 James H. Felce1,2
James H. Felce1,2 Simon J. Davis1,2*
Simon J. Davis1,2*- 1 T-cell Biology Group, Nuffield Department of Clinical Medicine, University of Oxford, Oxford, UK
- 2 MRC Human Immunology Unit, University of Oxford, John Radcliffe Hospital, Oxford, UK
The first and arguably most important question that could be asked about the biology of any protein is: does it function alone? Cell surface receptors present special problems for stoichiometric analysis because, being located within lipid bilayers, they are often very hydrophobic, which means that once isolated they can exhibit a strong tendency to aggregate. A very welcome development, therefore, has been the advent of in situ methods for probing receptor organization, the most important of which are presently based on resonance energy transfer. Our first bioluminescence resonance energy transfer (BRET) experiments were, however, inconclusive since both monomeric and dimeric receptors gave high levels of energy transfer (James et al., 2006). It was only with the application of theoretical principles first developed for (Fung and Stryer, 1978; Wolber and Hudson, 1979), and then used in (Kenworthy and Edidin, 1998), Förster resonance energy transfer (FRET) experiments that we could use BRET to confidently distinguish between monomers and dimers.
We were very keen to test G protein-coupled receptors (GPCRs) using the new approach given the great interest in these important proteins forming constitutive oligomeric complexes (Angers et al., 2000; Ramsay et al., 2002; Babcock et al., 2003). This seemed unlikely to us firstly because, structurally, GPCRs are ideally configured for functioning autonomously (Meng and Bourne, 2001) and, secondly, because functional autonomy explains the remarkable evolutionary success (Schiöth and Fredriksson, 2005) of this very large family of receptors. We were initially ignorant of the extent to which BRET was used to buttress the “GPCRs as oligomers” concept (Pfleger and Eidne, 2005), but when our initial analyses of human β2-adrenergic (β2AR) and mouse cannabinoid (mCannR2) receptors yielded the “BRET signatures” of monomers (James et al., 2006), we had to confront this body of data. The resulting controversy (Bouvier et al., 2007; James and Davis, 2007a,b; Salahpour and Masri, 2007) seems to have prompted the development of other, more complicated approaches. Here, we describe our experiences using BRET and briefly consider the merits of these alternative approaches.
Once is not Enough
Like all resonance energy transfer-based methods, BRET is based on the principle of non-radiative energy transfer (Förster, 1948). In this case excitation energy is passed from a luminescent donor (luciferase) to a fluorescent acceptor protein, typically a modified variant of green fluorescent protein (GFP) such as yellow fluorescent protein or GFP2. Many early studies of surface receptors, particularly GPCRs, employed “conventional” BRET assays developed for analyzing interacting soluble proteins, in which donor- and acceptor-fused receptors are expressed at a single, fixed ratio, and BRET efficiency (BRETeff) is measured as relative to controls (Angers et al., 2000; Ramsay et al., 2002; Babcock et al., 2003). These early studies were largely unanimous in concluding that the receptors in question form homo- and hetero-oligomeric interactions and were significant in establishing the oligomeric GPCR paradigm (Pfleger and Eidne, 2005). We initially used this assay to determine whether an immune protein, CD80, forms dimers at the cell surface as implied by our crystal structure (Ikemizu et al., 2000), and were pleased to see strong energy transfer in our first experiments. However, the closely related protein, CD86, which is a monomer, also yielded high levels of energy transfer – as much as 25% of the levels obtained for covalent homodimers (James et al., 2006). We suspected that this was “background” energy transfer arising from random interactions within the membrane, a view strengthened by analysis of a second monomer, CD2. We concluded that conventional BRET assays could be problematic for measuring receptor organization in membranes because, within the crowded two-dimensional plane of the cell membrane, the signal arising from random interactions can reach significant levels.
