

 Eric W.-F. Lam3
Eric W.-F. Lam3
- 1 Department of Veterinary Basic Sciences, The Royal Veterinary College, London, UK
- 2 Department of Craniofacial Development and Stem Cell Biology, King’s College London, Guy’s Hospital, London, UK
- 3 Department of Surgery and Cancer, Imperial College London, Hammersmith Hospital, London, UK
- 4 School of Veterinary Sciences, University of Bristol, Bristol, UK
The phosphoinositide 3-kinase and AKT (protein kinase B) signaling pathway (PI3K/AKT) plays a central role in the control of cell survival, growth, and proliferation throughout the body. With regard to bone, and particularly in osteoblasts, there is an increasing amount of evidence that the many signaling molecules exert some of their bone-specific effects in part via selectively activating some of the generic effects of the PI3K/AKT pathway in osteoblasts. There is further data demonstrating that PI3K/AKT has the capacity to specifically cross-talk with other signaling pathways and transcriptional networks controlling bone cells’ development in order to fine-tune the osteoblast phenotype. There is also evidence that perturbations in the PI3K/AKT pathway may well be responsible for certain bone pathologies. In this review, we discuss some of these findings and suggest that the PI3K/AKT pathway is a central nexus in the extensive network of extracellular signaling pathways that control the osteoblast.
Introduction
A fine balance between the bone forming activities of osteoblasts and the bone resorptive effects of osteoclasts, substantially influenced by osteocytes (Bonewald, 2011; Long, 2012) provides the skeleton with the correct architecture and strength to support everyday loads without fracture. Osteoblasts are derived from a pool of multipotent mesenchymal stem cells (MSCs) which are able to form bone, cartilage, muscle, adipose, and connective tissue via activation of distinct differentiation programs (Long, 2012). Thus, the phenotype of a mature osteoblast reflects its molecular history in terms of signaling molecule exposure, signal transduction pathway activation, and changes engendered in transcriptional networks, as well as epigenetic modifications. It is a reflection of this cellular context that a panoply of seemingly ubiquitous signaling molecules is capable of producing such a uniquely specific outcome, i.e., the transition from an MSC to a mature osteoblast. In this review, we will highlight the role of one pathway, the phosphoinositide 3-kinase and AKT signaling pathway (PI3K/AKT), in osteoblast differentiation and homeostasis.
The Phosphoinositide 3-Kinase and AKT Signaling Pathway
The PI3K pathway is activated through the receptor tyrosine kinase (RTK) class of receptors which include fibroblast growth factor receptors (FGFRs), insulin-like growth factor receptors (IGFRs), and insulin receptor (Figure 1). Engagement of the ligand with the RTK causes autophosphorylation of tyrosine residues in the cytoplasmic domain. These phosphorylated tyrosine residues then recruit docking proteins, most notably IRS1, which in turn recruits the p85 subunit of PI3K. PI3K itself consists of a regulatory p85 subunit and a catalytic p110 subunit. Recruitment of the PI3K complex to the inner surface of the plasma membrane juxtaposes it with its substrate phosphatidylinositol-4,5 diphosphate (PIP2) located in the inner lamina of the cell membrane. PIP2 is then phosphorylated by the p110 subunit to form phosphatidylinositol-3,4,5 trisphosphate (PIP3). The conversion of PIP3 to PIP2, and the subsequent inactivation of PI3K downstream signaling, is facilitated by the tumor suppressor and phosphatase; phosphatase and tensin homolog deleted on chromosome ten (PTEN) (Cantley and Neel, 1999).
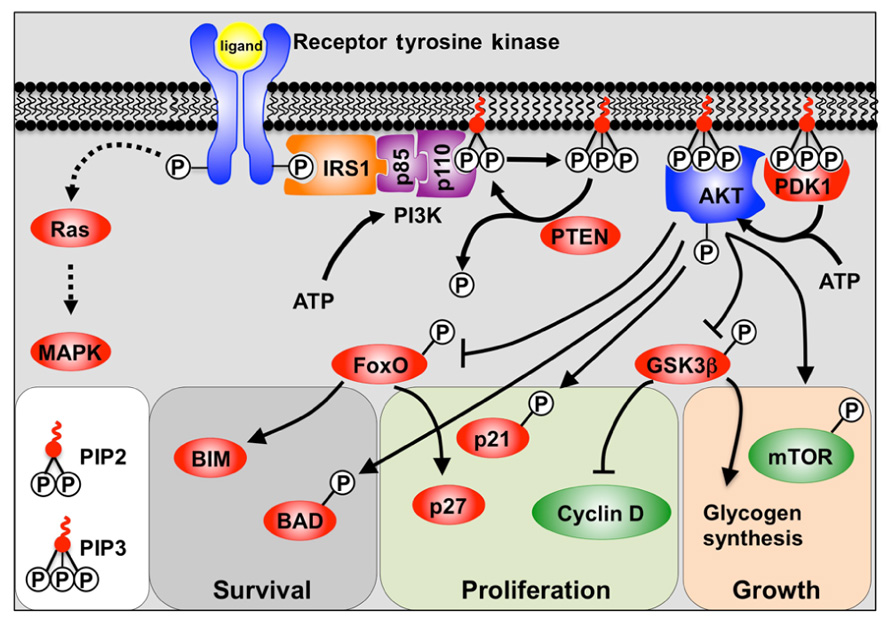
FIGURE 1. A model depicting PI3K signaling. Binding of the receptor tyrosine kinase ligand activates receptor auto-phosphorylation, leading to recruitment of substrate proteins such as IRS-1. This leads to recruitment of the regulatory (p85) and catalytic (p110) subunits of class 1a PI3K. PI3K phosphorylation of PIP2 to PIP3 allows PIP3 to act as a secondary messenger within the inner surface of the cell membrane. AKT and PDK1 bind to PIP3, and PDK1 and mTOR/Rictor activate AKT via phosphorylation. Active AKT is then able to promote cell survival, growth, and proliferation by phosphorylation of key substrates. Also shown is the alternative Ras pathway which can also be stimulated by receptor tyrosine kinases to activate MAPKs.
Phosphatidylinositol-3,4,5 trisphosphate then acts as a secondary messenger within the membrane, recruiting the binding of proteins containing pleckstrin homology (PH) domains to the inner surface of the cell membrane. Most notable amongst the PH domain containing proteins are AKT (also known as protein kinase B, PKB) and PDK1. The AKT/PKB family is comprised of three separate genes (AKT1–3) and all encode serine/threonine protein kinases. Upon translocation to the cell membrane, AKT is phosphorylated by PDK1, and the mTORC2/Rictor complex, which provides full activation of AKT (Cantley and Neel, 1999).
Whilst RTKs activate AKT via PI3K, they can also activate mitogen-activated protein kinases (MAPKs) such as ERK1/2 via the Ras/Raf pathway (Ramos, 2008).
