



- The Roslin Institute and Royal (Dick) School of Veterinary Studies, The University of Edinburgh, Midlothian, Scotland, UK
Vascular calcification has severe clinical consequences and is considered an accurate predictor of future adverse cardiovascular events, including myocardial infarction and stroke. Previously vascular calcification was thought to be a passive process which involved the deposition of calcium and phosphate in arteries and cardiac valves. However, recent studies have shown that vascular calcification is a highly regulated, cell-mediated process similar to bone formation. In this article, we outline the current understanding of key mechanisms governing vascular calcification and highlight the clinical consequences. By understanding better the molecular pathways and genetic circuitry responsible for the pathological mineralization process novel drug targets may be identified and exploited to combat and reduce the detrimental effects of vascular calcification on human health.
Introduction
Whilst it is tempting to presume that vascular calcification is a product of modern society, this pathological process was actually first documented in an autopsy of the mummy of an elderly Egyptian woman, which revealed calcific aortic atherosclerosis (Czermack, 1852). Following this report, Johann Georg Mönckeberg described medial calcific sclerosis, a form of arteriosclerosis or vessel hardening, where calcium deposits are found in the muscular middle layer of the walls of arteries (Mönckeberg, 1903). For many years, vascular calcification was regarded as a passive and degenerative disease without treatment options (Virchow, 1989). Over the past two decades, extensive research has conclusively shown that pathological vascular calcification is a tightly regulated process that shares many similarities with physiological bone mineralization (Shanahan et al., 1999; Moe and Chen, 2004; Vattikuti and Towler, 2004; Hruska et al., 2005; Demer and Tintut, 2008; Sage et al., 2011; Zhu et al., 2011). However, the precise mechanisms through which vascular calcification occurs still remains unclear. Progressively enlarging calcium deposits are seen in the major arteries of individuals older than 60 years of age (Allison et al., 2004). Extensive vascular calcification is also frequently observed in patients with atherosclerosis, chronic kidney disease (CKD), and diabetes (Fuchs et al., 1985; Sangiorgi et al., 1998; Allison et al., 2004; Giachelli, 2004; Reaven and Sacks, 2005; Okuno et al., 2007; Shroff and Shanahan, 2007; Kestenbaum et al., 2009). The incidence of vascular calcification is highly correlated with mortality and morbidity of cardiovascular disease (Arad et al., 2000; Rosenhek et al., 2000; Keelan et al., 2001; Wayhs et al., 2002), in the form of reduced aortic compliance, decreased cardiac efficiency, deterioration of coronary perfusion, and subendocardial ischemia (Kelly et al., 1992; Watanabe et al., 1992; Ohtsuka et al., 1994). Therefore the identification and characterization of novel mediators of vascular calcification will offer the potential for future therapeutics to inhibit progression or induce regression of vascular calcification. In this review, we will describe the mechanisms underpinning vascular calcification and discuss the risk factors, clinical consequences, and potential therapeutic targets for this pathological process. Abbreviations and acronyms are detailed in Table 1.
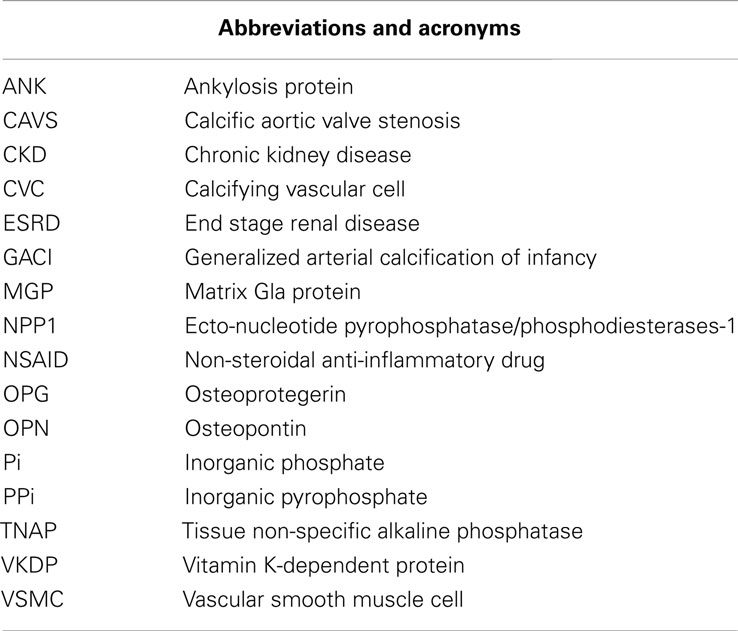
Table 1. Abbreviations and acronyms.
Types of Vascular Calcification
Vascular calcification can be categorized into four main types according to location: atherosclerotic intimal calcification, medial artery calcification (Mönckeberg’s sclerosis), cardiac valve calcification, and calcific uremic arteriolopathy. Histologically, calcified deposits may be amorphic, chondromorphic, or osteomorphic in structure, and may be characterized as metastatic or dystrophic.
Atheroslcerotic intimal calcification
Atherosclerosis is the development of plaques within the intimal layer of large vessels, and underlies coronary artery disease and cerebrovascular disease, the most common forms of life threatening cardiovascular disorders (Doherty et al., 2004). Atherosclerosis can by induced by chronic inflammation and lipid deposition, with dyslipidemia frequently linked to the severity of calcium deposition (Pohle et al., 2001; Schmermund et al., 2001). Atherosclerotic calcification is the most common form of calcific vasculopathy, and occurs as early as the second decade of life just after fatty streak formation (Stary et al., 1995). Small aggregates of crystalline calcium can be detected in developing lesions, and in adults past the fourth decade of life, greater lesion areas may be calcified (Stary, 2000). The degree of calcification correlates with the extent of atherosclerosis, with age, and hypertension as dominant risk factors for systemic calcified atherosclerosis (Allison et al., 2004). The predominant mineral form in calcified lesions is hydroxyapatite which may initially form in membrane bound matrix vesicles that bud from the membranes of chondrocytes and osteoblasts present within deposits of cartilage and bone tissue, respectively (Yu, 1974). In addition to mineral, these lesions also contain matrix vesicles as well as outright bone and cartilage (Tanimura et al., 1983; Mohler et al., 2001; Hunt et al., 2002).
Medial calcification
Medial calcification, also termed Mönckeberg’s sclerosis, occurs in the tunica media of blood vessels. It is a characteristic feature of Generalized Arterial Calcification of Infancy (GACI), diabetes, and CKD (Fuchs et al., 1985; Chen and Moe, 2003; Rutsch et al., 2003, 2008; London et al., 2005; Reaven and Sacks, 2005; Okuno et al., 2007; Kestenbaum et al., 2009), and is associated with increased cardiovascular mortality and amputation risk (Chantelau et al., 1995; Lehto et al., 1996; London et al., 2003). Medial calcification occurs independently of atherosclerotic calcification and is a process similar to intramembranous bone formation, with no cartilaginous precursor required (Towler et al., 1998; Vattikuti and Towler, 2004). Calcium deposition can be observed throughout most of the medial width in the early stage of disease. At later stages of disease, the media is filled with circumferential rings of mineral. In some cases, osteocytes and bone trabeculae can also be observed (Shanahan et al., 1999; Zhu et al., 2011).
Cardiac valve calcification
Heart valves allow unidirectional blood flow through the heart. The four main valves of the mammalian heart are: the two atrioventricular (AV) valves and the two semilunar (SL) valves. The AV valves including the mitral valve and the tricuspid valve are located between the atria and the ventricles. The SL valves are the aortic valve and the pulmonary valve and are located in the arteries leaving the heart. Calcific aortic valve disease is identified by thickening and calcification of the aortic valve leaflets (Figure 1) in the absence of rheumatic heart disease. It is divided into aortic sclerosis, in which the leaflets do not obstruct left ventricular outflow, and aortic stenosis, in which obstruction to the left ventricular outflow is present. A number of recent studies have shown that calcific aortic valve lesions have many features characteristic of an actively cell-regulated process, including lipoprotein deposition (O’Brien et al., 1996; Olsson et al., 1999), chronic inflammation (Olsson et al., 1994; Otto et al., 1994), and active calcification (Mohler et al., 2001; Rajamannan et al., 2003) and shares similar underlying mechanisms with atherosclerotic intimal calcification (Salhiyyah et al., 2011).
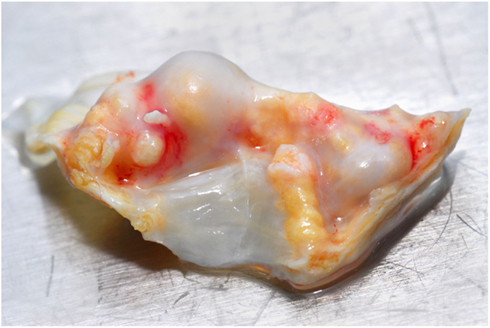
Figure 1. Extensive calcific aortic valve stenosis in the valve leaflets of a patient undergoing valve replacement surgery.
Calcific uremic arteriolopathy
Calcific uremic arteriolopathy is a severe type of widespread medial vascular calcification which occurs in blood vessels of patients with End stage renal disease (ESRD) often referred to as stage 5 CKD (Qunibi et al., 2002). It affects cutaneous and subcutaneous arteries and arterioles, leading to intimal proliferation, fibrosis, and thrombosis (Qunibi et al., 2002; Mwipatayi et al., 2007).
The Cellular Sources of Vascular Calcification
Cells that spontaneously produce calcified matrix and undergo a bone-like transdifferentiation include vascular smooth muscle cells (VSMCs), pericytes, and calcifying vascular cells (CVCs). These cell types are closely related and may be variant phenotypes of one another (Minasi et al., 2002; Tintut et al., 2003).
Vascular smooth muscle cells
Vascular smooth muscle cells normally reside in the media of blood vessels and are responsible for regulating vascular tone. VSMCs exhibit a contractile phenotype and highly express genes which are required for the maintenance of myofilament structure and function. These genes include α-smooth muscle-actin (SMA), SM22α, and SM-myosin heavy chain (Shanahan et al., 1993; Mackenzie et al., 2011). VSMCs can be activated from a quiescent, differentiated state into an actively proliferating and synthesizing phenotype (Hedin et al., 1999). This phenotypic change is associated with loss of smooth muscle cell markers and can be induced by various stimuli in vitro, including various growth factors, injury, or mechanical stress (Thyberg, 1996; Worth et al., 2001). VSMCs are thought to be the predominant cells associated with medial calcification (Essalihi et al., 2004; Narisawa et al., 2007; Zhu et al., 2011), in contrast to intimal calcification, which also involves lipids and inflammatory cells (Pohle et al., 2001; Schmermund et al., 2001). Vascular calcification is prevalent in patients with CKD, especially those with ESRD. ESRD patients typically have hyperphosphatemia compared to healthy control patients (Giachelli, 2009). A number of studies have shown that VSMCs cultured with high phosphate can undergo calcification in vitro (Figure 2) which involves the phenotypic transition to osteoblastic, chondrocytic, and osteocytic cells (Steitz et al., 2001; Johnson et al., 2005; Speer et al., 2009; Sage et al., 2011; Zavaczki et al., 2011; Zhu et al., 2011). This in vitro model has been widely used for investigating the cellular and molecular mechanisms responsible for vascular calcification.
