

- Division of Endocrinology, Diabetes and Metabolism, Department of Medicine, University of Miami Miller School of Medicine, Miami, FL, USA
Deiodinases constitute a group of thioredoxin fold-containing selenoenzymes that play an important function in thyroid hormone homeostasis and control of thyroid hormone action. There are three known deiodinases: D1 and D2 activate the pro-hormone thyroxine (T4) to T3, the most active form of thyroid hormone, while D3 inactivates thyroid hormone and terminates T3 action. A number of studies indicate that deiodinase expression is altered in several types of cancers, suggesting that (i) they may represent a useful cancer marker and/or (ii) could play a role in modulating cell proliferation – in different settings thyroid hormone modulates cell proliferation. For example, although D2 is minimally expressed in human and rodent skeletal muscle, its expression level in rhabdomyosarcoma (RMS)-13 cells is threefold to fourfold higher. In basal cell carcinoma (BCC) cells, sonic hedgehog (Shh)-induced cell proliferation is accompanied by induction of D3 and inactivation of D2. Interestingly a fivefold reduction in the growth of BCC in nude mice was observed if D3 expression was knocked down. A decrease in D1 activity has been described in renal clear cell carcinoma, primary liver cancer, lung cancer, and some pituitary tumors, while in breast cancer cells and tissue there is an increase in D1 activity. Furthermore D1 mRNA and activity were found to be decreased in papillary thyroid cancer while D1 and D2 activities were significantly higher in follicular thyroid cancer tissue, in follicular adenoma, and in anaplastic thyroid cancer. It is conceivable that understanding how deiodinase dysregulation in tumor cells affect thyroid hormone signaling and possibly interfere with tumor progression could lead to new antineoplastic approaches.
Introduction
Deiodinases constitute a group of thioredoxin fold-containing selenoenzymes that metabolize thyroid hormone via two distinct pathways, i.e., thyroid hormone activation through outer ring deiodination (ORD) or thyroid hormone inactivation through inner ring deiodination (IRD; Bianco et al., 2002; Callebaut et al., 2003). D2 and D3 are expressed in multiple tissues, representing the main deiodinases involved respectively in activation and inactivation of thyroid hormone. In contrast, D1 expression is mostly observed in liver and kidney where it catalyzes both ORD and IRD of conjugated thyroid hormone. Thus, it has been suggested that D1 is a scavenger enzyme that recycles iodine from the backbone of inactive iodothyronine en route to elimination via bile or urine (Galton et al., 2009). In addition, lower D1 activity levels are also detected in other tissues, including the thyroid gland itself (Pereira et al., 2011). However, given its very low Km(T4) and Km(T3), it is questionable whether D1 in these other tissues plays a significant physiological role in euthyroid healthy individuals.
The expression of D2 and D3 can be exquisitely cell-specific and change rapidly in response to a number of developmental, metabolic, and disease cues through different signaling pathways (Gereben et al., 2008). Because the expression of these enzymes can be turned on or off in discrete groups of cells, most of the time their actions do not affect circulating thyroid hormone levels, which are tightly controlled via the TRH/TSH axis. Thus, the actions of D2 and D3 are viewed as a cell-specific pre-receptor mechanism to control thyroid hormone signaling that cannot be predicted based on the levels of circulating thyroid hormone (Gereben et al., 2008). For example, stimulation of D2 expression in brown adipose tissue by the cAMP pathway accelerates transcription of T3-responsive genes such as UCP-1 and PGC-1, without elevating serum T3 levels (Hall et al., 2010). In fact, D2 has been shown to play a role in a number of systems by locally amplifying thyroid hormone action, e.g., interplay between astrocytes and neurons (Freitas et al., 2010), hypothalamus and the reproductive system (Yoshimura et al., 2003), and skeletal muscle (Dentice et al., 2011). In turn, ectopic D3 expression in the heart and brain during ischemia or hypoxia lifts the T3-dependent transcriptional footprint in these organs, in what can be seen as an adaptive mechanism to the disease state (Wassen et al., 2002; Olivares et al., 2007; Simonides et al., 2008; Pol et al., 2011).
Notably, a growing number of studies indicate that deiodinase expression is also altered in cancer (previously reviewed in Meyer et al., 2007; Piekielko-Witkowska and Nauman, 2011). While it is conceivable that deiodinase expression is unrelated to the cancer process, there is good indication that deiodinase reactivation could in some cases constitute a useful marker of the disease (Gereben and Bianco, 2009; Piekielko-Witkowska et al., 2009). In addition, it is also likely that the changes in thyroid hormone signaling resulting from deiodinase expression could play a role in cell proliferation and/or cell viability via affecting the expression of cycling D1, a protein factor that is part of a larger complex that controls G1-S transition in the mitotic cell cycle (Dentice et al., 2007). This is illustrated in studies of basal cell carcinoma (BCC) cells, in which sonic hedgehog (Shh)-induced cell proliferation is accompanied by induction of D3 and loss of D2 activity. In fact, the growth of BCC cells implanted in mice is dramatically reduced after D3 expression is knocked down, indicating that dampening of thyroid hormone signaling is important for BCC growth (Dentice et al., 2007).
Models of Altered Deiodinase Expression in Cancer Cells
Given the multiple signaling pathways that regulate deiodinase expression, it is not surprising that in cells that normally have deiodinase activity the expression of these enzymes would be affected by the cancerous transformation (Table 1).