AKT Targets
AKT activation is generally associated with the processes of cell survival, growth, and proliferation (Manning and Cantley, 2007). Cell survival is chiefly regulated by inhibitory phosphorylation of the FoxO family of transcription factors by AKT, and thus attenuation of their pro-apoptotic target gene Bim (Dijkers et al., 2002; Sunters et al., 2003), as well as by direct inactivation by phosphorylation of Bad (Datta et al., 2002). In contrast, cell growth is regulated by activation of mTOR (mammalian Target of Rapamycin) thereby promoting protein synthesis (Wullschleger et al., 2006). Proliferation is stimulated via a number of mechanisms including inhibition of the FoxO-mediated transcription of the cyclin-dependent kinase inhibitor p27Kip1 (Dijkers et al., 2000; Ho et al., 2008). AKT also inhibits both p27Kip1 and p21Cip1 function by phosphorylation (Zhou et al., 2001; Liang et al., 2002), as well as by regulating D-type cyclins (Liang and Slingerland, 2003).
Osteoblast Differentiation
Osteoblast differentiation begins with MSC commitment to the osteoblast lineage by expression of the osteoblast-specific transcription factor Runx2 (Otto et al., 1997). New osteoprogenitors proliferate then express the matrix promoting proteins: collagen 1a (Col-1a; Bellows et al., 1986; Lee et al., 2000), osteopontin (OPN; Hay et al., 1999; El-Tanani et al., 2004), alkaline phosphatase (ALP; Aronow et al., 1990), and bone morphogenic proteins (BMPs; van der Horst et al., 2002; Rosen, 2006). Integrin activation by the newly synthesized matrix promotes maturation into pre-osteoblasts, which express the transcription factor Osterix (Osx; Xiao et al., 1998; Meyers et al., 2004). As pre-osteoblasts mature, Runx2 and Col-1a expression declines, whilst Bone Sialoprotein (Chen et al., 1997; Lai and Cheng, 2005) and Osteocalcin expression increases (Hay et al., 1999).
Too Much PI3K/AKT – “Just Big Boned?”
One of the problems in delineating a truly osteoblastic role for PI3K/AKT signaling in bone biology is the dissection of the generic effects (survival, proliferation, and growth) from those that are limited to osteoblasts. However, genetic studies using transgenic and knockout mice targeting the PI3K/AKT pathway have yielded informative results. Original global deletions of the PTEN tumor suppressor resulted in cells with elevated PI3K/AKT signaling, but embryonic lethality (Di Cristofano et al., 1998; Suzuki et al., 1998). However, when mice containing Cre under the control of the Col2a1 promoter were crossed with mice containing a floxed PTEN allele, PTEN was knocked out in osteo-chondroprogenitor cells (Ford-Hutchinson et al., 2007). These mice exhibited a disorganized growth plate, excessive matrix production, and elevated AKT and S6K activation in growth plate chondrocytes and osteoblasts of the primary spongiosa (Ford-Hutchinson et al., 2007). The mice developed elongated long bones containing extensive trabeculation and increased cortical thickness, but interestingly no increase in chondrocyte proliferation (Ford-Hutchinson et al., 2007). Guntur et al. (2011) generated mice in which PTEN was ablated solely in osteoprogenitors. The osteoprogenitors in these mice had increased proliferation and survival, and this expanded pool of progenitors differentiated rapidly. However, accelerated osteoblast differentiation caused precocious osteoid deposition in the developing perichondrium, which was uncoupled from interaction with chondrocytes, resulting in shorter but broader bones (Guntur et al., 2011). Late stage osteoblast-specific PTEN knockout mice were obtained by crossing osteocalcin-Cre with floxed PTEN mice. These mice were of usual size, but exhibited increased bone formation throughout life due to elevated AKT-mediated cell survival (Liu et al., 2007). Whilst the PTEN knockout mice have hinted the consequence of elevated PI3K signaling to AKT, it is only recent studies using AKT knockout mice that have provided more detailed information regarding isoform specificity in osteoblast biology.
Not Enough PI3K/AKT – “Small Fry”
Global AKT-1 knockout mice are small with reduced bone mineral density, suggesting a potential osteoblast-specific effect (Kawamura et al., 2007). However, this may be a more generic effect, and indeed was attributed to an increase in apoptosis rates mediated by a failure of AKT1 to repress FoxO3a transcription of Bim (Kawamura et al., 2007). More specifically a reduction in Runx2 target gene expression was observed, including RANKL, which resulted in decreased osteoblast induced osteoclastogenesis and slow bone remodeling (Kawamura et al., 2007). Global AKT-2 knockouts develop severe diabetes, but skeletally they only display a very mild decrease in weight and length (Garofalo et al., 2003). AKT-1/AKT-2 double knockouts die shortly after birth and exhibit dwarfism (Peng et al., 2003). The osteoblastic phenotype is extreme, with embryos having negligible ossification (Peng et al., 2003) suggesting some functional redundancy between AKT1 and 2. AKT3 knockout mice are viable, have reduced brain size, but no discernable skeletal phenotype (Easton et al., 2005). Taken together, these genetic models suggest a specific role in bone is confined to AKT1 and/or 2, and occurs both in an osteoblast cell autonomous manner and via their interactions with chondrocytes and osteoclasts.
Stimulating PI3K/AKT in Bone
The AKT and PTEN knockouts indicate that skeletal development and functional integrity rely on fine-tuning of the AKT signaling pathway, itself controlled by other signaling molecules, the most important of which are fibroblast growth factor (FGF) and insulin-like growth factor (IGF).
The FGF family consists of at least 22 members coupled with at least 5 receptors (FGFRs) (Bottcher and Niehrs, 2005; Coutu and Galipeau, 2011). During endochondral ossification FGFR3 is the predominant receptor expressed by condensing mesenchymal chondrocytes and proliferating chondrocytes in the growth plate, whilst FGFR1 is expressed by hypertrophic chondrocytes, and thought to play a role in blocking proliferation. In osteoblasts FGFR-1 and -2 are both expressed, and FGF2 stimulates the proliferation of osteoblast precursors (Yu et al., 2003).
IGF-1 plays a crucial role in postnatal bone growth, especially during puberty. The majority of circulating IGF-1 is produced by the liver (DiGirolamo et al., 2007; Yakar et al., 2010), and mice with a conditional IGF-1 knockout in the liver are essentially normal, but have reduced circulating IGF-1 and a reduction in cortical bone deposition (Yakar et al., 2009). However, mice with a targeted IGF-1R deletion in osteoblasts exhibited a time lag between osteoid deposition and mineralization, and thus make poor quality bone (Zhang et al., 2002). Conversely, osteoblast-specific IGF-1 overexpression in mice increased trabecular bone volume without any associated hyperplasia (Zhao et al., 2000). These findings demonstrate that local IGF-1 signaling also influences bone architecture and mineralization. Insulin, like IGF-1, has been shown to be anabolic in bone, and induces osteocalcin expression, but lacks the mitogenic effects of IGF-1 (Zhang et al., 2012).
Whilst IGF and FGF signaling activates AKT via PI3K, they also have the ability to activate MAPKs (Ramos, 2008), and the relative contributions of these two pathways to osteoblast function remain unclear. Whilst the osteopenia observed in osteoblast-specific Gab1 knockout mice has been linked to impaired IGF-1/insulin signaling via both PI3K and Ras pathways (Weng et al., 2010), recent data suggests the mitogenic effect of IGF-1 requires both pathways, whilst differentiation relies on PI3K/AKT signaling (Raucci et al., 2008).