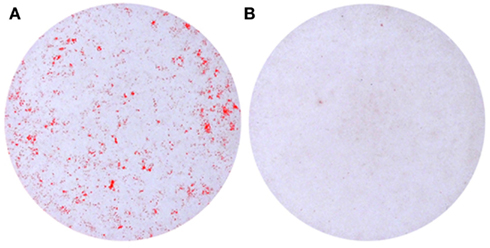
Figure 2. Calcification of murine vascular smooth muscle cells cultured in vitro in the presence of (A) high phosphate (2.5 mM) compared to (B) control medium.
Pericytes
Pericytes are elongated, contractile cells found wrapped about precapillary arterioles outside the basement membrane and are present in veins, arteries, and capillaries. Several pericyte markers have been identified, including SMA, non-muscle actin, non-muscle and muscle myosin, amino peptidase-N, amino peptidase-A, and a cell surface ganglioside (3G5; Andreeva et al., 1998). Previous studies have shown that pericytes can differentiate into osteoblasts and chondrocytes (Doherty and Canfield, 1999; Farrington-Rock et al., 2004), suggesting that pericytes may be central to the etiology of vascular calcification. Further studies have shown that signaling through the Wnt/beta-catenin pathway stimulates chondrogenic and inhibits adipogenic differentiation of pericytes (Kirton et al., 2007), which may directly contribute to the development and progression of calcium deposition. In addition, pericytes can form multicellular nodules that contain a mineralized matrix, similar to those found in calcified aortae (Doherty and Canfield, 1999; Cola et al., 2004). Molecules associated with bone development and formation have been observed in these mineralized nodules, emphasizing the regulatory similarities between vascular and bone calcification (Doherty et al., 1998; Canfield et al., 2000).
Calcifying vascular cells
Calcifying vascular cells are a subpopulation of smooth muscle cells which exhibit osteoblastic characteristics and undergo spontaneous calcification in vitro (Watson et al., 1994; Balica et al., 1997; Tintut et al., 1998; Radcliff et al., 2005). CVCs have features in common with pericytes including a similar morphology, osteoblastic characteristics, and 3G5 expression (Watson et al., 1994). During osteogenic differentiation, CVCs accumulate not only minerals but also lipids such as triglycerides. Indeed, the induction of de novo lipogenesis promotes the calcification of CVCs under pro-osteogenic conditions such as high phosphate levels (Ting et al., 2011). Studies characterizing the calcific nodules produced by CVCs in ApoE-null mice have revealed that the nodules resemble calcific atherosclerotic plaque and can be destabilized in the presence of active lipids and monocytes (Li et al., 2012), providing a novel animal model of vulnerable plaque dynamics.
Valve Interstitial Cells
Calcification of the aortic valve occurs following trans-differentiation of the valve interstitial cells (VICs) through a myofibroblast stage into osteoblast-like cells (Liu et al., 2007). VICs are present in all three layers of the aortic valve and can be induced to differentiate into myofibroblasts by inflammatory response (often caused by endothelial damage; Liu et al., 2007) and the release of Angiotensin, TGF-β, and matrix metalloproteinases (Zhou et al., 1996;Kaden et al., 2003, 2005). After further accumulation of lipids, changes in structure, and fibrosis, differentiation to an osteoblast phenotype is thought to occur via wnt3-Lrp5-β and osteoprotegerin (OPG)/receptor Activator of Nuclear Factor Kappa B (RANK) mediated signaling pathways (Cosmi et al., 2002; Osman et al., 2006; Rajamannan, 2009). Osteoblastic cells then mediate deposition of mineral by processes associated with bone formation (Rajamannan et al., 2003).
Mechanisms of Vascular Calcification
A series of clinical and basic science studies performed in the last several years underscored the biological complexity of the processes driving vascular calcification (Figure 3).
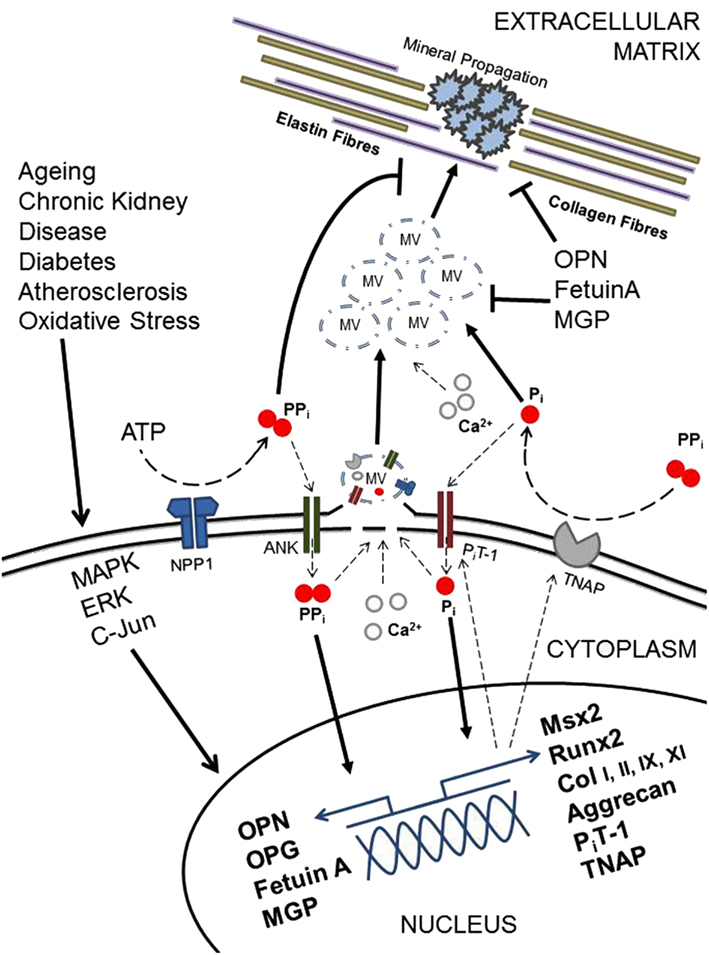
Figure 3. Diagramatic representation of selected regulatory factors and their potential roles in vascular calcification.
The pathological cell-mediated process of soft tissue calcification shares many similarities with that of the physiological matrix mineralization during skeletal development. Membrane-bound matrix vesicles nucleate hydroxyapatite crystals that contain calcium and inorganic phosphate (Anderson et al., 1990; Nahar et al., 2008) forming the first nidus for calcification. This occurs via a tightly controlled balance of inhibitors and inducers, including metabolic alterations (Chen et al., 2006; Kapustin et al., 2011; Sage et al., 2011; Sevinc Ok et al., 2012), inflammation (Tintut et al., 2000; Stompór et al., 2003; Lencel et al., 2011), drugs (Kirton et al., 2006; Helas et al., 2009; Beazley et al., 2012), and morphogens (Radcliff et al., 2005; Nakahara et al., 2010; Shimizu et al., 2011; Figure 4).
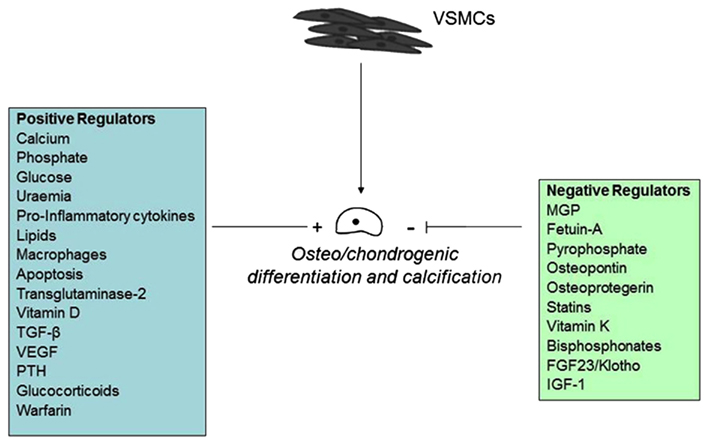
Figure 4. Positive and negative regulators of vascular calcification.
Matrix vesicles contain negative regulators of hydroxyapatite crystal nucleation and growth, such as fetuin-A and matrix gla protein (MGP; Reynolds et al., 2004; Murshed et al., 2005). In cooperation with local mediators such as pyrophosphate (PPi), these molecules protect the arteries from deposition and growth of minerals (Luo et al., 1997; Harmey et al., 2004; Jahnen-Dechent et al., 2011). In the absence of these inhibitors, or following the stimulation of cell death-related processes, together with the bone-like activity of vascular cells, calcification is readily induced (Canfield et al., 2002; Speer et al., 2002; Shroff et al., 2008).
Osteoblasts and chondrocytes are responsible for bone and cartilage formation and calcification within the skeleton. Normal VSMC populations contain cells that undergo phenotypic transition to osteocytic, osteoblastic, and chondrocytic cells in a calcified environment (Steitz et al., 2001; Johnson et al., 2005; Speer et al., 2009; Zhu et al., 2011). Chondro-osseous and calcification promoting genes reported in calcifying VSMCs include the transcription factor Msx2 which promotes osteoblastogenesis (Shao et al., 2005), the osteoblast master transcription factor Runx2 (Speer et al., 2009), the chondrocyte specific extracellular matrix constituent aggrecan and collagen Types I, II, IX, and XI (Johnson et al., 2008). The phosphate transporter PiT-1 is the predominant sodium-dependent phosphate co-transporter expressed in human VSMCs. Phosphate increases PiT-1 expression, which leads to increased levels of intracellular phosphate. This induces Runx2 expression and the osteogenic conversion of VSMCs (Li et al., 2006).
Tissue non-specific alkaline phosphatase (TNAP), a key enzyme for bone calcification, is also central to vascular calcification through the hydrolysis of the calcification inhibitor PPi and the generation of phosphate for hydroxyapatite formation in VSMCs (Narisawa et al., 2007; Lomashvili et al., 2008). Conversely, the ankylosis protein (ANK) and ecto-nucleotide pyrophosphatase/phosphodiesterases-1 (NPP1) inhibit vascular calcification through the promotion of extracellular PPi levels in VSMCs (Johnson et al., 2005; Narisawa et al., 2007), with mice lacking NPP1 developing severe aortic calcification (Figure 5). PPi inhibits calcium phosphate crystal growth and helps to prevent VSMC chondro-osseous differentiation and calcification (Rutsch et al., 2003; Johnson et al., 2005). Arterial calcification is also physiologically limited by VSMC expression of OPN, which is a recognized inhibitor of hydroxyapatite crystal formation and growth and promotes mineral resorption (Speer et al., 2002). OPG, the endogenous inhibitor MGP and the circulating inhibitor fetuin-A have also been shown to block VSMC calcification (Bucay et al., 1998; Canfield et al., 2002; Speer et al., 2002; Bennett et al., 2006; Matsui et al., 2009).
