
 Abdulrahman Al-Senaidy3
Abdulrahman Al-Senaidy3
- 1 Department of Molecular Pharmacology, Institute of Functional Genomics, CNRS UMR5203, Universities Montpellier 1 and 2, Montpellier, France
- 2 INSERM U661, Montpellier, France
- 3 Department of Biochemistry, College of Science, King Saud University, Riyadh, Kingdom of Saudi Arabia
Since its development, the bioluminescence resonance energy transfer (BRET) approach has been extensively applied to study G protein-coupled receptors (GPCRs) in real-time and in live cells. One of the major aspects of GPCRs investigated in considerable details is their physical coupling to the heterotrimeric G proteins. As a result, new concepts have emerged, but few questions are still a matter of debate illustrating the complexity of GPCR-G protein interactions and coupling. Here, we summarized the recent advances on our understanding of GPCR-G protein coupling based on BRET approaches and supported by other FRET-based studies. We essentially focused on our recent studies in which we addressed the concept of preassembly vs. the agonist-dependent interaction between the protease-activated receptor 1 (PAR1) and its cognate G proteins. We discussed the concept of agonist-induced conformational changes within the preassembled PAR1-G protein complexes as well as the critical question how the multiple coupling of PAR1 with two different G proteins, Gαi1 and Gα12, but also β-arrestin 1, can be regulated.
Introduction
G protein-coupled receptors (GPCRs) constitute one of the largest cell surface receptor family, and are involved in many cellular signaling and physiological responses (Bockaert, 1991; Gether, 2000). They are encoded by the largest gene family in the mammalian genomes and they constitute the site of binding and action of a large panel of natural mediators such as hormones and neurotransmitters (Bockaert and Pin, 1999). Thus, GPCRs are known to be the target of many drugs used to treat diseases (Schlyer and Horuk, 2006). Initially, the cellular signaling via GPCRs has been thought to occur only by their interaction with and activation of several types of guanine nucleotide binding proteins or G proteins (Limbird, 1983; Bockaert et al., 1987; Gilman, 1987). However, it is now obvious that in addition to G protein-dependent signaling, GPCRs also activate G protein-independent signaling pathways (Hermans, 2003; Lefkowitz and Whalen, 2004). Furthermore, GPCRs are now known to interact with many intracellular proteins other than G proteins and these proteins play a major role in promoting and regulating GPCR signaling (Brady and Limbird, 2002).
From their discovery until now, the coupling of GPCRs to the heterotrimeric G proteins and their activation has been extensively studied. The initial model explaining their functioning has considerably evolved (Bourne, 1997; Limbird, 2004; Strange, 2008) and new concepts have emerged such as, constitutive activity and precoupling (Leff and Scaramellini, 1998; Seifert and Wenzel-Seifert, 2002), multiple coupling (Hamm, 1998; Hermans, 2003; Perez and Karnik, 2005), functional selectivity (Rajagopal et al., 2011), and the role of GPCR oligomerization (Dean et al., 2001). The initial ternary model of GPCR/G protein activation postulated that agonist binding promotes the transition of the receptor from the inactive to the active state leading to the physical association of the receptor with the heterotrimeric Gαβγ protein, allowing the exchange of bound GDP for GTP in the Gα subunit (Limbird et al., 1980; Gether and Kobilka, 1998). GTP binding stabilizes the active state of the G protein leading to the dissociation of the receptor-G protein complex allowing Gα on one hand, and Gβγ on the other hand to act on their respective effectors and initiating signal transduction (Hamm, 1998; Oldham and Hamm, 2008). Moreover, it is now accepted that ligand binding to GPCRs promotes conformational changes in the receptor leading to the transition of the receptor to its active state. This evidence come from the functional studies of the downstream signaling as well as the biochemical, biophysical, and structural analysis of GPCRs themselves (Gether et al., 1995; Vilardaga et al., 2003; Bockenhauer et al., 2011).
Over the past 20 years, the question of how GPCR-G protein coupling occurs has been widely studied initially using radioligand binding (Stadel et al., 1981) and biochemical techniques (Smith and Limbird, 1981; Neumann et al., 2002) and recently through crystallographic analysis (Palczewski et al., 2000; Kobilka and Schertler, 2008) and energy transfer-based approaches, bioluminescence resonance energy transfer (BRET) and FRET (bioluminescence/fluorescence resonance energy transfer; Pin et al., 2008; Vilardaga et al., 2009; Lohse et al., 2012). BRET and FRET methods allow the determination of the proximity and/or relative orientation of two chromophores fused to the proteins being studied, such as between a GPCR and its cognate G protein, or between Gα and Gβγ subunits, directly in real-time and in live cells (Azpiazu and Gautam, 2004; Frank et al., 2005; Gales et al., 2005; Ayoub et al., 2010). As discussed in this review, these studies challenged the initial GPCR-G protein model to some extend and revealed new concepts with regard to receptor-G protein coupling as well as G protein subunit dissociation.
In this review, we focus on the recent studies using the BRET approach to investigate the physical and functional interaction between GPCRs and the heterotrimeric G proteins taking lessons from our observations on the interaction of the thrombin receptor, protease-activated receptor 1 (PAR1), with Gαi1 and Gα12 as well as β-arrestin 1 (Ayoub et al., 2007, 2009, 2010). PAR1 belongs to a particular GPCR family composed by three other subtypes, PAR2, PAR3, and PAR4, known to be activated by various and highly selective serine proteases such as thrombin, trypsin, plasmin, and the factor Xa (Cottrell et al., 2002; Hollenberg and Compton, 2002). The activation mechanism of PARs involved the cleavage of their N-terminal extremity by the protease, unmasking a new N terminus that acts as a tethered ligand, directly activating the transmembrane core of the receptor (Coughlin, 2000). Following activation, cleaved PARs are known to undergo a rapid desensitization, internalization, and degradation (Trejo, 2003). This desensitization and internalization processes involve the phosphorylation of the receptor by G protein-coupled receptor kinases and the recruitment of arrestins (Trejo, 2003). PARs have been reported to play crucial roles in a number of physiological processes such as thrombosis, vascular development, inflammation, cell proliferation, and tumorigenesis and therefore they are considered as interesting targets for the treatment of various pathologies (Coughlin, 2005). PAR1 is a prototype of the PARs family members, characterized by the diversity of its signaling pathways involving different G protein classes as well as arrestins. Indeed, PAR1 has been reported to couple to Gi/o, Gq as well as G12/13 proteins promoting multiple downstream signaling responses in various cellular models (Coughlin, 2000; Marinissen et al., 2003).
BRET to Study GPCR-G Protein Interaction
As mentioned above the initial GPCR/G protein activation model was based on elegant biochemical experiments using solubilized and purified proteins. Thus, for a long time and before the emergence of BRET and FRET techniques the detailed analysis of GPCRs and G protein activation in real-time and in live cells was very limited. Now such an analysis becomes feasible and indeed within the past 7 years a number of studies has examined the activation process of the heterotrimeric G proteins by various GPCRs, using either FRET or BRET techniques (Vilardaga et al., 2009; Lohse et al., 2012). The historically first energy transfer-based assay to study G protein activation by GPCRs was based on FRET approach using GFP variants as donor and acceptor and the pioneer study was in Dictyostelium discoideum using FRET between Gα and Gβγ subunits showing a direct evidence for G protein dissociation in live cells (Janetopoulos et al., 2001). Then other FRET studies on the activation and association/dissociation of the G protein subunits have been reported in yeast and various mammalian cell lines (Yi et al., 2003; Azpiazu and Gautam, 2004; Frank et al., 2005; Gibson and Gilman, 2006). These studies have reported contradictory conclusions with regard to the dissociation or non-dissociation of Gα and Gβγ subunits after receptor activation and this may depend on the GPCR-G protein pair.
Later, the investigation of the interaction and activation of GPCR-G protein complexes in real-time became possible through the measurement of FRET or BRET signals between the activating GPCRs themselves and either Gα, Gβ, or Gγ subunits (Gales et al., 2005; Hein et al., 2005; Nobles et al., 2005; Galés et al., 2006; Ayoub et al., 2007, 2010; Hasbi et al., 2007; Qin et al., 2008). These assays are based on the fusion of the energy donor and the energy acceptor with the receptor (generally on its C-terminus) and one of the G protein subunit (α or βγ at some specific position within the G protein subunit) and their co-expression and activation by the agonist (Figure 1A; Galés et al., 2006; Ayoub et al., 2007, 2010). Then receptor-G protein interaction and the activation of the complex are assessed either in real-time before and after agonist stimulation or after agonist preincubation depending on the model used (Figure 1B).
