 Yasuo Imanishi*
Yasuo Imanishi* Yuki Nagata and Masaaki Inaba
Yuki Nagata and Masaaki Inaba
- Department of Metabolism, Endocrinology and Molecular Medicine, Osaka City University Graduate School of Medicine, Osaka, Japan
Circulating calcium and phosphate are tightly regulated by three hormones: the active form of vitamin D (1,25-dihydroxyvitamin D), fibroblast growth factor (FGF)-23, and parathyroid hormone (PTH). PTH acts to stimulate a rapid increment in serum calcium and has a crucial role in calcium homeostasis. Major target organs of PTH are kidney and bone. The oversecretion of the hormone results in hypercalcemia, caused by increased intestinal calcium absorption, reduced renal calcium clearance, and mobilization of calcium from bone in primary hyperparathyroidism. In chronic kidney disease, secondary hyperparathyroidism of uremia is observed in its early stages, and this finally develops into the autonomous secretion of PTH during maintenance hemodialysis. Receptors in parathyroid cells, such as the calcium-sensing receptor, vitamin D receptor, and FGF receptor (FGFR)-Klotho complex have crucial roles in the regulation of PTH secretion. Genes such as Cyclin D1, RET, MEN1, HRPT2, and CDKN1B have been identified in parathyroid diseases. Genetically engineered animals with these receptors and the associated genes have provided us with valuable information on the patho-physiology of parathyroid diseases. The application of these animal models is significant for the development of new therapies.
Introduction
Hyperfunctioning parathyroid diseases such as primary hyperparathyroidism (PHPT) and secondary hyperparathyroidism of uremia (SHPT) are characterized by the abnormal metabolism of calcium (Ca) and phosphate (P). Parathyroid hormone (PTH), the active form of vitamin D (1,25-dihydroxyvitamin D, or 1,25-(OH)2D), and fibroblast growth factor (FGF)-23, are the principal physiological regulators of Ca and P homeostasis in humans (Imanishi et al., 2009a; Figure 1). There are feedback loops between ionized Ca (Ca2+), P, 1,25-(OH)2D, FGF-23, and PTH.
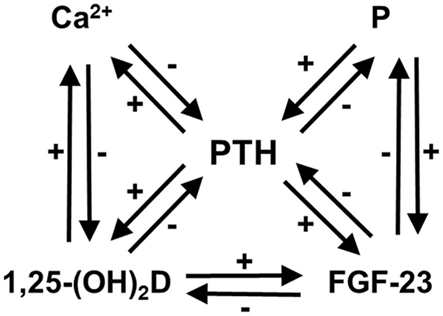
Figure 1. Feedback loops in Ca and P homeostasis, modified from previous reports (Imanishi et al., 2009a,b). There are feedback loops between Ca2+, P, 1,25-(OH)2D, FGF-23, and PTH. Ca2+, 1,25-(OH)2D, and FGF-23 suppress PTH secretion, whereas P overload accelerates it. The P overload does not always cause hyperphosphatemia, except for in some conditions such as chronic kidney disease.
Three receptors in parathyroid cells that are important in Ca and P homeostasis are the calcium-sensing receptor (CaR) and FGF receptor (FGFR-Klotho complex), which are located on the cell surface, and the vitamin D receptor (VDR) in the nucleus. Abnormal responses of these receptors by their ligands have been reported in the pathogenesis of PHPT and SHPT.
This review will focus on the animal models of parathyroid diseases, which exhibit abnormalities in Ca and P homeostasis (Table 1).