Theoretical Work-Arounds
Theoretical considerations (Fung and Stryer, 1978; Wolber and Hudson, 1979; Kenworthy and Edidin, 1998) have predicted that the dependence of FRET on total and relative donor and acceptor concentrations differs systematically for specific and non-specific energy transfer. Applied to BRET in “type 1”experiments, total protein concentration is held constant and the acceptor/donor ratio increased by replacing donors with acceptors (Figure 1A; James et al., 2006). In this context, BRETeff for monomers is independent of the acceptor/donor ratio above a certain threshold because donors always experience the same “acceptor environment.” For oligomers, however, replacing donors with acceptors reduces the fraction of donor–donor complexes, converting them into BRET-productive pairs and increasing BRETeff. In “type 2” experiments (Figure 1B; James et al., 2006) total protein density is varied at constant acceptor/donor ratio. For monomeric proteins BRETeff varies linearly with total surface density for low expression levels, tending to zero at very low densities. Conversely, for constitutive oligomeric proteins BRETeff is largely constant because expression itself is generally reliant on oligomerization. However, at high densities, BRETeff increases due to random interactions of the oligomers within the membrane. For this reason it is inappropriate to draw any conclusions from the gradient of the slope for BRETeff versus expression level as, e.g., in Ramsay et al. (2002).
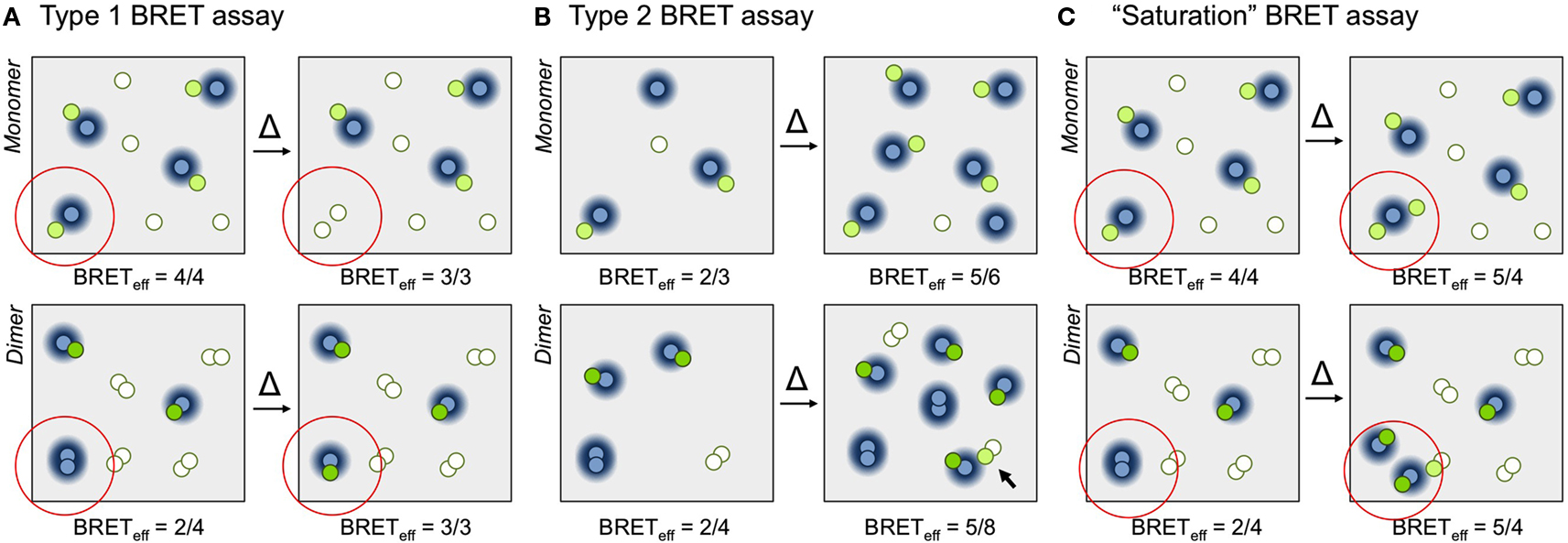
Figure 1. Principles of BRET assays. (A) In a type 1 BRET assay the acceptor/donor ratio is increased but surface density is kept constant. The increase in acceptor/donor ratio is obtained by exchanging a donor for an acceptor (the change is indicated within the red circle). For simplicity, BRETeff is defined here as the ratio of the numbers of fluorescent acceptors and luminescent donors. In the examples shown, for the monomer (top) BRETeff is unchanged (3/3 versus 4/4), whereas for the dimer (bottom) the ratio increases from 2/4 to 3/3 as the fraction of productive dimers increases. (B) In a type 2 BRET experiment, the acceptor/donor ratio is kept constant and surface density is varied, in this case by a factor of two. Due to the increase in monomer density (top) the likelihood of random collisions increases, and BRETeff increases from 2/3 to 5/6. For constitutive dimers (bottom), however, BRETeff is largely unchanged, increasing from 2/4 to 5/8, because the likelihood of dimerization doesn’t change. Only random interactions of dimers (arrow) contribute to increases in BRETeff (in reality, of course, these contributions will be significantly smaller than the effects of dimerization). (C) In the “saturation” BRET assay, the acceptor/donor ratio is increased by keeping donor numbers constant and increasing the numbers of acceptors. In the examples shown, BRETeff for monomers (top) and dimers (bottom) both increase upon addition of one or two extra acceptors, respectively (from 4/4 to 5/4 for the monomer, and from 2/4 to 5/4 for the dimer). This is due to the increased random interactions of monomers, and increased formation and random interactions of dimers. We expect assays in which BRETeff always increases to be more easily misinterpreted than assays in which changes in BRETeff vary systematically with receptor stoichiometry.Fluorescing and non-fluorescing acceptor molecules are shown as green and white circles, respectively, and donors as blue circles. The BRET-permissible area surrounding donors is represented as a blue halo.
