative proto-oncogenes are boxed in red and tumor-suppressor genes in blue. PI3K signaling is frequently activated by deletion of PTEN. PHLPP1 encodes a phosphatase that dephosphorylates activated Akt, and is frequently co-deleted with PTEN in metastatic tumors (Chen et al., 2011). Genomic rearrangements disrupt MAGI2, which encodes a scaffolding protein that stabilizes PTEN (Wu et al., 2000; Berger et al., 2011). Recurrent deletions inactivate the FOXO1A gene, which encodes a transcription factor substrate of Akt that mediates PI3K signaling. Although rare, oncogenic mutations in the receptor tyrosine kinase EGFR or AKT1 may activate the pathway upstream or downstream of PI3K (Cai et al., 2008; Boormans et al., 2010). The expression of most pathway members is dysregulated at the transcript level as well.
Amplification of PIK3CA, which encodes the catalytic subunit of PI3K, occurs in 13–39% of primary tumors and 50% of castration-resistant tumors (Edwards et al., 2003; Sun et al., 2009; Agell et al., 2011). Activating mutations have been observed in ~5% of primary tumors (Sun et al., 2009; Barbieri et al., 2012). PIK3CA activation and PTEN loss tend to be mutually exclusive, which suggests functional redundancy – although larger sample sizes are needed to assess this relationship robustly (Sun et al., 2009). Interestingly, PTEN loss and PIK3CA activation co-occur in endometrial cancer, suggesting that multiple lesions are required to activate the pathway, or that these events engage disparate oncogenic mechanisms (Oda et al., 2005). In support of the latter possibility, oncogenic Akt-independent signaling downstream of mutant PIK3CA has been observed in both primary tumors and cancer cell lines (Vasudevan et al., 2009).
The PI3K pathway may be activated by genomic alterations at additional pathway nodes and dysregulated expression of constituent genes (Figure 2; Dong et al., 2006; Cai et al., 2008; Taylor et al., 2010). Determining whether these lesions predict sensitivity or resistance to PI3K pathway inhibitors has become an active area of translational research.
The mitogen-activated protein kinase (MAPK) pathway also plays a role in prostate cancer pathogenesis, especially in advanced and castration-resistant tumors. MAPK pathway activation is associated with higher tumor stage and grade and recurrent disease (Gioeli et al., 1999). In the setting of castration resistance, PI3K and MAPK signaling are often coordinately dysregulated (Gao et al., 2006; Kinkade et al., 2008). Evidence for collaboration between these pathways continues to emerge. For instance, PTEN-induced senescence may be overcome by up-regulation of MAPK signaling induced by overexpression of HER2 (Ahmad et al., 2011).
Up-regulation of RAS family members, RAF1 and BRAF, or down-regulation of SPRY1 or SPRY2 genes, are common and enriched in prostate cancer metastases (Kwabi-Addo et al., 2004; McKie et al., 2005; Taylor et al., 2010). In some cases, expression of RAS, RAF1, and BRAF is activated by oncogenic fusions with highly expressed promoters (Palanisamy et al., 2010; Wang et al., 2011). Repression of the RAS-GAP gene DAB2IP by EZH2 may activate MAPK signaling and drive progression and metastasis (Min et al., 2010). Defining the relevant mechanisms of pathway activation in greater detail will likely inform strategies for targeting castration-resistant tumors.
Cell Cycle Regulatory Genes
Several cell cycle regulatory genes are disrupted in prostate cancer. Inactivation of cell cycle inhibitors appears to be required to avoid senescence induced by oncogenic signaling and possibly to bypass androgen-regulation of growth in metastatic or castration-resistant tumors.
Two critical cell cycle regulatory genes, TP53 and RB1, are commonly deleted or mutated in metastatic tumors (Bookstein et al., 1993; Heidenberg et al., 1995; Tricoli et al., 1996; Hyytinen et al., 1999). p53 activates expression of the p21WAF1 cyclin-dependent kinase inhibitor, and the Rb protein regulates transition from the G1 to S cell cycle phase. RB1 inactivation is common in castration-resistant tumors (Holcomb et al., 2009; Sharma et al., 2010). Likewise, inactivation of p53 is necessary to bypass cellular senescence mechanisms that are activated upon loss of PTEN (Chen et al., 2005).
Another key cell cycle regulator, CDKN1B, encodes the p27Kip1 cyclin-dependent kinase inhibitor, and resides within the 12p13 chromosomal region that is frequently deleted. Low p27Kip1 expression correlates with poor pathological prognostic markers (Vis et al., 2000; Dreher et al., 2004). Amplification of SKP2, which encodes a ubiquitin ligase that targets p27Kip1 for proteasomal degradation, may also serve to inactivate p27Kip1 (Taylor et al., 2010; Robbins et al., 2011). Disruption of CDKN1B promotes prostate cancer coordinately with hemizygous deletion of PTEN, suggesting an interaction between p27Kip1 and the PI3K pathway (Di Cristofano et al., 2001). Likewise, p27Kip1 induces senescence in PIN lesions driven by Akt1 in mice (Majumder et al., 2008).
Developmental and Androgen-Regulated Genes
Normal developmental and androgen-regulated processes appear to be co-opted during oncogenesis in the prostate. Several genes that participate in the development and differentiation of the prostate epithelium are dysregulated in prostate cancer (Prins and Putz, 2008).
The androgen receptor regulates cellular proliferation and differentiation in response to hormonal signals in the prostate epithelium. While androgen receptor is not mutated in primary tumors, the AR gene is frequently mutated or amplified in metastatic and castration-resistant disease (Visakorpi et al., 1995; Koivisto et al., 1997; Linja and Visakorpi, 2004). AR point mutations allow promiscuous activation by steroid hormones such as estrogens, progestins, glucocorticoids, and androgen antagonists in 10–30% of refractory cases (Gaddipati et al., 1994; Linja and Visakorpi, 2004). Alteration of androgen signaling may participate in localized disease as well: several AR-interacting genes are mutated or dysregulated in primary tumors, including NCOR2, NRIP1, TNK2, and EP300 (Taylor et al., 2010).
NKX3-1 encodes a prostate-specific transcription factor that is required for normal development of the prostate and is deleted or down-regulated in up to 90% of prostate cancers (Emmert-Buck et al., 1995; Vocke et al., 1996; Asatiani et al., 2005). Inactivation via hemizygous deletion of chromosome 8p appears to occur early and can be observed in PIN lesions (Emmert-Buck et al., 1995; Asatiani et al., 2005). NKX3-1-deficient mice exhibit defective branching morphogenesis of the prostate gland and develop PIN-like lesions with age (Bhatia-Gaur et al., 1999). In addition, NKX3-1 appears to protect the differentiated prostate epithelium from oxidative DNA damage (Ouyang et al., 2005; Bowen and Gelmann, 2010). Therefore, loss of NKX3-1 may both disrupt terminal differentiation and foster the mutational inactivation of collaborating cancer genes such as PTEN (Kim et al., 2002).