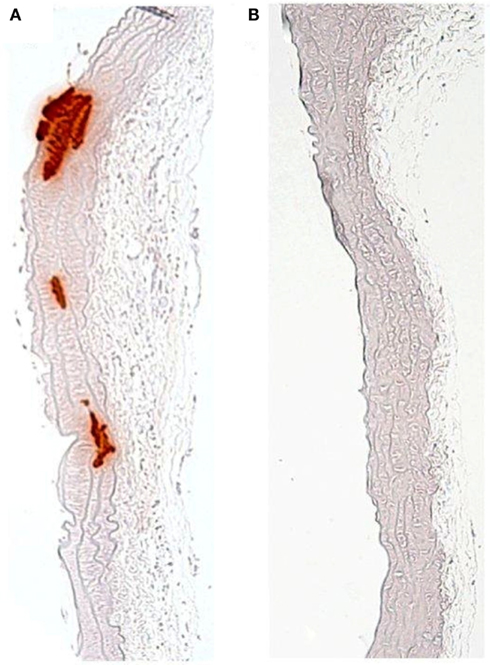
Figure 5. Medial calcification of the aorta due to depressed levels of the calcification inhibitor pyrophosphate in the (A) Enpp1-null mouse, compared to (B) wild-type control.
Changes to several intracellular signal transduction pathways have been reported during vascular calcification, including the induction of the extracellular signal-regulated kinase 1/2, c-Jun N-terminal kinase, and p38 Mitogen Activated Protein Kinase pathways (Simmons et al., 2004; Tanikawa et al., 2009). The growth arrest-specific gene 6 (Gas6)/Axl survival signal, which exerts an antiapoptotic effect through the Bcl2-mediated phosphatidylinositol 3-kinase/protein kinase b pathway has also been shown to be one of the key mechanisms for phosphate-induced calcification (Collett et al., 2007; Son et al., 2007). These studies suggest that therapeutics targeting the Axl receptor may open up new avenues for the prevention of vascular calcification in vivo.
Clinical Consequences of Vascular Calcification
Calcification of blood vessels is a common consequence of aging, atherosclerosis, CKD, and diabetes (Fuchs et al., 1985; Sangiorgi et al., 1998; Allison et al., 2004; Giachelli, 2004; Reaven and Sacks, 2005; Okuno et al., 2007; Shroff and Shanahan, 2007; Kestenbaum et al., 2009; Mackenzie and MacRae, 2011) and is associated with significant mortality and morbidity of cardiovascular disease (Arad et al., 2000; Rosenhek et al., 2000; Keelan et al., 2001; Wayhs et al., 2002). Indeed clinically, vascular calcification is now accepted as a valuable predictor of coronary heart disease (Greenland et al., 2007). The clinical ramifications of vascular calcification in CKD, atherosclerosis, and cardiac valve calcification are described here in more detail.
Chronic kidney disease
It has been reported that approximately 40% of patients with CKD have vascular calcification compared with 13% of control patients with normal renal function (Russo et al., 2004). Kramer et al. (2005) demonstrated a positive association between the presence of vascular calcification and renal failure, and that this association increased markedly in CKD diabetic patients. Converging evidence from clinical, epidemiological, and translational research studies has suggested that vascular calcification progresses inexorably during dialysis and may only partially reverse after successful transplantation (Ossareh, 2011; Shroff, 2011). Medial calcification leads to vascular stiffness and decreases the compliance of blood vessels. These changes result in both increased pulse pressure (Dao et al., 2005) and left ventricular hypertrophy (Speer and Giachelli, 2004). In dialysis patients, medial calcification contributes to calcific uremic arteriolopathy, a necrotizing skin condition with high mortality rates (Coates et al., 1998).
Generalized arterial calcification of infancy
Generalized Arterial Calcification of Infancy is a rare autosomal recessive disease which is characterized by the calcification of arteries, in conjunction with arterial stenosis caused by intimal proliferation. The majority of affected children die within the first 6 months of life as the result of end-organ damage. In a subset of patients, peri-articular calcification of joints also occurs (Rutsch et al., 2003, 2008).
Atherosclerosis
Previous studies have shown that intimal calcification is positively correlated with atherosclerotic plaque burden (Rumberger et al., 1995; Sangiorgi et al., 1998), increased risk of myocardial infarction (Beadenkopf et al., 1964; Loecker et al., 1992), and plaque instability (Fitzgerald et al., 1992; Burke et al., 2000). Furthermore, calcium deposits may directly alter atherosclerotic plaque stability (Wong et al., 2012). However, a major limitation of using calcium score progression as a marker of risk is that the positive predictive value appears to be low with substantial overlap among those with and without future adverse cardiovascular events (Greenland et al., 2007).
Cardiac valve calcification
In the aortic valve, calcification gives rise to life-threatening stenosis. Calcific aortic valve stenosis (CAVS) is the leading reason for valve replacement in Europe and North America, and is considered to be a major mode of failure of native as well as bioprosthetic valves (O’Keefe et al., 1991; Lindroos et al., 1993). CAVS is also correlated with a high risk of cardiovascular dysfunction, and is the third leading cause of cardiovascular disease (Ribeiro et al., 1998; Nkomo et al., 2006).
Risk Factors for Vascular Calcification
Elevated serum phosphate levels are recognized as a major risk factor for cardiovascular events in the general population (Dhingra et al., 2007; Kestenbaum et al., 2009) and in CKD (Young et al., 2005; Adeney et al., 2009). Serum phosphate levels greater than 5.5 mg/dL are strongly correlated with mortality in ESRD patients (Block et al., 2004; Tentori et al., 2008). Furthermore, relatively small increases in serum phosphate (3.5–4.5 mg/dL) have also been correlated with increased risk of cardiovascular and all-cause mortality in CKD patients (Kestenbaum et al., 2005) and the general population with normal renal function (Tonelli et al., 2005). Increased susceptibility of CKD patients to vascular calcification likely underlies this high risk of cardiovascular disease-related deaths in CKD patients.
A number of clinical studies have also shown an association between elevated serum calcium and increased risk of myocardial infarction and vascular calcification in both CKD patients and in the general population (Yamada et al., 2007; Kovesdy et al., 2010; Larsson et al., 2010; West et al., 2010). Furthermore, a recent meta-analysis has reported that dietary calcium supplementation is associated with a significantly increased risk of myocardial infarction (Bolland et al., 2010). Clinical studies investigating the patterns of systemic atherosclerotic calcification have further revealed age and hypertension as the dominant risk factors for calcification (Allison et al., 2004).
In recent years, several studies have demonstrated the positive relationship between vascular calcification and bone health (Frye et al., 1992; Kiel et al., 2001). Vascular calcification is often accompanied by either decreased bone mineral density or disturbed bone turnover. This association has been observed in general populations (Hyder et al., 2007) and also in patients with osteoporosis, Paget’s disease, and CKD (Laroche and Delmotte, 2005; Raggi et al., 2007; Toussaint et al., 2008; Osako et al., 2010; Bandeira et al., 2012). It appears that in patients with CKD that both extremes of bone remodeling, low turnover (adynamic bone), and hyperparathyroid bone, may accelerate vascular calcification by not allowing calcium or phosphorus into bone, or resorbing it out of bone, respectively (Moe and Chen, 2004). In genetically altered animals with deletions of OPG and klotho, a combined osteoporosis-arterial calcification phenotype has been observed (Bucay et al., 1998; Nabeshima, 2002). Furthermore, bone loss and vascular calcification share various common mechanisms, including estrogen deficiency, vitamin D and K abnormalities, chronic inflammation, and oxidative stress (Hofbauer et al., 2007).
Calcific aortic valve stenosis is associated with classic atherosclerotic risk factors, including hypercholesterolemia, hypertension, smoking, and male gender (Mohler et al., 1991; Stewart et al., 1997). A faster disease progression has also been reported in patients with a metabolic syndrome (Briand et al., 2006). Lifestyle modifications are therefore likely to be advantageous, however a beneficial effect of controlling cardiovascular risk factors has yet to be demonstrated in CAVS.
Potential Therapeutic Targets
Specific drugs capable of inhibiting vascular calcification have yet to be developed. Potential strategies that have recently been investigated include the administration of vitamin K, statins, bisphosphonates, TNAP inhibitors, and Non-Steroidal Anti-Inflammatory Drugs (NSAIDs).
Vitamin K
The vitamin K-dependent proteins (VKDPs) MGP and Gas-6 are produced by VSMCs and pericytes. The process of converting VKDPs to their biologically active forms requires the carboxylation of glutamic acid residues by vitamin K (Furie et al., 1999). In rats, inactivation of MGP by treatment with the vitamin K antagonist warfarin leads to rapid calcification of the arteries. This can be regressed by a vitamin K-rich diet (Schurgers et al., 2007). Specifically, Vitamin K2 supplementation prevents arterial calcification, yet vitamin K1 does not (Howe and Webster, 2000; Spronk et al., 2003). In the population based Rotterdam study, increased intake of vitamin K2, but not K1, was shown to be inversely related to all-cause mortality (relative risk = 0.91) and severe aortic calcification (relative risk = 0.74; Geleijnse et al., 2004). A more recent investigation examined the association of vitamin K1 and vitamin K2 intake with coronary calcification in a cross-sectional study among 564 post-menopausal women (Beulens et al., 2009) 0.62% of the women had coronary calcification. Vitamin K2 intake was again associated with decreased coronary calcification (relative risk = 0.80). Interestingly, one of the major dietary sources of vitamin K2 is cheese (Schurgers and Vermeer, 2000), which although is not related to a healthy lifestyle or diet, has yet to be established as a dietary risk factor for cardiovascular disease. It is therefore possible that cheese could exert a beneficial effect in the cardiovascular system and that the high cheese consumption in France and the Mediterranean countries may possibly account for the lower prevalence of cardiovascular disease.