Using these new types of BRET experiments we readily distinguished well-known monomeric and dimeric Type I membrane proteins, and even confirmed that CD80 forms apparently transient dimers at the cell surface, as implied by analytical ultracentrifugation (Ikemizu et al., 2000). Applied to two GPCRs, β2AR and mCannR2, these assays yielded the unambiguous “BRET signatures” of monomers (James et al., 2006). We also showed that the GABAβ receptor, a bona fide GPCR dimer, gave data characteristic of dimers and that transfer of the cytoplasmic domain of GABAβR2 to β2AR converted monomer-like into dimer-like behavior. As expected, since β2AR and other GPCRs were widely believed to form homo- and hetero-dimers (reviewed in Bouvier, 2001), these findings were controversial (Bouvier et al., 2007; James and Davis, 2007a,b; Salahpour and Masri, 2007).
Alternative Assays
Broadly speaking there is now consensus that conventional, single-ratio BRET experiments are inadequate to the task of assigning receptor stoichiometry. However, although type 1 and 2 BRET and FRET experiments are done occasionally (e.g., Kenworthy and Edidin, 1998; Meyer et al., 2006), these approaches are not widely used. Instead, the so-called BRET “saturation” assay first used in 2002 (Figure 1C; Mercier et al., 2002) remains popular (Contento et al., 2008; Ayoub and Pfleger, 2010). In this approach, donor numbers are kept constant and acceptor expression systematically increased. Under such conditions BRETeff for a monomeric protein is linearly related to acceptor expression level, whereas for oligomers the relationship is hyperbolic. The problem therefore becomes one of distinguishing between two increasing signals, which we would expect to be more difficult than distinguishing between increasing versus non-increasing signals, as in type 1 BRET assays (James et al., 2006). The problem becomes more acute for transient oligomers whose signals emerge from monomer/dimer equilibria, which is particularly relevant now that GPCRs are being claimed to transiently dimerize (Hern et al., 2010; Lambert, 2010; Kasai et al., 2011).
A second, newer assay, the “BRET competition” assay, presents subtler problems. In this assay, untagged “competitor” receptors are co-transfected with acceptor- and donor-tagged proteins, leading to reduced BRETeff for oligomers and unchanged BRETeff for monomers (Veatch and Stryer, 1977). In our experience, expression of untagged competitors often reduces expression of their tagged equivalents (Felce et al., unpublished data), including monomer control proteins, reducing BRETeff artifactually. In BRET competition assays of GPCR homo- and heterodimerization (e.g., Terrillon et al., 2003; Guo et al., 2008), reduced energy transfer in the presence of untagged competitors is always observed, yet the issue of surface density is never addressed. Such approaches have their place but the absolute levels of tagged protein must be factored in to avoid ambiguity.