The Wnt pathway regulates embryological development, and its contribution to prostate cancer is becoming increasingly recognized (Yardy and Brewster, 2005). Key pathway genes including APC, AXIN1 and the β-catenin gene CTNNB1 may be mutated at low frequency (Voeller et al., 1998; Chesire et al., 2000; Yardy et al., 2009). APC undergoes LOH in roughly 20% of primary cancers and promoter CpG methylation in up to 90% (Brewster et al., 1994; Phillips et al., 1994; Yegnasubramanian et al., 2004). β-Catenin may promote proliferation through co-activation of AR-mediated transcription (Truica et al., 2000; Cronauer et al., 2005). Additional mutations in Wnt pathway genes were recently documented in the progression to castration-resistant disease (Kumar et al., 2011). More pairs of pre- and post-relapse samples should be analyzed to clarify the importance of this pathway in refractory disease.
Genomic Heterogeneity of Prostate Cancer
Prostate cancer is a clinically and genetically heterogeneous disease. Independent cancerous foci with distinct morphological features often coexist in a single prostate. The course of disease also varies widely: some cancers remain indolent for decades while others rapidly progress to lethality. Distinct molecular features appear to underlie the clinical and histological differences. Identifying genomic determinants of aggressive disease might improve experimental modeling and stratification of patients with intermediate-risk prostate cancer.
Prostate cancer may arise in multiple foci from independent precursor cells that are driven to neoplastic transformation by carcinogenic exposures or genetic predisposition (Andreoiu and Cheng, 2010). The presence of genomic lesions can vary between foci, including TMPRSS2-ERG fusion, MYC amplification, and TP53 mutation (Mirchandani et al., 1995; Jenkins et al., 1997; Mehra et al., 2007). Multiple distinct clones can be identified in a single biopsy (Ruiz et al., 2011), but most metastatic prostate cancers appear to originate from a single clone within a primary tumor (Qian et al., 1995; Holcomb et al., 2009; Liu et al., 2009). Among other lesions, subclonal TP53 mutations may define cells in the primary tumor with metastatic potential (Mirchandani et al., 1995; Navone et al., 1999). Intratumoral heterogeneity complicates efforts to define prognostic mutations or expression signatures from primary tumors, because the subclone within a primary tumor that gives rise to metastatic disease must be adequately sampled (Sboner et al., 2010).
Despite the challenges posed by tumor heterogeneity, expression signatures have been proposed that delineate histologically aggressive disease or predict outcome independently of clinical variables (Singh et al., 2002; Glinsky et al., 2004; True et al., 2006; Febbo, 2009). However, the overlap between signatures from independent studies is moderate. Some genomic alterations appear to have prognostic value as well. The TMPRSS2-ERG fusion, MYC amplification, and PTEN or TP53 deletion predict cancer-specific death in at least some patient cohorts (Sato et al., 1999; Demichelis et al., 2007; Sircar et al., 2009). In some cases, a mutational signature may underlie expression-based sub-classifications (Lapointe et al., 2004, 2007).
Prostate Cancer in the ERA of Genomics-Driven Medicine
High-throughput genomic profiling has advanced the understanding, prognostication, and treatment of several tumor types. For example, identification of mutations in BAP1 in uveal melanoma (Harbour et al., 2010) or IDH1 in glioblastoma and acute myeloid leukemia (Parsons et al., 2008; Mardis et al., 2009) demonstrated the power of genome sequencing to pinpoint novel cancer-driving mutations. Risk-predictive transcriptional signatures have improved prognostication for patients with breast cancer (van’t Veer et al., 2002), while the mutational status of EGFR in non-small cell lung cancer predicts clinical response to inhibitors of this kinase (Paez et al., 2004). Prostate cancer may be similarly ripe for discovery of novel cancer genes and biomarkers as well, since genomic characterization of large cohorts of aggressive tumors has only recently become feasible.
Indeed, whole-exome sequencing of over 100 primary prostate tumor–normal pairs revealed that the ubiquitin ligase complex subunit gene SPOP is among the most frequently mutated genes in primary tumors, though its role in cancer was heretofore unrecognized (Barbieri et al., 2012). This study also identified novel recurrent mutations in the fork-head transcription factor gene FOXA1 and mediator complex gene MED12. Experimental study will be required to determine whether these mutations engage known molecular pathways relevant to prostate cancer or reflect novel mechanisms of oncogenesis.
Several hurdles must be overcome for prostate cancer genomics to impact the clinical management of this disease. For instance, biopsies produce scarce material for clinical genotyping and may not fully capture the relevant molecular heterogeneity within a tumor. Expression signatures have not yet demonstrated sufficient prognostic value to merit widespread use. In addition, recurrent genomic lesions identified thus far are largely not considered “druggable.”
These challenges can likely be surmounted by new approaches. For example, genomic characterization may identify opportunities to leverage synthetic lethality by inhibiting targets that are essential in the setting of a particular mutation, such as poly (ADP-ribose) polymerase in ETS-fusion positive prostate cancer (Brenner et al., 2011). The analysis of multiple samples from a primary tumor and perhaps from circulating tumor cells may allow aggressive tumor subclones to be identified. Ultimately, new paradigms for clinical trials may be required that incorporate cancer genomic information. In spite of these challenges, genomic profiling is likely to play an expanding role in the biological study of prostate cancer and ultimately in the clinical management of this malignancy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Agell, L., Hernandez, S., Salido, M., De Muga, S., Juanpere, N., Arumi-Uria, M., Menendez, S., Lorenzo, M., Lorente, J. A., Serrano, S., and Lloreta, J. (2011). PI3K signaling pathway is activated by PIK3CA mRNA overexpression and copy gain in prostate tumors, but PIK3CA, BRAF, KRAS and AKT1 mutations are infrequent events. Mod. Pathol. 24, 443–452.
Ahmad, I., Patel, R., Singh, L. B., Nixon, C., Seywright, M., Barnetson, R. J., Brunton, V. G., Muller, W. J., Edwards, J., Sansom, O. J., and Leung, H. Y. (2011). HER2 overcomes PTEN (loss)-induced senescence to cause aggressive prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 108, 16392–16397.
Alers, J. C., Rochat, J., Krijtenburg, P. J., Hop, W. C., Kranse, R., Rosenberg, C., Tanke, H. J., Schroder, F. H., and Van Dekken, H. (2000). Identification of genetic markers for prostatic cancer progression. Lab. Invest. 80, 931–942.
Andreoiu, M., and Cheng, L. (2010). Multifocal prostate cancer: biologic, prognostic, and therapeutic implications. Hum. Pathol. 41, 781–793.