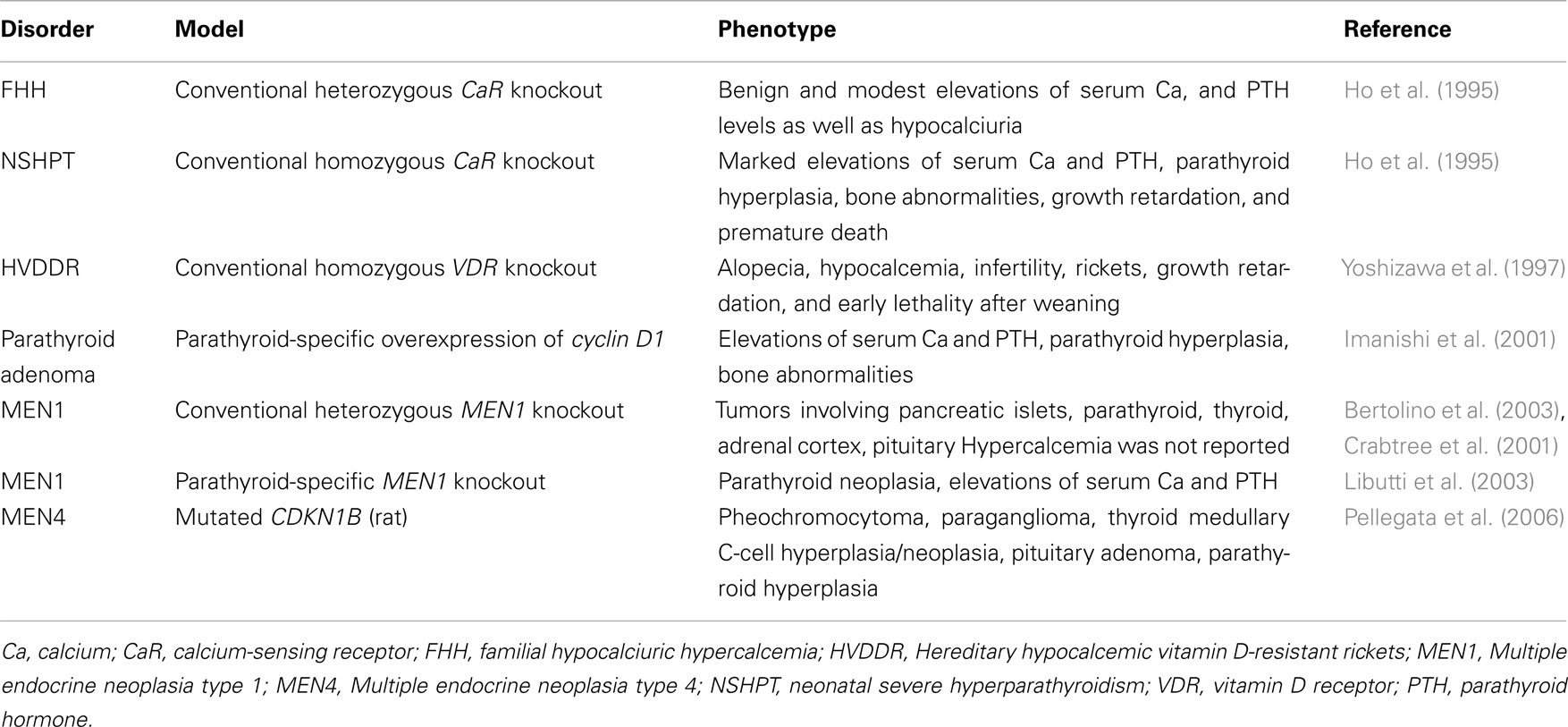
Table 1. Mouse models for parathyroid diseases.
Receptors in Parathyroid Cells
Calcium-Sensing Receptor
Positional cloning approaches have clarified that loss-of-function mutations in the CaR gene cause familial hypocalciuric hypercalcemia (heterozygous mutations) and neonatal severe hyperparathyroidism (homozygous mutations; Pollak et al., 1993). CaR has a crucial role in PTH secretion from parathyroid cells by sensing extracellular Ca2+ (Figure 2).