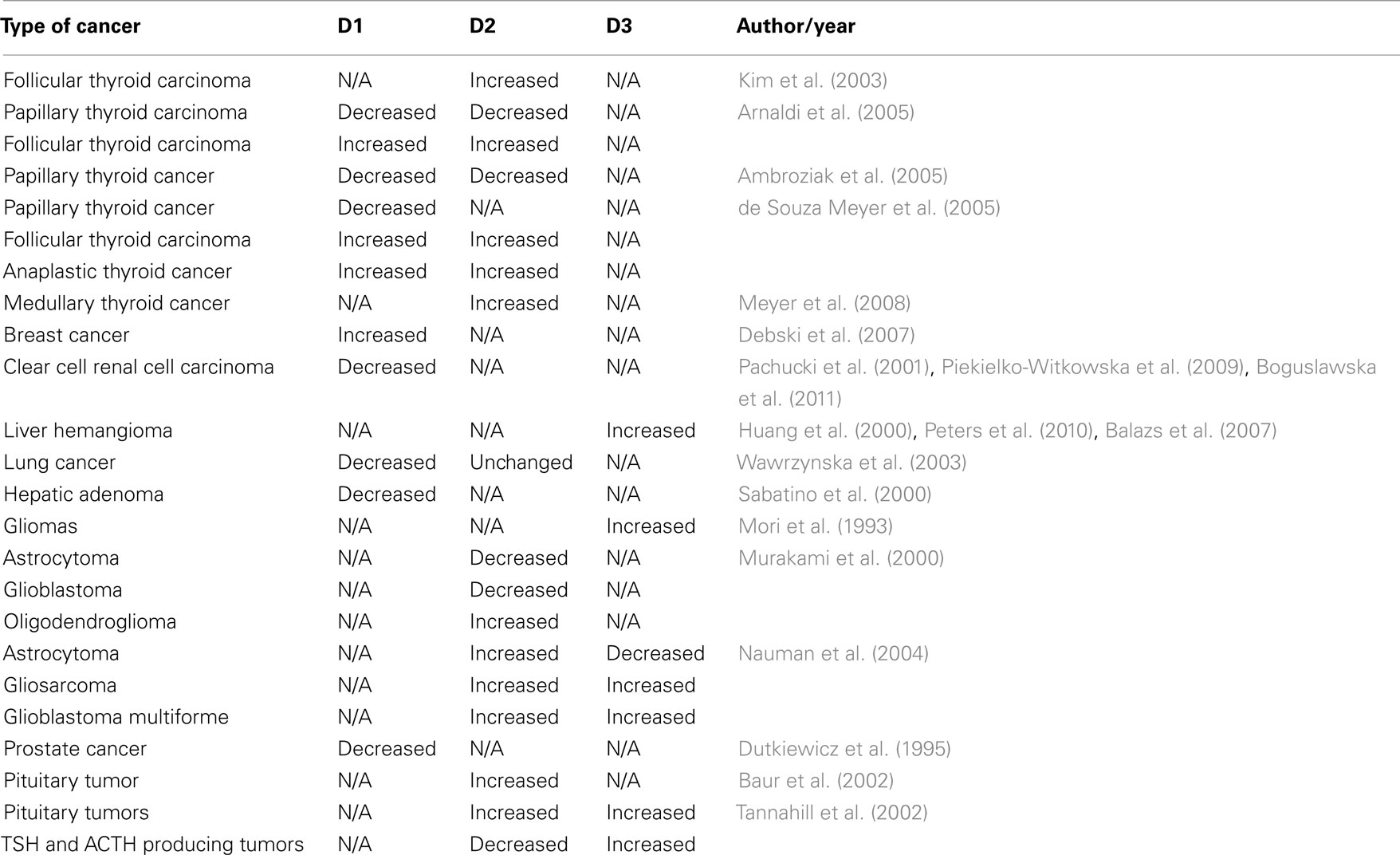
Table 1. Summary of deiodinases changes in human malignancies.
While D1 and D3 are transcriptionally regulated, D2 expression can be regulated both transcriptionally and post-translationally via ubiquitin-mediated D2 inactivation (Gereben et al., 2008). DIO1 is the human gene encoding D1, which consists of four exons and is located on chromosome 1 p32–p33 (Jakobs et al., 1997a). The gene is under the control of GC-rich SP1 promoters and contains two TREs in the 5′ flanking region (FR), both contributing to it T3 responsiveness (Toyoda et al., 1995; Jakobs et al., 1997b; Zhang et al., 1998). In turn, the DIO2 gene is located on the long arm of the 14th human chromosome in position 14q24.3 (Celi et al., 1998; Araki et al., 1999). The coding region is divided into two exons by a ∼7.4-kb intron (Celi et al., 1998). It has three transcriptional start sites (TSS), 707, 31, and 24 bp 5′ to the initiator ATG (Bartha et al., 2000). The human, mouse, and rat dio2 5′-FRs contain a functional cAMP responsive element (CRE; Bartha et al., 2000; Song et al., 2000; Gereben et al., 2001). In the human, dio2 5′ FR functional, thyroid transcription factor-1 (TTF-1 or Nkx-2.1), Nkx-2.5, AP-1, and NF-κB sites have also been described (Gereben et al., 2001; Dentice et al., 2003; Zeold et al., 2006). In addition, glucocorticoids also increase D2 expression transcriptionally as established in GC pituitary tumor cells and in the chicken brain (Kim et al., 1998; Van der Geyten et al., 2001) via an actinomycin-dependent mechanism, not affecting the half-life of D2 mRNA. Lastly, in animals and in some cell models, LPS, and the NF-κB pathway have been shown to potently increase D2 mRNA levels and enzymatic activity, indicating that pro-inflammatory signals might also upregulate D2 expression (Fekete et al., 2005).
DIO3 is localized on the human chromosome 14q32 (Hernandez et al., 1998). In the mouse, the coding regions and the 3′ UTR are contained in a single ∼1.9 kb long exon. The D3 promoter contains a TATA box, two CAAT boxes, and several GC boxes in the proximal 180-bp region of the 5′ FR (Hernandez et al., 1999). A conserved 180-bp-long enhancer was identified ∼6 kb 3′ to the dio3 TSS, and this region contains a consensus AP-1 site and serum response element (Hernandez and St Germain, 2003). A conserved Gli-2 (a member of the Gli transcription factor family that mediates Shh signaling) binding site, D3-A, is located in the mouse and human DIO3 5′ FR (Dentice et al., 2007). Human and mouse DIO3 genes map to chromosomal regions known to include imprinted genes and there is consensus that the DIO3 gene is imprinted, with preferential expression from the paternal chromosome (Hernandez, 2005). The expression of the DIO3 gene is regulated in vivo and in vitro by a number of different signaling pathways. Thyroid hormone up-regulates D3 activity in the rat brain (Peeters et al., 2001). In addition, D3 expression is also up-regulated by the action of serum, phorbol esters, and the epidermal and fibroblast growth factors (EGF, FGF; Hernandez and Obregon, 1995; Pallud et al., 1999; Hernandez and St Germain, 2002). High D3 activity can be found in human infant hemangiomas, a tumor enriched in blood vessels (Huang et al., 2000), indicating a relationship between D3 expression and angiogenic processes possibly through the basic FGF (bFGF) signaling pathway. In glial cells, induction of the DIO3 gene by growth factors appears to be mediated by the extracellular signal regulated kinases (ERKs; Pallud et al., 1999). In particular, D3 expression in astroglial cells is regulated by mitogens, growth factors, and hormones, and exposure to certain combinations of these agents results in synergistic induction of D3 mRNA levels and activity. The compounds that generate signals from the cell surface [tetradecanoyl phorbol acetate (TPA) and bFGF] induce rapid increases in D3 mRNA and activity, whereas treatment with ligands that interact with nuclear receptors (T3 and retinoic acid) result in slower effects. TGF-b stimulates transcription of the human DIO3 gene via a Smad-dependent pathway. Combinations of Smad2 or 3 with Smad4 stimulate the human DIO3 gene transcription only in cells that express endogenous D3 activity, indicating that Smads are necessary but not sufficient for D3 induction (Huang et al., 2005). TGF-b induces endogenous D3 in diverse human cell types, including fetal and adult fibroblasts from several tissues, hemangioma cells, and fetal epithelia. AT the same time, D3 promoter activity is induced threefold to fourfold by estradiol, a mechanism that could contribute to the increased T4 requirements during human pregnancy (Alexander et al., 2003; Huang et al., 2003). During embryonic development, secondary epithelia trans-differentiates into mature epithelia or, under the influence of TGF-b and other paracrine factors, undergoes epithelial-mesenchymal transition to produce the various cell types of connective tissue. Thus, D3 expression in fetal epithelia can be retained through the process of epithelial-mesenchymal transition or reactivated after terminal differentiation by the action of TGF-b (Huang et al., 2005).