Statins
The mechanism attributed to the pleiotrophic effects of statins involves the inhibition of RhoA/Rho-kinase (ROCK) activity (Rikitake and Liao, 2005). Inhibition of ROCK with the inhibitor Y-27632 or siRNA significantly increased ALP activity and calcification of bovine VSMCs and rat aorta organ cultures (Chen et al., 2010). Furthermore, matrix vesicles isolated from bovine VSMCs incubated with Y-27632, show increased ALP activity and increased ability of MVs to subsequently calcify collagen by 66% (Chen et al., 2010). Together these data clearly demonstrate that the RhoA/ROCK signaling pathway is an important negative regulator of vascular calcification. Exposure to fluvastatin has been shown to directly inhibit calcification in VSMCs in vitro, with warfarin treatment abolishing this beneficial effect (Nakano-Kurimoto et al., 2009). Atorvastatin has also been shown to protect cultured VSMCs from phosphate-induced calcification by inhibiting apoptosis via restoration of the Gas6-Axl pathway (Son et al., 2007). However, the clinical use of statins has yet to be shown to effectively inhibit vascular calcification, with neither fulvastatin (Forbat et al., 1998) nor atorvastatin (Schmermund et al., 2006) therapy able to attenuate coronary artery calcification progression. Furthermore a recent clinical trial focusing on changes in coronary artery plaque composition and plaque volume during aggressive dual lipid-lowering therapy with atorvastatin and ezetimibe demonstrated no significant differences in plaque calcification (Kovarnik et al., 2012). These clinical data may be due to statins inhibiting the initiation rather than the progression of vascular calcification.
Bisphosphonates
Bisphosphonates are used as standard therapy for osteoporosis. Studies in rats have shown that alendronate and ibandronate inhibit warfarin and uremia induced media calcification at doses that inhibit bone resorption (Price et al., 2001, 2006). However, it has recently been reported that whilst etidronate and pamidronate prevent the development of vascular calcification in rats with adenine-induced chronic renal failure, bone formation, and mineralization are adversely affected (Lomashvili et al., 2009). These findings support and extend previous results showing that the most effective etidronate dose for the prevention of arterial calcification also reduced bone mineral density in 5/6-nephrectomized rats (Tamura et al., 2007). In 2008, a multicenter genetic study and retrospective observational analysis of subjects affected by GACI revealed a positive association between survival and bisphosphonate treatment (Rutsch et al., 2008). More recently, the long-term survival of a severe case of GACI diagnosed prenatally and treated with etidronate over a 2-year period has been reported. Progressive resolution of arterial calcification was seen by 3 months of age, which was maintained until 2 years of age. Throughout the 2-year follow-up the patient developed mild hypophosphatemia, due to renal phosphate wasting, without signs of rickets (Edouard et al., 2011). This study supports the development of a formalized approach for the treatment of GACI with bisphosphonates.
TNAP inhibitors
Novel TNAP inhibitors which result in higher PPi levels and lower Pi levels have been reported (Narisawa et al., 2007). These compounds have been shown to be capable of reducing in vitro VSMC calcification, and will serve as scaffolds for future efforts to develop novel drugs for the treatment of soft tissue calcification.
Non-steroidal anti-inflammatory drugs
Non-Steroidal Anti-Inflammatory Drugs are commonly used for anti-inflammation and analgesia post-operatively in orthopedic patients. However, several studies have demonstrated that these drugs suppress bone growth, remodeling, and repair (Nilsson et al., 1986; Keller et al., 1987; Ho et al., 1995) through mechanisms including cell cycle arrest and cell death induction (Chang et al., 2005). The administration of Tanshinone IIA, one of the major lipophilic components extracted from the root of Salvia miltiorrhiza Bunge (Shang et al., 2012), attenuates atherosclerotic calcification in a rat model, through inhibition of oxidative stress (Tang et al., 2007). Furthermore, the natural antioxidants curcumin and silybin inhibit VSMC calcification in vitro (Roman-Garcia et al., 2011). However, the cyclooxygenase-2 inhibitor Celecoxib induced no significant changes in atherosclerotic calcification in a mouse model of atherosclerosis (Bea et al., 2003). Further studies are therefore required to more fully investigate the potential therapeutic applications of NSAIDs in suppressing vascular calcification.
Conclusion
Vascular calcification is associated with a number of human diseases including CKD, diabetes, and atherosclerosis, and is a significant independent risk factor for the development of cardiovascular disease. Whilst the molecular mechanisms of vascular calcification are similar to the process of bone mineralization, further insights are required to determine the precise pathways involved that will allow the identification of effective targets for the development of novel therapeutics.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by an Institute Strategic Program Grant and Institute Career Path Fellowship funding from the Biotechnology and Biological Sciences Research Council (BBSRC).
References
Adeney, K. L., Siscovick, D. S., Ix, J. H., Seliger, S. L., Shlipak, M. G., Jenny, N. S., and Kestenbaum, B. R. (2009). Association of serum phosphate with vascular and valvular calcification in moderate CKD. J. Am. Soc. Nephrol. 20, 381–387.
Allison, M. A., Criqui, M. H., and Wright, C. M. (2004). Patterns and risk factors for systemic calcified atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 24, 331–336.
Anderson, H. C., Stechschulte, D. J. Jr, Collins, D. E., Jacobs, D. H., Morris, D. C., Hsu, H. H., Redford, P. A., and Zeiger, S. (1990). Matrix vesicle biogenesis in vitro by rachitic and normal rat chondrocytes. Am. J. Pathol. 136, 391–398.
Andreeva, E. R., Pugach, I. M., Gordon, D., and Orekhov, A. N. (1998). Continuous subendothelial network formed by pericyte-like cells in human vascular bed. Tissue Cell 30, 127–135.
Arad, Y., Spadaro, L. A., Goodman, K., Newstein, D., and Guerci, A. D. (2000). Prediction of coronary events with electron beam computed tomography. J. Am. Coll. Cardiol. 36, 1253–1260.
Balica, M., Bostrom, K., Shin, V., Tillisch, K., and Demer, L. L. (1997). Calcifying subpopulation of bovine aortic smooth muscle cells is responsive to 17 beta-estradiol. Circulation 95, 1954–1960.
Bandeira, E., Neves, A. P., Costa, C., and Bandeira, F. (2012). Association between vascular calcification and osteoporosis in men with type 2 diabetes. J. Clin. Densitom. 15, 55–60.
Bea, F., Blessing, E., Bennett, B. J., Kuo, C. C., Campbell, L. A., Kreuzer, J., and Rosenfeld, M. E. (2003). Chronic inhibition of cyclooxygenase-2 does not alter plaque composition in a mouse model of advanced unstable atherosclerosis. Cardiovasc. Res. 60, 198–204.
Beadenkopf, W. G., Daoud, A. S., and Love, B. M. (1964). Calcification in the coronary arteries and its relationship to arteriosclerosis and myocardial infarction. Am. J. Roentgenol. Radium Ther. Nucl. Med. 92, 865–871.
Beazley, K. E., Deasey, S., Lima, F., and Nurminskaya, M. V. (2012). Transglutaminase 2-mediated activation of β-catenin signaling has a critical role in warfarin-induced vascular calcification. Arterioscler. Thromb. Vasc. Biol. 32, 123–130.
Bennett, B. J., Scatena, M., Kirk, E. A., Rattazzi, M., Varon, R. M., Averill, M., Schwartz, S. M., Giachelli, C. M., and Rosenfeld, M. E. (2006). Osteoprotegerin inactivation accelerates advanced atherosclerotic lesion progression and calcification in older ApoE−/− mice. Arterioscler. Thromb. Vasc. Biol. 26, 2117–2124.
Beulens, J. W., Bots, M. L., Atsma, F., Bartelink, M. L., Prokop, M., Geleijnse, J. M., Witteman, J. C., Grobbee, D. E., and van der Schouw, Y. T. (2009). High dietary menaquinone intake is associated with reduced coronary calcification. Atherosclerosis 203, 489–493.
Block, G. A., Klassen, P. S., Lazarus, J. M., Ofsthun, N., Lowrie, E. G., and Chertow, G. M. (2004). Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J. Am. Soc. Nephrol. 15, 2208–2218.
Bolland, M. J., Avenell, A., Baron, J. A., Grey, A., MacLennan, G. S., Gamble, G. D., and Reid, I. R. (2010). Effect of calcium supplements on risk of myocardial infarction and cardiovascular events: meta-analysis. BMJ 341, c3691.
Briand, M., Lemieux, I., Dumesnil, J. G., Mathieu, P., Cartier, A., Després, J. P., Arsenault, M., Couet, J., and Pibarot, P. (2006). Metabolic syndrome negatively influences disease progression and prognosis in aortic stenosis. J. Am. Coll. Cardiol. 47, 2229–2236.
Bucay, N., Sarosi, I., Dunstan, C. R., Morony, S., Tarpley, J., Capparelli, C., Scully, S., Tan, H. L., Xu, W., Lacey, D. L., Boyle, W. J., and Simonet, W. S. (1998). Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 12, 1260–1268.
Burke, A. P., Taylor, A., Farb, A., Malcom, G. T., and Virmani, R. (2000). Coronary calcification: insights from sudden coronary death victims. Z. Kardiol. 89, 49–53.
Canfield, A. E., Doherty, M. J., Wood, A. C., Farrington, C., Ashton, B., Begum, N., Harvey, B., Poole, A., Grant, M. E., and Boot-Handford, R. P. (2000). Role of pericytes in vascular calcification: a review. Z. Kardiol. 89, 20–27.
Canfield, A. E., Farrington, C., Dziobon, M. D., Boot-Handford, R. P., Heagerty, A. M., Kumar, S. N., and Roberts, I. S. (2002). The involvement of matrix glycoproteins in vascular calcification and fibrosis: an immunohistochemical study. J. Pathol. 196, 228–234.
Chang, J. K., Wang, G. J., Tsai, S. T., and Ho, M. L. (2005). Nonsteroidal anti-inflammatory drug effects on osteoblastic cell cycle, cytotoxicity, and cell death. Connect. Tissue Res. 46, 200–210.
Chantelau, E., Lee, K. M., and Jungblut, R. (1995). Association of below-knee atherosclerosis to medial arterial calcification in diabetes mellitus. Diabetes Res. Clin. Pract. 29, 169–172.
Chen, N. X., Chen, X., O’Neill, K. D., Atkinson, S. J., and Moe, S. M. (2010). RhoA/Rho kinase (ROCK) alters fetuin-A uptake and regulates calcification in bovine vascular smooth muscle cells (BVSMC). Am. J. Physiol. Renal Physiol. 299, F674–F680.
Chen, N. X., Duan, D., O’Neill, K. D., and Moe, S. M. (2006). High glucose increases the expression of Cbfa1 and BMP-2 and enhances the calcification of vascular smooth muscle cells. Nephrol. Dial. Transplant. 21, 3435–3442.
Coates, T., Kirkland, G. S., Dymock, R. B., Murphy, B. F., Brealey, J. K., Mathew, T. H., and Disney, A. P. (1998). Cutaneous necrosis from calcific uremic arteriolopathy. Am. J. Kidney Dis. 32, 384–391.
Cola, C., Almeida, M., Li, D., Romeo, F., and Mehta, J. L. (2004). Regulatory role of endothelium in the expression of genes affecting arterial calcification. Biochem. Biophys. Res. Commun. 320, 424–427.