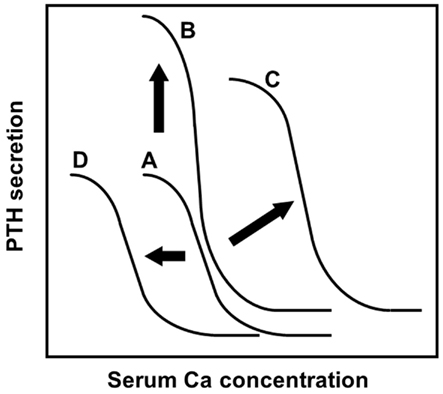
Figure 2. The sigmoidal curve of the PTH-Ca relationship, modified from a previous report (Imanishi, 2002). The analyses of PTH secretions inhibited by extracellular Ca in vitro revealed the sigmoidal relationship of the PTH-Ca relationship. Setpoint, the Ca concentration causing half-maximal inhibition of PTH secretion, is an indicator of sensitivity of parathyroid cells to extracellular Ca by CaR. (A) The relationship in healthy subjects was fitted to a symmetrical sigmoidal curve. (B) The normal sigmoidal curve will shift upward when the secretory cell number is increased, without changing its setpoint. (C) An altered sigmoidal curve is observed in human parathyroid adenomas, refractory SHPT, by changing the setpoint to the right. In the case of severe setpoint shift, PTH secretion is persistent even at high Ca concentration: so-called “autonomous” PTH secretion. An altered PTH-Ca relationship was also observed in PTH-cyclin D1 transgenic mice (Imanishi et al., 2001, 2009a). (D) Administration of cinacalcet or activating mutation of CaR observed in autosomal dominant hypocalcemia increases the CaR sensitivity to serum Ca. Activations of CaR result in the PTH-Ca relationship curve moving to the left.
Heterozygous knockout mice for CaR exhibited a similar phenotype of familial hypocalciuric hypercalcemia (Ho et al., 1995). Serum PTH levels were inappropriately elevated, but their parathyroid glands did not enlarge. Homozygous knockout mice had markedly elevated serum Ca, PTH, retarded growth, and premature death (Ho et al., 1995), symptoms that are concordant with human neonatal severe hyperparathyroidism. Double homozygous CaR- and PTH-deficient (CaR−/−PTH−/−) mice were rescued from early lethality and skeletal abnormalities, and exhibited normocalcemia with undetectable serum PTH (Liu et al., 2011), indicating that normocalcemia in patients with neonatal severe hyperparathyroidism may lengthen their lifespan and normalize skeletal growth and development.
Vitamin D Receptor
1,25(OH)2D3 is a steroid hormone that plays a crucial role in Ca and P homeostasis, which are mediated by the VDR. Hereditary hypocalcemic vitamin D-resistant rickets (HVDDR) is an autosomal recessive disorder, caused by inactivating mutations in the VDR gene, resulting in target tissue insensitivity to 1,25(OH)2D3 (Haussler et al., 1998). VDR knockout mice exhibit hypocalcemia, hypophosphatemia, rickets, alopecia, and hyperparathyroidism with enlarged parathyroid glands, a phenotype that is similar to HVDDR (Yoshizawa et al., 1997). Tissue-specific ablation of VDR in parathyroid tissue exhibits decreased parathyroid CaR expression and a moderate increment in basal PTH levels. However, no significant abnormalities in PTH-Ca sigmoidal curves were observed (Meir et al., 2009), suggesting a limited role for VDR in parathyroid patho-physiology.
FGF Receptor-Klotho Complex
Klotho, which is expressed in the kidney, and in the pituitary and parathyroid glands, converts FGFR1, a canonical receptor for various FGFs, into a specific receptor for FGF-23 (Urakawa et al., 2006). FGF-23 null mice exhibit various senescence-like phenotypes such as a short lifespan, infertility, atrophy of lymphopoietic and reproductive organs, decreased bone mineral density, and ectopic calcification, a phenotype that is similar to Klotho-deficient mice (Shimada et al., 2004), suggesting that FGF-23 signaling is Klotho dependent.