Interaction between PI3K/AKT and Osteogenic Factors
Recent evidence suggests that Runx2 functions more as a promoter organizer rather than a transcription factor, acting as a central hub to recruit transcriptional co-activators such as CBP (Schroeder et al., 2005) or inhibitors such as Sin3/histone deacetylases (Westendorf, 2006). Furthermore, Runx2 interacts with other transcription factors such as AP-1 and estrogen receptor alpha (ERα) (Westendorf, 2006; Khalid et al., 2008; Chen et al., 2012). The ability of Runx2 to function as a transcriptional activator or repressor is fine-tuned by phosphorylation, for example, ERK1/2 and p38-MAPK phosphorylation promote osteoblast differentiation (Xiao et al., 2002; Greenblatt et al., 2010). In contrast, JNK1 phosphorylation of Runx2 is inhibitory, blocking the early stages of differentiation (Huang et al., 2012), however, since JNK activity is associated with the terminal stages of differentiation, this inhibition of Runx2 might correlate with reduced Runx2 function in mature osteoblasts (Matsuguchi et al., 2009).
Although Runx2 target gene expression is reduced in AKT-1 knockout mice, Runx2–PI3K/AKT interactions are unlikely to occur via direct phosphorylation because Runx2 lacks an AKT consensus phosphorylation site (Kawamura et al., 2007). Notwithstanding, direct phosphorylation by AKT blocks the ability of GSK3β to inhibit Runx2 DNA binding (Kugimiya et al., 2007) and of FoxO1 to represses Runx2-dependent osteocalcin transcription (Yang et al., 2010; Zhang et al., 2011). A potential feed-forward loop between Runx2 and PI3K/AKT may also exist as Runx2 activates p85 and p110β PI3K subunit transcription (Fujita et al., 2004).
Osterix is a Runx2 target gene that stimulates osteoblast lineage commitment and promotes osteoblast maturation (Komori, 2006; Nishio et al., 2006; Zhou et al., 2010). Apart from the effects of PI3K/AKT on Runx2, there is little evidence of direct phosphorylation of Osx by AKT (Choi et al., 2011), but PI3K/AKT is required for BMP-induced Osx transcriptional activity (Mandal et al., 2010; Choi et al., 2011).
Osteogenic Signaling Pathways
Canonical Wnt Signaling
During absence of Wnt ligands, the kinase GSK3β phosphorylates the transcriptional co-activator β-catenin, thereby targeting it for proteosomal degradation. Engagement of Wnt ligands with the Frizzled/Lrp5 or -6 co-receptor inhibits GSK3β, leading to the accumulation of hypo-phosphorylated β-catenin, which translocates to the nucleus in order to stimulate Lef/TCF target gene expression. Wnt signaling functions during skeletal development partly by promoting osteoblastic commitment (Hill et al., 2005). Humans with inactivating or activating mutations in Lrp5 exhibit low or high bone mass, respectively, implicating Wnt in postnatal bone homeostasis (Whyte et al., 2004; Ai et al., 2005; Ferrari et al., 2005). Inhibitory phosphorylation of GSK3β by AKT results in the activation of β-catenin (Smith and Frenkel, 2005; Sunters et al., 2010). Furthermore, direct phosphorylation of β-catenin by AKT has been shown to increase its capacity for transcriptional activation of Lef/TCF target genes (Fang et al., 2007).
BMP Signaling
Bone morphogenic proteins are members of the TGFβ family and bind to tetrameric type I and II receptors on the cell surface which phosphorylate members of the SMAD family (SMAD 1, 5, and 8). Phospho-SMADs bind to SMAD4 and translocate to the nucleus to regulate gene expression. Blocking BMP signaling with the BMP antagonist Noggin reduces osteoblast differentiation, and results in mice with severe osteoporosis (Wu et al., 2003). Repression of BMP signaling by Twist 1/2 prevents osteoblast commitment by mesenchymal precursors by silencing Runx2, AP, and OPN transcription (Bialek et al., 2004; Hayashi et al., 2007). Mice lacking BMP2 and -4 in limb bud mesenchyme have impaired osteogenesis (Bandyopadhyay et al., 2006), and whilst BMP2 knockout mice do make bone, they exhibit a mineralization deficit making them susceptible to fractures (Tsuji et al., 2006). BMP3 is produced by mature osteoblasts and osteocytes to prevent osteoblast differentiation, providing a negative feedback loop to control osteoblast numbers (Kokabu et al., 2012). PI3K/AKT is required for BMP-induced Osx activation (Osyczka and Leboy, 2005; Mukherjee and Rotwein, 2009; Mandal et al., 2010; Choi et al., 2011), this dependency would also appear to be isoform specific, since recent work by Mukherjee et al. (2010) demonstrated that a unique function of AKT2 is required for BMP2-mediated osteoblast differentiation.
AKT has been also shown to be activated by BMPs via a mechanism that is not fully understood (Osyczka and Leboy, 2005; Mukherjee and Rotwein, 2009; Mukherjee et al., 2010). Intriguingly, osteogenic BMP targets include the Id family of genes which can inhibit the Twist blockade of BMP-induced transcription, and thus osteogenesis (Ogata et al., 1993; Miyazono and Miyazawa, 2002; Ying et al., 2003; Peng et al., 2004; Nakashima et al., 2005). Id1 transcription is repressed by FoxOs (Birkenkamp et al., 2007), and it is possible that PI3K/AKT inactivation of FoxOs could cooperate with SMADs in the BMP-mediated activation of Id1. Furthermore, BMP and Id1 can activate AKT via PTEN repression (Beck and Carethers, 2007; Chow et al., 2007,2008; Lee et al., 2009), however, this is unlikely to be the only mechanism responsible. It is attractive to speculate that these findings may represent reciprocal activation between PI3K/AKT and BMP signaling – which could potentially amplify osteogenic responses to BMP, IGF-1, and FGFs.
Pathologies Involving PI3K/AKT
The PI3K/AKT signaling pathway in osteoblasts is clearly important for normal skeletal development and homeostasis, however, it is also implicated in various pathological conditions.
Osteoporosis and Mechanical Strain
Bone loss and fracture susceptibility that characterize osteoporosis are often thought of as symptomatic of an attenuation of resident bone cells’ ability to use the mechanical strain engendered by normal load bearing activity as a stimulus to remodel both bone mass and architecture (Lanyon, 2008; Price et al., 2010). The initial detection of strain is thought to occur in osteocytes (Bonewald, 2007), but does also occur in osteoblasts, and involves signaling via ERα (Jessop et al., 2002; Lee et al., 2003; Zaman et al., 2010), production of prostaglandins (Zaman et al., 1997), nitric oxide (Pitsillides et al., 1995), ATP (Rumney et al., 2012), canonical Wnt signaling (Lau et al., 2006; Armstrong et al., 2007), IGFs (Zaman et al., 1997; Cheng et al., 1999), and the suppression of the soluble Wnt antagonist sclerostin (Galea et al., 2011). With regard to PI3K/AKT signaling, activation of β-catenin by strain requires AKT-mediated inhibition of GSK3β (Case et al., 2008; Sunters et al., 2010), which also occurs in osteocytes (Kitase et al., 2010). AKT activation occurs via PI3K and is Wnt independent, but depends on the formation of a complex between ERα and IGF-1R which increased the responsiveness of IGF-1R to ambient levels of IGF-1 (Sunters et al., 2010). Whilst administration of IGF-1 to osteoporosis patients has little positive effect on bone density (Friedlander et al., 2001), osteoblasts isolated from osteoporotic donors have an attenuated PI3K/AKT response to IGF-1, suggesting that IGF-1R responsiveness may play a role in osteoporosis (Perrini et al., 2008).