Collett, G. D., Sage, A. P., Kirkton, J. P., Alexander, M. Y., Gilmore, A. P., and Canfield, A. E. (2007). Axl/phosphatidylinositol 3-kinase signalling inhibits mineral deposition by vascular smooth muscle cells. Circ. Res. 100, 502–509.
Cosmi, J. E., Kort, S., Tunick, P. A., Rosenzweig, B. P., Freedberg, R. S., Katz, E. S., Applebaum, R. M., and Kronzon, I. (2002). The risk of the development of aortic stenosis in patients with “benign” aortic valve thickening. Arch. Intern. Med. 162, 2345–2347.
Czermack, J. (1852). Beschreibung und mikroskopische Untersuchung zweier ägyptischer Mumien. Sonder-Berichte Akad Wiss Wien 9, 427–469.
Dao, H. H., Essalihi, R., Bouvet, C., and Moreau, P. (2005). Evolution and modulation of age-related medial elastocalcinosis: impact on large artery stiffness and isolated systolic hypertension. Cardiovasc. Res. 66, 307–317.
Demer, L. L., and Tintut, Y. (2008). Vascular calcification: pathobiology of a multifaceted disease. Circulation 117, 2938–2948.
Dhingra, R., Sullivan, L. M., Fox, C. S., Wang, T. J., D’Agostino, R. S. Sr., Gaziano, J. M., and Vasan, R. S. (2007). Relations of serum phosphorus and calcium levels to the incidence of cardiovascular disease in the community. Arch. Intern. Med. 167, 879–885.
Doherty, M. J., Ashton, B. A., Walsh, S., Beresford, J. N., Grant, M. E., and Canfield, A. E. (1998). Vascular pericytes express osteogenic potential in vitro and in vivo. J. Bone Miner. Res. 13, 828–838.
Doherty, M. J., and Canfield, A. E. (1999). Gene expression during vascular pericyte differentiation. Crit. Rev. Eukaryot. Gene Expr. 9, 1–17.
Doherty, T. M., Fitzpatrick, L. A., Inoue, D., Qiao, J. H., Fishbein, M. C., Detrano, R. C., Shah, P. K., and Rajavashisth, T. B. (2004). Molecular, endocrine, and genetic mechanisms of arterial calcification. Endocr. Rev. 25, 629–672.
Edouard, T., Chabot, G., Miro, J., Buhas, D. C., Nitschke, Y., Lapierre, C., Rutsch, F., and Alos, N. (2011). Efficacy and safety of 2-year etidronate treatment in a child with generalized arterial calcification of infancy. Eur. J. Pediatr. 170, 1585–1590.
Essalihi, R., Ouellette, V., Dao, H. H., McKee, M. D., and Moreau, P. (2004). Phenotypic modulation of vascular smooth muscle cells during medial arterial calcification: a role for endothelin? J. Cardiovasc. Pharmacol. 44, S147–S150.
Farrington-Rock, C., Crofts, N. J., Doherty, M. J., Ashton, B. A., Griffin-Jones, C., and Canfield, A. E. (2004). Chondrogenic and adipogenic potential of microvascular pericytes. Circulation 110, 2226–2232.
Fitzgerald, P. J., Ports, T. A., and Yock, P. G. (1992). Contribution of localized calcium deposits to dissection after angioplasty. An observational study using intravascular ultrasound. Circulation 86, 64–70.
Forbat, S. M., Naoumova, R. P., Sidhu, P. S., Neuwirth, C., MacMahon, M., Thompson, G. R., and Underwood, S. R. (1998). The effect of cholesterol reduction with fluvastatin on aortic compliance, coronary calcification and carotid intimal-medial thickness: a pilot study. J. Cardiovasc. Risk 5, 1–10.
Frye, M. A., Melton, L. J., Bryant, S. C., Fitzpatrick, L. A., Wahner, H. W., Schwartz, R. S., and Riggs, B. L. (1992). Osteoporosis and calcification of the aorta. Bone Miner. 19, 185–194.
Fuchs, U., Caffier, P., Schulz, H. G., and Wieniecki, P. (1985). Arterial calcification in diabetics. Virchows Arch. A Pathol. Anat. Histopathol. 407, 431–439.
Furie, B., Bouchard, B. A., and Furie, B. C. (1999). Vitamin K-dependent biosynthesis of gamma-carboxyglutamic acid. Blood 93, 1798–1808.
Geleijnse, J. M., Vermeer, C., Grobbee, D. E., Schurgers, L. J., Knapen, M. H., van der Meer, I. M., Hofman, A., and Witteman, J. C. (2004). Dietary intake of menaquinone is associated with a reduced risk of coronary heart disease: the Rotterdam Study. J. Nutr. 134, 3100–3105.
Giachelli, C. M. (2009). The emerging role of phosphate in vascular calcification. Kidney Int. 75, 890–897.
Greenland, P., Bonow, R. O., Brundage, B. H., Budoff, M. J., Eisenberg, M. J., Grundy, S. M., Lauer, M. S., Post, W. S., Raggi, P., Redberg, R. F., Rodgers, G. P., Shaw, L. J., Taylor, A. J., Weintraub, W. S., Harrington, R. A., Abrams, J., Anderson, J. L., Bates, E. R., Grines, C. L., Hlatky, M. A., Lichtenberg, R. C., Lindner, J. R., Pohost, G. M., Schofield, R. S., Shubrooks, S. J. Jr, Stein, J. H., Tracy, C. M., Vogel, R. A., and Wesley, D. J. (2007). ACCF/AHA 2007 clinical expert consensus document on coronary artery calcium scoring by computed tomography in global cardiovascular risk assessment and in evaluation of patients with chest pain: a report of the American College of Cardiology Foundation Clinical Expert Consensus Task Force. Circulation 115, 402–426.
Harmey, D., Hessle, L., Narisawa, S., Johnson, K. A., Terkeltaub, R., and Millán, J. L. (2004). Concerted regulation of inorganic pyrophosphate and osteopontin by akp2, enpp1, and ank: an integrated model of the pathogenesis of mineralization disorders. Am. J. Pathol. 164, 1199–1209.
Hedin, U., Roy, J., Tran, P. K., Lundmark, K., and Rahman, A. (1999). Control of smooth muscle cell proliferation – the role of the basement membrane. Thromb. Haemost. 82, 23–26.
Helas, S., Goettsch, C., Schoppet, M., Zeitz, U., Hempel, U., Morawietz, H., Kostenuik, P. J., Erben, R. G., and Hofbauer, L. C. (2009). Inhibition of receptor activator of NF-kappaB ligand by denosumab attenuates vascular calcium deposition in mice. Am. J. Pathol. 175, 473–478.
Ho, M. L., Chang, J. K., and Wang, G. J. (1995). Antiinflammatory drug effects on bone repair and remodeling in rabbits. Clin. Orthop. 313, 270–278.
Hofbauer, L. C., Brueck, C. C., Shanahan, C. M., Schoppet, M., and Dobnig, H. (2007). Vascular calcification and osteoporosis – from clinical observation towards molecular understanding. Osteoporos. Int. 18, 251–259.
Howe, A. M., and Webster, W. S. (2000). Warfarin exposure and calcification of the arterial system in the rat. Int. J. Exp. Pathol. 81, 51–56.
Hruska, K. A., Mathew, S., and Saab, G. (2005). Bone morphogenetic proteins in vascular calcification. Circ. Res. 97, 105–114.
Hunt, J. L., Fairman, R., Mitchell, M. E., Carpenter, J. P., Golden, M., Khalapyan, T., Wolfe, M., Neschis, D., Milner, R., Scoll, B., Cusack, A., and Mohler, E. R. (2002). Bone formation in carotid plaques: a clinicopathological study. Stroke 33, 1214–1219.
Hyder, J. A., Allison, M. A., Criqui, M. H., and Wright, C. M. (2007). Association between systemic calcified atherosclerosis and bone density. Calcif. Tissue Int. 80, 301–306.
Jahnen-Dechent, W., Heiss, A., Schäfer, C., and Ketteler, M. (2011). Fetuin-A regulation of calcified matrix metabolism. Circ. Res. 108, 1494–1509.
Johnson, K., Polewski, M., van Etten, D., and Terkeltaub, R. (2005). Chondrogenesis mediated by PPi depletion promotes spontaneous aortic calcification in NPP1−/− mice. Arterioscler. Thromb. Vasc. Biol. 25, 686–691.
Johnson, K. A., Polewski, M., and Terkeltaub, R. A. (2008). Transglutaminase 2 is central to induction of the arterial calcification program by smooth muscle cells. Circ. Res. 102, 529–537.
Kaden, J. J., Dempfle, C. E., Grobholz, R., Tran, H. T., Kiliç, R., Sarikoç, A., Brueckmann, M., Vahl, C., Hagl, S., Haase, K. K., and Borggrefe, M. (2003). Interleukin-1 beta promotes matrix metalloproteinase expression and cell proliferation in calcific aortic valve stenosis. Atherosclerosis 170, 205–211.
Kaden, J. J., Dempfle, C. E., Grobholz, R., Fischer, C. S., Vocke, D. C., Kiliç, R., Sarikoç, A., Piñol, R., Hagl, S., Lang, S., Brueckmann, M., and Borggrefe, M. (2005). Inflammatory regulation of extracellular matrix remodeling in calcific aortic valve stenosis. Cardiovasc. Pathol. 14, 80–87.
Kapustin, A. N., Davies, J. D., Reynolds, J. L., McNair, R., Jones, G. T., Sidibe, A., Schurgers, L. J., Skepper, J. N., Proudfoot, D., Mayr, M., and Shanahan, C. M. (2011). Calcium regulates key components of vascular smooth muscle cell-derived matrix vesicles to enhance mineralization. 109, e1–e12.
Keelan, P. C., Bielak, L. F., Ashai, K., Jamjoum, L. S., Denktas, A. E., Rumberger, J. A., Sheedy, I. P., Peyser, P. A., and Schwartz, R. S. (2001). Long-term prognostic value of coronary calcification detected by electron-beam computed tomography in patients undergoing coronary angiography. Circulation 104, 412–417.
Keller, J., Bünger, C., Andreassen, T. T., Bak, B., and Lucht, U. (1987). Bone repair inhibited by indomethacin. Effects on bone metabolism and strength of rabbit osteotomies. Acta Orthop. Scand. 58, 379–383.
Kelly, R. P., Tunin, R., and Kass, D. A. (1992). Effect of reduced aortic compliance on cardiac efficiency and contractile function of in situ canine left ventricle. Circ. Res. 71, 490–502.
Kestenbaum, B., Sampson, J. N., Rudser, K. D., Patterson, D. J., Seliger, S. L., Young, B., Sherrard, D. J., and Andress, D. L. (2005). Serum phosphate levels and mortality risk among people with chronic kidney disease. J. Am. Soc. Nephrol. 16, 520–528.