The parathyroid cells expressing Klotho and FGFR1 are responsive to FGF-23, both in vivo and in vitro (Ben-Dov et al., 2007). Reduced expressions of Klotho and FGFR1 in hyperplastic parathyroid glands from SHPT patients (Komaba et al., 2010), suggesting reduced signaling by FGF-23 to parathyroid cells, have a role in the development of SHPT. However, studies on Klotho expression in uremic animals show conflicting results (Canalejo et al., 2010; Hofman-Bang et al., 2010). Further studies are necessary to clarify the role of FGFR-Klotho signaling in uremic parathyroid glands.
Genes Identified in Parathyroid Diseases
Cyclin D1
Cyclin D1 was identified from parathyroid adenomas which harbored a DNA rearrangement that separated the PTH gene’s 5′ flanking region from PTH coding exons, and the DNA recombined with cyclin D1 proto-oncogene (Arnold, 1993). The parathyroid tissue-specific enhancer in the PTH 5′ flanking region drives the cyclin D1 expression located downstream of the enhancer by the rearrangement (Mallya et al., 2010).
To define the role of cyclin D1 in parathyroid neoplasia, transgenic mice that overexpress the cyclin D1 oncogene in parathyroid glands were generated using a transgene that mimics the human PTH-cyclin D1 gene rearrangement (Imanishi et al., 2001). PTH-cyclin D1 transgenic mice not only developed abnormal parathyroid cell proliferation, but also developed biochemical hyperparathyroidism with characteristic abnormalities in bone. Specifically, the transgenic mice had an altered PTH-Ca relationship, which was shifted upward and to the right, and was steeper relative to that in the wild-type mice (Figure 2), due to reduced CaR expression in the parathyroid glands of transgenic animals.
Cinacalcet is an allosteric modulator that activates the CaR and inhibits PTH secretion from parathyroid cells. Administration of cinacalcet shifts the sigmoidal curve left in the PTH-Ca relationship (Figure 2), because cinacalcet increases the sensitivity of CaR to Ca in parathyroid cells. A single administration of cinacalcet significantly suppressed serum Ca levels in PTH-cyclin D1 transgenic mice with moderate biochemical hyperparathyroidism (Kawata et al., 2005). In older transgenic mice with advanced hyperparathyroidism caused by severe hypo-expression of CaR, serum Ca, and PTH levels were not suppressed by the same doses of cinacalcet administered to mice with moderate biochemical hyperparathyroidism. These levels were, however, significantly suppressed by increasing cinacalcet, suggesting that higher doses of this compound could overcome severe hyperparathyroidism.
Cinacalcet also successfully suppressed parathyroid proliferation in the PTH-cyclin D1 transgenic mice (Imanishi et al., 2011). This mouse is thought to be a suitable model for PHPT and refractory SHPT in drug evaluations.
RET
Multiple endocrine neoplasia type 2A (MEN2A) is an autosomal dominant syndrome of multiple endocrine neoplasms, including medullary thyroid carcinoma, pheochromocytoma, and multiglandular parathyroid tumors. The gene responsible for MEN2A was identified as germline mutations of the RET proto-oncogene, which encodes a tyrosine kinase receptor with a cadherin-like and cystein-rich extracellular domain (Mulligan et al., 1993). The MEN2A mutation leads to the dimerization of RET even in the absence of its ligand, with consequent constitutive activation of the intracellular signaling pathways (Santoro et al., 1995).
Transgenic mice expressing the RET proto-oncogene with an MEN2A mutation (cysteine 6343arginine) developed thyroid C-cell hyperplasia or medullary carcinoma (Kawai et al., 2000). Despite the widespread transgene expression, however, transgenic mice displayed a very peculiar tissue-restricted phenotype, such as mammary or parotid gland adenocarcinoma. The role of RET should be elucidated in the pathogenesis of parathyroid tumorigenesis.
MEN1
Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominant familial endocrine neoplasm syndrome characterized by tumors in parathyroids, enteropancreatic endocrine tissues, and the anterior pituitary. MEN1, encoding menin, is a tumor suppressor gene, contributing to a mutated cell’s selective advantage for growth through its bi-allelic inactivation. Menin interacts with Smad3 and enhance the TGF-β signaling pathway to inhibit cell proliferation (Kaji et al., 2001). Menin also interacts with histone modifying enzymes, transcription factors including nuclear receptors to suppress cell proliferation.