Osteosarcoma
Osteosarcoma is the most common malignancy affecting the skeleton, and although relatively rare in humans (Broadhead et al., 2011), is much more common in dogs, especially large and giant breeds (Tjalma, 1966; Chun and de Lorimier, 2003; Chun, 2005). Since susceptible large breed dogs have higher IGF-1 levels (Burrow et al., 1998), and IGF-1R is often overexpressed in canine osteosarcomas (Eigenmann et al., 1984; Sutter et al., 2007), a link between IGF-1 signaling and osteosarcoma has been proposed. In support of this, PTEN is commonly down regulated, mutated, or deleted in many canine and human osteosarcomas (Levine et al., 2002; MacEwen et al., 2004) and is associated with elevated AKT activation, suggesting that PI3K/AKT may play a causative role in osteosarcoma formation.
It is possible that other conditions associated with increased or decreased bone formation may have perturbed PI3K/AKT function as a common event. For example, the PTEN bone-specific knockout mouse, which results in an increase in AKT signaling, has increased bone formation, leading to osteopetrosis (Liu et al., 2007). Moreover, mutations in Irs1 resulting in reduced AKT phosphorylation lead to reduced bone formation (DeMambro et al., 2010). However, effects need not be manifest globally in the whole skeleton as demonstrated by the Twist haploinsufficiency model, where down regulation of the ubiquitin ligase Cbl promotes AKT signaling through a reduction in PI3K degradation. The result is increased bone formation specifically in the coronal suture in Saethre-Chotzen syndrome (Guenou et al., 2006).
Summary
Given the generic roles of the PI3K/AKT pathway, it is not unexpected that it would play a role in osteoblasts. However recent evidence suggests that some of these generic effects are selectively activated in osteoblasts during normal physiology. Additional specificity is introduced when one considerers the highly selective downstream interactions between the PI3K/AKT pathway and other pathways controlling osteoblast differentiation and function. Our contention is that the PI3K/AKT pathway may well be a central nexus in the networks of signaling pathways that helps to fine-tune osteoblast differentiation and homeostasis to produce a normal skeleton. Thus AKT represents a viable therapeutic target in multiple skeletal diseases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Imelda M. McGonnell, Joanna S. Price, and Andrew Sunters are supported by grants from the Wellcome Trust, Agamemnon E. Grigoriadis is supported by grants from Guy’s and St Thomas’ Charity, and Eric W.-F. Lam is supported by grants from Breast Cancer Campaign and Cancer Research-UK. The authors would also like to acknowledge the Bone Research Society for funding the publication costs of this article.
References
Ai, M., Heeger, S., Bartels, C. F., and Schelling, D. K. (2005). Clinical and molecular findings in osteoporosis-pseudoglioma syndrome. Am. J. Hum. Genet. 77, 741–753.
Armstrong, V. J., Muzylak, M., Sunters, A., Zaman, G., Saxon, L. K., Price, J. S., and Lanyon, L. E. (2007). Wnt/beta-catenin signaling is a component of osteoblastic bone cell early responses to load-bearing and requires estrogen receptor alpha. J. Biol. Chem. 282, 20715–20727.
Aronow, M. A., Gerstenfeld, L. C., Owen, T. A., Tassinari, M. S., Stein, G. S., and Lian, J. B. (1990). Factors that promote progressive development of the osteoblast phenotype in cultured fetal rat calvaria cells. J. Cell Physiol. 143, 213–221.
Bandyopadhyay, A., Tsuji, K., Cox, K., Harfe, B. D., Rosen, V., and Tabin, C. J. (2006). Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS Genet. 2, e216. doi: 10.1371/journal.pgen.0020216
Beck, S. E., and Carethers, J. M. (2007). BMP suppresses PTEN expression via RAS/ERK signaling. Cancer Biol. Ther. 6, 1313–1317.
Bellows, C. G., Aubin, J. E., Heersche, J. N., and Antosz, M. E. (1986). Mineralized bone nodules formed in vitro from enzymatically released rat calvaria cell populations. Calcif. Tissue Int. 38, 143–154.
Bialek, P., Kern, B., Yang, X., Schrock, M., Sosic, D., Hong, N., Wu, H., Yu, K., Ornitz, D. M., Olson, E. N., Justice, M. J., and Karsenty, G. (2004). A twist code determines the onset of osteoblast differentiation. Dev. Cell 6, 423–435.
Birkenkamp, K. U., Essafi, A., Van Der Vos, K. E., Da Costa, M., Hui, R. C., Holstege, F., Koenderman, L., Lam, E. W., and Coffer, P. J. (2007). FOXO3a induces differentiation of Bcr-Abl-transformed cells through transcriptional down-regulation of Id1. J. Biol. Chem. 282, 2211–2220.
Bonewald, L. F. (2007). Osteocytes as dynamic multifunctional cells. Ann. N.Y. Acad. Sci. 1116, 281–290.
Bottcher, R. T., and Niehrs, C. (2005). Fibroblast growth factor signaling during early vertebrate development. Endocr. Rev. 26, 63–77.
Broadhead, M. L., Clark, J. C., Myers, D. E., Dass, C. R., and Choong, P. F. (2011). The molecular pathogenesis of osteosarcoma: a review. Sarcoma 2011, 959248.
Burrow, S., Andrulis, I. L., Pollak, M., and Bell, R. S. (1998). Expression of insulin-like growth factor receptor, IGF-1, and IGF-2 in primary and metastatic osteosarcoma. J. Surg. Oncol. 69, 21–27.
Cantley, L. C., and Neel, B. G. (1999). New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc. Natl. Acad. Sci. U.S.A. 96, 4240–4245.
Case, N., Ma, M., Sen, B., Xie, Z., Gross, T. S., and Rubin, J. (2008). Beta-catenin levels influence rapid mechanical responses in osteoblasts. J. Biol. Chem. 283, 29196–29205.
Chen, D., Harris, M. A., Rossini, G., Dunstan, C. R., Dallas, S. L., Feng, J. Q., Mundy, G. R., and Harris, S. E. (1997). Bone morphogenetic protein 2 (BMP-2) enhances BMP-3, BMP-4, and bone cell differentiation marker gene expression during the induction of mineralized bone matrix formation in cultures of fetal rat calvarial osteoblasts. Calcif. Tissue Int. 60, 283–290.
Chen, J., Yuan, K., Mao, X., Miano, J. M., Wu, H., and Chen, Y. (2012). Serum response factor regulates bone formation via IGF-1 and Runx2 signals. J. Bone Miner. Res. doi: 10.1002/jbmr.1607 [Epub ahead of print].