Type 1 Deiodinase
D1 activity is readily detectable in the liver, kidney, and thyroid gland (Larsen et al., 1981). Notably, D1 expression is often suppressed in cancer cells compared with the healthy tissue. This is the case for example in the renal clear cell carcinoma (RCCC) where both D1 expression and activity were found to be undetectable compared with normal kidney cells (Pachucki et al., 2001). Furthermore D1 activity was studies in 44 patients with lung cancer (23 squamous cell and 21 adenocarcinoma) and found to be significantly lower as compared to peripheral lung tissue (Wawrzynska et al., 2003). Additionally, DI activity is decreased in hepatic adenocarcinoma (Sabatino et al., 2000) and in prostate cancer tissue (Dutkiewicz et al., 1995).
Interestingly, an opposite pattern has been detected in different histological types of mammary gland tumors induced in Sprague-Dawley rats by injections of 1-methyl-1-nitrosourea (MNU). D1 activity was twofold higher in malignant mammary gland tumors compared with non-lactating mammary gland (Macejova et al., 2001). D1-mediated ORD was also tested in two breast cancer cell lines: MCF-7 (ovarian hormone-dependent) and MDA-MB-231 (ovarian hormone-independent). While D1 activity was present in MCF-7, which was stimulated only by retinoic acid treatments but not by T3 or the beta-adrenergic agonist isoproterenol, in MDA-MB-231 cells, no deiodinase activity could be detected in control conditions or under any of these treatments. These results suggest that D1 expression could represent a sensitive differentiation marker of breast cancer cells (Garcia-Solis and Aceves, 2003). More recently, the D1 activity was evaluated in 36 samples of breast cancers (grade G1 to G3) and in non-cancerous breast tissue taken from the opposite side to the location of the tumor. D1 activity in non-cancerous breast tissues was found to be very low or non-measurable. In contrast, in cancer tissues from the same breasts – especially in G1 and G2 tumors – D1 activity was significantly increased (Debski et al., 2007).
There are several studies assessing D1 expression in thyroid cancer. D1 mRNA and enzyme activity were noted to be significantly decreased in papillary thyroid cancer compared with the normal thyroid tissue, regardless of the histological subtype and/or the clinical stage (de Souza Meyer et al., 2005). Additionally, D1 gene expression was significantly lower in papillary thyroid cancer as assessed from a cDNA analysis of three thyroid carcinoma cell lines using 1807 open reading frame expressed sequence tags (ORESTES) previously recognized as cancer related genes (Arnaldi et al., 2005). In one study, even though both D1 activity and mRNA levels were found to be decreased in papillary thyroid cancer compared with healthy thyroid tissue, there was no correlation between protein expression and enzymatic activity, possibly due to both transcriptional and posttranslational mechanisms; it was also observed that there was a statistically significant correlation between D1 and Pax-8 expression in papillary thyroid tissue (Ambroziak et al., 2005).
In contrast, D1 expression was found to be higher in follicular thyroid cancer tissue and in follicular adenoma (de Souza Meyer et al., 2005). However, in follicular thyroid cancer cell lines D1 activity appeared to be present and to have a normal response to retinoic acid but lost the physiologic responsiveness to TSH and T3 (Schreck et al., 1994). D1 activity was evaluated in anaplastic cell carcinoma cell line as well and found to be undetectable, even after retinoic acid stimulation (Schreck et al., 1994); the opposite finding was obtained in other study (de Souza Meyer et al., 2005),with D1 activity significantly higher in one sample of anaplastic cancer compared with normal tissue. This opposite result, if confirmed, may be due to the different histological characteristic present before the dedifferentiation process took place.
Type 1 Deiodinase Expression as a Marker of Human Renal Cancer
As mentioned above, D1 is highly expressed in the normal kidney. However, DIO1 expression is reduced in the most common subtype of renal cancer, i.e., the RCCC. Both D1 expression and activity were undetectable in RCCC tissues (Pachucki et al., 2001). Additionally, there seems to be a loss of the normal correlation between D1 mRNA and activity in these tissues, possibly as a result of post-transcriptional regulation. Studies from the same group expanded their initial findings reporting interesting results related to alternative splicing of DIO1 (Piekielko-Witkowska et al., 2009). They cloned and identified a total of 11 D1mRNA transcripts, seven of which were previously unreported. Of the 11 variants, all were expressed in the RCCC samples, even if in significantly lower amount compared with the control groups, while only eight were present in healthy renal tissue. These findings lead to the conclusion that three new splicing variants were expressed exclusively in the cancer cells suggesting that they could potentially be used as unique molecular markers for kidney tumors. All the putative D1 protein encoded by these three new variants are truncated products of 111, 115, and 14 amino acids. In the last two alternative splice forms the premature termination codon (PTC) is located more than 50 nucleotides upstream of the final exon–exon junction and usually they are degraded by the nonsense-mediated mRNA decay (NMD) mechanism. Additionally all of them would be inactive since they lack the exon 2 region that encodes the enzyme’s active center. These observations could explain the original finding of undetectable D1 expression and activity in RCCC tissue.
Recently the same authors investigate microRNAs (miRNAs) as alternative regulators of DIO1 expression and function (Boguslawska et al., 2011). MicroRNAs bind to complementary sequences of target mRNAs and behave as post-transcriptional regulators interfering with translation or causing target degradation. Using bioinformatic analysis they identify seven potential miRNA targeting regions of the 3′ untranslated region (3′ UTR) of DIO1 mRNA, two of which (miR-224 and miR-383) were significantly over expressed in RCCC compared with normal tissue. They also observed that there was a significant reduction of DIO1 transcript in the clear cell carcinoma cell line Caki-2 which was previously transfected with miR-224 precursor. Additionally the introduction of anti-miR-224 in these cells increased DIO1 expression by 45%. Furthermore in miR-224 and miR-383 transfected Hela cells a decrease of the activity of a luciferase reporter containing the 3′ UTR of DIO1 was observed. This decrease was abolished when mutated constructs were used instead, suggesting that these miRNAs directly bind to DIO1 3′ UTR. Finally miR-224 expression in RCCC cells was found to correlate negatively with DIO1 mRNA content and T3 concentration suggesting that miR-224 induce intracellular hypothyroidism via a loss of DIO1 function. Taken together these results open the possibility of an important regulatory role of microRNAs in deiodinase activity particularly in cancer cells.
Type 2 Deiodinase
D2 is highly expressed in the brain with its mRNA and activity normally present in astrocytes and other glial cells where it participates in the paracrine control of T3-responsive genes in neurons (Freitas et al., 2010). However, D2 expression is much higher in most brain tumors such as astrocytoma and glioblastoma with the highest D2 activity in gliosarcomas and oligodendrogliomas (Mori et al., 1993; Murakami et al., 2000; Nauman et al., 2004). Remarkably, even neuroblastomas express D2, given that normal neurons are not known for exhibiting D2 activity (St Germain, 1986). Still within the central nervous system, the pituitary gland is another structure that normally expresses D2, specifically in the TSH-producing cells, participating in the normal TSH feed-back mechanism (Christoffolete et al., 2006). D2 mRNA levels in 105 pituitary tumors were found to be 2.6-fold increased in all pituitary tumors with the highest expression observed in non-functional adenoma when compared with normal pituitary tissue. The only exceptions were the TSH and ACTH producing tumors where D2 mRNA was actually reduced (0.1-fold; Tannahill et al., 2002). A higher D2 activity in TSH- and PRL-producing adenomas was also reported, with variable D1/D2 ratios among patients with similar types of tumors (Baur et al., 2002).