Kestenbaum, B. R., Adeney, K. L., de Boer, I. H., Ix, J. H., Shlipak, M. G., and Siscovick, D. S. (2009). Incidence and progression of coronary calcification in chronic kidney disease: the Multi-Ethnic Study of Atherosclerosis. Kidney Int. 76, 991–998.
Kiel, D. P., Kauppila, L. I., Cupples, L. A., Hannan, M. T., O’Donnell, C. J., and Wilson, P. W. (2001). Bone loss and the progression of abdominal aortic calcification over a 25 year period: the Framingham Heart Study. Calcif. Tissue Int. 68, 271–276.
Kirton, J. P., Crofts, N. J., George, S. J., Brennan, K., and Canfield, A. E. (2007). Wnt/beta-catenin signaling stimulates chondrogenic and inhibits adipogenic differentiation of pericytes: potential relevance to vascular disease? Circ. Res. 101, 581–589.
Kirton, J. P., Wilkinson, F. L., Canfield, A. E., and Alexander, M. Y. (2006). Dexamethasone downregulates calcification-inhibitor molecules and accelerates osteogenic differentiation of vascular pericytes: implications for vascular calcification. Circ. Res. 98, 1264–1272.
Kovarnik, T., Mintz, G. S., Skalicka, H., Kral, A., Horak, J., Skulec, R., Uhrova, J., Martasek, P., Downe, R. W., Wahle, A., Sonka, M., Mrazek, V., Aschermann, M., and Linhart, A. (2012). Virtual histology evaluation of atherosclerosis regression during atorvastatin and ezetimibe administration: HEAVEN study. Circ. J. 76, 176–183.
Kovesdy, C. P., Kuchmak, O., Lu, J. L., and Kalantar-Zadeh, K. (2010). Outcomes associated with serum calcium level in men with non-dialysis-dependent chronic kidney disease. Clin. J. Am. Soc. Nephrol. 5, 468–476.
Kramer, H., Toto, R., Peshock, R., Cooper, R., and Victor, R. (2005). Association between chronic kidney disease and coronary artery calcification: the Dallas Heart Study. J. Am. Soc. Nephrol. 16, 507–513.
Laroche, M., and Delmotte, A. (2005). Increased arterial calcification in Paget’s disease of bone. Calcif. Tissue Int. 77, 129–133.
Larsson, T. E., Olauson, H., Hagstrom, E., Ingelsson, E., Arnlov, J., Lind, L., and Sundstrom, J. (2010). Conjoint effects of serum calcium and phosphate on risk of total, cardiovascular, and noncardiovascular mortality in the community. Arterioscler. Thromb. Vasc. Biol. 30, 333–339.
Lehto, S., Niskanen, L., Suhonen, M., Ronnemaa, T., and Laakso, M. (1996). Medial artery calcification: a neglected harbinger of cardiovascular complications in non-insulin-dependent diabetes mellitus. Arterioscler. Thromb. Vasc. Biol. 16, 978–983.
Lencel, P., Delplace, S., Pilet, P., Leterme, D., Miellot, F., Sourice, S., Caudrillier, A., Hardouin, P., Guicheux, J., and Magne, D. (2011). Cell-specific effects of TNF-α and IL-1β on alkaline phosphatase: implication for syndesmophyte formation and vascular calcification. Lab. Invest. 91, 1434–1442.
Li, R., Mittelstein, D., Lee, J., Fang, K., Majumdar, R., Tintut, Y., Demer, L. L., and Hsiai, T. K. (2012). A dynamic model of calcific nodule destabilization in response to monocyte- and oxidized lipid-induced matrix metalloproteinases. Am. J. Physiol. Cell Physiol. 302, C658–C665.
Li, X., Yang, H. Y., and Giachelli, C. M. (2006). Role of the sodium-dependent phosphate cotransporter, Pit-1, in vascular smooth muscle cell calcification. Circ. Res. 98, 905–912.
Lindroos, M., Kupari, M., Heikkilä, J., and Tilvis, R. (1993). Prevalence of aortic valve abnormalities in the elderly: an echocardiographic study of a random population sample. J. Am. Coll. Cardiol. 21, 1220–1225.
Liu, A. C., Joag, V. R., and Gotlieb, A. I. (2007). The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology. Am. J. Pathol. 171, 1407–1418.
Loecker, T. H., Schwartz, R. S., Cotta, C. W., and Hickman, J. R. (1992). Fluoroscopic coronary artery calcification and associated coronary disease in asymptomatic young men. J. Am. Coll. Cardiol. 19, 1167–1172.
Lomashvili, K. A., Garg, P., Narisawa, S., Millan, J. L., and O’Neill, W. C. (2008). Upregulation of alkaline phosphatase and pyrophosphate hydrolysis: potential mechanism for uremic vascular calcification. Kidney Int. 73, 1024–1030.
Lomashvili, K. A., Monier-Faugere, M. C., Wang, X., Malluche, H. H., and O’Neill, W. C. (2009). Effect of bisphosphonates on vascular calcification and bone metabolism in experimental renal failure. Kidney Int. 75, 617–625.
London, G. M., Guerin, A. P., Marchais, S. J., Metivier, F., Pannier, B., and Adda, H. (2003). Arterial media calcification in end-stage renal disease: impact on all-cause and cardiovascular mortality. Nephrol. Dial. Transplant. 18, 1731–1740.
London, G. M., Marchais, S. J., Guerin, A. P., and Metivier, F. (2005). Arteriosclerosis, vascular calcifications and cardiovascular disease in uremia. Curr. Opin. Nephrol. Hypertens. 14, 525–531.
Luo, G., Ducy, P., McKee, M. D., Pinero, G. J., Loyer, E., Behringer, R. R., and Karsenty, G. (1997). Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 386, 78–81.
Mackenzie, N. C., and MacRae, V. E. (2011). The role of cellular senescence during vascular calcification: a key paradigm in aging research. Curr. Aging Sci. 4, 128–136.
Mackenzie, N. C., Zhu, D., Longley, L., Patterson, C. S., Kommareddy, S., and MacRae, V. E. (2011). MOVAS-1 cell line: a new in vitro model of vascular calcification. Int. J. Mol. Med. 27, 663–668.
Matsui, I., Hamano, T., Mikami, S., Fujii, N., Takabatake, Y., Nagasawa, Y., Kawada, N., Ito, T., Rakugi, H., Imai, E., and Isaka, Y. (2009). Fully phosphorylated fetuin-A forms a mineral complex in the serum of rats with adenine-induced renal failure. Kidney Int. 75, 915–928.
Minasi, M. G., Riminucci, M., De Angelis, L., Borello, U., Berarducci, B., Innocenzi, A., Caprioli, A., Sirabella, D., Baiocchi, M., De Maria, R., Boratto, R., Jaffredo, T., Broccoli, V., Bianco, P., and Cossu, G. (2002). The meso-angioblast: a multipotent, self-renewing cell that originates from the dorsal aorta and differentiates into most mesodermal tissues. Development 129, 2773–2783.
Moe, S. M., and Chen, N. X. (2004). Pathophysiology of vascular calcification in chronic kidney disease. Circ. Res. 95, 560–567.
Mohler, E. B. P. R., Gannon, F., Reynolds, C., Zimmerman, R., Keane, M. G., and Kaplan, F. S. (2001). Bone formation and inflammation in cardiac valves. Circulation 103, 1522–1528.
Mohler, E. R., Sheridan, M. J., Nichols, R., Harvey, W. P., and Waller, B. F. (1991). Development and progression of aortic valve stenosis: atherosclerosis risk factors – a causal relationship? A clinical morphologic study. Clin. Cardiol. 14, 995–999.
Mönckeberg, J. G. (1903). Über die reine Mediaverkalkung der Extremitätenarterien und ihr Verhalten zur Arteriosklerose. Virchows Arch. Pathol. Anat. Physiol. Klin. Med. 171, 141–167.
Murshed, M., Harmey, D., Millán, J. L., McKee, M. D., and Karsenty, G. (2005). Unique coexpression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes Dev. 19, 1093–1104.
Mwipatayi, B. P., Cooke, C., Sinniah, R. H., Abbas, M., Angel, D., and Sieunarine, K. (2007). Calciphylaxis: emerging concept in vascular patients. Eur. J. Dermatol. 17, 73–78.
Nahar, N. N., Missana, L. R., Garimella, R., Tague, S. E., and Anderson, H. C. (2008). Matrix vesicles are carriers of bone morphogenetic proteins (BMPs), vascular endothelial growth factor (VEGF), and noncollagenous matrix proteins. J. Bone Miner. Metab. 26, 514–519.
Nakahara, T., Sato, H., Shimizu, T., Tanaka, T., Matsui, H., Kawai-Kowase, K., Sato, M., Iso, T., Arai, M., and Kurabayashi, M. (2010). Fibroblast growth factor-2 induces osteogenic differentiation through a Runx2 activation in vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 394, 243–248.
Nakano-Kurimoto, R., Ikeda, K., Uraoka, M., Nakagawa, Y., Yutaka, K., Koide, M., Takahashi, T., Matoba, S., Yamada, H., Okigaki, M., and Matsubara, H. (2009). Replicative senescence of vascular smooth muscle cells enhances the calcification through initiating the osteoblastic transition. Am. J. Physiol. Heart Circ. Physiol. 297, 1673–1684.
Narisawa, S., Harmey, D., Yadav, M. C., O’Neill, W. C., Hoylaerts, M. F., and Millán, J. L. (2007). Novel inhibitors of alkaline phosphatase suppress vascular smooth muscle cell calcification. J. Bone Miner. Res. 22, 1700–1710.
Nilsson, O. S., Bauer, H. C., Brosjo, O., and Tornkvist, H. (1986). Influence of indomethacin on induced heterotopic bone formation in rats. Importance of length of treatment and of age. Clin. Orthop. 207. 239–245.
Nkomo, V. T., Gardin, J. M., Skelton, T. N., Gottdiener, J. S., Scott, C. G., and Enriquez-Sarano, M. (2006). Burden of valvular heart diseases: a population-based study. Lancet 368, 1005–1011.
O’Brien, K. D., Reichenbach, D. D., Marcovina, S. M., Kuusisto, J., Alpers, C. E., and Otto, C. M. (1996). Apolipoproteins B, (a), and E accumulate in the morphologically early lesion of ‘degenerative’ valvular aortic stenosis. Arterioscler. Thromb. Vasc. Biol. 16, 523–532.
Ohtsuka, S., Kakihana, M., Watanabe, H., and Sugishita, Y. (1994). Chronically decreased aortic distensibility causes deterioration of coronary perfusion during increased left ventricular contraction. J. Am. Coll. Cardiol. 24, 1406–1414.