Cheng, M., Zaman, G., Rawlinson, S. C., Mohan, S., Baylink, D. J., and Lanyon, L. E. (1999). Mechanical strain stimulates ROS cell proliferation through IGF-II and estrogen through IGF-I. J. Bone Miner. Res. 14, 1742–1750.
Choi, Y. H., Jeong, H. M., Jin, Y. H., Li, H., Yeo, C. Y., and Lee, K. Y. (2011). Akt phosphorylates and regulates the osteogenic activity of Osterix. Biochem. Biophys. Res. Commun. 411, 637–641.
Chow, J. Y., Dong, H., Quach, K. T., Van Nguyen, P. N., Chen, K., and Carethers, J. M. (2008). TGF-beta mediates PTEN suppression and cell motility through calcium-dependent PKC-alpha activation in pancreatic cancer cells. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G899–G905.
Chow, J. Y., Quach, K. T., Cabrera, B. L., Cabral, J. A., Beck, S. E., and Carethers, J. M. (2007). RAS/ERK modulates TGFbeta-regulated PTEN expression in human pancreatic adenocarcinoma cells. Carcinogenesis 28, 2321–2327.
Chun, R. (2005). Common malignant musculoskeletal neoplasms of dogs and cats. Vet. Clin. North Am. Small Anim. Pract. 35, 1155–1167, vi.
Chun, R., and de Lorimier, L. P. (2003). Update on the biology and management of canine osteosarcoma. Vet. Clin. North Am. Small Anim. Pract. 33, 491–516, vi.
Coutu, D. L., and Galipeau, J. (2011). Roles of FGF signaling in stem cell self-renewal, senescence and aging. Aging (Albany, NY) 3, 920–933.
Datta, S. R., Ranger, A. M., Lin, M. Z., Sturgill, J. F., Ma, Y. C., Cowan, C. W., Dikkes, P., Korsmeyer, S. J., and Greenberg, M. E. (2002). Survival factor-mediated BAD phosphorylation raises the mitochondrial threshold for apoptosis. Dev. Cell 3, 631–643.
DeMambro, V. E., Kawai, M., Clemens, T. L., Fulzele, K., Maynard, J. A., Marin De Evsikova, C., Johnson, K. R., Canalis, E., Beamer, W. G., Rosen, C. J., and Donahue, L. R. (2010). A novel spontaneous mutation of Irs1 in mice results in hyperinsulinemia, reduced growth, low bone mass and impaired adipogenesis. J. Endocrinol. 204, 241–253.
Di Cristofano, A., Pesce, B., Cordon-Cardo, C., and Pandolfi, P. P. (1998). Pten is essential for embryonic development and tumour suppression. Nat. Genet. 19, 348–355.
DiGirolamo, D. J., Mukherjee, A., Fulzele, K., Gan, Y., Cao, X., Frank, S. J., and Clemens, T. L. (2007). Mode of growth hormone action in osteoblasts. J. Biol. Chem. 282, 31666–31674.
Dijkers, P. F., Birkenkamp, K. U., Lam, E. W., Thomas, N. S., Lammers, J. W., Koenderman, L., and Coffer, P. J. (2002). FKHR-L1 can act as a critical effector of cell death induced by cytokine withdrawal: protein kinase B-enhanced cell survival through maintenance of mitochondrial integrity. J. Cell Biol. 156, 531–542.
Dijkers, P. F., Medema, R. H., Pals, C., Banerji, L., Thomas, N. S., Lam, E. W., Burgering, B. M., Raaijmakers, J. A., Lammers, J. W., Koenderman, L., and Coffer, P. J. (2000). Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional regulation of p27(KIP1). Mol. Cell. Biol. 20, 9138–9148.
Easton, R. M., Cho, H., Roovers, K., Shineman, D. W., Mizrahi, M., Forman, M. S., Lee, V. M., Szabolcs, M., De Jong, R., Oltersdorf, T., Ludwig, T., Efstratiadis, A., and Birnbaum, M. J. (2005). Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Mol. Cell. Biol. 25, 1869–1878.
Eigenmann, J. E., Patterson, D. F., Zapf, J., and Froesch, E. R. (1984). Insulin-like growth factor I in the dog: a study in different dog breeds and in dogs with growth hormone elevation. Acta Endocrinol. (Copenh) 105, 294–301.
El-Tanani, M., Platt-Higgins, A., Rudland, P. S., and Campbell, F. C. (2004). Ets gene PEA3 cooperates with beta-catenin-Lef-1 and c-Jun in regulation of osteopontin transcription. J. Biol. Chem. 279, 20794–20806.
Fang, D., Hawke, D., Zheng, Y., Xia, Y., Meisenhelder, J., Nika, H., Mills, G. B., Kobayashi, R., Hunter, T., and Lu, Z. (2007). Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J. Biol. Chem. 282, 11221–11229.
Ferrari, S. L., Deutsch, S., and Antonarakis, S. E. (2005). Pathogenic mutations and polymorphisms in the lipoprotein receptor-related protein 5 reveal a new biological pathway for the control of bone mass. Curr. Opin. Lipidol. 16, 207–214.
Ford-Hutchinson, A. F., Ali, Z., Lines, S. E., Hallgrimsson, B., Boyd, S. K., and Jirik, F. R. (2007). Inactivation of Pten in osteo-chondroprogenitor cells leads to epiphyseal growth plate abnormalities and skeletal overgrowth. J. Bone Miner. Res. 22, 1245–1259.
Friedlander, A. L., Butterfield, G. E., Moynihan, S., Grillo, J., Pollack, M., Holloway, L., Friedman, L., Yesavage, J., Matthias, D., Lee, S., Marcus, R., and Hoffman, A. R. (2001). One year of insulin-like growth factor I treatment does not affect bone density, body composition, or psychological measures in postmenopausal women. J. Clin. Endocrinol. Metab. 86, 1496–1503.
Fujita, T., Azuma, Y., Fukuyama, R., Hattori, Y., Yoshida, C., Koida, M., Ogita, K., and Komori, T. (2004). Runx2 induces osteoblast and chondrocyte differentiation and enhances their migration by coupling with PI3K-Akt signaling. J. Cell Biol. 166, 85–95.
Galea, G. L., Sunters, A., Meakin, L. B., Zaman, G., Sugiyama, T., Lanyon, L. E., and Price, J. S. (2011). Sost down-regulation by mechanical strain in human osteoblastic cells involves PGE2 signaling via EP4. FEBS Lett. 585, 2450–2454.
Garofalo, R. S., Orena, S. J., Rafidi, K., Torchia, A. J., Stock, J. L., Hildebrandt, A. L., Coskran, T., Black, S. C., Brees, D. J., Wicks, J. R., Mcneish, J. D., and Coleman, K. G. (2003). Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. J. Clin. Invest. 112, 197–208.
Greenblatt, M. B., Shim, J. H., Zou, W., Sitara, D., Schweitzer, M., Hu, D., Lotinun, S., Sano, Y., Baron, R., Park, J. M., Arthur, S., Xie, M., Schneider, M. D., Zhai, B., Gygi, S., Davis, R., and Glimcher, L. H. (2010). The p38 MAPK pathway is essential for skeletogenesis and bone homeostasis in mice. J. Clin. Invest. 120, 2457–2473.