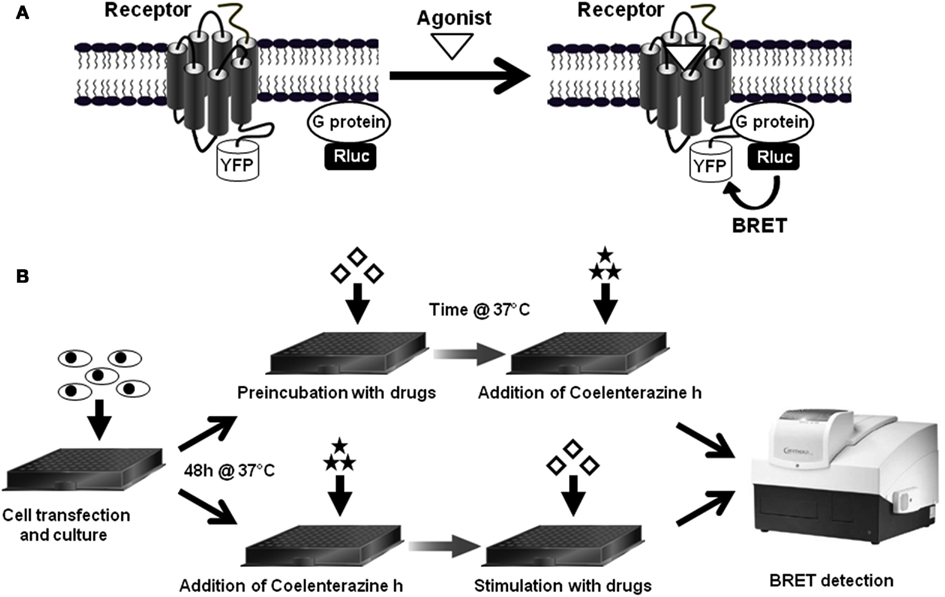
Figure 1. BRET assay to study receptor-G protein interactions in live cells. (A) First, to study the interaction between a GPCR and its cognate heterotrimeric G protein, the G protein subunit (α, β, or γ) is fused to the energy donor, Renilla luciferase (Rluc) and the receptor is fused to the energy acceptor, YFP, and then both fusion protein are co-expressed and BRET signal is measured before and after receptor activation, as described previously (Ayoub et al., 2007, 2010). (B) The standard BRET protocol is based on cell transfection and culture in BRET compatible 96-well plates and then BRET assay can be performed in two different ways depending on the specificities of the model studied. In the first way, cells are first preincubated with drugs (agonist, antagonist, inhibitor etc.,) and then BRET is measured straightaway after the addition of Rluc substrate, Coelenterazine h. This method can be used for slow and sustained ligand-induced interactions, such as a stable β-arrestin recruitment or to detect irreversible BRET changes within constitutive protein complexes. The second way consists to add Coelenterazine h and measure BRET before any cell stimulation (basal BRET) and then stimulate cells with drugs in the aim to detect any rapid and transient BRET change resulted from the activation of the protein complexes. This method is recommended to detect rapid and reversible conformational changes within receptor-G protein complexes.
We will here illustrate such studies based on our recent finding using PAR1 and different effectors. In these studies we used proteins fused to either the energy donor Renilla luciferase (Rluc) or the energy acceptor yellow fluorescent protein (YFP). The energy transfer process between Rluc and YFP mainly depends on the distance between the two proteins of interest and/or their relative orientation within the protein complexes (Pin et al., 2008). Thus, the intimate interaction which is supposed to occur between GPCRs and their specific heterotrimeric G proteins constitutes an exciting field of investigation using BRET as a proximity- and conformational-based approach.
To monitor GPCR-G protein interaction and activation, three different assay configurations can be used: (i) the fusion of the receptor with YFP (Receptor-YFP) and the Gα subunit with Rluc (Gα-Rluc) in the presence of untagged Gβ and Gγ subunits (Figure 2A), (ii) the fusion of the Receptor-YFP and the Gβ or Gγ subunits with Rluc (Gβ/Gγ-Rluc) in the presence of untagged Gα subunit (Figure 2B), and (iii) the fusion of Gα subunit with Rluc (Gα-Rluc) and the Gβ or Gγ subunits with YFP (Gβ/Gγ-YFP) in the presence of untagged GPCR (Figure 2C). For each BRET assay configuration, the fusion proteins are transiently co-expressed in cell lines and then the basal BRET signal as well as the agonist-promoted BRET changes are measured in real-time and live cells as previously described (Ayoub et al., 2007, 2009, 2010). Therefore, real-time kinetic and dose-response analysis as well as the application of specific GPCR antagonists or agents targeting G proteins can be performed.
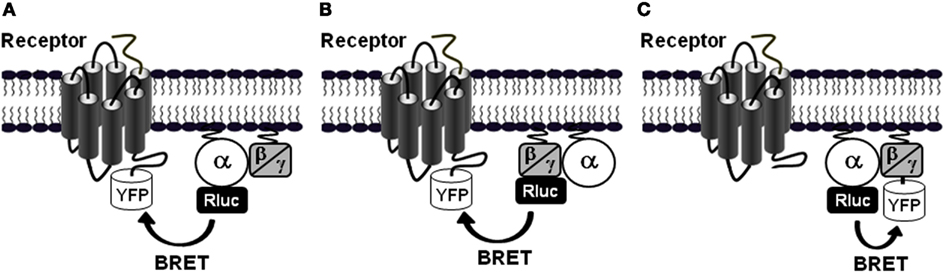
Figure 2. Different BRET assay configurations to study receptor-G protein interactions. To investigate the interaction between GPCRs and the heterotrimeric G proteins and their activation in live cells using BRET, at least three configurations of the assay can be used. (A) BRET between the receptor fused to YFP and the Gα subunit, which can be Gαs, Gαi/o, Gαq, or Gα12/13, fused to Rluc in the presence of the untagged β and γ subunits. In this configuration, either BRET increase or decrease can be expected depending on the receptor-G protein pair and the nature of ligand-induced effect, conformational changes or G protein recruitment (Galés et al., 2006; Ayoub et al., 2010). (B) BRET between the receptor fused to YFP and either the Gβ or Gγ subunit fused to Rluc in the presence of a specific untagged Gα subunit which could be Gαs, Gαi/o, Gαq, or Gα12/13 (Gales et al., 2005; Ayoub et al., 2009). Similarly to the configuration A, either BRET increase or decrease can be expected depending on the receptor-G protein pair and the ligand-induced conformational changes. (C) BRET between a specific Gα subunit fused to Rluc and either Gβ or Gγ fused to YFP in the presence of the untagged receptor. In this configuration, in most cases a BRET decrease is expected but a BRET increase is also possible (Galés et al., 2006).
Receptor-G Protein Precoupling/Preassembly
From the initial studies performed 30 years ago, it has become well accepted that agonist activation of a GPCR allows its physical association with a heterotrimeric G protein, promoting the GDP/GTP exchange on Gα subunit and G protein activation (De Lean et al., 1980; Limbird et al., 1980; Gether and Kobilka, 1998). It is evident that the study of receptor/G protein interaction has considerably evolved during the recent last years. However, some key questions around this subject are still unresolved and constitute a matter of debate. The main question concerns the dynamics of the receptor-G protein interaction and through the literature two different models have been proposed. The first model called “free collision coupling” model postulates that both GPCRs and G proteins diffuse freely within the plasma membrane, and only the active receptor (i.e., agonist-activated receptor) couple to and specifically activates G proteins (Leff, 1995; Hein and Bunemann, 2009; Lohse et al., 2012). This model corroborates the initial GPCR-G protein model postulating that agonist binding is the prerequisite to GPCR-G protein physical interaction and activation and once activated, the receptor and G proteins dissociate. In addition, the collision model is compatible to some degree with the ternary complex model, where agonist–receptor–G protein complex is stabilized in the absence of guanine nucleotides and GTP addition is supposed to dissociate the ternary complex. This dogma was well accepted for a long time by most of the GPCR scientists. The second model assumes that GPCRs and G proteins are “precoupled” which means here the receptor and G proteins form stable complexes regardless of the activation state of the receptor (Hein and Bunemann, 2009; Lohse et al., 2012). Thus, according to this model the agonist binding promotes receptor activation which leads to conformational changes within the pre-existing receptor-G protein complex resulting in G protein activation without physical dissociation between the receptor and G proteins. Of note, this model has been proposed to explain the high constitutive activity observed with some GPCRs (Lachance et al., 1999; Roka et al., 1999; Seifert and Wenzel-Seifert, 2002). However, other possibilities cannot be excluded such as; (i) preassociated receptors and G proteins which dissociate upon agonist-induced receptor activation, or even (ii) the separated inactive receptors and G proteins which form a stable complex only when the receptor is activated. Therefore, it is possible that GPCR-G protein coupling may be differentially structured; depending upon the specific GPCR, G protein subtype, their expression levels and the cellular system used as discussed previously (Vilardaga et al., 2009; Qin et al., 2011). This will be further discussed later in this review.
Our recent data using BRET to study the physical association of PAR1, with different G proteins revealed the existence of preassociated complex between PAR1 and Gαi1 protein (Ayoub et al., 2007, 2010), and PAR1 and Gαo (Ayoub et al., 2009) when both partners are transiently co-expressed in COS-7 cells. Indeed, a significant and saturable basal BRET signal was measured between Gαi1-Rluc and PAR1-YFP in the absence of receptor activation with thrombin or PAR1 peptide agonists. This finding was in fact not unexpected as it can be reconciled with the “precoupling” model of GPCRs but of course, the observation of a basal BRET signal between two different proteins raises the question with regard to the specificity of such a BRET signal measured under the resting condition. Our different assays and controls using BRET and also time-resolved FRET (TR-FRET) led us to accumulate several lines of evidence demonstrating the specificity of PAR1-Gαi1 association and the constitutive energy transfer signals measured. First, in contrast to the large BRET signal between PAR1 and Gαi1, no significant BRET signal could be detected between PAR1 and Gαs used a negative control and expressed at similar levels than Gαi1 (Ayoub et al., 2007, 2010). Although the absence of BRET does not mean the absence of interaction, since the lack of energy transfer can be the consequence of a perpendicular orientation of the dipoles, the absence of interaction was confirmed using a TR-FRET approach that only relies on the distance since the donor fluorophore cannot be constraint in its orientation (Pin et al., 2008). In addition, under similar conditions the basal BRET signal measured between Gα12- or Gα13-Rluc and PAR1-YFP was also very weak compared to Gαi1-Rluc (Ayoub et al., 2010) and this constitutes an interesting observation since PAR1 is also known to activate Gα12/13 proteins and this point will be discussed later. Second, the quantitative analysis of the expression levels of PAR1 and Gαi1 clearly indicated that the constitutive BRET signal measured cannot be explained by the overexpression of the BRET partners since 100,000 receptor molecules were expressed at the cell surface of COS-7 cells consistent with PAR1 expression in platelets (Ayoub et al., 2007). Third, the basal BRET signal measured between Gαi1-Rluc and PAR1-YFP was nicely saturable and blocked by the overexpression of an untagged Gαi1 protein demonstrating the specificity of the signal observed (Ayoub et al., 2007). Finally, the basal BRET signal between PAR1 and Gαi1 was also confirmed by TR-FRET developed for the first time to study GPCR-G protein interaction using Flag- and Myc-tagged proteins and antibodies conjugated with homogeneous TR-FRET-compatible fluorophores (Ayoub et al., 2010). Together, these observations rule out the possibility of an artifactual signal resulting from the overexpression of the fusion proteins because of the heterologous system used.