Asatiani, E., Huang, W. X., Wang, A., Rodriguez Ortner, E., Cavalli, L. R., Haddad, B. R., and Gelmann, E. P. (2005). Deletion, methylation, and expression of the NKX3.1 suppressor gene in primary human prostate cancer. Cancer Res. 65, 1164–1173.
Barbieri, C. E., Baca, S. C., Lawrence, M. S., Demichelis, F., Blattner, M., Theurillat, J.-P., White, T. A., Stojanov, P., Van Allen, E., Stransky, N., Nickerson, E., Chae, S.-S., Boysen, G., Auclair, D., Onofrio, R. C., Park, K., Kitabayashi, N., MacDonald, T. Y., Sheikh, K., Vuong, T., Guiducci, C., Cibulskis, K., Sivachenko, A., Carter, S. L., Saksena, G., Voet, D., Hussain, W. M., Ramos, A. H., Winckler, W., Redman, M. C., Ardlie, K., Tewari, A. K., Mosquera, J. M., Rupp, N., Wild, P. J., Moch, H., Morrissey, C., Nelson, P. S., Kantoff, P. W., Gabriel, S. B., Golub, T. R., Meyerson, M., Lander, E. S., Getz, G., Rubin, M. A., and Garraway, L. A. (2012). Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. (in press).
Berger, M. F., Lawrence, M. S., Demichelis, F., Drier, Y., Cibulskis, K., Sivachenko, A. Y., Sboner, A., Esgueva, R., Pflueger, D., Sougnez, C., Onofrio, R., Carter, S. L., Park, K., Habegger, L., Ambrogio, L., Fennell, T., Parkin, M., Saksena, G., Voet, D., Ramos, A. H., Pugh, T. J., Wilkinson, J., Fisher, S., Winckler, W., Mahan, S., Ardlie, K., Baldwin, J., Simons, J. W., Kitabayashi, N., Macdonald, T. Y., Kantoff, P. W., Chin, L., Gabriel, S. B., Gerstein, M. B., Golub, T. R., Meyerson, M., Tewari, A., Lander, E. S., Getz, G., Rubin, M. A., and Garraway, L. A. (2011). The genomic complexity of primary human prostate cancer. Nature 470, 214–220.
Beroukhim, R., Getz, G., Nghiemphu, L., Barretina, J., Hsueh, T., Linhart, D., Vivanco, I., Lee, J. C., Huang, J. H., Alexander, S., Du, J., Kau, T., Thomas, R. K., Shah, K., Soto, H., Perner, S., Prensner, J., Debiasi, R. M., Demichelis, F., Hatton, C., Rubin, M. A., Garraway, L. A., Nelson, S. F., Liau, L., Mischel, P. S., Cloughesy, T. F., Meyerson, M., Golub, T. A., Lander, E. S., Mellinghoff, I. K., and Sellers, W. R. (2007). Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc. Natl. Acad. Sci. U.S.A. 104, 20007–20012.
Bhatia-Gaur, R., Donjacour, A. A., Sciavolino, P. J., Kim, M., Desai, N., Young, P., Norton, C. R., Gridley, T., Cardiff, R. D., Cunha, G. R., Abate-Shen, C., and Shen, M. M. (1999). Roles for Nkx3.1 in prostate development and cancer. Genes Dev. 13, 966–977.
Bookstein, R., Macgrogan, D., Hilsenbeck, S. G., Sharkey, F., and Allred, D. C. (1993). p53 is mutated in a subset of advanced-stage prostate cancers. Cancer Res. 53, 3369–3373.
Boormans, J. L., Korsten, H., Ziel-Van Der Made, A. C., Van Leenders, G. J., Verhagen, P. C., and Trapman, J. (2010). E17K substitution in AKT1 in prostate cancer. Br. J. Cancer 102, 1491–1494.
Bowen, C., and Gelmann, E. P. (2010). NKX3.1 activates cellular response to DNA damage. Cancer Res. 70, 3089–3097.
Brenner, J. C., Ateeq, B., Li, Y., Yocum, A. K., Cao, Q., Asangani, I. A., Patel, S., Wang, X., Liang, H., Yu, J., Palanisamy, N., Siddiqui, J., Yan, W., Cao, X., Mehra, R., Sabolch, A., Basrur, V., Lonigro, R. J., Yang, J., Tomlins, S. A., Maher, C. A., Elenitoba-Johnson, K. S., Hussain, M., Navone, N. M., Pienta, K. J., Varambally, S., Feng, F. Y., and Chinnaiyan, A. M. (2011). Mechanistic rationale for inhibition of poly(ADP-ribose) polymerase in ETS gene fusion-positive prostate cancer. Cancer Cell 19, 664–678.
Brewster, S. F., Browne, S., and Brown, K. W. (1994). Somatic allelic loss at the DCC, APC, nm23-H1 and p53 tumor suppressor gene loci in human prostatic carcinoma. J. Urol. 151, 1073–1077.
Brothman, A. R., Maxwell, T. M., Cui, J., Deubler, D. A., and Zhu, X. L. (1999). Chromosomal clues to the development of prostate tumors. Prostate 38, 303–312.
Cai, C. Q., Peng, Y., Buckley, M. T., Wei, J., Chen, F., Liebes, L., Gerald, W. L., Pincus, M. R., Osman, I., and Lee, P. (2008). Epidermal growth factor receptor activation in prostate cancer by three novel missense mutations. Oncogene 27, 3201–3210.
Cairns, P., Okami, K., Halachmi, S., Halachmi, N., Esteller, M., Herman, J. G., Jen, J., Isaacs, W. B., Bova, G. S., and Sidransky, D. (1997). Frequent inactivation of PTEN/MMAC1 in primary prostate cancer. Cancer Res. 57, 4997–5000.
Chen, M., Pratt, C. P., Zeeman, M. E., Schultz, N., Taylor, B. S., O’Neill, A., Castillo-Martin, M., Nowak, D. G., Naguib, A., Grace, D. M., Murn, J., Navin, N., Atwal, G. S., Sander, C., Gerald, W. L., Cordon-Cardo, C., Newton, A. C., Carver, B. S., and Trotman, L. C. (2011). Identification of PHLPP1 as a tumor suppressor reveals the role of feedback activation in PTEN-mutant prostate cancer progression. Cancer Cell 20, 173–186.
Chen, Y., Wang, J., Fraig, M. M., Metcalf, J., Turner, W. R., Bissada, N. K., Watson, D. K., and Schweinfest, C. W. (2001). Defects of DNA mismatch repair in human prostate cancer. Cancer Res. 61, 4112–4121.
Chen, Z., Trotman, L. C., Shaffer, D., Lin, H. K., Dotan, Z. A., Niki, M., Koutcher, J. A., Scher, H. I., Ludwig, T., Gerald, W., Cordon-Cardo, C., and Pandolfi, P. P. (2005). Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 436, 725–730.
Chesire, D. R., Ewing, C. M., Sauvageot, J., Bova, G. S., and Isaacs, W. B. (2000). Detection and analysis of beta-catenin mutations in prostate cancer. Prostate 45, 323–334.