Control Problems
An important factor complicating some BRET experiments is the heterogeneity of protein distribution, emphasizing the importance of the careful choice of controls. The cell membrane is a highly complex environment (Kusumi et al., 2011), and evidence is mounting that complex regulatory processes may control the localization and movement of integral membrane proteins, including GPCRs (Meyer et al., 2006; Nikolaev et al., 2010; Weigel et al., 2011). The potential for proteins to be localized to different areas of the cell surface, or to have different constraints on their trafficking, has important implications for data interpretation. This applies especially to “irrelevant” controls, which should have similar hydrodynamic diameter to the protein of interest but be sufficiently unrelated to not form specific associations (Angers et al., 2000; Mercier et al., 2002). However, such proteins may not be similarly localized at the membrane. For example, if the control protein exhibits strong association with the cytoskeleton but the protein of interest does not, BRETeff will be lower in the control experiment than it would be if the two proteins co-localized but randomly interacted. Similarly, control proteins may be expressed at different total densities or have different stoichiometries, adding further complications. Without knowing their behavior and expression characteristics in detail, it is difficult to select appropriate controls.
Approaches in which acceptors are recruited to donor-tagged proteins of interest are especially dependent on control choice. In “Third-party BRET” (Kuravi et al., 2010), a membrane-associated acceptor is chemically recruited to an untagged receptor of interest and BRETeff increases if the untagged receptor is a dimer that brings with it a donor-tagged receptor, the goal being to avoid the complication of varying expression levels. However, if the receptors are co-localized but do not interact, then acceptor/untagged receptor dimerization could recruit the acceptor to an area of greater donor concentration, increasing BRETeff without genuine association. Similar arguments apply to GPCR-Heteromer Identification Technology (GPCR-HIT; Pfleger, 2009; Mustafa and Pfleger, 2011). For this reason, no conclusively reliable BRET-based assay for heterodimers presently exists. Despite these difficulties, conventional (Pfleger and Eidne, 2005), saturation (Sohy et al., 2009), and competition (Terrillon et al., 2003) BRET assays have all been used to support claims for GPCR heterodimerization.
Concluding Remarks
There is now implicit agreement that single measurements of BRETeff are unhelpful because the contribution of random interactions to the signal is not easily discerned. Similarly, the notion that varying expression levels can also give potentially misleading changes in BRETeff is taking root, prompting new methods such as “Third-part BRET,” which seek to control for background effects in single measurements. The problem with these approaches is their heavy reliance on negative controls, which as we have discussed are often difficult to choose. We are surprised that the relatively simple approaches involving systematic variations of the acceptor/donor ratio, or of expression level alone, are not more widely used. We emphasize once again that the key to these methods is their exclusive reliance on the measurable, intrinsic behavior of populations of receptors diffusing in the plane of the membrane, and that an important advantage is that the assays are effectively control-independent.
Overall, the question of whether or not GPCRs generally form oligomers remains unsettled. The notion that they do is driven not only by BRET experiments, but also by FRET (Albizu et al., 2010; Cunningham et al., 2012), photon-counting analyses (Kilpatrick et al., 2012), and single-molecule microscopy (Hern et al., 2010; Kasai et al., 2011). We are seeking to test our BRET-based conclusions using super-resolution imaging, and to address GPCR stoichiometry at the family level using type 1 BRET and other experiments implemented in a high throughput setting.