Mice with heterozygous deletion of the MEN1 gene exon 3–8 developed tumors involving pancreatic islets, and the parathyroid, thyroid, adrenal cortex, and pituitary, with loss of the wild-type MEN1 allele (Crabtree et al., 2001). Another mouse knockout model has been generated by deleting exon 3, and its heterozygous mice developed parathyroid adenomas and carcinomas, insulinomas, gastrinomas, glucagonomas, prolactinomas, or somatotrophinomas (Bertolino et al., 2003). All these features seem to be compatible with human MEN1 syndrome. Although mice with heterozygous MEN1 inactivation developed parathyroid neoplasia, hypercalcemia was not reported (Crabtree et al., 2001; Bertolino et al., 2003).
The mice with parathyroid-specific deletion of the MEN1 gene exhibited not only parathyroid neoplasia but also biochemical hyperparathyroidism such as hypercalcemia with elevated PTH concentration (Libutti et al., 2003). It is still unknown why only the mice with parathyroid-specific deletion of the MEN1 gene but not conventional MEN1 mice exhibited biochemical hyperparathyroidism.
HRPT2
Hyperparathyroidism-jaw tumor (HPT-JT) syndrome is a rare autosomal dominant disorder, characterized by cystic parathyroid tumors and fibro-osseous lesions of the mandible and maxilla. The gene responsible for HPT-JT encodes parafibromin, a ubiquitously expressed 531-amino acid protein (Carpten et al., 2002). The inactivated mutations were observed in the encoded parafibromin protein, suggesting the gene is a tumor suppressor.
To determine the role of parafibromin in parathyroid tumorigenesis, a transcription factor encoded by the Hrpt2 gene, conventional, and conditional knockout mice were generated (Wang et al., 2008). Homozygous knockout mice were embryonic lethal. Controlled deletion of the gene after embryonic day 8.5 resulted in apoptosis and growth retardation. Deletion of the gene in the adult led to severe cachexia and early death. These results revealed the important role of parafibromin in development and survival, but its role in parathyroid tumorigenesis is still unknown.
CDKN1B
Recently, an MEN1-like recessive multiple endocrine neoplasia-like syndrome was identified (named MEN4) in rats and humans, which is due to mutations in the CDKN1B gene, encoding for p27kip1, a cyclin-dependent kinase (Cdk) inhibitor that regulates the transition of cells from G1 to S phase (Pellegata et al., 2006). Mutated CDKN1B, encoding the p27Kip1, was also identified in MENX rats with juvenile cataracts (Pellegata et al., 2006). These rats exhibited neoplasia of multiple endocrine tissues such as pheochromocytoma, paraganglioma, thyroid medullary C-cell hyperplasia/neoplasia, adenoma of the anterior pituitary gland, and hyperplasia of the parathyroid gland (Fritz et al., 2002), which were compatible to human MEN4.
Interestingly, p27-null mice developed pituitary adenomas as the sole tumor phenotype, although the MENX rats developed a broader spectrum of neuroendocrine tumors (Pellegata et al., 2006). The altered sensitivity to p27 loss in various tissues by species may lead to the altered tissue expression patterns and phenotypes.
Conclusion
In this review, the animal models exhibiting abnormal Ca and P homeostasis were discussed. Many kinds of animal models can be generated by manipulating genes relating to Ca and P homeostasis, and genes identified in parathyroid diseases. Uremic animals such as 5/6-nephrectomized rats are also good models for SHPT, which is not discussed in this review. These models are the best tool not only for understanding the pathogenesis of parathyroid diseases, but also for developing new therapies for these diseases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Arnold, A. (1993). Genetic basis of endocrine disease 5. Molecular genetics of parathyroid gland neoplasia. J. Clin. Endocrinol. Metab. 77, 1108–1112.