Cho, N. Y., Choi, M., Kim, B. H., Cho, Y. M., Moon, K. C., and Kang, G. H. (2006). BRAF and KRAS mutations in prostatic adenocarcinoma. Int. J. Cancer 119, 1858–1862.
Cronauer, M. V., Schulz, W. A., Ackermann, R., and Burchardt, M. (2005). Effects of WNT/beta-catenin pathway activation on signaling through T-cell factor and androgen receptor in prostate cancer cell lines. Int. J. Oncol. 26, 1033–1040.
Dahiya, R., Lee, C., Mccarville, J., Hu, W., Kaur, G., and Deng, G. (1997). High frequency of genetic instability of microsatellites in human prostatic adenocarcinoma. Int. J. Cancer 72, 762–767.
Demichelis, F., Fall, K., Perner, S., Andren, O., Schmidt, F., Setlur, S. R., Hoshida, Y., Mosquera, J. M., Pawitan, Y., Lee, C., Adami, H. O., Mucci, L. A., Kantoff, P. W., Andersson, S. O., Chinnaiyan, A. M., Johansson, J. E., and Rubin, M. A. (2007). TMPRSS2:ERG gene fusion associated with lethal prostate cancer in a watchful waiting cohort. Oncogene 26, 4596–4599.
Di Cristofano, A., De Acetis, M., Koff, A., Cordon-Cardo, C., and Pandolfi, P. P. (2001). Pten and p27KIP1 cooperate in prostate cancer tumor suppression in the mouse. Nat. Genet. 27, 222–224.
Dong, X. Y., Chen, C., Sun, X., Guo, P., Vessella, R. L., Wang, R. X., Chung, L. W., Zhou, W., and Dong, J. T. (2006). FOXO1A is a candidate for the 13q14 tumor suppressor gene inhibiting androgen receptor signaling in prostate cancer. Cancer Res. 66, 6998–7006.
Dreher, T., Zentgraf, H., Abel, U., Kappeler, A., Michel, M. S., Bleyl, U., and Grobholz, R. (2004). Reduction of PTEN and p27kip1 expression correlates with tumor grade in prostate cancer. Analysis in radical prostatectomy specimens and needle biopsies. Virchows Arch. 444, 509–517.
Eastham, J. A., Stapleton, A. M., Gousse, A. E., Timme, T. L., Yang, G., Slawin, K. M., Wheeler, T. M., Scardino, P. T., and Thompson, T. C. (1995). Association of p53 mutations with metastatic prostate cancer. Clin. Cancer Res. 1, 1111–1118.
Edwards, J., Krishna, N. S., Witton, C. J., and Bartlett, J. M. (2003). Gene amplifications associated with the development of hormone-resistant prostate cancer. Clin. Cancer Res. 9, 5271–5281.
Emmert-Buck, M. R., Vocke, C. D., Pozzatti, R. O., Duray, P. H., Jennings, S. B., Florence, C. D., Zhuang, Z., Bostwick, D. G., Liotta, L. A., and Linehan, W. M. (1995). Allelic loss on chromosome 8p12–21 in microdissected prostatic intraepithelial neoplasia. Cancer Res. 55, 2959–2962.
Febbo, P. G. (2009). Genomic approaches to outcome prediction in prostate cancer. Cancer 115, 3046–3057.
Gaddipati, J. P., Mcleod, D. G., Heidenberg, H. B., Sesterhenn, I. A., Finger, M. J., Moul, J. W., and Srivastava, S. (1994). Frequent detection of codon 877 mutation in the androgen receptor gene in advanced prostate cancers. Cancer Res. 54, 2861–2864.
Gao, H., Ouyang, X., Banach-Petrosky, W. A., Gerald, W. L., Shen, M. M., and Abate-Shen, C. (2006). Combinatorial activities of Akt and B-Raf/Erk signaling in a mouse model of androgen-independent prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 103, 14477–14482.
Gioeli, D., Mandell, J. W., Petroni, G. R., Frierson, H. F. Jr., and Weber, M. J. (1999). Activation of mitogen-activated protein kinase associated with prostate cancer progression. Cancer Res. 59, 279–284.
Glinsky, G. V., Glinskii, A. B., Stephenson, A. J., Hoffman, R. M., and Gerald, W. L. (2004). Gene expression profiling predicts clinical outcome of prostate cancer. J. Clin. Invest. 113, 913–923.
Gray, I. C., Stewart, L. M., Phillips, S. M., Hamilton, J. A., Gray, N. E., Watson, G. J., Spurr, N. K., and Snary, D. (1998). Mutation and expression analysis of the putative prostate tumour-suppressor gene PTEN. Br. J. Cancer 78, 1296–1300.
Greenman, C., Stephens, P., Smith, R., Dalgliesh, G. L., Hunter, C., Bignell, G., Davies, H., Teague, J., Butler, A., Stevens, C., Edkins, S., O’Meara, S., Vastrik, I., Schmidt, E. E., Avis, T., Barthorpe, S., Bhamra, G., Buck, G., Choudhury, B., Clements, J., Cole, J., Dicks, E., Forbes, S., Gray, K., Halliday, K., Harrison, R., Hills, K., Hinton, J., Jenkinson, A., Jones, D., Menzies, A., Mironenko, T., Perry, J., Raine, K., Richardson, D., Shepherd, R., Small, A., Tofts, C., Varian, J., Webb, T., West, S., Widaa, S., Yates, A., Cahill, D. P., Louis, D. N., Goldstraw, P., Nicholson, A. G., Brasseur, F., Looijenga, L., Weber, B. L., Chiew, Y. E., Defazio, A., Greaves, M. F., Green, A. R., Campbell, P., Birney, E., Easton, D. F., Chenevix-Trench, G., Tan, M. H., Khoo, S. K., Teh, B. T., Yuen, S. T., Leung, S. Y., Wooster, R., Futreal, P. A., and Stratton, M. R. (2007). Patterns of somatic mutation in human cancer genomes. Nature 446, 153–158.
Haffner, M. C., Aryee, M. J., Toubaji, A., Esopi, D. M., Albadine, R., Gurel, B., Isaacs, W. B., Bova, G. S., Liu, W., Xu, J., Meeker, A. K., Netto, G., De Marzo, A. M., Nelson, W. G., and Yegnasubramanian, S. (2010). Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat. Genet. 42, 668–675.
Harbour, J. W., Onken, M. D., Roberson, E. D., Duan, S., Cao, L., Worley, L. A., Council, M. L., Matatall, K. A., Helms, C., and Bowcock, A. M. (2010). Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 330, 1410–1413.
Heidenberg, H. B., Sesterhenn, I. A., Gaddipati, J. P., Weghorst, C. M., Buzard, G. S., Moul, J. W., and Srivastava, S. (1995). Alteration of the tumor suppressor gene p53 in a high fraction of hormone refractory prostate cancer. J. Urol. 154, 414–421.