Several neoplastic cell lines were found to exhibit high D2 expression as compared with their normal counterparts. For example, D2 is usually expressed in placenta and is also present in JEG3, a choriocarcinoma cell line (Canettieri et al., 2000). In these cells, D2 has been shown to be highly responsive to cAMP treatment that involves the binding of transcription factor CRE binding protein (CREB) to the CRE located in the hD2 promoter (Canettieri et al., 2000). Similar levels of D2 activity were reported in normal lung tissue as well as in lung cancers (squamous cell cancer and adenocarcinoma; Wawrzynska et al., 2003). At the same time, D2, which is expressed in mesothelial cells, has 40-fold higher expression in the mesothelioma cell line (MSTO-211H), with the highest levels of D2 ever seen in cultured cells (Curcio et al., 2001). Furthermore, D2 is only minimally expressed in human and rodent skeletal muscle or in primary cultures (Grozovsky et al., 2009; Ramadan et al., 2011), however its expression is much higher in rhabdomyosarcoma (RMS)-13 cells (da-Silva et al., 2007).
The expression of D2 mRNA and the presence of D2 activity were detected in human osteoblast-like osteosarcoma (SaOS-2) cell line but this time in lower amount compared with the normal human osteoblast (NHOst) cells (Gouveia et al., 2005). Interestingly, TSH was able to increase equally D2 mRNA expression and activity in both cell lines via a TSH receptor-cAMP mediated pathway suggesting that transcriptional regulation of D2 may play an important role in the homeostasis of human osteoblasts (Morimura et al., 2005).
D2 is also expressed in normal human thyroid tissue but its expression changes in thyroid adenomas and cancer. D2 mRNA and activity was found to be significantly increased in hyperfunctioning thyroid adenoma compared with the normal tissue (Murakami et al., 2001). In follicular carcinoma, D2 has increased activity as well. In three patients with large or widely metastatic follicular thyroid carcinoma, there was a persistent increment of the ratio of serum T3 to T4 in the absence of autonomous production of T3 by the tumor. D2 activity was analyzed in one of these patient and was found to be eightfold up-regulated compared with the normal tissue. Resection of the tumor normalized the serum T3 to T4 ratio (Kim et al., 2003). Similarly in anaplastic thyroid cancer D2 activity was found to be higher than normal thyroid tissue (de Souza Meyer et al., 2005). In contrast D2 mRNA and activity are decreased in papillary thyroid cancer compared with the normal thyroid cells (Arnaldi et al., 2005; de Souza Meyer et al., 2005). In one study they observed poor correlation between the low D2 mRNA level and the enzymatic activity in papillary thyroid cancer but a statistically significant correlation between D2, Pax-8, and Titf1/Nkx-2 mRNA expression suggesting a potential role in the impairment of deiodinase expression (Ambroziak et al., 2005). D2 is also highly expressed in human medullary thyroid carcinoma (MTC), with activity that was comparable to those found in surrounding normal follicular tissue (Meyer et al., 2008).
Type 3 Deiodinase
Type 3 iodothyronine deiodinase (D3), the main physiological inactivation mechanism of thyroid hormone, is widely expressed during embryonic life. However, after birth D3 expression subsides in most tissues while remaining present in the human placenta, endometrium, skin, and brain of healthy adults (Bates et al., 1999; Huang, 2005). Interestingly, D3 activity can be reactivated in many tissues in disease states by signals such as hypoxia or ischemia, as well as in tumoral tissues (Huang and Bianco, 2008). For example, D3 activity was evaluated in different brain tumors and compared with normal tissue and found to be increased in all the eight cases of gliosarcoma and in 9 out of 10 cases of glioblastoma multiforme. Additionally the concentration of T3 and T4 were significantly lower in glioma than in the non-tumoral brain tissue samples, indicating that D3 expression was decreasing thyroid hormone signaling locally. On the contrary, the activity of D3 was found to be decreased in all cases of astrocytoma regardless of their grade (Nauman et al., 2004).
D3 expression was also evaluated in 105 pituitary tumors and 10 normal pituitaries. In the tumors, there was significant increase in D3 mRNA compared with the normal tissue, especially in the tumors producing TSH (13-fold), ACTH (sevenfold), GH (sixfold), and the non-functional ones (sevenfold). However, despite the increase in D3 mRNA, D3 enzymatic activity was increased in only three non-functional tumors of the 16 analyzed in total (Tannahill et al., 2002).
D3 expression was evaluated as well in several neoplastic cell lines. D3 mRNA was detected in endometrium carcinoma (ECC-1), mamma carcinoma (MCF-7), and neuroblastoma (SH-SY5Y), but not in the hepatocarcinoma (HepG2), choriocarcinoma, or astrocytoma cell line. Phorbol ester 12-O-tetradecanoylphorbol-13-acetate, a tumor promoter, increased D3 activity twofold to ninefold in ECC-1, MCF-7, and SH-SY5Y cells. In turn, estradiol increased D3 activity threefold only in ECC-1, suggesting its potential role in regulating D3 expression in endometrium during pregnancy. Incubation with retinoids increased D3 activity twofold to threefold in ECC-1 and MCF-7 cells but decreased D3 activity in SH-SY5Y cells. Finally, they also observed in all the cell lines a loss of D3 response to known physiologic stimuli such as T3, possibly due to the underling neoplastic process (Kester et al., 2006).
High levels of D3 activity have been reported in vascular benign tumors like infantile hemangiomas and hepatic hemangioendothelioma (Huang et al., 2000; Ruppe et al., 2005). In many cases the D3 activity level is so high that may result in thyroid function abnormalities due to the accelerated rate of thyroid hormone degradation. This results in subclinical hypothyroidism and even in clinically relevant hypothyroidism (Murakami et al., 2001). The first patient with consumptive hypothyroidism described was a 3-month-old infant with hepatic hemangiomas and severe hypothyroidism refractory to the standard dose of thyroid hormones replacement (Huang et al., 2000). Such patients may improve with T3 replacement treatment (Peters et al., 2010) or by the surgical removal of the tumor as suggested by a case of a patient with consumptive hypothyroidism and liver hemangioendothelioma cured by liver transplantation (Balazs et al., 2007).
Recently, a cluster of 23 up-regulated miRNAs was identified in mice liver tumors and encoded within the Dlk1-Gtl2 imprinted locus on chromosome 12qF1. This region maps to the human DLK1-DIO3 region on chromosome 14q32.2. The expression of DLK1-DIO3 miRNA was examined in 97 patients with hepatocellular carcinoma (HCC) associated with hepatitis B infection. Eighteen of such patients exhibited a strong overexpression of miRNAs which was not observed in other previously tested cancers such as breast, lung, kidney, stomach, or colon. Furthermore, the increased expression of the DLK1-DIO3 miRNA was found to be correlated with some HCC stem cell markers, with a high level of serum α-fetoprotein and a poor survival rate suggesting that DLK1-DIO3 miRNA may be used as a marker for those subtypes of HCC associated with poor prognosis (Luk et al., 2011).