Ben-Dov, I. Z., Galitzer, H., Lavi-Moshayoff, V., Goetz, R., Kuro-O, M., Mohammadi, M., Sirkis, R., Naveh-Many, T., and Silver, J. (2007). The parathyroid is a target organ for FGF23 in rats. J. Clin. Invest. 117, 4003–4008.
Bertolino, P., Tong, W. M., Galendo, D., Wang, Z. Q., and Zhang, C. X. (2003). Heterozygous Men1 mutant mice develop a range of endocrine tumors mimicking multiple endocrine neoplasia type 1. Mol. Endocrinol. 17, 1880–1892.
Canalejo, R., Canalejo, A., Martinez-Moreno, J. M., Rodriguez-Ortiz, M. E., Estepa, J. C., Mendoza, F. J., Munoz-Castaneda, J. R., Shalhoub, V., Almaden, Y., and Rodriguez, M. (2010). FGF23 fails to inhibit uremic parathyroid glands. J. Am. Soc. Nephrol. 21, 1125–1135.
Carpten, J. D., Robbins, C. M., Villablanca, A., Forsberg, L., Presciuttini, S., Bailey-Wilson, J., Simonds, W. F., Gillanders, E. M., Kennedy, A. M., Chen, J. D., Agarwal, S. K., Sood, R., Jones, M. P., Moses, T. Y., Haven, C., Petillo, D., Leotlela, P. D., Harding, B., Cameron, D., Pannett, A. A., Hoog, A., Heath, H. III, James-Newton, L. A., Robinson, B., Zarbo, R. J., Cavaco, B. M., Wassif, W., Perrier, N. D., Rosen, I. B., Kristoffersson, U., Turnpenny, P. D., Farnebo, L. O., Besser, G. M., Jackson, C. E., Morreau, H., Trent, J. M., Thakker, R. V., Marx, S. J., Teh, B. T., Larsson, C., and Hobbs, M. R. (2002). HRPT2, encoding parafibromin, is mutated in hyperparathyroidism-jaw tumor syndrome. Nat. Genet. 32, 676–680.
Crabtree, J. S., Scacheri, P. C., Ward, J. M., Garrett-Beal, L., Emmert-Buck, M. R., Edgemon, K. A., Lorang, D., Libutti, S. K., Chandrasekharappa, S. C., Marx, S. J., Spiegel, A. M., and Collins, F. S. (2001). A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. Proc. Natl. Acad. Sci. U.S.A. 98, 1118–1123.
Fritz, A., Walch, A., Piotrowska, K., Rosemann, M., Schaffer, E., Weber, K., Timper, A., Wildner, G., Graw, J., Hofler, H., and Atkinson, M. J. (2002). Recessive transmission of a multiple endocrine neoplasia syndrome in the rat. Cancer Res. 62, 3048–3051.
Haussler, M. R., Whitfield, G. K., Haussler, C. A., Hsieh, J. C., Thompson, P. D., Selznick, S. H., Dominguez, C. E., and Jurutka, P. W. (1998). The nuclear vitamin D receptor: biological and molecular regulatory properties revealed. J. Bone Miner. Res. 13, 325–349.
Ho, C., Conner, D. A., Pollak, M. R., Ladd, D. J., Kifor, O., Warren, H. B., Brown, E. M., Seidman, J. G., and Seidman, C. E. (1995). A mouse model of human familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Nat. Genet. 11, 389–394.
Hofman-Bang, J., Martuseviciene, G., Santini, M. A., Olgaard, K., and Lewin, E. (2010). Increased parathyroid expression of klotho in uremic rats. Kidney Int. 78, 1119–1127.
Imanishi, Y. (2002). Molecular pathogenesis of tumorigenesis in sporadic parathyroid adenomas. J. Bone Miner. Metab. 20, 190–195.
Imanishi, Y., Hosokawa, Y., Yoshimoto, K., Schipani, E., Mallya, S., Papanikolaou, A., Kifor, O., Tokura, T., Sablosky, M., Ledgard, F., Gronowicz, G., Wang, T. C., Schmidt, E. V., Hall, C., Brown, E. M., Bronson, R., and Arnold, A. (2001). Primary hyperparathyroidism caused by parathyroid-targeted overexpression of cyclin D1 in transgenic mice. J. Clin. Invest. 107, 1093–1102.