Despite the controversies over its use BRET still has a very bright future. New luciferases, such as Rluc2 and Rluc8 (De et al., 2007), and acceptor fluorophores, such as Venus (Kocan et al., 2008), mOrange (De et al., 2009), and Renilla GFP (RGFP; Kamal et al., 2009), are brighter and offer up the possibility of in vivo studies (De et al., 2009). Future developments in BRET-quantum dot (Wu et al., 2011; Quiñones et al., 2012) and BRET-FRET (Carriba et al., 2008) assays will also advance the technique. The effective resolution of resonance energy transfer methods in live cells, i.e., ∼10 nm, is presently significantly better than that of in situ single-molecule imaging techniques, which, even in fixed cells, is limited to ∼20 nm (Moerner, 2012). We think that it will be some time before BRET, rigorously applied, is surpassed as a probe of receptor stoichiometry.
Acknowledgments
The authors acknowledge the critical contributions of J.R. James to this work, and thank R. Knox for helpful comments on the manuscript. This work was funded by the Wellcome Trust and UK Medical Research Council.
References
Albizu, L., Cottet, M., Kralikova, M., Stoev, S., Seyer, R., Brabet, I., Roux, T., Bazin, H., Bourrier, E., Lamarque, L., Breton, C., Rives, M. L., Newman, A., Javitch, J., Trinquet, E., Manning, M., Pin, J. P., Mouillac, B., and Durroux, T. (2010). Time-resolved FRET between GPCR ligands reveals oligomers in native tissues. Nat. Chem. Biol. 6, 587–594.
Angers, S., Salahpour, A., Joly, E., Hilairet, S., Chelsky, D., Dennis, M., and Bouvier, M. (2000). Detection of β2-adrenergic receptor dimerisation in living cells using bioluminescence resonance energy transfer (BRET). Proc. Natl. Acad. Sci. U.S.A. 97, 3684–3689.
Ayoub, M. A., and Pfleger, K. D. G. (2010). Recent advances in bioluminescence resonance energy transfer technologies to study GPCR heteromerisation. Curr. Opin. Pharmacol. 10, 44–52.
Babcock, G., Farzan, M., and Sodroski, J. (2003). Ligand-independent dimerisation of CXCR4, a principal HIV-1 coreceptor. J. Biol. Chem. 278, 3378–3385.
Bouvier, M. (2001). Oligomerization of G protein-coupled transmitter receptors. Nat. Rev. 2, 274–286.
Bouvier, M., Heveker, N., Jockers, R., Marullo, S., and Milligan, G. (2007). BRET analysis of GPCR oligomerisation: newer does not mean better. Nat. Methods 4, 3–4.
Carriba, P., Navarro, G., Ciruela, F., Ferré, S., Casadó, V., Agnati, L., Cortés, A., Mallol, J., Fuxe, K., Canela, E. I., Lluís, C., and Franco, R. (2008). Detection of heteromerization of more than two proteins by sequential BRET-FRET. Nat. Methods 5, 727–733.
Contento, R. L., Molon, B., Boularan, C., Pozzan, T., Manes, S., Marullo, S., and Viola, A. (2008). CXCR4-CCR5: a couple modulating T cell functions. Proc. Natl. Acad. Sci. U.S.A. 105, 10101–10106.
Cunningham, M. R., McIntosh, K. A., Pediani, J. D., Robben, J., Cooke, A. E., Nilsson, M., Gould, G. W., Mundell, S., Milligan, G., and Plevin, R. (2012). Novel role for proteinase-activated receptor 2 (PAR2) in membrane trafficking of proteinase-activated receptor 4 (PAR4). J. Biol. Chem. 287, 16656–16669.
De, A., Loening, A. M., and Gambhir, S. S. (2007). An improved bioluminescence resonance energy transfer strategy for imagine intracellular events in single cells and living subjects. Cancer Res. 67, 7175–7183.
De, A., Ray, P., Loening, A. M., and Gambhir, S. S. (2009). BRET3: a red-shifted bioluminescence resonance energy transfer (BRET)-based integrated platform for imagine protein-protein interactions from single live cells and living animals. FASEB J. 23, 2702–2709.
Fung, B. K., and Stryer, L. (1978). Surface density determination in membranes by fluorescence energy transfer. Biochemistry 17, 5241–5248.
Guo, W., Urizar, E., Kralikova, M., Mobarec, J. C., Shi, L., Filizola, M., and Javitch, J. A. (2008). Dopamine D2 receptors form higher order oligomers at physiological expression levels. EMBO J. 27, 2293–2304.