What is the Result of Deiodinase-Mediated Changes in Thyroid Hormone Signaling in Cancer?
Given that in some settings (e.g., BCC) thyroid hormone reduces cell proliferation (Dentice et al., 2007), could a deiodinase dysregulation in tumor cells affect thyroid hormone signaling and thus interfere with tumor progression? This question stems from the modern paradigm that thyroid hormone signaling can be regulated relatively independent of plasma thyroid hormone levels, in a time- and tissue-specific fashion by the deiodinases (Gereben et al., 2008). In fact, in the developing chicken growth plate, loss of D2 activity via Shh-induced D2 ubiquitination has been linked to parathyroid hormone-related peptide (PTHrP) expression and chondrocyte proliferation (Dentice et al., 2005).
These studies prompted investigators to look into other settings in which the Shh signaling pathway is active, such as the BCC, the most common human malignancy characterized by a constitutively active Shh pathway (Dentice et al., 2007). In these cells, Shh increases D3 expression via a Gli-2-mediated transcriptional mechanism, which reduces intracellular T3 concentrations. This effect synergizes with the Shh-stimulated ubiquitin inactivation of D2, which further decreases the intracellular levels of T3. The decrease in thyroid hormone signaling results in an increase in cyclin D1, increasing cell proliferation. Subsequently, a specific D3 shRNAi construct (iD3) was transfected into BCC cells and thymidine uptake experiments showed that D3 depletion significantly reduced cyclin D1 levels and therefore proliferation. Furthermore, a rescue experiment by reintroducing a functional human D3 gene in D3-depleted cells resulted once again in increased cyclin D1 levels and cell proliferation, confirming that D3 plays a key role in cell cycle in these cells. Interestingly, a fivefold reduction in the growth of BCC in nude mice was observed if nude mice receiving BCC xenografts in which D3 expression was knocked down (Dentice et al., 2007). At this writing, it is unclear that such a mechanism would operate in other tumors that also express deiodinases.
At the same time, an increase in thyroid hormone signaling accelerates the metabolic rate and the oxidation of energy substrates such as glucose and fatty acids in most cells types (Bianco et al., 2005). Cancer cells are known to have accelerated metabolism and increased glycolysis (Koppenol et al., 2011). Thus, it is conceivable that, by affecting thyroid hormone signaling, deiodinases could interfere with the metabolism of cancerous cells. If confirmed, this could also constitute a potential therapeutic strategy for certain types of cancers that depend on a high metabolic rate.
Conclusion
Deiodinases are enzymes that can up- or down-regulate thyroid hormone signaling on a cell-specific basis, independently of circulating thyroid hormone levels. Several types of cancers and cancerous cell lines express high (low) levels of deiodinases that could contribute to the loss in control of cell division and consequently tumor development; this could also potentially affect their metabolic rate and selection of oxidative substrates. Understanding the mechanisms underlying the dysregulation in deiodinase expression in tumor cells as well as the downstream impact of changes in thyroid hormone signaling could potentially lead to the development of new antineoplastic approaches.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alexander, E. K., Marqusee, E., Lawrence, J., Jarolim, P., Fischer, G. A., and Larsen, P. R. (2003). “Time of onset and magnitude of increase in levothyroxine requirements during pregnancy in women with hypothyroidism,” in 75th Annual Meeting of the American Thyroid Association, Palm Beach, FL.
Ambroziak, M., Pachucki, J., Stachlewska-Nasfeter, E., Nauman, J., and Nauman, A. (2005). Disturbed expression of type 1 and type 2 iodothyronine deiodinase as well as titf1/nkx2-1 and pax-8 transcription factor genes in papillary thyroid cancer. Thyroid 15, 1137–1146.
Araki, O., Murakami, M., Morimura, T., Kamiya, Y., Hosoi, Y., Kato, Y., and Mori, M. (1999). Assignment of type II iodothyronine deiodinase gene (DIO2) to human chromosome band 14q24.2 – >q24.3 by in situ hybridization. Cytogenet. Cell Genet. 84, 73–74.
Arnaldi, L. A., Borra, R. C., Maciel, R. M., and Cerutti, J. M. (2005). Gene expression profiles reveal that DCN, DIO1, and DIO2 are underexpressed in benign and malignant thyroid tumors. Thyroid 15, 210–221.
Balazs, A. E., Athanassaki, I., Gunn, S. K., Tatevian, N., Huang, S. A., Haymond, M. W., and Karaviti, L. P. (2007). Rapid resolution of consumptive hypothyroidism in a child with hepatic hemangioendothelioma following liver transplantation. Ann. Clin. Lab. Sci. 37, 280–284.
Bartha, T., Kim, S. W., Salvatore, D., Gereben, B., Tu, H. M., Harney, J. W., Rudas, P., and Larsen, P. R. (2000). Characterization of the 5′-flanking and 5′-untranslated regions of the cyclic adenosine 3′,5′-monophosphate-responsive human type 2 iodothyronine deiodinase gene. Endocrinology 141, 229–237.
Bates, J. M., St Germain, D. L., and Galton, V. A. (1999). Expression profiles of the three iodothyronine deiodinases, D1, D2, and D3, in the developing rat. Endocrinology 140, 844–851.
Baur, A., Buchfelder, M., and Kohrle, J. (2002). Expression of 5′-deiodinase enzymes in normal pituitaries and in various human pituitary adenomas. Eur. J. Endocrinol. 147, 263–268.
Bianco, A. C., Maia, A. L., da Silva, W. S., and Christoffolete, M. A. (2005). Adaptive activation of thyroid hormone and energy expenditure. Biosci. Rep. 25, 191–208.
Bianco, A. C., Salvatore, D., Gereben, B., Berry, M. J., and Larsen, P. R. (2002). Biochemistry, cellular and molecular biology and physiological roles of the iodothyronine selenodeiodinases. Endocr. Rev. 23, 38–89.
Boguslawska, J., Wojcicka, A., Piekielko-Witkowska, A., Master, A., and Nauman, A. (2011). MiR-224 targets the 3′UTR of type 1 5′-iodothyronine deiodinase possibly contributing to tissue hypothyroidism in renal cancer. PLoS ONE 6, e24541. doi:10.1371/journal.pone.0024541
Callebaut, I., Curcio-Morelli, C., Mornon, J. P., Gereben, B., Buettner, C., Huang, S., Castro, B., Fonseca, T. L., Harney, J. W., Larsen, P. R., and Bianco, A. C. (2003). The iodothyronine selenodeiodinases are thioredoxin-fold family proteins containing a glycoside hydrolase clan GH-A-like structure. J. Biol. Chem. 278, 36887–36896.
Canettieri, G., Celi, F. S., Baccheschi, G., Salvatori, L., Andreoli, M., and Centanni, M. (2000). Isolation of human type 2 deiodinase gene promoter and characterization of a functional cyclic adenosine monophosphate response element. Endocrinology 141, 1804–1813.
Celi, F. S., Canettieri, G., Yarnall, D. P., Burns, D. K., Andreoli, M., Shuldiner, A. R., and Centanni, M. (1998). Genomic characterization of the coding region of the human type II 5′-deiodinase gene. Mol. Cell. Endocrinol. 141, 49–52.