Holcomb, I. N., Young, J. M., Coleman, I. M., Salari, K., Grove, D. I., Hsu, L., True, L. D., Roudier, M. P., Morrissey, C. M., Higano, C. S., Nelson, P. S., Vessella, R. L., and Trask, B. J. (2009). Comparative analyses of chromosome alterations in soft-tissue metastases within and across patients with castration-resistant prostate cancer. Cancer Res. 69, 7793–7802.
Hyytinen, E. R., Frierson, H. F. Jr., Boyd, J. C., Chung, L. W., and Dong, J. T. (1999). Three distinct regions of allelic loss at 13q14, 13q21–22, and 13q33 in prostate cancer. Genes Chromosomes Cancer 25, 108–114.
Jemal, A., Bray, F., Center, M. M., Ferlay, J., Ward, E., and Forman, D. (2011). Global cancer statistics. CA Cancer J. Clin. 61, 69–90.
Jenkins, R. B., Qian, J., Lieber, M. M., and Bostwick, D. G. (1997). Detection of c-myc oncogene amplification and chromosomal anomalies in metastatic prostatic carcinoma by fluorescence in situ hybridization. Cancer Res. 57, 524–531.
Kan, Z., Jaiswal, B. S., Stinson, J., Janakiraman, V., Bhatt, D., Stern, H. M., Yue, P., Haverty, P. M., Bourgon, R., Zheng, J., Moorhead, M., Chaudhuri, S., Tomsho, L. P., Peters, B. A., Pujara, K., Cordes, S., Davis, D. P., Carlton, V. E., Yuan, W., Li, L., Wang, W., Eigenbrot, C., Kaminker, J. S., Eberhard, D. A., Waring, P., Schuster, S. C., Modrusan, Z., Zhang, Z., Stokoe, D., De Sauvage, F. J., Faham, M., and Seshagiri, S. (2010). Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 466, 869–873.
Kim, J., Roh, M., Doubinskaia, I., Algarroba, G. N., Eltoum, I. E., and Abdulkadir, S. A. (2012). A mouse model of heterogeneous, c-MYC-initiated prostate cancer with loss of Pten and p53. Oncogene 31, 322–332.
Kim, M. J., Cardiff, R. D., Desai, N., Banach-Petrosky, W. A., Parsons, R., Shen, M. M., and Abate-Shen, C. (2002). Cooperativity of Nkx3.1 and Pten loss of function in a mouse model of prostate carcinogenesis. Proc. Natl. Acad. Sci. U.S.A. 99, 2884–2889.
King, J. C., Xu, J., Wongvipat, J., Hieronymus, H., Carver, B. S., Leung, D. H., Taylor, B. S., Sander, C., Cardiff, R. D., Couto, S. S., Gerald, W. L., and Sawyers, C. L. (2009). Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat. Genet. 41, 524–526.
Kinkade, C. W., Castillo-Martin, M., Puzio-Kuter, A., Yan, J., Foster, T. H., Gao, H., Sun, Y., Ouyang, X., Gerald, W. L., Cordon-Cardo, C., and Abate-Shen, C. (2008). Targeting AKT/mTOR and ERK MAPK signaling inhibits hormone-refractory prostate cancer in a preclinical mouse model. J. Clin. Invest. 118, 3051–3064.
Koivisto, P., Kononen, J., Palmberg, C., Tammela, T., Hyytinen, E., Isola, J., Trapman, J., Cleutjens, K., Noordzij, A., Visakorpi, T., and Kallioniemi, O. P. (1997). Androgen receptor gene amplification: a possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. 57, 314–319.
Konishi, N., Hiasa, Y., Tsuzuki, T., Tao, M., Enomoto, T., and Miller, G. J. (1997). Comparison of ras activation in prostate carcinoma in Japanese and American men. Prostate 30, 53–57.
Kumar, A., White, T. A., Mackenzie, A. P., Clegg, N., Lee, C., Dumpit, R. F., Coleman, I., Ng, S. B., Salipante, S. J., Rieder, M. J., Nickerson, D. A., Corey, E., Lange, P. H., Morrissey, C., Vessella, R. L., Nelson, P. S., and Shendure, J. (2011). Exome sequencing identifies a spectrum of mutation frequencies in advanced and lethal prostate cancers. Proc. Natl. Acad. Sci. U.S.A. 108, 17087–17092.
Kumar-Sinha, C., Tomlins, S. A., and Chinnaiyan, A. M. (2008). Recurrent gene fusions in prostate cancer. Nat. Rev. Cancer 8, 497–511.
Kwabi-Addo, B., Wang, J., Erdem, H., Vaid, A., Castro, P., Ayala, G., and Ittmann, M. (2004). The expression of Sprouty1, an inhibitor of fibroblast growth factor signal transduction, is decreased in human prostate cancer. Cancer Res. 64, 4728–4735.
Lapointe, J., Li, C., Giacomini, C. P., Salari, K., Huang, S., Wang, P., Ferrari, M., Hernandez-Boussard, T., Brooks, J. D., and Pollack, J. R. (2007). Genomic profiling reveals alternative genetic pathways of prostate tumorigenesis. Cancer Res. 67, 8504–8510.
Lapointe, J., Li, C., Higgins, J. P., Van De Rijn, M., Bair, E., Montgomery, K., Ferrari, M., Egevad, L., Rayford, W., Bergerheim, U., Ekman, P., Demarzo, A. M., Tibshirani, R., Botstein, D., Brown, P. O., Brooks, J. D., and Pollack, J. R. (2004). Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 101, 811–816.
Linja, M. J., and Visakorpi, T. (2004). Alterations of androgen receptor in prostate cancer. J. Steroid Biochem. Mol. Biol. 92, 255–264.
Liu, W., Laitinen, S., Khan, S., Vihinen, M., Kowalski, J., Yu, G., Chen, L., Ewing, C. M., Eisenberger, M. A., Carducci, M. A., Nelson, W. G., Yegnasubramanian, S., Luo, J., Wang, Y., Xu, J., Isaacs, W. B., Visakorpi, T., and Bova, G. S. (2009). Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nat. Med. 15, 559–565.
Majumder, P. K., Grisanzio, C., O’Connell, F., Barry, M., Brito, J. M., Xu, Q., Guney, I., Berger, R., Herman, P., Bikoff, R., Fedele, G., Baek, W. K., Wang, S., Ellwood-Yen, K., Wu, H., Sawyers, C. L., Signoretti, S., Hahn, W. C., Loda, M., and Sellers, W. R. (2008). A prostatic intraepithelial neoplasia-dependent p27 Kip1 checkpoint induces senescence and inhibits cell proliferation and cancer progression. Cancer Cell 14, 146–155.