Hern, J. A., Baig, A. H., Mashanov, G. I., Birdsall, B., Corrie, J. E. T., Lazareno, S., Molloy, J. E., and Birdsall, N. J. M. (2010). Formation and dissociation of M1 muscarinic receptor dimers seen by total internal reflection fluorescence imaging of single molecules. Proc. Natl. Acad. Sci. U.S.A. 107, 2693–2698.
Ikemizu, S., Gilbert, R. J., Fennelly, J. A., Collins, A. V., Harlos, K., Jones, E. Y., Stuart, D. I., and Davis, S. J. (2000). Structure and dimerization of a soluble form of B7-1. Immunity 12, 51–60.
James, J. R., and Davis, S. J. (2007a). Reply to: BRET analysis of GPCR oligomerisation: newer does not mean better. Nat. Methods 4, 4.
James, J. R., and Davis, S. J. (2007b). Reply to: experimental challenge to a “rigorous” BRET analysis of GPCR oligomerisation. Nat. Methods 4, 601.
James, J. R., Oliveira, M. I., Carmo, A. M., Iaboni, A., and Davis, S. J. (2006). A rigorous experimental framework for detecting protein oligomerization using bioluminescence resonance energy transfer. Nat. Methods 3, 1001–1006.
Kamal, M., Marquez, M., Vauthier, V., Leloire, A., Froguel, P., Jockers, R., and Couturier, C. (2009). Improved donor/acceptor BRET couples for monitoring beta-arrestin recruitment to G protein-coupled receptors. Biotechnol. J. 4, 1337–1344.
Kasai, R. S., Suzuki, K. G. N., Prossnitz, E. R., Koyama-Honda, I., Nakada, C., Fujiwara, T. K., and Kusumi, A. (2011). Full characterization of GPCR monomer-dimer dynamic equilibrium by single molecule imaging. J. Cell Biol. 192, 463–480.
Kenworthy, A. K., and Edidin, M. (1998). Distribution of a glycosylphosphatidylinositol anchored protein at the apical surface of MDCK cells examined at a resolution of <100 A using imaging fluorescence resonance energy transfer. J. Cell Biol. 142, 69–84.
Kilpatrick, L. E., Briddon, S. J., and Holliday, N. D. (2012). Fluorescence correlation spectroscopy, combined with bimolecular fluorescence complementation, reveals the effects of β-arrestin complexes and endocytic targeting on the membrane mobility of neuropeptide Y receptors. Biochim. Biophys. Acta 1823, 1068–1081.
Kocan, M., See, H. B., Seeber, R. M., Eidne, K. A., and Pfleger, K. D. G. (2008). Demonstration of improvements to the bioluminescence resonance energy transfer (BRET) technology for the monitoring of G protein-coupled receptors in live cells. J. Biomol. Screen 13, 888–898.
Kuravi, S., Lan, T. H., Barik, A., and Lambert, N. A. (2010). Third-party bioluminescence resonance energy transfer indicates constitutive association of membrane proteins: application to class A G-protein-coupled receptor and G proteins. Biophys. J. 98, 2391–2399.
Kusumi, A., Suzuki, K. G. N., Kasai, R. S., Ritchie, K., and Fujiwara, T. K. (2011). Hierarchical mesoscale domain organization of the plasma membrane. Trends Biol. Sci. 36, 604–615.
Meng, E. C., and Bourne, H. R. (2001). Receptor activation: what does the rhodopsin structure tell us? Trends Pharmacol. Sci. 22, 587–593.
Mercier, J. F., Salahpour, A., Angers, A., Breit, A., and Bouvier, M. (2002). Quantitative assessment of β1- and β2-adrenergic receptor homo- and heterodimerization by bioluminescence resonance energy transfer. J. Biol. Chem. 277, 44925–44931.