Christoffolete, M. A., Ribeiro, R., Singru, P., Fekete, C., da Silva, W. S., Gordon, D. F., Huang, S. A., Crescenzi, A., Harney, J. W., Ridgway, E. C., Larsen, P. R., Lechan, R. M., and Bianco, A. C. (2006). Atypical expression of type 2 iodothyronine deiodinase in thyrotrophs explains the thyroxine-mediated pituitary thyrotropin feedback mechanism. Endocrinology 147, 1735–1743.
Curcio, C., Baqui, M. M., Salvatore, D., Rihn, B. H., Mohr, S., Harney, J. W., Larsen, P. R., and Bianco, A. C. (2001). The human type 2 iodothyronine deiodinase is a selenoprotein highly expressed in a mesothelioma cell line. J. Biol. Chem. 276, 30183–30187.
da-Silva, W. S., Harney, J. W., Kim, B. W., Li, J., Bianco, S. D., Crescenzi, A., Christoffolete, M. A., Huang, S. A., and Bianco, A. C. (2007). The small polyphenolic molecule kaempferol increases cellular energy expenditure and thyroid hormone activation. Diabetes 56, 767–776.
de Souza Meyer, E. L., Dora, J. M., Wagner, M. S., and Maia, A. L. (2005). Decreased type 1 iodothyronine deiodinase expression might be an early and discrete event in thyroid cell dedifferentiation towards papillary carcinoma. Clin. Endocrinol. (Oxf.) 62, 672–678.
Debski, M. G., Pachucki, J., Ambroziak, M., Olszewski, W., and Bar-Andziak, E. (2007). Human breast cancer tissue expresses high level of type 1 5′-deiodinase. Thyroid 17, 3–10.
Dentice, M., Bandyopadhyay, A., Gereben, B., Callebaut, I., Christoffolete, M. A., Kim, B. W., Nissim, S., Mornon, J. P., Zavacki, A. M., Zeold, A., Capelo, L. P., Curcio-Morelli, C., Ribeiro, R., Harney, J. W., Tabin, C. J., and Bianco, A. C. (2005). The Hedgehog-inducible ubiquitin ligase subunit WSB-1 modulates thyroid hormone activation and PTHrP secretion in the developing growth plate. Nat. Cell Biol. 7, 698–705.
Dentice, M., Luongo, C., Huang, S., Ambrosio, R., Elefante, A., Mirebeau-Prunier, D., Zavacki, A. M., Fenzi, G., Grachtchouk, M., Hutchin, M., Dlugosz, A. A., Bianco, A. C., Missero, C., Larsen, P. R., and Salvatore, D. (2007). Sonic hedgehog-induced type 3 deiodinase blocks thyroid hormone action enhancing proliferation of normal and malignant keratinocytes. Proc. Natl. Acad. Sci. U.S.A. 104, 14466–14471.
Dentice, M., Marsili, A., Ambrosio, R., Guardiola, O., Sibilio, A., Paik, J. H., Minchiotti, G., DePinho, R. A., Fenzi, G., Larsen, P. R., and Salvatore, D. (2011). The FoxO3/type 2 deiodinase pathway is required for normal mouse myogenesis and muscle regeneration. J. Clin. Invest. 120, 4021–4030.
Dentice, M., Morisco, C., Vitale, M., Rossi, G., Fenzi, G., and Salvatore, D. (2003). The different cardiac expression of the type 2 iodothyronine deiodinase gene between human and rat is related to the differential response of the Dio2 genes to Nkx-2.5 and GATA-4 transcription factors. Mol. Endocrinol. 17, 1508–1521.
Dutkiewicz, S., Witeska, A., and Nauman, A. (1995). The deiodination of thyroxine to triiodothyronine in the testes of patients with prostate cancer. Int. Urol. Nephrol. 27, 81–85.
Fekete, C., Sarkar, S., Christoffolete, M. A., Emerson, C. H., Bianco, A. C., and Lechan, R. M. (2005). Bacterial lipopolysaccharide (LPS)-induced type 2 iodothyronine deiodinase (D2) activation in the mediobasal hypothalamus (MBH) is independent of the LPS-induced fall in serum thyroid hormone levels. Brain Res. 1056, 97–99.
Freitas, B. C., Gereben, B., Castillo, M., Kallo, I., Zeold, A., Egri, P., Liposits, Z., Zavacki, A. M., Maciel, R. M., Jo, S., Singru, P., Sanchez, E., Lechan, R. M., and Bianco, A. C. (2010). Paracrine signaling by glial cell-derived triiodothyronine activates neuronal gene expression in the rodent brain and human cells. J. Clin. Invest. 120, 2206–2217.
Galton, V. A., Schneider, M., Clark, A. S., and Germain, D. L. (2009). Life without T4 to T3 conversion: studies in mice devoid of the 5′-deiodinases. Endocrinology 150, 2957–2963.
Garcia-Solis, P., and Aceves, C. (2003). 5′Deiodinase in two breast cancer cell lines: effect of triiodothyronine, isoproterenol and retinoids. Mol. Cell. Endocrinol. 201, 25–31.
Gereben, B., and Bianco, A. C. (2009). Covering the base-pairs in iodothyronine deiodinase-1 biology: holes remain in the lineup. Thyroid 19, 1027–1029.
Gereben, B., Salvatore, D., Harney, J. W., Tu, H. M., and Larsen, P. R. (2001). The human, but not rat, dio2 gene is stimulated by thyroid transcription factor-1 (TTF-1). Mol. Endocrinol. 15, 112–124.
Gereben, B., Zavacki, A. M., Ribich, S., Kim, B. W., Huang, S. A., Simonides, W. S., Zeold, A., and Bianco, A. C. (2008). Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endocr. Rev. 29, 898–938.
Gouveia, C. H., Christoffolete, M. A., Zaitune, C. R., Dora, J. M., Harney, J. W., Maia, A. L., and Bianco, A. C. (2005). Type 2 iodothyronine selenodeiodinase is expressed throughout the mouse skeleton and in the MC3T3-E1 mouse osteoblastic cell line during differentiation. Endocrinology 146, 195–200.
Grozovsky, R., Ribich, S., Rosene, M. L., Mulcahey, M. A., Huang, S. A., Patti, M. E., Bianco, A. C., and Kim, B. W. (2009). Type 2 deiodinase expression is induced by peroxisomal proliferator-activated receptor-gamma agonists in skeletal myocytes. Endocrinology 150, 1976–1983.
Hall, J. A., Ribich, S., Cristoffolete, M. A., Simovic, G., Correa, M., Patti, M. E., and Bianco, A. C. (2010). Absence of thyroid hormone activation during development underlies a permanent defect in adaptive thermogenesis. Endocrinology 151, 4573–4582.
Hernandez, A., Lyon, G. J., Schneider, M. J., and St Germain, D. L. (1999). Isolation and characterization of the mouse gene for the type 3 iodothyronine deiodinase. Endocrinology 140, 124–130.
Hernandez, A., and Obregon, M. J. (1995). Presence of growth factors-induced type III iodothyronine 5-deiodinase in cultured rat brown adipocytes. Endocrinology 136, 4543–4550.