Furthermore, the other aspect that we addressed is the plausible link between the basal BRET measured between PAR1 and Gαi1 and any constitutive activity of the receptor-G protein complex. This is important since the precoupling model was proposed following the observation that many GPCRs display constitutive activity, a phenomena that would be consistent with the assembly of G proteins with the non-activated GPCR. We found that the constitutive BRET signal was completely insensitive to pertussis toxin (PTX) treatment which inhibits the Gαi1 protein activation (Ayoub et al., 2007, 2010), such that the assembly observed had nothing to do with G protein activation. Similarly, the basal BRET signal measured between PAR1 and Gαi1 was only partially diminished by BIM46187, a synthetic compound reported to inhibit GPCR-mediated signaling mostly by disrupting the physical association between GPCRs and the Gα subunit (Ayoub et al., 2009). Accordingly, our observation of the preassembly between PAR1 and Gαi1 was not related to any basal activity of the receptor. To clarify this issue, we considered the basal BRET observed as the consequence of a preassembly, rather than a precoupling that would instead be associated with a basal activation of the G protein.
The data obtained with PAR1 using BRET and TR-FRET are consistent with other BRET studies reporting a pre-association of other GPCRs such as α2A-adrenergic (Galés et al., 2006), δ-opioid (Audet et al., 2008), chemokine CXCR4 and CXCR7 (Levoye et al., 2009) receptors and their cognate G proteins in the absence of receptor activation. Similar observations using other techniques to investigate the interaction between other GPCRs and G protein types have been reported including FRET between various GPCRs (adrenergic α2A, Muscarinic M4, Dopamine 2S, and Adenosine A1) with the Gαo protein (Nobles et al., 2005; Philip et al., 2007) or FRAP between the muscarininc M3 receptor and Gαq protein (Qin et al., 2011) that all support GPCR-G protein preassembly. In this context, we should also include our data using TR-FRET technique showing a preassembly of Gα12 with the serotonin 5HT2c, vasopressin V1a, and muscarinic M1 receptors, but not PAR1, in COS-7 cells (Ayoub et al., 2010). Moreover, the GPCR-Gα preassembly concept can also be supported to some extent by other studies again based on BRET and FRET between Gα and Gβγ revealing a constant proximity between the G protein subunits (Bunemann et al., 2003; Galés et al., 2006). In these studies, it has been reported that GPCR activation promotes a relative movement of βγ and α subunits associated with G protein activation which is illustrated by either a partial increase or a partial decrease in the energy transfer efficiency following the receptor activation, depending on the position where BRET/FRET chromophores are inserted (Bunemann et al., 2003; Galés et al., 2006; Gibson and Gilman, 2006). This hypothesis of a non-dissociation between Gα and Gβγ is supported by the recent report that Gαq and Gβγ are still in proximity after binding of GRK2 as observed in the crystal structure of the Gαq-GRK2 complex (Tesmer et al., 2005).
All these data on GPCR-G protein preassembly in live cells using both BRET and different FRET-based assays are in fact supported by previous biochemical studies using coimmunoprecipitation between receptors and G proteins in the absence of receptor agonists and performed in different cellular backgrounds. This is true for D2 receptor and Gαi (Senogles et al., 1987), β2-adrenergic receptor and Gαs (Lachance et al., 1999), δ-opioid receptor, and Gαi (Law and Reisine, 1997), somatostatin receptor and Gαi and Gαo (Law et al., 1993), AT2 receptor and Gαi (Zhang and Pratt, 1996), melatonin MT1 receptor, and Gαi (Roka et al., 1999). Pharmacological studies have also been useful to demonstrate a tight association between a GPCR and its G protein as shown for the serotonin 5-HT7 preassociated with Gαs even in the absence of agonist (Andressen et al., 2006). As discussed later, such a preassembly is not observed with all GPCR-G protein couples, indicating that this cannot be considered as a general phenomena involved in receptor-G protein coupling, then illustrating the importance and the specificity of such association when it can be observed.
These data then raised the question of the functional significance of the basal BRET observed between a GPCR and its target G protein. The basal BRET signal indicating a close proximity (≤10 nm) between the receptor and the G protein may result from their direct physical interaction even though we were unable to get both PAR1 and Gαi1 coimmunoprecipitated when they are co-expressed in COS-7 or HEK293 cells. This possibility is supported by our data using BIM46187 reported to inhibit GPCR-mediated signaling (Ayoub et al., 2009). Indeed, we have shown by both BRET in live cells and also FRET on purified GPCR and G proteins that BIM46187 inhibits the activation of G proteins by GPCRs by binding to the Gα subunit and thereby blocks its physical interaction with the receptor (Ayoub et al., 2009). BIM46187 partially diminished the basal BRET measured between PAR1 and Gαi1 or Gαo indicating that part of the basal BRET signal reflects the direct or indirect association between the receptor and the G proteins. The other explanation is the colocalization of PAR1 and Gαi1 in specific membrane microdomains where the limited number of G proteins are in close vicinity to the receptor, consistent with a saturable basal BRET signal. In fact, the high density of both the receptor and G proteins in such microdomains (Insel et al., 2005) may favor the energy transfer to occur between the BRET partners. This cannot be completely excluded since we observed that the treatment of cells with Methyl-β-cyclodextrin, which is known to extract cholesterol from the membrane and thereby increases membrane fluidity and disrupts microdomains, significantly increased the basal BRET signal between PAR1 and Gαi1 (unpublished data). Finally, this emerging preassembly theory to explain some specific GPCR-G protein association are also supported by a recent study reporting that large complexes forming by GPCRs, G proteins but also specific effectors are formed early in the endoplasmic reticulum (Dupre et al., 2006). Together, the data obtained with PAR1 and Gαi1 and with other GPCR-G protein pairs are therefore more consistent with the preassembly theory postulating that this specific molecular organization of GPCRs and G protein would allow a faster process of G protein activation since there is no time needed for the receptor and G protein to collide. This is nicely illustrated by our data showing a differential nature of the interaction of PAR1 with either Gαi1 (preassembly) or Gα12 (recruitment; Ayoub et al., 2010). In addition, a slow activation of Gαq by a mutant of the muscarinic M3 receptor which does not preassemble with the G protein has been reported suggesting the importance of GPCR-G protein preassembly in accelerating signaling (Qin et al., 2011). In fact, Qin et al. (2011) proposed that preassembly may have a crucial functional role in native tissues where the expression levels of GPCRs and/or G proteins is lower compared to the overexpression in cell lines. Moreover, GPCR-G protein preassembly may allow a better control of the selectivity of the signaling cascades since one would argue that a preassembled receptor has a limited availability in space (same tissue or cell) and in time (simultaneously) to interact and couple to different G proteins as we nicely demonstrated for PAR1 and Gαi1 and Gα12 (Ayoub et al., 2010). The GPCR-G protein preassembly may also control the efficiency of signaling by sequestering or limiting access to a common G protein pool as this has been shown for CXCR7 attenuating β-adrenergic-mediated Gαs/adenylate cyclase activation (Andressen et al., 2006).
Agonist-Promoted Conformational Changes within the Preassembled Receptor-G Protein Complex
The preassembly concept raises many key questions related to the activation mechanism of the preassembled GPCR-G protein complexes and the consequences of the agonist activation on such complexes. What would be the dynamics of the receptor-G protein complexes after receptor activation? Is there any change in the complex number after activation or do GPCR and G proteins associate further or dissociate as a consequence of agonist-promoted activation? All these questions continue to be a matter of controversy in the GPCR community and this is true for both receptor-G protein interaction and the interaction between the different G protein subunits (α, β, and γ; Hein and Bunemann, 2009; Vilardaga et al., 2009; Lohse et al., 2012). In fact, as mentioned above according to the “precoupling” model if GPCRs are preassembled with their cognate G proteins the number of G proteins available for one receptor is only one or at least very limited assuming that there is no reversible dissociation of the complex. This is determined either during the early stage of protein synthesis and/or somehow during their life in the plasma membrane of cells. However, in the “free collision” model, a receptor has a possibility to interact and activate many G proteins in different coupling/uncoupling cycles as long as the agonist is available to maintain the receptor in its active state at the cell surface.