Imanishi, Y., Inaba, M., Kawata, T., and Nishizawa, Y. (2009a). Animal models of hyperfunctioning parathyroid diseases for drug development. Expert Opin. Drug Discov. 4, 727–740.
Imanishi, Y., Inaba, M., Kawata, T., and Nishizawa, Y. (2009b). Cinacalcet in hyperfunctioning parathyroid diseases. Ther. Apher. Dial. 13(Suppl. 1), S7–S11.
Imanishi, Y., Kawata, T., Kenko, T., Wada, M., Nagano, N., Miki, T., Arnold, A., and Inaba, M. (2011). Cinacalcet HCl suppresses cyclin D1 oncogene-derived parathyroid cell proliferation in a murine model for primary hyperparathyroidism. Calcif. Tissue Int. 89, 29–35.
Kaji, H., Canaff, L., Lebrun, J. J., Goltzman, D., and Hendy, G. N. (2001). Inactivation of menin, a Smad3-interacting protein, blocks transforming growth factor type beta signaling. Proc. Natl. Acad. Sci. U.S.A. 98, 3837–3842.
Kawai, K., Iwashita, T., Murakami, H., Hiraiwa, N., Yoshiki, A., Kusakabe, M., Ono, K., Iida, K., Nakayama, A., and Takahashi, M. (2000). Tissue-specific carcinogenesis in transgenic mice expressing the RET proto-oncogene with a multiple endocrine neoplasia type 2A mutation. Cancer Res. 60, 5254–5260.
Kawata, T., Imanishi, Y., Kobayashi, K., Kenko, T., Wada, M., Ishimura, E., Miki, T., Nagano, N., Inaba, M., Arnold, A., and Nishizawa, Y. (2005). Relationship between parathyroid calcium-sensing receptor expression and potency of the calcimimetic, cinacalcet, in suppressing parathyroid hormone secretion in an in vivo murine model of primary hyperparathyroidism. Eur. J. Endocrinol. 153, 587–594.
Komaba, H., Goto, S., Fujii, H., Hamada, Y., Kobayashi, A., Shibuya, K., Tominaga, Y., Otsuki, N., Nibu, K., Nakagawa, K., Tsugawa, N., Okano, T., Kitazawa, R., Fukagawa, M., and Kita, T. (2010). Depressed expression of Klotho and FGF receptor 1 in hyperplastic parathyroid glands from uremic patients. Kidney Int. 77, 232–238.
Libutti, S. K., Crabtree, J. S., Lorang, D., Burns, A. L., Mazzanti, C., Hewitt, S. M., O’Connor, S., Ward, J. M., Emmert-Buck, M. R., Remaley, A., Miller, M., Turner, E., Alexander, H. R., Arnold, A., Marx, S. J., Collins, F. S., and Spiegel, A. M. (2003). Parathyroid gland-specific deletion of the mouse Men1 gene results in parathyroid neoplasia and hypercalcemic hyperparathyroidism. Cancer Res. 63, 8022–8028.
Liu, J., Lv, F., Sun, W., Tao, C., Ding, G., Karaplis, A., Brown, E., Goltzman, D., and Miao, D. (2011). The abnormal phenotypes of cartilage and bone in calcium-sensing receptor deficient mice are dependent on the actions of calcium, phosphorus, and PTH. PLoS Genet. 7, e1002294. doi: 10.1371/journal.pgen.1002294
Mallya, S. M., Wu, H. I., Saria, E. A., Corrado, K. R., and Arnold, A. (2010). Tissue-specific regulatory regions of the PTH gene localized by novel chromosome 11 rearrangement breakpoints in a parathyroid adenoma. J. Bone Miner. Res. 25, 2606–2612.