Hernandez, A., Park, J. P., Lyon, G. J., Mohandas, T. K., and St Germain, D. L. (1998). Localization of the type 3 iodothyronine deiodinase (DIO3) gene to human chromosome 14q32 and mouse chromosome 12F1. Genomics 53, 119–121.
Hernandez, A., and St Germain, D. L. (2002). Dexamethasone inhibits growth factor-induced type 3 deiodinase activity and mRNA expression in a cultured cell line derived from rat neonatal brown fat vascular-stromal cells. Endocrinology 143, 2652–2658.
Hernandez, A., and St Germain, D. L. (2003). Activity and response to serum of the mammalian thyroid hormone deiodinase 3 gene promoter: identification of a conserved enhancer. Mol. Cell. Endocrinol. 206, 23–32.
Huang, S. A. (2005). Physiology and pathophysiology of type 3 deiodinase in humans. Thyroid 15, 875–881.
Huang, S. A., and Bianco, A. C. (2008). Reawakened interest in type III iodothyronine deiodinase in critical illness and injury. Nat. Clin. Pract. Endocrinol. Metab. 4, 148–155.
Huang, S. A., Dorfman, D. M., Genest, D. R., Salvatore, D., and Larsen, P. R. (2003). Type 3 iodothyronine deiodinase is highly expressed in the human uteroplacental unit and in fetal epithelium. J. Clin. Endocrinol. Metab. 88, 1384–1388.
Huang, S. A., Mulcahey, M. A., Crescenzi, A., Chung, M., Kim, B., Barnes, C. A., Kuijt, W., Tu, H. M., Harney, J. W., and Larsen, P. R. (2005). TGF-B promotes inactivation of extracellular thyroid hormones via transcriptional stimulation of type 3 iodothyronine deiodinase. Mol. Endocrinol. 19, 3126–3136.
Huang, S. A., Tu, H. M., Harney, J. W., Venihaki, M., Butte, A. J., Kozakewich, H. P., Fishman, S. J., and Larsen, P. R. (2000). Severe hypothyroidism caused by type 3 iodothyronine deiodinase in infantile hemangiomas. N. Engl. J. Med. 343, 185–189.
Jakobs, T. C., Koehler, M. R., Schmutzler, C., Glaser, F., Schmid, M., and Kohrle, J. (1997a). Structure of the human type I iodothyronine 5′-deiodinase gene and localization to chromosome 1p32-p33. Genomics 42, 361–363.
Jakobs, T. C., Schmutzler, C., Meissner, J., and Kohrle, J. (1997b). The promoter of the human type I 5′-deiodinase gene – mapping of the transcription start site and identification of a DR+4 thyroid-hormone- responsive element. Eur. J. Biochem. 247, 288–297.
Kester, M. H., Kuiper, G. G., Versteeg, R., and Visser, T. J. (2006). Regulation of type III iodothyronine deiodinase expression in human cell lines. Endocrinology 147, 5845–5854.
Kim, B. W., Daniels, G. H., Harrison, B. J., Price, A., Harney, J. W., Larsen, P. R., and Weetman, A. P. (2003). Overexpression of type 2 iodothyronine deiodinase in follicular carcinoma as a cause of low circulating free thyroxine levels. J. Clin. Endocrinol. Metab. 88, 594–598.
Kim, S. W., Harney, J. W., and Larsen, P. R. (1998). Studies of the hormonal regulation of type 2 5′-iodothyronine deiodinase messenger ribonucleic acid in pituitary tumor cells using semiquantitative reverse transcription-polymerase chain reaction. Endocrinology 139, 4895–4905.
Koppenol, W. H., Bounds, P. L., and Dang, C. V. (2011). Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 11, 325–337.
Larsen, P. R., Silva, J. E., and Kaplan, M. M. (1981). Relationships between circulating and intracellular thyroid hormones: physiological and clinical implications. Endocr. Rev. 2, 87–102.
Luk, J. M., Burchard, J., Zhang, C., Liu, A. M., Wong, K. F., Shek, F. H., Lee, N. P., Fan, S. T., Poon, R. T., Ivanovska, I., Philippar, U., Cleary, M. A., Buser, C. A., Shaw, P. M., Lee, C. N., Tenen, D. G., Dai, H., and Mao, M. (2011). DLK1-DIO3 genomic imprinted microRNA cluster at 14q32.2 defines a stemlike subtype of hepatocellular carcinoma associated with poor survival. J. Biol. Chem. 286, 30706–30713.
Macejova, D., Liska, J., and Brtko, J. (2001). Mammary gland carcinoma-related increase of type I iodothyronine 5′-deiodinase activity in Sprague-Dawley rats. Gen. Physiol. Biophys. 20, 293–302.
Meyer, E. L., Goemann, I. M., Dora, J. M., Wagner, M. S., and Maia, A. L. (2008). Type 2 iodothyronine deiodinase is highly expressed in medullary thyroid carcinoma. Mol. Cell. Endocrinol. 289, 16–22.
Meyer, E. L., Wagner, M. S., and Maia, A. L. (2007). Iodothyronine deiodinases expression in thyroid neoplasias. Arq. Bras. Endocrinol. Metabol. 51, 690–700.
Mori, K., Yoshida, K., Kayama, T., Kaise, N., Fukazawa, H., Kiso, Y., Kikuchi, K., Aizawa, Y., and Abe, K. (1993). Thyroxine 5-deiodinase in human brain tumors. J. Clin. Endocrinol. Metab. 77, 1198–1202.
Morimura, T., Tsunekawa, K., Kasahara, T., Seki, K., Ogiwara, T., Mori, M., and Murakami, M. (2005). Expression of type 2 iodothyronine deiodinase in human osteoblast is stimulated by thyrotropin. Endocrinology 146, 2077–2084.
Murakami, M., Araki, O., Hosoi, Y., Kamiya, Y., Morimura, T., Ogiwara, T., Mizuma, H., and Mori, M. (2001). Expression and regulation of type II iodothyronine deiodinase in human thyroid gland. Endocrinology 142, 2961–2967.
Murakami, M., Araki, O., Morimura, T., Hosoi, Y., Mizuma, H., Yamada, M., Kurihara, H., Ishiuchi, S., Tamura, M., Sasaki, T., and Mori, M. (2000). Expression of type II iodothyronine deiodinase in brain tumors. J. Clin. Endocrinol. Metab. 85, 4403–4406.
Nauman, P., Bonicki, W., Michalik, R., Warzecha, A., and Czernicki, Z. (2004). The concentration of thyroid hormones and activities of iodothyronine deiodinases are altered in human brain gliomas. Folia Neuropathol. 42, 67–73.
Olivares, E. L., Marassi, M. P., Fortunato, R. S., da Silva, A. C., Costa-e-Sousa, R. H., Araujo, I. G., Mattos, E. C., Masuda, M. O., Mulcahey, M. A., Huang, S. A., Bianco, A. C., and Carvalho, D. P. (2007). Thyroid function disturbance and type 3 iodothyronine deiodinase induction after myocardial infarction in rats a time course study. Endocrinology 148, 4786–4792.
Pachucki, J., Ambroziak, M., Tanski, Z., Luczak, J., Nauman, J., and Nauman, A. (2001). Type I 5′-iodothyronine deiodinase activity and mRNA are remarkably reduced in renal clear cell carcinoma. J. Endocrinol. Invest. 24, 253–261.