For PAR1 and Gαi1 (and also Gαo), we observed that despite the constitutive high BRET, the activation of PAR1 with thrombin or PAR1 selective agonist peptides largely increased the BRET signal in a time- and dose-dependent manner (Ayoub et al., 2007, 2009, 2010). This agonist-increased BRET was completely blocked by PTX (Ayoub et al., 2007, 2010), protease inhibitors and PAR1 antagonist, SCH79797 (Ayoub et al., 2007), and a non-selective G protein inhibitor, BIM46187 (Ayoub et al., 2009) clearly demonstrating that agonist-induced BRET increase between PAR1 and Gαi1 reflects the activation of their preassembled complex. The EC50 values of thrombin (∼0.5 – 6 nM) and PAR1 selective agonist peptides (∼6 μM) measured in BRET assay are consistent with the activation of PAR1 in a functional assay (Ayoub et al., 2007). Moreover, the kinetic analysis showed a rapid and transient BRET increase between PAR1 and Gαi1 after agonist application (t1/2 = 4.3 ± 0.6 s for thrombin; Ayoub et al., 2007, 2010). Indeed, thrombin-induced BRET increase persists for a few minutes before returning to the basal level and the decay kinetic of thrombin-induced BRET between PAR1 and Gαi1 (t1/2 = 6.9 ± 1.7 min) paralleled the kinetic of thrombin-promoted β-arrestin 1 recruitment also measured by BRET (t1/2 = 5.4 ± 0.9 min; Ayoub et al., 2007) or TR-FRET (t1/2 = 7.5 ± 1.5 min; Ayoub et al., 2010) indicating the desensitization of the activated PAR1-Gαi1 complex. Furthermore, the decay of the induced BRET signal did not go beyond the basal level (Ayoub et al., 2007, 2010) further supports the concept of a preassembled receptor-G protein complex being formed even in the absence of activation and non-dissociated after receptor activation. It is important to point out here that the agonist-promoted PAR1-Gαi1 activation measured in BRET assay is still slower compared to what has been reported in the others studies (Hein et al., 2005; Galés et al., 2006) as well as what was expected for GPCR-G protein activation process. This may be due to the limitation of our BRET assay and/or the luminescence reader used for such a kinetic analysis. A difference in the activation kinetic between different GPCRs as reported between the PTH and α2-adrenergic receptors (Vilardaga et al., 2003) or a difference in the feature of the interaction observed between various GPCR-G protein pairs as we found between PAR1 and Gα12 (Ayoub et al., 2010) may be other reasonable explanations. More interestingly, BRET assay allowed us to detect the transition of PAR1-Gαi1 complex from the inactive to the active state in an agonist-dependent manner and this supports the different models implying that GPCRs and G proteins exist in at least two different states (De Lean et al., 1980; Leff, 1995).
How can the rapid and transient agonist-induced BRET increase between PAR1 and Gαi1 be interpreted? Our studies and others clearly agree that such BRET changes reflect conformational changes within the preassembled receptor-G protein complex rather than a further recruitment of G proteins to the activated receptors, resulting from a change of either the distance or the orientation of the chromophores (Galés et al., 2006; Ayoub et al., 2007, 2010; Levoye et al., 2009; Figure 3A). First, in BRET saturation assay between PAR1 and Gαi1 no difference in the BRET50 value, corresponding to 50% of the BRET saturation value, was observed when cells were stimulated with thrombin (Ayoub et al., 2007), consistent with an absence of any change in their relative affinity, and then arguing against an increase BRET due to further recruitment of new G proteins. However, the maximal BRET signal was largely increased consistent with a movement occurring between Rluc and YFP during activation of the preassembled complex. Second, we observed that depending on the insertion position of BRET donor and acceptor (Rluc and YFP) in the receptor and/or G protein, the BRET increase observed after PAR1 activation was not always detectable (Ayoub et al., 2007). Similarly, Galés et al. (2006) have nicely shown that depending on the position of Rluc within the G protein either an increase or a decrease in BRET signal after receptor activation was observed, not consistent with the recruitment of new G proteins (Galés et al., 2006). Third, a significant basal BRET signal between Gαi1 and a wild-type CXCR4 as well as its mutant (CXCR4-N119K), which cannot activate G proteins, has been reported, indicating that the preassembly between Gαi1 and CXCR4 does not require the activation of the G protein (Levoye et al., 2009). In parallel, the wild-type CXCR4 showed a high basal BRET which can be largely increased after receptor activation (Levoye et al., 2009). Finally, in TR-FRET assay using large fluorophore-conjugated antibodies against Flag and Myc tags, no FRET increase was measured between PAR1-Myc and Gαi1-Flag after stimulation of cells with thrombin whereas thrombin nicely and specifically promoted Gα12-Flag as well as Flag-β-arrestin 1 recruitment to the activated PAR1-Myc (Ayoub et al., 2010). Together, these observations are a demonstration that the agonist-induced BRET changes within PAR1-Gαi1 protein complex cannot be a consequence of further G protein recruitment by the activated receptor, but rather reflects a change in the relative position of the interacting proteins associated with the activation process, leading to a change of either the distance or the orientation of the chromophores (Figure 3A).
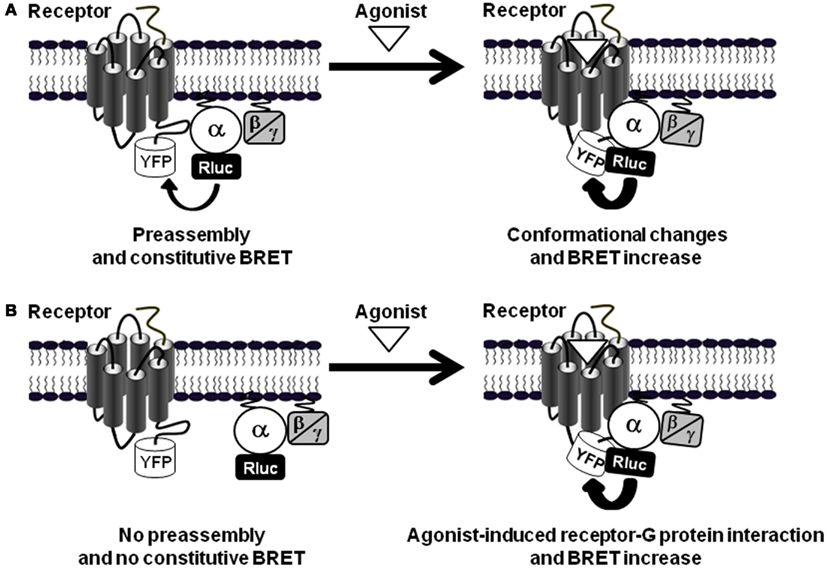
Figure 3. GPCR-G protein preassembly or agonist-induced association. (A) In the preassembly model, as shown for PAR1-Gαi1 interaction (Ayoub et al., 2007, 2010), a basal BRET signal is measured between the receptor-YFP and Gα-Rluc fusion proteins. Agonist stimulation induces BRET changes (here indicated by an increase) without changing the receptor-G protein interaction supporting agonist-induced conformational changes within the preassembled complexes. (B) The other situation is illustrated by the data obtained with PAR1-Gα12 interaction (Ayoub et al., 2010) where there is no significant basal BRET signal indicating no preassembly and only receptor activation promotes receptor-G protein association and thereby induces BRET signal increase.
Agonist-Induced Receptor-G Protein Association
However, some other studies have reported opposite observations with regard to the constitutive association between GPCRs and G proteins suggesting that preassembly is not a general feature of all GPCR and G protein couples. Indeed, a recent study using a variant of BRET-based assay with FKBP-Rapamycin system failed to detect constitutive BRET signal between various class A GPCRs and different G proteins (Kuravi et al., 2010). Similarly, Hein et al. (2005) have reported no specific basal FRET signal between the α2A-adrenergic receptor and Gi in the absence of receptor activation. In this study and in agreement with the “free collision” model, the expression levels of the G proteins was found to determine the kinetics of receptor-G protein interaction (Hein et al., 2005). Moreover, Qin et al. (2011) have demonstrated that in their inactive state M3R-Gαq complexes are transient and become more stable after receptor activation. Thus, these data clearly sustain the debate around the nature of GPCR-G protein interaction and activation since they support more the “free collision” model than the “precoupling” one. These opposite observations however may have different reasons: (i) GPCRs might differ in their coupling properties due to the differences in their active conformations and the way how they activate different G proteins (Leff and Scaramellini, 1998; Hermans, 2003; Ayoub et al., 2010), (ii) the cellular model used and the expression level of GPCRs and G proteins, and of course (iii) the sensitivities of the different energy transfer-based assays or other techniques used (Lohse et al., 2012).