Meir, T., Levi, R., Lieben, L., Libutti, S., Carmeliet, G., Bouillon, R., Silver, J., and Naveh-Many, T. (2009). Deletion of the vitamin D receptor specifically in the parathyroid demonstrates a limited role for the receptor in parathyroid physiology. Am. J. Physiol. Renal Physiol. 297, F1192–F1198.
Mulligan, L. M., Kwok, J. B., Healey, C. S., Elsdon, M. J., Eng, C., Gardner, E., Love, D. R., Mole, S. E., Moore, J. K., Papi, L., Ponder, M. A., Telenius, H., Tunnacliffe, A., and Ponder, B. A. J. (1993). Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature 363, 458–460.
Pellegata, N. S., Quintanilla-Martinez, L., Siggelkow, H., Samson, E., Bink, K., Hofler, H., Fend, F., Graw, J., and Atkinson, M. J. (2006). Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc. Natl. Acad. Sci. U.S.A. 103, 15558–15563.
Pollak, M. R., Brown, E. M., Chou, Y. H., Hebert, S. C., Marx, S. J., Steinmann, B., Levi, T., Seidman, C. E., and Seidman, J. G. (1993). Mutations in the human Ca2+-sensing receptor gene cause familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Cell 75, 1297–1303.
Santoro, M., Carlomagno, F., Romano, A., Bottaro, D. P., Dathan, N. A., Grieco, M., Fusco, A., Vecchio, G., Matoskova, B., Kraus, M. H., and Di Fiore, P. P. (1995). Activation of RET as a dominant transforming gene by germline mutations of MEN2A and MEN2B. Science 267, 381–383.
Shimada, T., Kakitani, M., Yamazaki, Y., Hasegawa, H., Takeuchi, Y., Fujita, T., Fukumoto, S., Tomizuka, K., and Yamashita, T. (2004). Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J. Clin. Invest. 113, 561–568.
Urakawa, I., Yamazaki, Y., Shimada, T., Iijima, K., Hasegawa, H., Okawa, K., Fujita, T., Fukumoto, S., and Yamashita, T. (2006). Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444, 770–774.
Wang, P., Bowl, M. R., Bender, S., Peng, J., Farber, L., Chen, J., Ali, A., Zhang, Z., Alberts, A. S., Thakker, R. V., Shilatifard, A., Williams, B. O., and Teh, B. T. (2008). Parafibromin, a component of the human PAF complex, regulates growth factors and is required for embryonic development and survival in adult mice. Mol. Cell. Biol. 28, 2930–2940.
Yoshizawa, T., Handa, Y., Uematsu, Y., Takeda, S., Sekine, K., Yoshihara, Y., Kawakami, T., Arioka, K., Sato, H., Uchiyama, Y., Masushige, S., Fukamizu, A., Matsumoto, T., and Kato, S. (1997). Mice lacking the vitamin D receptor exhibit impaired bone formation, uterine hypoplasia and growth retardation after weaning. Nat. Genet. 16, 391–396.
Keywords: calcitriol, CaR, cinacalcet, CKD, FGF-23, hyperparathyroidism, Klotho, PTH
Citation: Imanishi Y, Nagata Y and Inaba M (2012) Parathyroid diseases and animal models. Front. Endocrin. 3:78. doi: 10.3389/fendo.2012.00078
Received: 21 April 2012; Accepted: 31 May 2012;
Published online: 27 June 2012.
Edited by:
Francesca Carlomagno, Universita’ degli Studi di Napoli Federico II, ItalyReviewed by:
Caroline Kim, Perelman School of Medicine at the University of Pennsylvania, USANikolaos Stathatos, Massachusetts General Hospital, USA
Copyright: © 2012 Imanishi, Nagata and Inaba. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Yasuo Imanishi, Department of Metabolism, Endocrinology and Molecular Medicine, Osaka City University Graduate School of Medicine, 1-4-3, Asahi-machi, Abeno-ku, Osaka 545-8585, Japan. e-mail:aW1hbmlzaGlAbWVkLm9zYWthLWN1LmFjLmpw