Guenou, H., Kaabeche, K., Dufour, C., Miraoui, H., and Marie, P. J. (2006). Down-regulation of ubiquitin ligase Cbl induced by twist haploinsufficiency in Saethre-Chotzen syndrome results in increased PI3K/Akt signaling and osteoblast proliferation. Am. J. Pathol. 169, 1303–1311.
Guntur, A. R., Reinhold, M. I., Cuellar, J. Jr., and Naski, M. C. (2011). Conditional ablation of Pten in osteoprogenitors stimulates FGF signaling. Development 138, 1433–1444.
Hay, E., Hott, M., Graulet, A. M., Lomri, A., and Marie, P. J. (1999). Effects of bone morphogenetic protein-2 on human neonatal calvaria cell differentiation. J. Cell Biochem. 72, 81–93.
Hayashi, M., Nimura, K., Kashiwagi, K., Harada, T., Takaoka, K., Kato, H., Tamai, K., and Kaneda, Y. (2007). Comparative roles of Twist-1 and Id1 in transcriptional regulation by BMP signaling. J. Cell Sci. 120, 1350–1357.
Hill, T. P., Spater, D., Taketo, M. M., Birchmeier, W., and Hartmann, C. (2005). Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev. Cell 8, 727–738.
Ho, K. K., Myatt, S. S., and Lam, E. W. (2008). Many forks in the path: cycling with FoxO. Oncogene 27, 2300–2311.
Huang, Y. F., Lin, J. J., Lin, C. H., Su, Y., and Hung, S. C. (2012). c-Jun N-terminal kinase 1 negatively regulates osteoblastic differentiation induced by BMP-2 via phosphorylation of Runx2 at Ser104. J. Bone Miner. Res. 27, 1093–1105.
Jessop, H. L., Rawlinson, S. C., Pitsillides, A. A., and Lanyon, L. E. (2002). Mechanical strain and fluid movement both activate extracellular regulated kinase (ERK) in osteoblast-like cells but via different signaling pathways. Bone 31, 186–194.
Kawamura, N., Kugimiya, F., Oshima, Y., Ohba, S., Ikeda, T., Saito, T., Shinoda, Y., Kawasaki, Y., Ogata, N., Hoshi, K., Akiyama, T., Chen, W. S., Hay, N., Tobe, K., Kadowaki, T., Azuma, Y., Tanaka, S., Nakamura, K., Chung, U. I., and Kawaguchi, H. (2007). Akt1 in osteoblasts and osteoclasts controls bone remodeling. PLoS ONE 2, e1058. doi: 10.1371/journal.pone.0001058
Khalid, O., Baniwal, S. K., Purcell, D. J., Leclerc, N., Gabet, Y., Stallcup, M. R., Coetzee, G. A., and Frenkel, B. (2008). Modulation of Runx2 activity by estrogen receptor-alpha: implications for osteoporosis and breast cancer. Endocrinology 149, 5984–5995.
Kitase, Y., Barragan, L., Qing, H., Kondoh, S., Jiang, J. X., Johnson, M. L., and Bonewald, L. F. (2010). Mechanical induction of PGE2 in osteocytes blocks glucocorticoid-induced apoptosis through both the beta-catenin and PKA pathways. J. Bone Miner. Res. 25, 2657–2668.
Kokabu, S., Gamer, L., Cox, K., Lowery, J., Tsuji, K., Raz, R., Economides, A., Katagiri, T., and Rosen, V. (2012). BMP3 suppresses osteoblast differentiation of bone marrow stromal cells via interaction with Acvr2b. Mol. Endocrinol. 26, 87–94.
Komori, T. (2006). Regulation of osteoblast differentiation by transcription factors. J. Cell Biochem. 99, 1233–1239.
Kugimiya, F., Kawaguchi, H., Ohba, S., Kawamura, N., Hirata, M., Chikuda, H., Azuma, Y., Woodgett, J. R., Nakamura, K., and Chung, U. I. (2007). GSK-3beta controls osteogenesis through regulating Runx2 activity. PLoS ONE 2, e837. doi: 10.1371/journal.pone.0000837
Lai, C. F., and Cheng, S. L. (2005). AlphaVbeta integrins play an essential role in BMP-2 induction of osteoblast differentiation. J. Bone Miner. Res. 20, 330–340.
Lanyon, L. (2008). Strain-related control of bone (re)modeling: objectives, mechanisms and failures. J. Musculoskelet. Neuronal. Interact. 8, 298–300.
Lau, K. H., Kapur, S., Kesavan, C., and Baylink, D. J. (2006). Up-regulation of the Wnt, estrogen receptor, insulin-like growth factor-I, and bone morphogenetic protein pathways in C57BL/6J osteoblasts as opposed to C3H/HeJ osteoblasts in part contributes to the differential anabolic response to fluid shear. J. Biol. Chem. 281, 9576–9588.
Lee, J. Y., Kang, M. B., Jang, S. H., Qian, T., Kim, H. J., Kim, C. H., Kim, Y., and Kong, G. (2009). Id-1 activates Akt-mediated Wnt signaling and p27(Kip1) phosphorylation through PTEN inhibition. Oncogene 28, 824–831.
Lee, K., Jessop, H., Suswillo, R., Zaman, G., and Lanyon, L. (2003). Endocrinology: bone adaptation requires oestrogen receptor-alpha. Nature 424, 389.
Lee, K. S., Kim, H. J., Li, Q. L., Chi, X. Z., Ueta, C., Komori, T., Wozney, J. M., Kim, E. G., Choi, J. Y., Ryoo, H. M., and Bae, S. C. (2000). Runx2 is a common target of transforming growth factor beta1 and bone morphogenetic protein 2, and cooperation between Runx2 and Smad5 induces osteoblast-specific gene expression in the pluripotent mesenchymal precursor cell line C2C12. Mol. Cell. Biol. 20, 8783–8792.
Levine, R. A., Forest, T., and Smith, C. (2002). Tumor suppressor PTEN is mutated in canine osteosarcoma cell lines and tumors. Vet. Pathol. 39, 372–378.
Liang, J., and Slingerland, J. M. (2003). Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle 2, 339–345.
Liang, J., Zubovitz, J., Petrocelli, T., Kotchetkov, R., Connor, M. K., Han, K., Lee, J. H., Ciarallo, S., Catzavelos, C., Beniston, R., Franssen, E., and Slingerland, J. M. (2002). PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat. Med. 8, 1153–1160.
Liu, X., Bruxvoort, K. J., Zylstra, C. R., Liu, J., Cichowski, R., Faugere, M. C., Bouxsein, M. L., Wan, C., Williams, B. O., and Clemens, T. L. (2007). Lifelong accumulation of bone in mice lacking Pten in osteoblasts. Proc. Natl. Acad. Sci. U.S.A. 104, 2259–2264.
Long, F. (2012). Building strong bones: molecular regulation of the osteoblast lineage. Nat. Rev. Mol. Cell. Biol. 13, 27–38.
MacEwen, E. G., Pastor, J., Kutzke, J., Tsan, R., Kurzman, I. D., Thamm, D. H., Wilson, M., and Radinsky, R. (2004). IGF-1 receptor contributes to the malignant phenotype in human and canine osteosarcoma. J. Cell Biochem. 92, 77–91.