In this context, our data with PAR1 and its interaction with Gα12 monitored by BRET and TR-FRET have shed some light on understanding the GPCR-G protein coupling (Ayoub et al., 2010). Indeed, in contrast to the PAR1-Gαi1 pair for which we observed preassembly, we found that PAR1 and Gα12 are not preassembled in COS-7 cells as illustrated by very low basal BRET and TR-FRET signals (Ayoub et al., 2010). This was not due to the differences in the expression levels between Gαi1 and Gα12 or the position where the fluorophores were fused within the G proteins. Moreover, the short term activation (up to 2 min) of PAR1 had no effect on the basal BRET signal between PAR1 and Gα12 while a large increase was observed between PAR1 and Gαi1 as mentioned above (Ayoub et al., 2010). However, a long-term activation of PAR1 (≥10–60 min) largely increased both BRET and TR-FRET signals between PAR1 and Gα12, in a time- and dose-dependent manner (Ayoub et al., 2010). Thrombin- as well as PAR1 agonist peptide-induced BRET/TR-FRET increase was specific for PAR1-Gα12 association and it was not observed with Gαs or even Gα13 also known to be activated by PAR1 in many other models. Together, BRET, TR-FRET, and coimmunoprecipitation experiments confirmed the physical recruitment of Gα12 to PAR1 in an agonist-dependent way (Figure 3B). In addition, the persistence of PAR1-Gα12 association under Triton X-100 condition used in coimmunorecipitation and TR-FRET assays clearly illustrates the strength of such a physical interaction. These observations suggest that the stable interaction between PAR1 and Gα12 is more compatible with the “free collision model” as observed with other GPCRs using energy-transfer approaches (Hein et al., 2005; Qin et al., 2008; Kuravi et al., 2010). However, this recruitment is very slow compared to what was really expected for GPCR activation kinetics and it is also very stable in time since it was observed even 1 h after receptor activation. Gα12 recruitment to the activated PAR1 is in fact slower than Gα12 activation monitored by p115-RhoGEF translocation to the plasma membrane (Tanabe et al., 2004). However, the activation of the small G protein RhoA mediated by Gα12/13 proteins has also been reported to occur a few minutes after G protein activation (Tanabe et al., 2004). Unfortunately, we were unable to measure Gα12 activation by any specific readout or signaling assay in a fast enough way to compare the kinetics of Gα12 activation and recruitment to PAR1. It is then still unclear if this slow and sustained recruitment of Gα12 to the activated PAR1 is really associated with its activation in our model, or has other functional meanings. Moreover, the stability of PAR1-Gα12 complex after its formation may be explained by either a non-dissociation of the complex or many rapid association-dissociation cycles which cannot be detected by BRET and as a result a continuous BRET or TR-FRET signal. Therefore, we speculated about the significance of our BRET and TR-FRET data with Gα12 and we argued that the only reason for Gα12 to be recruited to the receptor is indeed its activation by this activated receptor. This is consistent with the strong and sustained developing effects of thrombin and PAR1 on cell morphology and proliferation where stabilizing PAR1-Gα12 interaction and maintaining their long term activation may be crucial for the control of cell proliferation, differentiation, migration, or oncogenesis (Dhanasekaran and Dermott, 1996; Riobo and Manning, 2005). However, one would exclude that the stable Gα12 association with PAR1 may inhibits subsequent receptor activation and via a sequestration process this limits the pool of Gα12 available, thereby reduces the action of Gα12-dependent signaling promoted by other GPCRs as observed with the preassembly of 5-HT7 with Gαs inhibiting this G protein to be activated by the β-adrenergic receptor (Andressen et al., 2006).
Interestingly, the agonist-promoted Gα12 recruitment is a particular feature of PAR1 and cannot be generalized to other receptors reported to couple to Gα12. Indeed, when other GPCRs such as serotonin 5-HT2c, vasopressin V1a, and muscarinic M3 receptors were tested, high constitutive TR-FRET (Ayoub et al., 2010) and BRET (unpublished data) signals was measured between Gα12 and these receptors. Agonist stimulation had no effect on the basal signals again illustrates that the preassembly or agonist-induced G protein recruitment depend on receptor-G protein pairs. Taken together with the data obtained with PAR1-Gαi1 preassembly, these observations illustrate the specificity, diversity and complexity of GPCR-G protein coupling and show how one GPCR (PAR1) can associate differently with two distinct classes of G protein (Gαi1 and Gα12; Figure 4A), and on the other hand how a given G protein (Gα12) is differently interacting with distinct GPCRs (PAR1 and 5-HT2c, V1aR or M3R; Figure 4B).
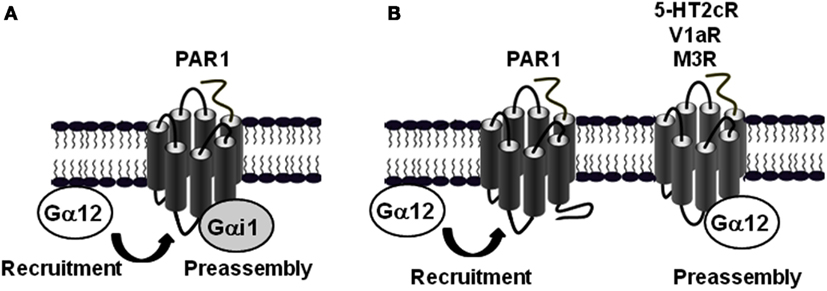
Figure 4. Differential association mode of Gα12 protein with PAR1 and other GPCRs. (A) BRET and TR-FRET data demonstrated that PAR1 is preassembled with Gαi1, but not Gα12 which is only recruited after receptor activation (Ayoub et al., 2010). (B) In contrast to PAR1, Gα12 can be preassembled with other GPCRs such as 5-HT2c, V1aR, and M3R (Ayoub et al., 2010).
The Multiple Coupling of PAR1 to Gαi1, Gα12, and β-Arrestin 1
It is now evident that GPCRs are able to control various physiological responses by promoting diverse signaling pathways via their coupling to different classes of G proteins and other intracellular proteins (Figure 5; Hamm, 1998; Hermans, 2003). As elegantly discussed by Hermans the multiple coupling abilities of GPCRs is selectively controlled and regulated at different levels (Table 1; Hermans, 2003) and our data on PAR1, Gαi1, Gα12, and β-arrestin 1 and others came to complement the discussion with the new concepts of preassembly and agonist-dependent G protein interaction with some specificities for PAR1.
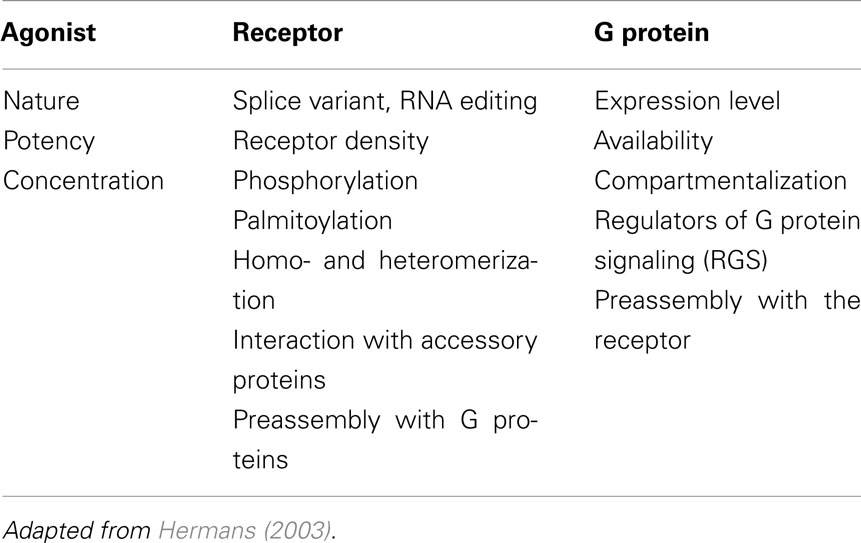
Table 1. The different levels of regulation of the multiple of GPCR-G protein coupling.
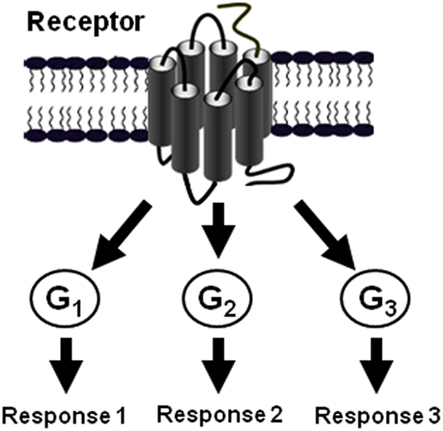
Figure 5. The multiple coupling of GPCRs. The diversity of GPCR signaling as a result of the possibility of a given GPCR to couple and activate G protein of various classes (Hermans, 2003).
PAR1 can be considered as an ideal model to study such a multiple coupling since it has been shown to couple to different G proteins including Gαi/o, Gαq, and Gα12/13 as well as to arrestins (Coughlin, 2000; Trejo, 2003). Indeed, our studies using BRET and TR-FRET approaches in live COS-7 cells moved one step further in understanding how the multiple coupling of PAR1 with Gαi1, Gα12, and β-arrestin 1 and its signaling can be regulated and integrated. We found two main differences between the interaction of PAR1 with Gαi1 and Gα12. The first one consists of the nature of the interaction, constitutive (for Gαi1; Figure 3A) or agonist-induced (for Gα12; Figure 3B), and consequently this results in a second difference in terms of the kinetic of their association with the receptor, rapid, and transient (for Gαi1) or slow and stable (for Gα12; Figure 6). Nevertheless, another interesting difference between PAR1-Gαi1 and PAR1-Gα12 coupling has been found at the molecular level. Indeed, we have shown that the last eight C-terminal residues of Gα12 are crucial for its association with PAR1 in an agonist-dependent manner whereas the corresponding region in Gαi1 does not seem to be important for preassembly. Moreover, even the mutation of the cysteine at position-4 in Gαi1, known to be crucial for PTX-induced Gαi protein inhibition, had no effect on PAR1-Gαi1 preassembly indicating a secondary role of the C-terminal region of the Gα subunit in the preassembly and suggesting the importance of other molecular determinants within the alpha subunit. Therefore, we propose that the preassembly between PAR1 and Gαi1 does not involve the C-terminal of the Gα subunit, in contrast to PAR1-Gαi1 coupling (or activation) as shown by PTX inhibiting the agonist-induced BRET increase (Ayoub et al., 2007). Furthermore, we observed that the deletion of a large part of PAR1 C-terminus completely inhibited the agonist-induced BRET increase between PAR1 and Gαi1 without affecting the high basal BRET suggesting that the C-terminus of PAR1 likely plays a role in the functional coupling of PAR1 with Gαi1, but not in their preassembly (Ayoub et al., 2007).