Mardis, E. R., Ding, L., Dooling, D. J., Larson, D. E., Mclellan, M. D., Chen, K., Koboldt, D. C., Fulton, R. S., Delehaunty, K. D., McGrath, S. D., Fulton, L. A., Locke, D. P., Magrini, V. J., Abbott, R. M., Vickery, T. L., Reed, J. S., Robinson, J. S., Wylie, T., Smith, S. M., Carmichael, L., Eldred, J. M., Harris, C. C., Walker, J., Peck, J. B., Du, F., Dukes, A. F., Sanderson, G. E., Brummett, A. M., Clark, E., McMichael, J. F., Meyer, R. J., Schindler, J. K., Pohl, C. S., Wallis, J. W., Shi, X., Lin, L., Schmidt, H., Tang, Y., Haipek, C., Wiechert, M. E., Ivy, J. V., Kalicki, J., Elliott, G., Ries, R. E., Payton, J. E., Westervelt, P., Tomasson, M. H., Watson, M. A., Baty, J., Heath, S., Shannon, W. D., Nagarajan, R., Link, D. C., Walter, M. J., Graubert, T. A., Dipersio, J. F., Wilson, R. K., and Ley, T. J. (2009). Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 361, 1058–1066.
McKie, A. B., Douglas, D. A., Olijslagers, S., Graham, J., Omar, M. M., Heer, R., Gnanapragasam, V. J., Robson, C. N., and Leung, H. Y. (2005). Epigenetic inactivation of the human sprouty2 (hSPRY2) homologue in prostate cancer. Oncogene 24, 2166–2174.
McMenamin, M. E., Soung, P., Perera, S., Kaplan, I., Loda, M., and Sellers, W. R. (1999). Loss of PTEN expression in paraffin-embedded primary prostate cancer correlates with high Gleason score and advanced stage. Cancer Res. 59, 4291–4296.
Mehra, R., Han, B., Tomlins, S. A., Wang, L., Menon, A., Wasco, M. J., Shen, R., Montie, J. E., Chinnaiyan, A. M., and Shah, R. B. (2007). Heterogeneity of TMPRSS2 gene rearrangements in multifocal prostate adenocarcinoma: molecular evidence for an independent group of diseases. Cancer Res. 67, 7991–7995.
Min, J., Zaslavsky, A., Fedele, G., Mclaughlin, S. K., Reczek, E. E., De Raedt, T., Guney, I., Strochlic, D. E., Macconaill, L. E., Beroukhim, R., Bronson, R. T., Ryeom, S., Hahn, W. C., Loda, M., and Cichowski, K. (2010). An oncogene-tumor suppressor cascade drives metastatic prostate cancer by coordinately activating Ras and nuclear factor-kappaB. Nat. Med. 16, 286–294.
Mirchandani, D., Zheng, J., Miller, G. J., Ghosh, A. K., Shibata, D. K., Cote, R. J., and Roy-Burman, P. (1995). Heterogeneity in intratumor distribution of p53 mutations in human prostate cancer. Am. J. Pathol. 147, 92–101.
Navone, N. M., Labate, M. E., Troncoso, P., Pisters, L. L., Conti, C. J., Von Eschenbach, A. C., and Logothetis, C. J. (1999). p53 mutations in prostate cancer bone metastases suggest that selected p53 mutants in the primary site define foci with metastatic potential. J. Urol. 161, 304–308.
Oda, K., Stokoe, D., Taketani, Y., and Mccormick, F. (2005). High frequency of coexistent mutations of PIK3CA and PTEN genes in endometrial carcinoma. Cancer Res. 65, 10669–10673.
Osborne, C. S., Chakalova, L., Brown, K. E., Carter, D., Horton, A., Debrand, E., Goyenechea, B., Mitchell, J. A., Lopes, S., Reik, W., and Fraser, P. (2004). Active genes dynamically colocalize to shared sites of ongoing transcription. Nat. Genet. 36, 1065–1071.
Ouyang, X., Deweese, T. L., Nelson, W. G., and Abate-Shen, C. (2005). Loss-of-function of Nkx3.1 promotes increased oxidative damage in prostate carcinogenesis. Cancer Res. 65, 6773–6779.
Paez, J. G., Janne, P. A., Lee, J. C., Tracy, S., Greulich, H., Gabriel, S., Herman, P., Kaye, F. J., Lindeman, N., Boggon, T. J., Naoki, K., Sasaki, H., Fujii, Y., Eck, M. J., Sellers, W. R., Johnson, B. E., and Meyerson, M. (2004). EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304, 1497–1500.
Palanisamy, N., Ateeq, B., Kalyana-Sundaram, S., Pflueger, D., Ramnarayanan, K., Shankar, S., Han, B., Cao, Q., Cao, X., Suleman, K., Kumar-Sinha, C., Dhanasekaran, S. M., Chen, Y. B., Esgueva, R., Banerjee, S., Lafargue, C. J., Siddiqui, J., Demichelis, F., Moeller, P., Bismar, T. A., Kuefer, R., Fullen, D. R., Johnson, T. M., Greenson, J. K., Giordano, T. J., Tan, P., Tomlins, S. A., Varambally, S., Rubin, M. A., Maher, C. A., and Chinnaiyan, A. M. (2010). Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nat. Med. 16, 793–798.
Parsons, D. W., Jones, S., Zhang, X., Lin, J. C., Leary, R. J., Angenendt, P., Mankoo, P., Carter, H., Siu, I. M., Gallia, G. L., Olivi, A., Mclendon, R., Rasheed, B. A., Keir, S., Nikolskaya, T., Nikolsky, Y., Busam, D. A., Tekleab, H., Diaz, L. A. Jr., Hartigan, J., Smith, D. R., Strausberg, R. L., Marie, S. K., Shinjo, S. M., Yan, H., Riggins, G. J., Bigner, D. D., Karchin, R., Papadopoulos, N., Parmigiani, G., Vogelstein, B., Velculescu, V. E., and Kinzler, K. W. (2008). An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812.
Phillips, S. M., Morton, D. G., Lee, S. J., Wallace, D. M., and Neoptolemos, J. P. (1994). Loss of heterozygosity of the retinoblastoma and adenomatous polyposis susceptibility gene loci and in chromosomes 10p, 10q and 16q in human prostate cancer. Br. J. Urol. 73, 390–395.
Pleasance, E. D., Cheetham, R. K., Stephens, P. J., Mcbride, D. J., Humphray, S. J., Greenman, C. D., Varela, I., Lin, M. L., Ordonez, G. R., Bignell, G. R., Ye, K., Alipaz, J., Bauer, M. J., Beare, D., Butler, A., Carter, R. J., Chen, L., Cox, A. J., Edkins, S., Kokko-Gonzales, P. I., Gormley, N. A., Grocock, R. J., Haudenschild, C. D., Hims, M. M., James, T., Jia, M., Kingsbury, Z., Leroy, C., Marshall, J., Menzies, A., Mudie, L. J., Ning, Z., Royce, T., Schulz-Trieglaff, O. B., Spiridou, A., Stebbings, L. A., Szajkowski, L., Teague, J., Williamson, D., Chin, L., Ross, M. T., Campbell, P. J., Bentley, D. R., Futreal, P. A., and Stratton, M. R. (2010a). A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 463, 191–196.