Mandal, C. C., Drissi, H., Choudhury, G. G., and Ghosh-Choudhury, N. (2010). Integration of phosphatidylinositol 3-kinase, Akt kinase, and Smad signaling pathway in BMP-2-induced osterix expression. Calcif. Tissue Int. 87, 533–540.
Manning, B. D., and Cantley, L. C. (2007). AKT/PKB signaling: navigating downstream. Cell 129, 1261–1274.
Matsuguchi, T., Chiba, N., Bandow, K., Kakimoto, K., Masuda, A., and Ohnishi, T. (2009). JNK activity is essential for Atf4 expression and late-stage osteoblast differentiation. J. Bone Miner. Res. 24, 398–410.
Meyers, V. E., Zayzafoon, M., Gonda, S. R., Gathings, W. E., and Mcdonald, J. M. (2004). Modeled microgravity disrupts collagen I/integrin signaling during osteoblastic differentiation of human mesenchymal stem cells. J. Cell Biochem. 93, 697–707.
Mukherjee, A., and Rotwein, P. (2009). Akt promotes BMP2-mediated osteoblast differentiation and bone development. J. Cell Sci. 122, 716–726.
Mukherjee, A., Wilson, E. M., and Rotwein, P. (2010). Selective signaling by Akt2 promotes bone morphogenetic protein 2-mediated osteoblast differentiation. Mol. Cell. Biol. 30, 1018–1027.
Nakashima, A., Katagiri, T., and Tamura, M. (2005). Cross-talk between Wnt and bone morphogenetic protein 2 (BMP-2) signaling in differentiation pathway of C2C12 myoblasts. J. Biol. Chem. 280, 37660–37668.
Nishio, Y., Dong, Y., Paris, M., O’keefe, R. J., Schwarz, E. M., and Drissi, H. (2006). Runx2-mediated regulation of the zinc finger Osterix/Sp7 gene. Gene 372, 62–70.
Ogata, T., Wozney, J. M., Benezra, R., and Noda, M. (1993). Bone morphogenetic protein 2 transiently enhances expression of a gene, Id (inhibitor of differentiation), encoding a helix-loop-helix molecule in osteoblast-like cells. Proc. Natl. Acad. Sci. U.S.A. 90, 9219–9222.
Osyczka, A. M., and Leboy, P. S. (2005). Bone morphogenetic protein regulation of early osteoblast genes in human marrow stromal cells is mediated by extracellular signal-regulated kinase and phosphatidylinositol 3-kinase signaling. Endocrinology 146, 3428–3437.
Otto, F., Thornell, A. P., Crompton, T., Denzel, A., Gilmour, K. C., Rosewell, I. R., Stamp, G. W., Beddington, R. S., Mundlos, S., Olsen, B. R., Selby, P. B., and Owen, M. J. (1997). Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 89, 765–771.
Peng, X. D., Xu, P. Z., Chen, M. L., Hahn-Windgassen, A., Skeen, J., Jacobs, J., Sundararajan, D., Chen, W. S., Crawford, S. E., Coleman, K. G., and Hay, N. (2003). Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev. 17, 1352–1365.
Peng, Y., Kang, Q., Luo, Q., Jiang, W., Si, W., Liu, B. A., Luu, H. H., Park, J. K., Li, X., Luo, J., Montag, A. G., Haydon, R. C., and He, T. C. (2004). Inhibitor of DNA binding/differentiation helix-loop-helix proteins mediate bone morphogenetic protein-induced osteoblast differentiation of mesenchymal stem cells. J. Biol. Chem. 279, 32941–32949.
Perrini, S., Natalicchio, A., Laviola, L., Cignarelli, A., Melchiorre, M., De Stefano, F., Caccioppoli, C., Leonardini, A., Martemucci, S., Belsanti, G., Miccoli, S., Ciampolillo, A., Corrado, A., Cantatore, F. P., Giorgino, R., and Giorgino, F. (2008). Abnormalities of insulin-like growth factor-I signaling and impaired cell proliferation in osteoblasts from subjects with osteoporosis. Endocrinology 149, 1302–1313.
Pitsillides, A. A., Rawlinson, S. C., Suswillo, R. F., Bourrin, S., Zaman, G., and Lanyon, L. E. (1995). Mechanical strain-induced NO production by bone cells: a possible role in adaptive bone (re)modeling? FASEB J. 9, 1614–1622.
Price, J. S., Sugiyama, T., Galea, G. L., Meakin, L. B., Sunters, A., and Lanyon, L. E. (2010). Role of endocrine and paracrine factors in the adaptation of bone to mechanical loading. Curr. Osteoporos. Rep. 9, 76–82.
Ramos, J. W. (2008). The regulation of extracellular signal-regulated kinase (ERK) in mammalian cells. Int. J. Biochem. Cell Biol. 40, 2707–2719.
Raucci, A., Bellosta, P., Grassi, R., Basilico, C., and Mansukhani, A. (2008). Osteoblast proliferation or differentiation is regulated by relative strengths of opposing signaling pathways. J. Cell Physiol. 215, 442–451.
Rumney, R. M., Sunters, A., Reilly, G. C., and Gartland, A. (2012). Application of multiple forms of mechanical loading to human osteoblasts reveals increased ATP release in response to fluid flow in 3D cultures and differential regulation of immediate early genes. J. Biomech. 45, 549–554.
Schroeder, T. M., Jensen, E. D., and Westendorf, J. J. (2005). Runx2: a master organizer of gene transcription in developing and maturing osteoblasts. Birth Defects Res. C Embryo Today 75, 213–225.
Smith, E., and Frenkel, B. (2005). Glucocorticoids inhibit the transcriptional activity of LEF/TCF in differentiating osteoblasts in a glycogen synthase kinase-3beta-dependent and -independent man-ner. J. Biol. Chem. 280, 2388–2394.
Sunters, A., Armstrong, V. J., Zaman, G., Kypta, R. M., Kawano, Y., Lanyon, L. E., and Price, J. S. (2010). Mechano-transduction in osteoblastic cells involves strain-regulated estrogen receptor alpha-mediated control of insulin-like growth factor (IGF) I receptor sensitivity to Ambient IGF, leading to phosphatidylinositol 3-kinase/AKT-dependent Wnt/LRP5 receptor-independent activation of beta-catenin signaling. J. Biol. Chem. 285, 8743–8758.
Sunters, A., Fernandez De Mattos, S., Stahl, M., Brosens, J. J., Zoumpoulidou, G., Saunders, C. A., Coffer, P. J., Medema, R. H., Coombes, R. C., and Lam, E. W. (2003). FoxO3a transcriptional regulation of Bim controls apoptosis in paclitaxel-treated breast cancer cell lines. J. Biol. Chem. 278, 49795–49805.
Sutter, N. B., Bustamante, C. D., Chase, K., Gray, M. M., Zhao, K., Zhu, L., Padhukasahasram, B., Karlins, E., Davis, S., Jones, P. G., Quignon, P., Johnson, G. S., Parker, H. G., Fretwell, N., Mosher, D. S., Lawler, D. F., Satyaraj, E., Nordborg, M., Lark, K. G., Wayne, R. K., and Ostrander, E. A. (2007). A single IGF1 allele is a major determinant of small size in dogs. Science 316, 112–115.