O’Keefe, J. H., Lavie, C. J., Nishimura, R. A., and Edwards, W. D. (1991). Degenerative aortic stenosis. One effect of the graying of America. Postgrad. Med. 89, 143–146.
Okuno, S., Ishimura, E., Kitatani, K., Fujino, Y., Kohno, K., Maeno, Y., Maekawa, K., Yamakawa, T., Imanishi, Y., Inaba, M., and Nishizawa, Y. (2007). Presence of abdominal aortic calcification is significantly associated with all-cause and cardiovascular mortality in maintenance hemodialysis patients. Am. J. Kidney Dis. 49, 417–425.
Olsson, M., Dalsgaard, C. J., Haegerstrand, A., Rosenqvist, M., Rydén, L., and Nilsson, J. (1994). Accumulation of T lymphocytes and expression of interleukin-2 receptors in nonrheumatic stenotic aortic valves. J. Am. Coll. Cardiol. 23, 1162–1170.
Olsson, M., Thyberg, J., and Nilsson, J. (1999). Presence of oxidized low density lipoprotein in nonrheumatic stenotic aortic valves. Arterioscler. Thromb. Vasc. Biol. 19, 1218–1222.
Osako, M. K., Nakagami, H., Koibuchi, N., Shimizu, H., Nakagami, F., Koriyama, H., Shimamura, M., Miyake, T., Rakugi, H., and Morishita, R. (2010). Estrogen inhibits vascular calcification via vascular RANKL system: common mechanism of osteoporosis and vascular calcification. Circ. Res. 107, 466–475.
Osman, L., Yacoub, M. H., Latif, N., Amrani, M., and Chester, A. H. (2006). Role of human valve interstitial cells in valve calcification and their response to atorvastatin. Circulation 114, I547–I552.
Ossareh, S. (2011). Vascular calcification in chronic kidney disease: mechanisms and clinical implications. Iran J. Kidney Dis. 5, 285–299.
Otto, C. M., Kuusisto, J., Reichenbach, D. D., Gown, A. M., and O’Brien, K. D. (1994). Characterization of the early lesion of ‘degenerative’ valvular aortic stenosis. Histological and immunohistochemical studies. Circulation 90, 844–853.
Pohle, K., Maffert, R., Ropers, D., Moshage, W., Stilianakis, N., Daniel, W. G., and Achenbach, S. (2001). Progression of aortic valve calcification: association with coronary atherosclerosis and cardiovascular risk factors. Circulation 104, 1927–1932.
Price, P. A., Faus, S. A., and Williamson, M. K. (2001). Bisphosphonates alendronate and ibandronate inhibit artery calcification at doses comparable to those that inhibit bone resorption. Arterioscler. Thromb. Vasc. Biol. 21, 817–824.
Price, P. A., Roublick, A. M., and Williamson, M. K. (2006). Artery calcification in uremic rats is increased by a low protein diet and prevented by treatment with ibandronate. Kidney Int. 70, 1577–1583.
Qunibi, W. Y., Nolan, C. A., and Ayus, J. C. (2002). Cardiovascular calcification in patients with end-stage renal disease: a century-old phenomenon. Kidney Int. 82, S73–S80.
Radcliff, K., Tang, T. B., Lim, J., Zhang, Z., Abedin, M., Demer, L. L., and Tintut, Y. (2005). Insulin-like growth factor-I regulates proliferation and osteoblastic differentiation of calcifying vascular cells via extracellular signal-regulated protein kinase and phosphatidylinositol 3-kinase pathways. Circ. Res. 96, 398–400.
Raggi, P., Bellasi, A., Ferramosca, E., Islam, T., Muntner, P., and Block, G. A. (2007). Association of pulse wave velocity with vascular and valvular calcification in hemodialysis patients. Kidney Int. 71, 802–807.
Rajamannan, N. M. (2009). Calcific aortic stenosis: lessons learned from experimental and clinical studies. Arterioscler. Thromb. Vasc. Biol. 29, 162–168.
Rajamannan, N. M., Subramaniam, M., Rickard, D., Stock, S. R., Donovan, J., Springett, M., Orszulak, T., Fullerton, D. A., Tajik, A. J., Bonow, R. O., and Spelsberg, T. (2003). Human aortic valve calcification is associated with an osteoblast phenotype. Circulation 107, 2181–2184.
Reaven, P. D., and Sacks, J. (2005). Coronary artery and abdominal aortic calcification are associated with cardiovascular disease in type 2 diabetes. Diabetologia 48, 379–385.
Reynolds, J. L., Joannides, A. J., Skepper, J. N., McNair, R., Schurgers, L. J., Proudfoot, D., Jahnen-Dechent, W., Weissberg, P. L., and Shanahan, C. M. (2004). Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J. Am. Soc. Nephrol. 15, 2857–2867.
Ribeiro, S., Ramos, A., Brandao, A., Rebelo, J. R., Guerra, A., Resina, C., Vila-Lobos, A., Carvalho, F., Remedio, F., and Ribeiro, F. (1998). Cardiac valve calcification in haemodialysis patients: role of calcium-phosphate metabolism. Nephrol. Dial. Transplant. 13, 2037–2040.
Rikitake, Y., and Liao, J. K. (2005). Rho GTPases, statins, and nitric oxide. Circ. Res. 97, 1232–1235.
Roman-Garcia, P., Barrio-Vazquez, S., Fernandez-Martin, J. L., Ruiz-Torres, M. P., and Cannata-Andia, J. B. (2011). Natural antioxidants and vascular calcification: a possible benefit. J. Nephrol. 24, 669–672.
Rosenhek, R., Binder, T., Porenta, G., Lang, I., Christ, G., Schemper, M., Maurer, G., and Baumgartner, H. (2000). Predictors of outcome in severe, asymptomatic aortic stenosis. N. Engl. J. Med. 343, 611–617.
Rumberger, J. A., Simons, D. B., Fitzpatrick, L. A., Sheedy, P. F., and Schwartz, R. S. (1995). Coronary artery calcium area by electron-beam computed tomography and coronary atherosclerotic plaque area. A histopathologic correlative study. Circulation 92, 2157–2162.
Russo, D., Palmiero, G., De Blasio, A. P., Balletta, M. M., and Andreucci, V. E. (2004). Coronary artery calcification in patients with CRF not undergoing dialysis. Am. J. Kidney Dis. 44, 1024–1030.
Rutsch, F., Böyer, P., Nitschke, Y., Ruf, N., Lorenz-Depierieux, B., Wittkampf, T., Weissen-Plenz, G., Fischer, R. J., Mughal, Z., Gregory, J. W., Davies, J. H., Loirat, C., Strom, T. M., Schnabel, D., Nürnberg, P., and Terkeltaub, R. (2008). Hypophosphatemia, hyperphosphaturia, and bisphosphonate treatment are associated with survival beyond infancy in generalized arterial calcification of infancy. Circ. Cardiovasc. Genet. 1, 133–140.
Rutsch, F., Ruf, N., Vaingankar, S., Toliat, M. R., Suk, A., Höhne, W., Schauer, G., Lehmann, M., Roscioli, T., Schnabel, D., Epplen, J. T., Knisely, A., Superti-Furga, A., McGill, J., Filippone, M., Sinaiko, A. R., Vallance, H., Hinrichs, B., Smith, W., Ferre, M., Terkeltaub, R., and Nürnberg, P. (2003). Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification. Nat. Genet. 34, 379–381.
Sage, A. P., Lu, J., Tintut, Y., and Demer, L. L. (2011). Hyperphosphatemia-induced nanocrystals upregulate the expression of bone morphogenetic protein-2 and osteopontin genes in mouse smooth muscle cells in vitro. Kidney Int. 79, 414–422.
Salhiyyah, K., Yacoub, M. H., and Chester, A. H. (2011). Cellular mechanisms in mitral valve disease. J. Cardiovasc. Transl. Res. 4, 702–709.
Sangiorgi, G., Rumberger, J. A., Severson, A., Edwards, W. D., Gregoire, J., Fitzpatrick, L. A., and Schwartz, R. S. (1998). Arterial calcification and not lumen stenosis is highly correlated with atherosclerotic plaque burden in humans: a histologic study of 723 coronary artery segments using nondecalcifying methodology. J. Am. Coll. Cardiol. 31, 126–133.
Schmermund, A., Achenbach, S., Budde, T., Buziashvili, Y., Förster, A., Friedrich, G., Henein, M., Kerkhoff, G., Knollmann, F., Kukharchuk, V., Lahiri, A., Leischik, R., Moshage, W., Schartl, M., Siffert, W., Steinhagen-Thiessen, E., Sinitsyn, V., Vogt, A., Wiedeking, B., and Erbel, R. (2006). Effect of intensive versus standard lipid-lowering treatment with atorvastatin on the progression of calcified coronary atherosclerosis over 12 months: a multicenter, randomized, double-blind trial. Circulation 113, 427–337.
Schmermund, A., Baumgart, D., Mohlenkamp, S., Kriener, P., Pump, H., Gronemeyer, D., Seibel, R., and Erbel, R. (2001). Natural history and topographic pattern of progression of coronary calcification in symptomatic patients: an electron-beam CT study. Arterioscler. Thromb. Vasc. Biol. 21, 421–426.
Schurgers, L. J., Shearer, M. J., Hamulyák, K., Stöcklin, E., and Vermeer, C. (2007). Vitamin K-containing dietary supplements: comparison of synthetic vitamin K1and natto-derived menaquinone-7. Blood 109, 3279–3283.
Schurgers, L. J., and Vermeer, C. (2000). Determination of phylloquinone and menaquinones in food. Effect of food matrix on circulating vitamin K concentrations. Haemostasis 30, 298–307.
Sevinc Ok, E., Asci, G., Kircelli, F., Duman, S., Dheir, H., Sezis Demirci, M., Ozkahya, M., Toz, H., and Ok, E. (2012). Relationship between glucose exposure via peritoneal dialysis solutions and coronary artery calcification in non-diabetic peritoneal dialysis patients. Int. Urol. Nephro. doi: 10.1007/s11255-012-0138-8
Shanahan, C. M., Cary, N. R., Salisbury, J. R., Proudfoot, D., Weissberg, P. L., and Edmonds, M. E. (1999). Medial localization of mineralization-regulating proteins in association with Mönckeberg sclerosis: evidence for smooth muscle cell-mediated vascular calcification. Circulation 100, 2168–2176.
Shanahan, C. M., Weissberg, P. L., and Metcalfe, J. C. (1993). Isolation of gene markers of differentiated and proliferating vascular smooth muscle cells. Circ. Res. 73, 193–204.
Shang, Q., Xu, H., and Huang, L. (2012). Tanshinone IIA: a promising natural cardioprotective agent. Evid. Based Complement. Alternat. Med. 2012, 716459. doi: 10.1155/2012/716459
Shao, J. S., Cheng, S. L., Pingsterhaus, J. M., Charlton-Kachigian, N., Loewy, A. P., and Towler, D. A. (2005). Msx2 promotes cardiovascular calcification by activating paracrine Wnt signals. J. Clin. Invest. 115, 1210–1220.