Pleasance, E. D., Stephens, P. J., O’Meara, S., Mcbride, D. J., Meynert, A., Jones, D., Lin, M. L., Beare, D., Lau, K. W., Greenman, C., Varela, I., Nik-Zainal, S., Davies, H. R., Ordonez, G. R., Mudie, L. J., Latimer, C., Edkins, S., Stebbings, L., Chen, L., Jia, M., Leroy, C., Marshall, J., Menzies, A., Butler, A., Teague, J. W., Mangion, J., Sun, Y. A., Mclaughlin, S. F., Peckham, H. E., Tsung, E. F., Costa, G. L., Lee, C. C., Minna, J. D., Gazdar, A., Birney, E., Rhodes, M. D., Mckernan, K. J., Stratton, M. R., Futreal, P. A., and Campbell, P. J. (2010b). A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature 463, 184–190.
Prins, G. S., and Putz, O. (2008). Molecular signaling pathways that regulate prostate gland development. Differentiation 76, 641–659.
Qian, J., Bostwick, D. G., Takahashi, S., Borell, T. J., Herath, J. F., Lieber, M. M., and Jenkins, R. B. (1995). Chromosomal anomalies in prostatic intraepithelial neoplasia and carcinoma detected by fluorescence in situ hybridization. Cancer Res. 55, 5408–5414.
Robbins, C. M., Tembe, W. A., Baker, A., Sinari, S., Moses, T. Y., Beckstrom-Sternberg, S., Beckstrom-Sternberg, J., Barrett, M., Long, J., Chinnaiyan, A., Lowey, J., Suh, E., Pearson, J. V., Craig, D. W., Agus, D. B., Pienta, K. J., and Carpten, J. D. (2011). Copy number and targeted mutational analysis reveals novel somatic events in metastatic prostate tumors. Genome Res. 21, 47–55.
Roychowdhury, S., Iyer, M. K., Robinson, D. R., Lonigro, R. J., Wu, Y. M., Cao, X., Kalyana-Sundaram, S., Sam, L., Balbin, O. A., Quist, M. J., Barrette, T., Everett, J., Siddiqui, J., Kunju, L. P., Navone, N., Araujo, J. C., Troncoso, P., Logothetis, C. J., Innis, J. W., Smith, D. C., Lao, C. D., Kim, S. Y., Roberts, J. S., Gruber, S. B., Pienta, K. J., Talpaz, M., and Chinnaiyan, A. M. (2011). Personalized oncology through integrative high-throughput sequencing: a pilot study. Sci. Transl. Med. 3, 111ra121.
Ruiz, C., Lenkiewicz, E., Evers, L., Holley, T., Robeson, A., Kiefer, J., Demeure, M. J., Hollingsworth, M. A., Shen, M., Prunkard, D., Rabinovitch, P. S., Zellweger, T., Mousses, S., Trent, J. M., Carpten, J. D., Bubendorf, L., Von Hoff, D., and Barrett, M. T. (2011). Advancing a clinically relevant perspective of the clonal nature of cancer. Proc. Natl. Acad. Sci. U.S.A. 108, 12054–12059.
Sato, K., Qian, J., Slezak, J. M., Lieber, M. M., Bostwick, D. G., Bergstralh, E. J., and Jenkins, R. B. (1999). Clinical significance of alterations of chromosome 8 in high-grade, advanced, nonmetastatic prostate carcinoma. J. Natl. Cancer Inst. 91, 1574–1580.
Sboner, A., Demichelis, F., Calza, S., Pawitan, Y., Setlur, S. R., Hoshida, Y., Perner, S., Adami, H. O., Fall, K., Mucci, L. A., Kantoff, P. W., Stampfer, M., Andersson, S. O., Varenhorst, E., Johansson, J. E., Gerstein, M. B., Golub, T. R., Rubin, M. A., and Andren, O. (2010). Molecular sampling of prostate cancer: a dilemma for predicting disease progression. BMC Med. Genomics 3, 8. doi: 10.1186/1755-8794-3-8
Sharma, A., Yeow, W. S., Ertel, A., Coleman, I., Clegg, N., Thangavel, C., Morrissey, C., Zhang, X., Comstock, C. E., Witkiewicz, A. K., Gomella, L., Knudsen, E. S., Nelson, P. S., and Knudsen, K. E. (2010). The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J. Clin. Invest. 120, 4478–4492.
Singh, D., Febbo, P. G., Ross, K., Jackson, D. G., Manola, J., Ladd, C., Tamayo, P., Renshaw, A. A., D’Amico, A. V., Richie, J. P., Lander, E. S., Loda, M., Kantoff, P. W., Golub, T. R., and Sellers, W. R. (2002). Gene expression correlates of clinical prostate cancer behavior. Cancer Cell 1, 203–209.
Sircar, K., Yoshimoto, M., Monzon, F. A., Koumakpayi, I. H., Katz, R. L., Khanna, A., Alvarez, K., Chen, G., Darnel, A. D., Aprikian, A. G., Saad, F., Bismar, T. A., and Squire, J. A. (2009). PTEN genomic deletion is associated with p-Akt and AR signalling in poorer outcome, hormone refractory prostate cancer. J. Pathol. 218, 505–513.
Sun, X., Huang, J., Homma, T., Kita, D., Klocker, H., Schafer, G., Boyle, P., and Ohgaki, H. (2009). Genetic alterations in the PI3K pathway in prostate cancer. Anticancer Res. 29, 1739–1743.
Taylor, B. S., Schultz, N., Hieronymus, H., Gopalan, A., Xiao, Y., Carver, B. S., Arora, V. K., Kaushik, P., Cerami, E., Reva, B., Antipin, Y., Mitsiades, N., Landers, T., Dolgalev, I., Major, J. E., Wilson, M., Socci, N. D., Lash, A. E., Heguy, A., Eastham, J. A., Scher, H. I., Reuter, V. E., Scardino, P. T., Sander, C., Sawyers, C. L., and Gerald, W. L. (2010). Integrative genomic profiling of human prostate cancer. Cancer Cell 18, 11–22.
Tomlins, S. A., Laxman, B., Dhanasekaran, S. M., Helgeson, B. E., Cao, X., Morris, D. S., Menon, A., Jing, X., Cao, Q., Han, B., Yu, J., Wang, L., Montie, J. E., Rubin, M. A., Pienta, K. J., Roulston, D., Shah, R. B., Varambally, S., Mehra, R., and Chinnaiyan, A. M. (2007). Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature 448, 595–599.