Suzuki, A., De La Pompa, J. L., Stambolic, V., Elia, A. J., Sasaki, T., Del Barco Barrantes, I., Ho, A., Wakeham, A., Itie, A., Khoo, W., Fukumoto, M., and Mak, T. W. (1998). High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr. Biol. 8, 1169–1178.
Tjalma, R. A. (1966). Canine bone sarcoma: estimation of relative risk as a function of body size. J. Natl. Cancer Inst. 36, 1137–1150.
Tsuji, K., Bandyopadhyay, A., Harfe, B. D., Cox, K., Kakar, S., Gerstenfeld, L., Einhorn, T., Tabin, C. J., and Rosen, V. (2006). BMP2 activity, although dispensable for bone formation, is required for the initiation of fracture healing. Nat. Genet. 38, 1424–1429.
van der Horst, G., Van Bezooijen, R. L., Deckers, M. M., Hoogendam, J., Visser, A., Lowik, C. W., and Karperien, M. (2002). Differentiation of murine preosteoblastic KS483 cells depends on autocrine bone morphogenetic protein signaling during all phases of osteoblast formation. Bone 31, 661–669.
Weng, T., Mao, F., Wang, Y., Sun, Q., Li, R., Yang, G., Zhang, X., Luo, J., Feng, G. S., and Yang, X. (2010). Osteoblastic molecular scaffold Gab1 is required for maintaining bone homeostasis. J. Cell Sci. 123, 682–689.
Whyte, M. P., Reinus, W. H., and Mumm, S. (2004). High-bone-mass disease and LRP5. New Engl. J. Med. 350, 2096–2099; author reply 2096–2099.
Wu, X. B., Li, Y., Schneider, A., Yu, W., Rajendren, G., Iqbal, J., Yamamoto, M., Alam, M., Brunet, L. J., Blair, H. C., Zaidi, M., and Abe, E. (2003). Impaired osteoblastic differentiation, reduced bone formation, and severe osteoporosis in noggin-overexpressing mice. J. Clin. Invest. 112, 924–934.
Wullschleger, S., Loewith, R., and Hall, M. N. (2006). TOR signaling in growth and metabolism. Cell 124, 471–484.
Xiao, G., Jiang, D., Gopalakrishnan, R., and Franceschi, R. T. (2002). Fibroblast growth factor 2 induction of the osteocalcin gene requires MAPK activity and phosphorylation of the osteoblast transcription factor, Cbfa1/Runx2. J. Biol. Chem. 277, 36181–36187.
Xiao, G., Wang, D., Benson, M. D., Karsenty, G., and Franceschi, R. T. (1998). Role of the alpha2-integrin in osteoblast-specific gene expression and activation of the Osf2 transcription factor. J. Biol. Chem. 273, 32988–32994.
Yakar, S., Canalis, E., Sun, H., Mejia, W., Kawashima, Y., Nasser, P., Courtland, H. W., Williams, V., Bouxsein, M., Rosen, C., and Jepsen, K. J. (2009). Serum IGF-1 determines skeletal strength by regulating sub-periosteal expansion and trait interactions. J. Bone Miner. Res. 24, 1481–1492.
Yakar, S., Courtland, H. W., and Clemmons, D. (2010). IGF-1 and bone: New discoveries from mouse models. J. Bone Miner. Res. 25, 2543–2552.
Yang, S., Xu, H., Yu, S., Cao, H., Fan, J., Ge, C., Fransceschi, R. T., Dong, H. H., and Xiao, G. (2010). Foxo1 mediates insulin-like growth factor 1 (IGF1)/insulin regulation of osteocalcin expression by antagonizing Runx2 in osteoblasts. J. Biol. Chem. 286, 19149–19158.
Ying, Q. L., Nichols, J., Chambers, I., and Smith, A. (2003). BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell 115, 281–292.
Yu, K., Xu, J., Liu, Z., Sosic, D., Shao, J., Olson, E. N., Towler, D. A., and Ornitz, D. M. (2003). Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development 130, 3063–3074.
Zaman, G., Saxon, L. K., Sunters, A., Hilton, H., Underhill, P., Williams, D., Price, J. S., and Lanyon, L. E. (2010). Loading-related regulation of gene expression in bone in the contexts of estrogen deficiency, lack of estrogen receptor alpha and disuse. Bone 46, 628–642.
Zaman, G., Suswillo, R. F., Cheng, M. Z., Tavares, I. A., and Lanyon, L. E. (1997). Early responses to dynamic strain change and prostaglandins in bone-derived cells in culture. J. Bone Miner. Res. 12, 769–777.
Zhang, H., Pan, Y., Zheng, L., Choe, C., Lindgren, B., Jensen, E. D., Westendorf, J. J., Cheng, L., and Huang, H. (2011). FOXO1 inhibits Runx2 transcriptional activity and prostate cancer cell migration and invasion. Cancer Res. 71, 3257–3267.
Zhang, M., Xuan, S., Bouxsein, M. L., Von Stechow, D., Akeno, N., Faugere, M. C., Malluche, H., Zhao, G., Rosen, C. J., Efstratiadis, A., and Clemens, T. L. (2002). Osteoblast-specific knockout of the insulin-like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization. J. Biol. Chem. 277, 44005–44012.
Zhang, W., Shen, X., Wan, C., Zhao, Q., Zhang, L., Zhou, Q., and Deng, L. (2012). Effects of insulin and insulin-like growth factor 1 on osteoblast proliferation and differentiation: differential signalling via Akt and ERK. Cell Biochem. Funct. 30, 297–302.
Zhao, G., Monier-Faugere, M. C., Langub, M. C., Geng, Z., Nakayama, T., Pike, J. W., Chernausek, S. D., Rosen, C. J., Donahue, L. R., Malluche, H. H., Fagin, J. A., and Clemens, T. L. (2000). Targeted overexpression of insulin-like growth factor I to osteoblasts of transgenic mice: increased trabecular bone volume without increased osteoblast proliferation. Endocrinology 141, 2674–2682.
Zhou, B. P., Liao, Y., Xia, W., Spohn, B., Lee, M. H., and Hung, M. C. (2001). Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat. Cell Biol. 3, 245–252.
Keywords: osteoblast, phosphoinositide 3-kinase, PI3K, AKT, protein kinase B
Citation: McGonnell IM, Grigoriadis AE, Lam EW-F, Price JS and Sunters A (2012) A specific role for phosphoinositide 3-kinase and AKT in osteoblasts? Front. Endocrin. 3:88. doi: 10.3389/fendo.2012.00088
Received: 30 April 2012; Accepted: 29 June 2012;
Published online: 20 July 2012.
Edited by:
Alison Gartland, The University of Sheffield, UKReviewed by:
Florent Elefteriou, Vanderbilt University, USARobert Hipskind, Centre Nationale de la Recherche Scientifique, France
Copyright: © 2012 McGonnell, Grigoriadis, Lam, Price and Sunters. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution, and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Andrew Sunters, Department of Veterinary Basic Sciences, The Royal Veterinary College, Royal College Street, Camden, London NW1 0TU, UK. e-mail:YXN1bnRlcnNAcnZjLmFjLnVr