Pallud, S., Ramauge, M., Gavaret, J. M., Lennon, A. M., Munsch, N., St Germain, D. L., Pierre, M., and Courtin, F. (1999). Regulation of type 3 iodothyronine deiodinase expression in cultured rat astrocytes: role of the Erk cascade. Endocrinology 140, 2917–2923.
Peeters, R., Fekete, C., Goncalves, C., Legradi, G., Tu, H. M., Harney, J. W., Bianco, A. C., Lechan, R. M., and Larsen, P. R. (2001). Regional physiological adaptation of the central nervous system deiodinases to iodine deficiency. Am. J. Physiol. Endocrinol. Metab. 281, E54–E61.
Pereira, V. S., Marassi, M. P., Rosenthal, D., Vaisman, M., and Correa da Costa, V. M. (2011). Positive correlation between type 1 and 2 iodothyronine deiodinases activities in human goiters. Endocrine 41, 532–538.
Peters, C., Langham, S., Mullis, P. E., and Dattani, M. T. (2010). Use of combined liothyronine and thyroxine therapy for consumptive hypothyroidism associated with hepatic haemangiomas in infancy. Horm. Res. Paediatr. 74, 149–152.
Piekielko-Witkowska, A., Master, A., Wojcicka, A., Boguslawska, J., Brozda, I., Tanski, Z., and Nauman, A. (2009). Disturbed expression of type 1 iodothyronine deiodinase splice variants in human renal cancer. Thyroid 19, 1105–1113.
Piekielko-Witkowska, A., and Nauman, A. (2011). Iodothyronine deiodinases and cancer. J. Endocrinol. Invest. 34, 716–728.
Pol, C. J., Muller, A., Zuidwijk, M. J., van Deel, E. D., Kaptein, E., Saba, A., Marchini, M., Zucchi, R., Visser, T. J., Paulus, W. J., Duncker, D. J., and Simonides, W. S. (2011). Left-ventricular remodeling after myocardial infarction is associated with a cardiomyocyte-specific hypothyroid condition. Endocrinology 152, 669–679.
Ramadan, W., Marsili, A., Huang, S., Larsen, P. R., and Silva, J. E. (2011). Type-2 iodothyronine 5′deiodinase in skeletal muscle of C57BL/6 mice. I. Identity, subcellular localization, and characterization. Endocrinology 152, 3082–3092.
Ruppe, M. D., Huang, S. A., and Jan de Beur, S. M. (2005). Consumptive hypothyroidism caused by paraneoplastic production of type 3 iodothyronine deiodinase. Thyroid 15, 1369–1372.
Sabatino, L., Iervasi, G., Ferrazzi, P., Francesconi, D., and Chopra, I. J. (2000). A study of iodothyronine 5′-monodeiodinase activities in normal and pathological tissues in man and their comparison with activities in rat tissues. Life Sci. 68, 191–202.
Schreck, R., Schnieders, F., Schmutzler, C., and Kohrle, J. (1994). Retinoids stimulate type I iodothyronine 5′-deiodinase activity in human follicular thyroid carcinoma cell lines. J. Clin. Endocrinol. Metab. 79, 791–798.
Simonides, W. S., Mulcahey, M. A., Redout, E. M., Muller, A., Zuidwijk, M. J., Visser, T. J., Wassen, F. W., Crescenzi, A., da-Silva, W. S., Harney, J., Engel, F. B., Obregon, M. J., Larsen, P. R., Bianco, A. C., and Huang, S. A. (2008). Hypoxia-inducible factor induces local thyroid hormone inactivation during hypoxic-ischemic disease in rats. J. Clin. Invest. 118, 975–983.
Song, S., Adachi, K., Katsuyama, M., Sorimachi, K., and Oka, T. (2000). Isolation and characterization of the 5′-upstream and untranslated regions of the mouse type II iodothyronine deiodinase gene. Mol. Cell. Endocrinol. 165, 189–198.
St Germain, D. L. (1986). Hormonal control of a low Km (type II) iodothyronine 5′-deiodinase in cultured NB41A3 mouse neuroblastoma cells. Endocrinology 119, 840–846.
Tannahill, L. A., Visser, T. J., McCabe, C. J., Kachilele, S., Boelaert, K., Sheppard, M. C., Franklyn, J. A., and Gittoes, N. J. (2002). Dysregulation of iodothyronine deiodinase enzyme expression and function in human pituitary tumours. Clin. Endocrinol. (Oxf.) 56, 735–743.
Toyoda, N., Zavacki, A. M., Maia, A. L., Harney, J. W., and Larsen, P. R. (1995). A novel retinoid X receptor-independent thyroid hormone response element is present in the human type 1 deiodinase gene. Mol. Cell. Biol. 15, 5100–5112.
Van der Geyten, S., Segers, I., Gereben, B., Bartha, T., Rudas, P., Larsen, P. R., Kuhn, E. R., and Darras, V. M. (2001). Transcriptional regulation of iodothyronine deiodinases during embryonic development. Mol. Cell. Endocrinol. 183, 1–9.
Wassen, F. W., Schiel, A. E., Kuiper, G. G., Kaptein, E., Bakker, O., Visser, T. J., and Simonides, W. S. (2002). Induction of thyroid hormone-degrading deiodinase in cardiac hypertrophy and failure. Endocrinology 143, 2812–2815.
Wawrzynska, L., Sakowicz, A., Rudzinski, P., Langfort, R., and Kurzyna, M. (2003). The conversion of thyroxine to triiodothyronine in the lung: comparison of activity of type I iodothyronine 5′ deiodinase in lung cancer with peripheral lung tissues. Monaldi Arch. Chest Dis. 59, 140–145.
Yoshimura, T., Yasuo, S., Watanabe, M., Iigo, M., Yamamura, T., Hirunagi, K., and Ebihara, S. (2003). Light-induced hormone conversion of T4 to T3 regulates photoperiodic response of gonads in birds. Nature 426, 178–181.
Zeold, A., Doleschall, M., Haffner, M. C., Capelo, L. P., Menyhert, J., Liposits, Z., da Silva, W. S., Bianco, A. C., Kacskovics, I., Fekete, C., and Gereben, B. (2006). Characterization of the nuclear factor-kappa B responsiveness of the human dio2 gene. Endocrinology 147, 4419–4429.
Keywords: thyroid, deiodinase, cancer
Citation: Casula S and Bianco AC (2012) Thyroid hormone deiodinases and cancer. Front. Endocrin. 3:74. doi: 10.3389/fendo.2012.00074
Received: 15 February 2012; Accepted: 15 May 2012;
Published online: 01 June 2012.
Edited by:
Carmelo Nucera, Beth Israel Deaconess Medical Center/Harvard Medical School, USAReviewed by:
Francesco Saverio Celi, National Institutes of Health, USAJosef Köhrle, Charité-Universitätsmedizin Berlin, Germany
Copyright: © 2012 Casula and Bianco. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Antonio C. Bianco, University of Miami Miller School of Medicine, Batchelor Research Building, 1400 N.W. 10th Avenue, Suite 601, Miami, FL 33136, USA. e-mail:YWJpYW5jb0BkZWlvZGluYXNlLm9yZw==