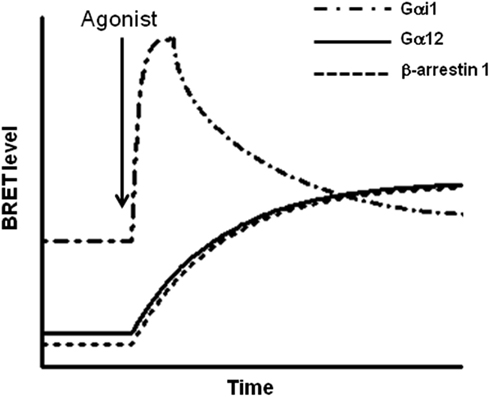
Figure 6. Kinetic profiles of the interaction of PAR1 with Gαi1, Gα12, and β-arrestin 1. Based on BRET analysis, the kinetic of the interaction of PAR1 with Gαi1, Gα12, and β-arrestin 1 has been found to be interestingly different. Indeed, PAR1-Gαi1 interaction is constitutive and agonist stimulation leads to a rapid and transient transition to the activated state of the preassembled complex. The deactivation phase of the preassembled PAR1-Gαi1 complex appears to be parallel to the slow and stable recruitment of both Gα12 and β-arrestin 1 to the activated PAR1. This difference in BRET kinetic analysis clearly suggests a differential mode of association of PAR1 with these different proteins. Furthermore, the data obtained with PAR1 nicely illustrate how the multiple coupling of one GPCR to different signaling proteins can be regulated at the level of the kinetic of the interaction/activation between the receptor and the G proteins.
Together our observations led us to propose a speculative model based on our observations in COS-7 cells where the key element in PAR1-G protein interaction and regulation, when Gαi1, Gα12, and arrestins are considered in the system, is actually the existence of at least two different populations of receptors (Figure 7; Ayoub et al., 2010). Indeed, the first population is exclusively preassembled with Gαi1, but not Gα12, and this is supported by a basal BRET and TR-FRET signals between PAR1 and Gαi1 and the absence of any specific signal between Gαi1 and Gα12 (unpublished data). This preassembled PAR1-Gαi1 population is rapidly and transiently activated probably to control cAMP-dependent signaling in a faster way and the deactivation process which is very important appears to be parallel to the recruitment of β-arrestin 1. Interestingly, we found that PAR1 and Gαi1 do not dissociate even after β-arrestin 1 being recruited to the preassembled complex. Indeed, for the first time we were able to detect very high BRET and TR-FRET signals between Gαi1 and β-arrestin 1 when PAR1 was activated, bringing clear evidence that β-arrestin can be recruited by a receptor still associated with Gαi1 (Ayoub et al., 2010). This was observed only when β-arrestin 1 is recruited to PAR1 since a mutant of PAR1 unable to interact with β-arrestin 1 did not show any specific BRET signal between Gαi1 and β-arrestin1 despite its preassembly with Gαi1. The co-existence of PAR1, Gαi1, and β-arrestin 1, but not Gα12, in a same complex has nicely been confirmed for the first time using a multi-complex assay based on TR-FRET approach (Ayoub et al., 2010). Having observed that the agonist-promoted BRET increase disappeared in parallel to β-arrestin 1 recruitment, this finding indicates that the preassembled PAR1-Gαi1 complex is desensitized following β-arrestin 1 recruitment to the preassembled complex, but this does not result in a physical dissociation between PAR1 and Gαi1. Thus, our findings essentially reconcile the GPCR-G protein preassembly concluded from BRET studies in live cells. Although the preassembly may appear consistent with the “precoupling” model, we would like to point out here that our proposed preassembly model does not include basal activation of the G protein, as suggested in the precoupling model. Thus, receptor-G protein “preassembly” does not necessarily mean their “precoupling.”
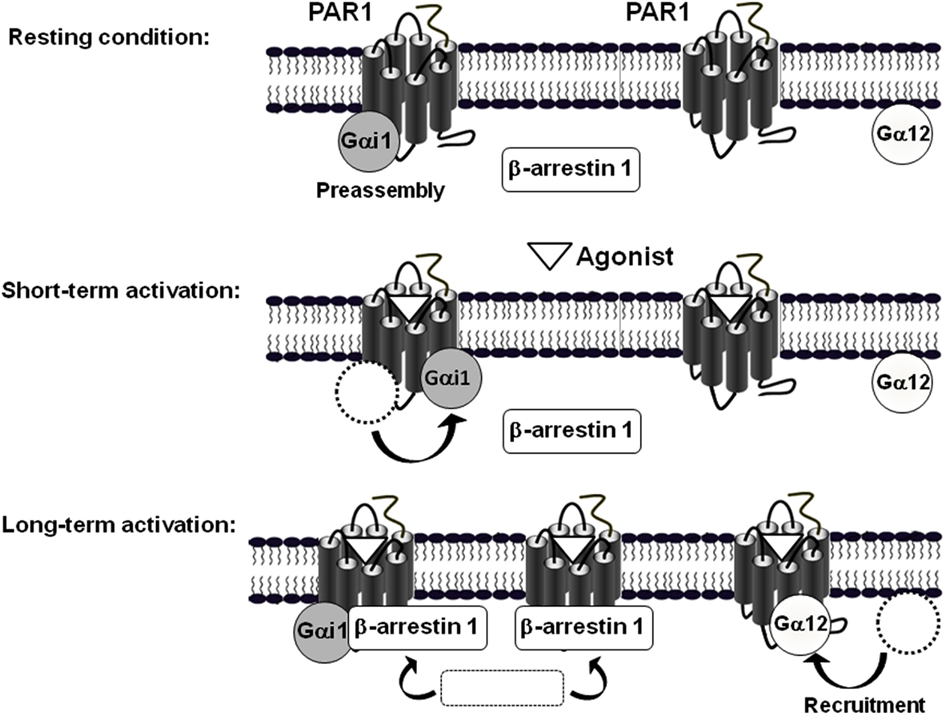
Figure 7. Model of the differential mode of association of PAR1 with Gαi1, Gα12, and β-arrestin 1. The model is based on BRET observations and suggests the existence of two populations of PAR1, at least in COS-7 cells (Ayoub et al., 2010). In the absence of receptor activation, one population of PAR1 is preassembled with Gαi1 and another one would be non-associated with any of Gαi1, Gα12, or β-arrestin 1. A short term activation of PAR1 induces a rapid activation of the preassembled PAR1-Gαi1 complex characterized by a transient change in the relative position of PAR1 and Gαi1 without any change on free PAR1. In contrast, long term activation first results in the deactivation of the preassembled PAR1-Gαi1 complex and in parallel to the concomitant recruitment of both Gα12 and β-arrestin 1 to the activated PAR1. Interestingly, even though their similar kinetic of recruitment, Gα12 and β-arrestin 1 are not co-recruited to the same population of PAR1. Indeed, it has been clearly demonstrated that β-arrestin 1, but not Gα12, can be recruited by the activated PAR1-Gαi1 complex. In addition, it is not excluded that β-arrestin 1 is also recruited by the activated free PAR1. In contrast, after long term activation of PAR1-Gα12 seems to be translocated to the no assembled population of PAR1 only. (Adapted from Ayoub et al. (2010) Differential association modes of the thrombin receptor PAR1 with Gαi1, Gα12, and β-arrestin 1. The FASEB Journal (2010), 24(9): 3522-3535).
The second population of PAR1 would not be preassembled with any of Gαi1, Gα12, β-arrestin 1. Following PAR1 activation Gα12 is recruited to the activated receptor in slow and prolonged kinetics (Figure 6). In contrast to what observed with Gαi1, Gα12 recruitment (t1/2 = 8.8 ± 1.9 min) was concomitant to β-arrestin 1 recruitment (t1/2 = 7.5 ± 1.5 min; Ayoub et al., 2010) suggesting a co-recruitment of Gα12 and β-arrestin 1 to the same activated receptor. This is unlikely because, neither BRET nor TR-FRET signal was measured between Gα12 and β-arrestin 1 after PAR1 activation (Ayoub et al., 2010). Furthermore, in a BRET competition-based assay we found that the overexpression of β-arrestin 1 significantly reduced PAR1-Gα12 association. In contrast, when Gα12 was overexpressed the agonist-promoted β-arrestin 1 recruitment was not affected at all. In addition, a mutant of PAR1 unable to interact with β-arrestin 1 was still able to recruit Gα12 in an agonist-dependent manner and the inhibition of thrombin-promoted PAR1 internalization, which seems to be arrestin-dependent, did not affect Gα12 recruitment. Together, these observations indicate a competitive and exclusive recruitment of Gα12 or β-arrestin 1 to the same pool of activated PAR1. In another word, Gα12 and β-arrestin 1 cannot be recruited simultaneously to the same receptor and among the free population of PAR1 molecules there are some receptor interacting with Gα12 only and others with β-arrestin 1 only but not with both. This constitutes an interesting finding which in fact raises many other questions regarding (i) the desensitization of PAR1-Gα12 complex if such a complex cannot recruit arrestins while Gα12 is there, (ii) the significance of the recruitment of β-arrestin 1 to a free PAR1 (is this population non-associated with any G protein?), and finally (iii) the molecular mechanisms involved in such a competitive and exclusive recruitment of Gα12 or β-arrestin 1. Of course this needs further investigations to be clarified.