Meyer, B., Segura, J. M., Martinez, K. L., Hovius, R., George, N., Johnsson, K., and Vogel, H. (2006). FRET imaging reveals that functional neurokinin-1 receptors are monomeric and reside in membrane microdomains of live cells. Proc. Natl. Acad. Sci. U.S.A. 103, 2138–2143.
Moerner, W. E. (2012). Microscopy beyond the diffraction limit using actively controlled single molecules. J. Microsc. 246, 213–220.
Mustafa, S., and Pfleger, K. D. G. (2011). G protein-coupled receptor heteromer identification technology: identification and profiling of GPCR heteromers. J. Lab. Autom. 16, 285–291.
Nikolaev, V. O., Moshkov, A., Lyon, A. R., Miragoli, M., Novak, P., Paur, H., Lohse, M. J., Korchev, Y. E., Harding, S. E., and Gorelik, J. (2010). β2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science 327, 1653–1657.
Pfleger, K. D. G. (2009). New technology to identify potential novel G protein-coupled receptor heterodimers and heterodimer-specific compounds. J. Biomol. Screen 14, 890.
Pfleger, K. D. G., and Eidne, K. A. (2005). Monitoring the formation of dynamic G protein-coupled receptor-protein complexes in living cells. Biochem. J. 285, 625–637.
Quiñones, G. A., Miller, S. C., Bhattacharyya, S., Sobek, D., and Stephan, J. P. (2012). Ultrasensitive detection of cellular protein interactions using bioluminescence resonance energy transfer quatum dot-based nanoprobes. J. Cell Biol. 113, 2397–2405.
Ramsay, D., Kellett, E., McVey, M., Rees, S., and Milligan, G. (2002). Homo- and hetero-oligomeric interactions between G protein-coupled receptors in living cells monitored by two variants of bioluminescence resonance energy transfer (BRET): hetero-oligomers between receptor subtypes form more efficiently than between less closely related sequences. Biochem. J. 365, 429–440.
Salahpour, A., and Masri, B. (2007). Experimental challenge to a “rigorous” BRET analysis of GPCR oligomerisation. Nat. Methods 4, 599–600.
Schiöth, H. B., and Fredriksson, R. (2005). The GRAFS classification system of G-protein coupled receptors in comparative perspective. Gen. Comp. Endocrinol. 142, 94–101.
Sohy, D., Yano, H., de Nadai, P., Urizar, E., Guillabert, A., Javitch, J. A., Parmentier, M., and Springael, J. Y. (2009). Hetero-oligomerization of CCR2, CCR5, and CXCR4 and the protean effects of “selective” antagonists. J. Biol. Chem. 284, 31270–31279.
Terrillon, S., Durroux, T., Mouillac, B., Breit, A., Ayoub, M. A., Taulan, M., Jockers, R., Barberis, C., and Bouvier, M. (2003). Oxytocin and vasopressin V1a and V2 receptors form constitutive homo- and heterodimers during biosynthesis. Mol. Endocrinol. 17, 677–691.
Veatch, W., and Stryer, L. (1977). The dimeric nature of the gramicidin A transmembrane channel: conductance and fluorescence energy transfer studies of hybrid channels. J. Mol. Biol. 133, 89–102.
Weigel, A. V., Simon, B., Tamkun, M. M., and Krapf, D. (2011). Ergodic and nonergodic processes coexist in the plasma membrane as observed by single-molecule tracking. Proc. Natl. Acad. Sci. U.S.A. 108, 6438–6443.
Wolber, P. K., and Hudson, B. S. (1979). An analytic solution to the Forster energy transfer problem in two dimensions. Biophys. J. 28, 197–210.
Citation: Felce JH and Davis SJ (2012) Unraveling receptor stoichiometry using BRET. Front. Endocrin. 3:86. doi: 10.3389/fendo.2012.00086
Received: 18 June 2012; Accepted: 23 June 2012;
Published online: 12 July 2012.
Edited by:
Milka Vrecl, University of Ljubljana, SloveniaReviewed by:
Pierre De Meyts, Novo Nordisk A/S, DenmarkCopyright: © 2012 Felce and Davis. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence:c2ltb24uZGF2aXNAbmRtLm94LmFjLnVr