Tomlins, S. A., Rhodes, D. R., Perner, S., Dhanasekaran, S. M., Mehra, R., Sun, X. W., Varambally, S., Cao, X., Tchinda, J., Kuefer, R., Lee, C., Montie, J. E., Shah, R. B., Pienta, K. J., Rubin, M. A., and Chinnaiyan, A. M. (2005). Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 310, 644–648.
Tricoli, J. V., Gumerlock, P. H., Yao, J. L., Chi, S. G., D’Souza, S. A., Nestok, B. R., and Devere White, R. W. (1996). Alterations of the retinoblastoma gene in human prostate adenocarcinoma. Genes Chromosomes Cancer 15, 108–114.
True, L., Coleman, I., Hawley, S., Huang, C. Y., Gifford, D., Coleman, R., Beer, T. M., Gelmann, E., Datta, M., Mostaghel, E., Knudsen, B., Lange, P., Vessella, R., Lin, D., Hood, L., and Nelson, P. S. (2006). A molecular correlate to the Gleason grading system for prostate adenocarcinoma. Proc. Natl. Acad. Sci. U.S.A. 103, 10991–10996.
Truica, C. I., Byers, S., and Gelmann, E. P. (2000). Beta-catenin affects androgen receptor transcriptional activity and ligand specificity. Cancer Res. 60, 4709–4713.
van ’t Veer, L. J., Dai, H., Van De Vijver, M. J., He, Y. D., Hart, A. A., Mao, M., Peterse, H. L., Van Der Kooy, K., Marton, M. J., Witteveen, A. T., Schreiber, G. J., Kerkhoven, R. M., Roberts, C., Linsley, P. S., Bernards, R., and Friend, S. H. (2002). Gene expression profiling predicts clinical outcome of breast cancer. Nature 415, 530–536.
Vasudevan, K. M., Barbie, D. A., Davies, M. A., Rabinovsky, R., Mcnear, C. J., Kim, J. J., Hennessy, B. T., Tseng, H., Pochanard, P., Kim, S. Y., Dunn, I. F., Schinzel, A. C., Sandy, P., Hoersch, S., Sheng, Q., Gupta, P. B., Boehm, J. S., Reiling, J. H., Silver, S., Lu, Y., Stemke-Hale, K., Dutta, B., Joy, C., Sahin, A. A., Gonzalez-Angulo, A. M., Lluch, A., Rameh, L. E., Jacks, T., Root, D. E., Lander, E. S., Mills, G. B., Hahn, W. C., Sellers, W. R., and Garraway, L. A. (2009). AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell 16, 21–32.
Vis, A. N., Noordzij, M. A., Fitoz, K., Wildhagen, M. F., Schroder, F. H., and Van Der Kwast, T. H. (2000). Prognostic value of cell cycle proteins p27(kip1) and MIB-1, and the cell adhesion protein CD44s in surgically treated patients with prostate cancer. J. Urol. 164, 2156–2161.
Visakorpi, T., Kallioniemi, A. H., Syvanen, A. C., Hyytinen, E. R., Karhu, R., Tammela, T., Isola, J. J., and Kallioniemi, O. P. (1995). Genetic changes in primary and recurrent prostate cancer by comparative genomic hybridization. Cancer Res. 55, 342–347.
Vocke, C. D., Pozzatti, R. O., Bostwick, D. G., Florence, C. D., Jennings, S. B., Strup, S. E., Duray, P. H., Liotta, L. A., Emmert-Buck, M. R., and Linehan, W. M. (1996). Analysis of 99 microdissected prostate carcinomas reveals a high frequency of allelic loss on chromosome 8p12–21. Cancer Res. 56, 2411–2416.
Voeller, H. J., Truica, C. I., and Gelmann, E. P. (1998). Beta-catenin mutations in human prostate cancer. Cancer Res. 58, 2520–2523.
Wang, X. S., Shankar, S., Dhanasekaran, S. M., Ateeq, B., Sasaki, A. T., Jing, X., Robinson, D., Cao, Q., Prensner, J. R., Yocum, A. K., Wang, R., Fries, D. F., Han, B., Asangani, I. A., Cao, X., Li, Y., Omenn, G. S., Pflueger, D., Gopalan, A., Reuter, V. E., Kahoud, E. R., Cantley, L. C., Rubin, M. A., Palanisamy, N., Varambally, S., and Chinnaiyan, A. M. (2011). Characterization of KRAS rearrangements in metastatic prostate cancer. Cancer Discov. 1, 35–43.
Watanabe, M., Shiraishi, T., Yatani, R., Nomura, A. M., and Stemmermann, G. N. (1994). International comparison on ras gene mutations in latent prostate carcinoma. Int. J. Cancer 58, 174–178.
Wu, X., Hepner, K., Castelino-Prabhu, S., Do, D., Kaye, M. B., Yuan, X. J., Wood, J., Ross, C., Sawyers, C. L., and Whang, Y. E. (2000). Evidence for regulation of the PTEN tumor suppressor by a membrane-localized multi-PDZ domain containing scaffold protein MAGI-2. Proc. Natl. Acad. Sci. U.S.A. 97, 4233–4238.
Yardy, G. W., Bicknell, D. C., Wilding, J. L., Bartlett, S., Liu, Y., Winney, B., Turner, G. D., Brewster, S. F., and Bodmer, W. F. (2009). Mutations in the AXIN1 gene in advanced prostate cancer. Eur. Urol. 56, 486–494.
Yardy, G. W., and Brewster, S. F. (2005). Wnt signalling and prostate cancer. Prostate Cancer Prostatic Dis. 8, 119–126.
Yegnasubramanian, S., Kowalski, J., Gonzalgo, M. L., Zahurak, M., Piantadosi, S., Walsh, P. C., Bova, G. S., De Marzo, A. M., Isaacs, W. B., and Nelson, W. G. (2004). Hypermethylation of CpG islands in primary and metastatic human prostate cancer. Cancer Res. 64, 1975–1986.
Keywords: prostate cancer, genomic, genome sequencing
Citation: Baca SC and Garraway LA (2012) The genomic landscape of prostate cancer. Front. Endocrin. 3:69. doi:10.3389/fendo.2012.00069
Received: 21 March 2012; Paper pending published: 13 April 2012;
Accepted: 30 April 2012; Published online: 16 May 2012.
Edited by:
Carmen Priolo, Brigham and Women’s Hospital, Harvard Medical School, USAReviewed by:
Andrea Lenzi, Sapienza University of Rome, ItalyAlessandra Mosca, University Hospital “Maggiore della Carità”, Italy
Copyright: © 2012 Baca and Garraway. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Levi A. Garraway, Department of Medical Oncology, Dana-Farber Cancer Institute, 44 Binney Street, Boston, MA 02115, USA. e-mail:bGV2aV9nYXJyYXdheUBkZmNpLmhhcnZhcmQuZWR1