The existence of two different populations of PAR1 is the only explanation of our BRET and TR-FRET data. This may imply that in the same cell the preassembled PAR1-Gαi1 complex and PAR1 susceptible to recruit Gα12 or β-arrestin 1 exist in different membrane domains. Our data using methyl-β-cyclodextrin support this hypothesis to some extent since we observed that treatment of cells with methyl-β-cyclodextrin largely increased both basal and agonist-induced BRET signal between PAR1 and Gαi1, but had no effect on agonist-induced Gα12 recruitment (unpublished data). The differential recruitment of β-arrestin 1 when PAR1-Gαi1 and PAR1-Gα12 complexes are compared clearly demonstrates the co-existence of at least two populations of PAR1 in COS-7 cells. Of course, the existence of these two different populations may be specific for our cellular model using transient expression in COS-7 cells, but one would hypothesize that this may also occur in native tissues. Thus, the co-expression of both Gαi1 and Gα12 proteins with PAR1 in a same cell type and at the same time, their relative expression levels and the involvement of other accessory intracellular proteins could be the major factors controlling the existence of the two populations of PAR1 and the preassembly or not with the G proteins.
Together, our studies on the physical interaction of PAR1 with Gαi1, Gα12, and β-arrestin 1 using BRET and TR-FRET approaches constitute one step further to better understand GPCR-G protein coupling and again illustrates the complexity of GPCR-G protein coupling. It nicely illustrates that many different processes can be involved in this coupling mechanism, depending on the receptor-G protein couple examined. Therefore, we conclude that the nature of the molecular association between GPCRs and G proteins characterized by either the preassembly or the agonist-dependent recruitment depends on the receptor-G protein pair and this differential association between GPCR and G proteins may constitute a novel way to control the multiple coupling of GPCRs.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Cisbio Bioassays for continuous strong support of this work. This work was supported by grants from the CNRS, INSERM, French Ministry of Research, Cisbio International, and the Agence Nationale pour la Recherche (contract ANR-05-PRIB-02502).
References
Andressen, K. W., Norum, J. H., Levy, F. O., and Krobert, K. A. (2006). Activation of adenylyl cyclase by endogenous G(s)-coupled receptors in human embryonic kidney 293 cells is attenuated by 5-HT(7) receptor expression. Mol. Pharmacol. 69, 207–215.
Audet, N., Gales, C., Archer-Lahlou, E., Vallieres, M., Schiller, P. W., Bouvier, M., and Pineyro, G. (2008). Bioluminescence resonance energy transfer assays reveal ligand-specific conformational changes within preformed signaling complexes containing delta-opioid receptors and heterotrimeric G proteins. J. Biol. Chem. 283, 15078–15088.
Ayoub, M. A., Damian, M., Gespach, C., Ferrandis, E., Lavergne, O., De Wever, O., Baneres, J. L., Pin, J. P., and Prevost, G. P. (2009). Inhibition of heterotrimeric G-protein signaling by a small molecule acting on Galpha subunit. J. Biol. Chem. 284, 29136–29145.
Ayoub, M. A., Maurel, D., Binet, V., Fink, M., Prezeau, L., Ansanay, H., and Pin, J. P. (2007). Real-time analysis of agonist-induced activation of protease-activated receptor 1/Galphai1 protein complex measured by bioluminescence resonance energy transfer in living cells. Mol. Pharmacol. 71, 1329–1340.
Ayoub, M. A., Trinquet, E., Pfleger, K. D., and Pin, J. P. (2010). Differential association modes of the thrombin receptor PAR1 with Galphai1, Galpha12, and beta-arrestin 1. FASEB J. 24, 3522–3535.
Azpiazu, I., and Gautam, N. (2004). A fluorescence resonance energy transfer-based sensor indicates that receptor access to a G protein is unrestricted in a living mammalian cell. J. Biol. Chem. 279, 27709–27718.
Bockaert, J. (1991). G proteins and G-protein-coupled receptors: structure, function and interactions. Curr. Opin. Neurobiol. 1, 32–42.
Bockaert, J., Homburger, V., and Rouot, B. (1987). GTP binding proteins: a key role in cellular communication. Biochimie 69, 329–338.
Bockaert, J., and Pin, J. P. (1999). Molecular tinkering of G protein-coupled receptors: an evolutionary success. EMBO J. 18, 1723–1729.
Bockenhauer, S., Furstenberg, A., Yao, X. J., Kobilka, B. K., and Moerner, W. E. (2011). Conformational dynamics of single G protein-coupled receptors in solution. J. Phys. Chem. B 115, 13328–13338.
Brady, A. E., and Limbird, L. E. (2002). G protein-coupled receptor interacting proteins: emerging roles in localization and signal transduction. Cell Signal. 14, 297–309.
Bunemann, M., Frank, M., and Lohse, M. J. (2003). Gi protein activation in intact cells involves subunit rearrangement rather than dissociation. Proc. Natl. Acad. Sci. U.S.A. 100, 16077–16082.
Cottrell, G. S., Coelho, A. M., and Bunnett, N. W. (2002). Protease-activated receptors: the role of cell-surface proteolysis in signalling. Essays Biochem. 38, 169–183.
Coughlin, S. R. (2005). Protease-activated receptors in hemostasis, thrombosis and vascular biology. J. Thromb. Haemost. 3, 1800–1814.
De Lean, A., Stadel, J. M., and Lefkowitz, R. J. (1980). A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J. Biol. Chem. 255, 7108–7117.
Dean, M. K., Higgs, C., Smith, R. E., Bywater, R. P., Snell, C. R., Scott, P. D., Upton, G. J., Howe, T. J., and Reynolds, C. A. (2001). Dimerization of G-protein-coupled receptors. J. Med. Chem. 44, 4595–4614.
Dhanasekaran, N., and Dermott, J. M. (1996). Signaling by the G12 class of G proteins. Cell Signal. 8, 235–245.
Dupre, D. J., Robitaille, M., Ethier, N., Villeneuve, L. R., Mamarbachi, A. M., and Hebert, T. E. (2006). Seven transmembrane receptor core signaling complexes are assembled prior to plasma membrane trafficking. J. Biol. Chem. 281, 34561–34573.
Frank, M., Thumer, L., Lohse, M. J., and Bunemann, M. (2005). G protein activation without subunit dissociation depends on a G{alpha}i-specific region. J. Biol. Chem. 280, 24584–24590.
Gales, C., Rebois, R. V., Hogue, M., Trieu, P., Breit, A., Hebert, T. E., and Bouvier, M. (2005). Real-time monitoring of receptor and G-protein interactions in living cells. Nat. Methods 2, 177–184.
Galés, C., Van Durm, J. J., Schaak, S., Pontier, S., Percherancier, Y., Audet, M., Paris, H., and Bouvier, M. (2006). Probing the activation-promoted structural rearrangements in preassembled receptor-G protein complexes. Nat. Struct. Mol. Biol. 13, 778–786.
Gether, U. (2000). Uncovering molecular mechanisms involved in activation of G protein-coupled receptors. Endocr. Rev. 21, 90–113.
Gether, U., and Kobilka, B. K. (1998). G protein-coupled receptors. II. Mechanism of agonist activation. J. Biol. Chem. 273, 17979–17982.
Gether, U., Lin, S., and Kobilka, B. K. (1995). Fluorescent labeling of purified beta 2 adrenergic receptor. Evidence for ligand-specific conformational changes. J. Biol. Chem. 270, 28268–28275.
Gibson, S. K., and Gilman, A. G. (2006). Gialpha and Gbeta subunits both define selectivity of G protein activation by alpha2-adrenergic receptors. Proc. Natl. Acad. Sci. U.S.A. 103, 212–217.
Gilman, A. G. (1987). G proteins: transducers of receptor-generated signals. Annu. Rev. Biochem. 56, 615–649.
Hasbi, A., Nguyen, T., Fan, T., Cheng, R., Rashid, A., Alijaniaram, M., Rasenick, M. M., O’Dowd, B. F., and George, S. R. (2007). Trafficking of preassembled opioid mu-delta heterooligomer-Gz signaling complexes to the plasma membrane: coregulation by agonists. Biochemistry 46, 12997–13009.
Hein, P., and Bunemann, M. (2009). Coupling mode of receptors and G proteins. Naunyn Schmiedebergs Arch. Pharmacol. 379, 435–443.
Hein, P., Frank, M., Hoffmann, C., Lohse, M. J., and Bunemann, M. (2005). Dynamics of receptor/G protein coupling in living cells. EMBO J. 24, 4106–4114.
Hermans, E. (2003). Biochemical and pharmacological control of the multiplicity of coupling at G-protein-coupled receptors. Pharmacol. Ther. 99, 25–44.
Hollenberg, M. D., and Compton, J. S. (2002). International union of pharmacology. XXVIII. Proteinase-activated receptors. Pharmacol. Rev. 54, 203–217.
Insel, P. A., Head, B. P., Patel, H. H., Roth, D. M., Bundey, R. A., and Swaney, J. S. (2005). Compartmentation of G-protein-coupled receptors and their signalling components in lipid rafts and caveolae. Biochem. Soc. Trans. 33, 1131–1134.
Janetopoulos, C., Jin, T., and Devreotes, P. (2001). Receptor-mediated activation of heterotrimeric G-proteins in living cells. Science 291, 2408–2411.
Kobilka, B., and Schertler, G. F. (2008). New G-protein-coupled receptor crystal structures: insights and limitations. Trends Pharmacol. Sci. 29, 79–83.
Kuravi, S., Lan, T. H., Barik, A., and Lambert, N. A. (2010). Third-party bioluminescence resonance energy transfer indicates constitutive association of membrane proteins: application to class a g-protein-coupled receptors and g-proteins. Biophys. J. 98, 2391–2399.
Lachance, M., Ethier, N., Wolbring, G., Schnetkamp, P. P., and Hebert, T. E. (1999). Stable association of G proteins with beta 2AR is independent of the state of receptor activation. Cell Signal. 11, 523–533.
Law, S. F., and Reisine, T. (1997). Changes in the association of G protein subunits with the cloned mouse delta opioid receptor on agonist stimulation. J. Pharmacol. Exp. Ther. 281, 1476–1486.
Law, S. F., Yasuda, K., Bell, G. I., and Reisine, T. (1993). Gi alpha 3 and G(o) alpha selectively associate with the cloned somatostatin receptor subtype SSTR2. J. Biol. Chem. 268, 10721–10727.
Leff, P., and Scaramellini, C. (1998). Promiscuity, pre-coupling and instability. Trends Pharmacol. Sci. 19, 13.
Lefkowitz, R. J., and Whalen, E. J. (2004). Beta-arrestins: traffic cops of cell signaling. Curr. Opin. Cell Biol. 16, 162–168.
Levoye, A., Balabanian, K., Baleux, F., Bachelerie, F., and Lagane, B. (2009). CXCR7 heterodimerizes with CXCR4 and regulates CXCL12-mediated G protein signaling. Blood 113, 6085–6093.
Limbird, L. E. (1983). Beta-adrenergic stimulation of adenylate cyclase and alpha-adrenergic inhibition of adenylate cyclase: GTP-binding proteins as macromolecular messengers. Adv. Exp. Med. Biol. 161, 91–111.
Limbird, L. E., Gill, D. M., and Lefkowitz, R. J. (1980). Agonist-promoted coupling of the beta-adrenergic receptor with the guanine nucleotide regulatory protein of the adenylate cyclase system. Proc. Natl. Acad. Sci. U.S.A. 77, 775–779.
Lohse, M. J., Nuber, S., and Hoffmann, C. (2012). Fluorescence/Bioluminescence resonance energy transfer techniques to study g-protein-coupled receptor activation and signaling. Pharmacol. Rev. 64, 299–336.
Marinissen, M. J., Servitja, J. M., Offermanns, S., Simon, M. I., and Gutkind, J. S. (2003). Thrombin protease-activated receptor-1 signals through Gq– and G13-initiated MAPK cascades regulating c-Jun expression to induce cell transformation. J. Biol. Chem. 278, 46814–46825.
Neumann, L., Wohland, T., Whelan, R. J., Zare, R. N., and Kobilka, B. K. (2002). Functional immobilization of a ligand-activated G-protein-coupled receptor. Chembiochem 3, 993–998.
Nobles, M., Benians, A., and Tinker, A. (2005). Heterotrimeric G proteins precouple with G protein-coupled receptors in living cells. Proc. Natl. Acad. Sci. U.S.A. 102, 18706–18711.
Oldham, W. M., and Hamm, H. E. (2008). Heterotrimeric G protein activation by G-protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 9, 60–71.
Palczewski, K., Kumasaka, T., Hori, T., Behnke, C. A., Motoshima, H., Fox, B. A., Le Trong, I., Teller, D. C., Okada, T., Stenkamp, R. E., Yamamoto, M., and Miyano, M. (2000). Crystal structure of rhodopsin: a G protein-coupled receptor. Science 289, 739–745.
Perez, D. M., and Karnik, S. S. (2005). Multiple signaling states of G-protein-coupled receptors. Pharmacol. Rev. 57, 147–161.
Philip, F., Sengupta, P., and Scarlata, S. (2007). Signaling through a G Protein-coupled receptor and its corresponding G protein follows a stoichiometrically limited model. J. Biol. Chem. 282, 19203–19216.
Pin, J. P., Ayoub, M. A., Maurel, D., Perroy, J., and Trinquet, E. (2008) “energy transfer technologies to monitor the dynamics and signaling properties of G – protein – coupled receptors in living cells,” in Biophysical Analysis of Membrane Proteins – Investigating Structure and Function, ed. E. Pebay-Peyroula (Weinheim: Wiley-VCH) 13, 311–334.
Qin, K., Dong, C., Wu, G., and Lambert, N. A. (2011). Inactive-state preassembly of G(q)-coupled receptors and G(q) heterotrimers. Nat. Chem. Biol. 7, 740–747.
Qin, K., Sethi, P. R., and Lambert, N. A. (2008). Abundance and stability of complexes containing inactive G protein-coupled receptors and G proteins. FASEB J. 22, 2920–2927.
Rajagopal, S., Ahn, S., Rominger, D. H., Gowen-MacDonald, W., Lam, C. M., Dewire, S. M., Violin, J. D., and Lefkowitz, R. J. (2011). Quantifying ligand bias at seven-transmembrane receptors. Mol. Pharmacol. 80, 367–377.
Riobo, N. A., and Manning, D. R. (2005). Receptors coupled to heterotrimeric G proteins of the G12 family. Trends Pharmacol. Sci. 26, 146–154.
Roka, F., Brydon, L., Waldhoer, M., Strosberg, A. D., Freissmuth, M., Jockers, R., and Nanoff, C. (1999). Tight association of the human Mel(1a)-melatonin receptor and G(i): precoupling and constitutive activity. Mol. Pharmacol. 56, 1014–1024.
Schlyer, S., and Horuk, R. (2006). I want a new drug: G-protein-coupled receptors in drug development. Drug Discov. Today 11, 481–493.
Seifert, R., and Wenzel-Seifert, K. (2002). Constitutive activity of G-protein-coupled receptors: cause of disease and common property of wild-type receptors. Naunyn Schmiedebergs Arch. Pharmacol. 366, 381–416.
Senogles, S. E., Benovic, J. L., Amlaiky, N., Unson, C., Milligan, G., Vinitsky, R., Spiegel, A. M., and Caron, M. G. (1987). The D2-dopamine receptor of anterior pituitary is functionally associated with a pertussis toxin-sensitive guanine nucleotide binding protein. J. Biol. Chem. 262, 4860–4867.
Smith, S. K., and Limbird, L. E. (1981). Solubilization of human platelet alpha-adrenergic receptors: evidence that agonist occupancy of the receptor stabilizes receptor – effector interactions. Proc. Natl. Acad. Sci. U.S.A. 78, 4026–4030.
Stadel, J. M., Shorr, R. G., Limbird, L. E., and Lefkowitz, R. J. (1981). Evidence that a beta-adrenergic receptor-associated guanine nucleotide regulatory protein conveys guanosine 5’-O-(3-thiotriphosphate) – dependent adenylate cyclase activity. J. Biol. Chem. 256, 8718–8723.
Strange, P. G. (2008). Signaling mechanisms of GPCR ligands. Curr. Opin. Drug Discov. Devel. 11, 196–202.
Tanabe, S., Kreutz, B., Suzuki, N., and Kozasa, T. (2004). Regulation of RGS-RhoGEFs by Galpha12 and Galpha13 Proteins. Meth. Enzymol. 390, 285–294.
Tesmer, V. M., Kawano, T., Shankaranarayanan, A., Kozasa, T., and Tesmer, J. J. (2005). Snapshot of activated G proteins at the membrane: the Galphaq-GRK2-Gbetagamma complex. Science 310, 1686–1690.
Trejo, J. (2003). Protease-activated receptors: new concepts in regulation of G protein-coupled receptor signaling and trafficking. J. Pharmacol. Exp. Ther. 307, 437–442.
Vilardaga, J. P., Bunemann, M., Feinstein, T. N., Lambert, N., Nikolaev, V. O., Engelhardt, S., Lohse, M. J., and Hoffmann, C. (2009). GPCR and G proteins: drug efficacy and activation in live cells. Mol. Endocrinol. 23, 590–599.
Vilardaga, J. P., Bunemann, M., Krasel, C., Castro, M., and Lohse, M. J. (2003). Measurement of the millisecond activation switch of G protein-coupled receptors in living cells. Nat. Biotechnol. 21, 807–812.
Yi, T. M., Kitano, H., and Simon, M. I. (2003). A quantitative characterization of the yeast heterotrimeric G protein cycle. Proc. Natl. Acad. Sci. U.S.A. 100, 10764–10769.
Keywords: BRET, PAR1, G proteins, preassembly, precoupling, protein interactions
Citation: Ayoub MA, Al-Senaidy A and Pin J-P (2012) Receptor-G protein interaction studied by bioluminescence resonance energy transfer: lessons from protease-activated receptor 1. Front. Endocrin. 3:82. doi: 10.3389/fendo.2012.00082
Received: 25 April 2012; Accepted: 04 June 2012;
Published online: 22 June 2012.
Edited by:
Milka Vrecl, University of Ljubljana, SloveniaReviewed by:
Leigh Stoddart, University of Nottingham, UKMilka Vrecl, University of Ljubljana, Slovenia
Copyright: © 2012 Ayoub, Al-Senaidy and Pin. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Mohammed Akli Ayoub, Department of Biochemistry, College of Science, King Saud University P.O. Box: 2455, Riyadh – 11451 Kingdom of Saudi Arabia. e-mail:bWF5b3ViQGtzdS5lZHUuc2E=; Jean-Philippe Pin, Department of Molecular Pharmacology, Institute of Functional Genomics, CNRS UMR5203, INSERM U661, Universities Montpellier 1 and 2 – 141, rue de la cardonille, 34094 Montpellier Cedex 05, France. e-mail:anBwaW5AaWdmLmNucnMuZnI=