Shimizu, T., Tanaka, T., Iso, T., Matsui, H., Ooyama, Y., Kawai-Kowase, K., Arai, M., and Kurabayashi, M. (2011). Notch signaling pathway enhances bone morphogenetic protein 2 (BMP2) responsiveness of Msx2 gene to induce osteogenic differentiation and mineralization of vascular smooth muscle cells. J. Biol. Chem. 286, 19138–11948.
Shroff, R. (2011). Dysregulated mineral metabolism in children with chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 20, 233–240.
Shroff, R. C., McNair, R., Figg, N., Skepper, J. N., Schurgers, L., Gupta, A., Hiorns, M., Donald, A. E., Deanfield, J., Rees, L., and Shanahan, C. M. (2008). Dialysis accelerates medial vascular calcification in part by triggering smooth muscle cell apoptosis. Circulation 118, 1748–1757.
Shroff, R. C., and Shanahan, C. M. (2007). The vascular biology of calcification. Semin. Dial. 20, 103–109.
Simmons, C. A., Nikolovski, J., Thornton, A. J., Matlis, S., and Mooney, D. J. (2004). Mechanical stimulation and mitogen-activated protein kinase signaling independently regulate osteogenic differentiation and mineralization by calcifying vascular cells. J. Biomech. 37, 1531–1541.
Son, B. K., Kozaki, K., Iijima, K., Eto, M., Nakano, T., Akishita, M., and Ouchi, Y. (2007). Gas6/Axl-PI3K/Akt pathway plays a central role in the effect of statins on inorganic phosphate-induced calcification of vascular smooth muscle cells. Eur. J. Pharmacol. 556, 1–8.
Speer, M. Y., and Giachelli, C. M. (2004). Regulation of cardiovascular calcification. Cardiovasc. Pathol. 13, 63–70.
Speer, M. Y., McKee, M. D., Guldberg, R. E., Liaw, L., Yang, H. Y., Tung, E., Karsenty, G., and Giachelli, C. M. (2002). Inactivation of the osteopontin gene enhances vascular calcification of matrix Gla protein-deficient mice: evidence for osteopontin as an inducible inhibitor of vascular calcification in vivo. J. Exp. Med. 196, 1047–1055.
Speer, M. Y., Yang, H. Y., Brabb, T., Leaf, E., Look, A., Lin, W. L., Frutkin, A., Dichek, D., and Giachelli, C. M. (2009). Smooth muscle cells give rise to osteochondrogenic precursors and chondrocytes in calcifying arteries. Circ. Res. 104, 733–741.
Spronk, H. M., Soute, B. A., Schurgers, L. J., Thijssen, H. H., De Mey, J. G., and Vermeer, C. (2003). Tissue-specific utilization of menaquinone-4 results in the prevention of arterial calcification in warfarin-treated rats. J. Vasc. Res. 40, 531–537.
Stary, H. C. (2000). Natural history of calcium deposits in atherosclerosis progression and regression. Z. Kardiol. 89, 28–35.
Stary, H. C., Chandler, A. B., Dinsmore, R. E., Fuster, V., Glagov, S., Insull, W., Rosenfeld, M. E., Schwartz, C. J., Wagner, W. D., and Wissler, R. W. (1995). A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 92, 1355–1374.
Steitz, S. A., Speer, M. Y., Curinga, G., Yang, H. Y., Haynes, P., Aebersold, R., Schinke, T., Karsenty, G., and Giachelli, C. M. (2001). Smooth muscle cell phenotypic transition associated with calcification: upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ. Res. 89, 1147–1154.
Stewart, B. F., Siscovick, D., Lind, B. K., Gardin, J. M., Gottdiener, J. S., Smith, V. E., Kitzman, D. W., and Otto, C. M. (1997). Clinical factors associated with calcific aortic valve disease. Cardiovascular Health Study. J. Am. Coll. Cardiol. 29, 630–634.
Stompór, T., Rajzer, M., Sulowicz, W., Dembinska-Kiec, A., Janda, K., Kawecka-Jaszcz, K., Wójcik, K., Tabor, B., Zdzienicka, A., and Janusz-Grzybowska, E. (2003). An association between aortic pulse wave velocity, blood pressure and chronic inflammation in ESRD patients on peritoneal dialysis. Int. J. Artif. Organs 26, 188–195.
Tamura, K., Suzuki, Y., Matsushita, M., Fujii, H., Miyaura, C., Aizawa, S., and Kogo, H. (2007). Prevention of aortic calcification by etidronate in the renal failure rat model. Eur. J. Pharmacol. 558, 159–166.
Tang, F., Wu, X., Wang, T., Wang, P., Li, R., Zhang, H., Gao, J., Chen, S., Bao, L., Huang, H., and Liu, P. (2007). Tanshinone II A attenuates atherosclerotic calcification in rat model by inhibition of oxidative stress. Vascul. Pharmacol. 46, 427–438.
Tanikawa, T., Okada, Y., Tanikawa, R., and Tanaka, Y. (2009). Advanced glycation end products induce calcification of vascular smooth muscle cells through RAGE/p38 MAPK. J. Vasc. Res. 46, 572–580.
Tanimura, A., McGregor, D. H., and Anderson, H. C. (1983). Matrix vesicles in atherosclerotic calcification. Proc. Soc. Exp. Biol. Med. 172, 173–177.
Tentori, F., Blayney, M. J., Albert, J. M., Gillespie, B. W., Kerr, P. G., Bommer, J., Young, E. W., Akizawa, T., Akiba, T., Pisoni, R. L., Robinson, B. M., and Port, F. K. (2008). Mortality risk for dialysis patients with different levels of serum calcium, phosphorus, and PTH: the Dialysis Outcomes and Practice Patterns Study (DOPPS). Am. J. Kidney Dis. 52, 519–530.
Thyberg, J. (1996). Differentiated properties and proliferation of arterial smooth muscle cells in culture. Int. Rev. Cytol. 169, 183–265.
Ting, T. C., Miyazaki-Anzai, S., Masuda, M., Levi, M., Demer, L. L., Tintut, Y., and Miyazaki, M. (2011). Increased lipogenesis and stearate accelerate vascular calcification in calcifying vascular cells. J. Biol. Chem. 286, 23938–23949.
Tintut, Y., Alfonso, Z., Saini, T., Radcliff, K., Watson, K., Bostrom, K., and Demer, L. L. (2003). Multilineage potential of cells from the artery wall. Circulation 108, 2505–2510.
Tintut, Y., Parhami, F., Boström, K., Jackson, S. M., and Demer, L. L. (1998). cAMP stimulates osteoblast-like differentiation of calcifying vascular cells. Potential signaling pathway for vascular calcification. J. Biol. Chem. 27, 7547–7553.
Tintut, Y., Patel, J., Parhami, F., and Demer, L. L. (2000). Tumor necrosis factor-alpha promotes in vitro calcification of vascular cells via the cAMP pathway. Circulation 102, 2636–2642.
Tonelli, M., Sacks, F., Pfeffer, M., Gao, Z., and Curhan, G. (2005). Relation between serum phosphate level and cardiovascular event rate in people with coronary disease. Circulation 112, 2627–2633.
Toussaint, N. D., Lau, K. K., Strauss, B. J., Polkinghorne, K. R., and Kerr, P. G. (2008). Associations between vascular calcification, arterial stiffness and bone mineral density in chronic kidney disease. Nephrol. Dial. Transplant. 23, 586–593.
Towler, D. A., Bidder, M., Latifi, T., Coleman, T., and Semenkovich, C. F. (1998). Diet induced diabetes activates an osteogenic gene regulatory program in the aortas of low density lipoprotein receptor-deficient mice. J. Biol. Chem. 273, 30427–30434.
Vattikuti, R., and Towler, D. A. (2004). Osteogenic regulation of vascular calcification: an early perspective. Am. J. Physiol. Endocrinol. Metab. 286, E686–E696.
Virchow, R. (1989). Cellular pathology. As based upon physiological and pathological histology. Lecture XVI–Atheromatous affection of arteries. Nutr. Rev. 47, 23–25.
Watanabe, H., Ohtsuka, S., Kakihana, M., and Sugishita, Y. (1992). Decreased aortic compliance aggravates subendocardial ischaemia in dogs with stenosed coronary artery. Cardiovasc. Res. 26, 1212–1218.
Watson, K. E., Bostrom, K., Ravindranath, R., Lam, T., Norton, B., and Demer, L. L. (1994). TGF-beta 1 and 25-hydroxycholesterol stimulate osteoblast-like vascular cells to calcify. J. Clin. Invest. 93, 2106–2113.
Wayhs, R., Zelinger, A., and Raggi, P. (2002). High coronary artery calcium scores pose an extremely elevated risk for hard events. J. Am. Coll. Cardiol. 39, 225–230.
West, S. L., Swan, V. J., and Jamal, S. A. (2010). Effects of calcium on cardiovascular events in patients with kidney disease and in a healthy population. Clin. J. Am. Soc. Nephrol. 5, S41–S47.
Wong, K. K., Thavornpattanapong, P., Cheung, S. C., Sun, Z., and Tu, J. (2012). Effect of calcification on the mechanical stability of plaque based on a three-dimensional carotid bifurcation model. BMC Cardiovasc. Disord. 12, 7. doi: 10.1186/1471-2261-12-7
Worth, N. F., Rolfe, B. E., Song, J., and Campbell, G. R. (2001). Vascular smooth muscle cell phenotypic modulation in culture is associated with reorganisation of contractile and cytoskeletal proteins. Cell Motil. Cytoskeleton 49, 130–145.
Yamada, K., Fujimoto, S., Nishiura, R., Komatsu, H., Tatsumoto, M., Sato, Y., Hara, S., Hisanaga, S., Ochiai, H., Nakao, H., and Eto, T. (2007). Risk factors of the progression of abdominal aortic calcification in patients on chronic haemodialysis. Nephrol. Dial. Transplant. 22, 2032–2037.
Young, E. W., Albert, J. M., Satayathum, S., Goodkin, D. A., Pisoni, R. L., Akiba, T., Akizawa, T., Kurokawa, K., Bommer, J., Piera, L., and Port, F. K. (2005). Predictors and consequences of altered mineral metabolism: the Dialysis Outcomes and Practice Patterns Study. Kidney Int. 67, 1179–1187.
Zavaczki, E., Jeney, V., Agarwal, A., Zarjou, A., Oros, M., Katko, M., Varga, Z., Balla, G., and Balla, J. (2011). Hydrogen sulfide inhibits the calcification and osteoblastic differentiation of vascular smooth muscle cells. Kidney Int. 80, 731–739. doi:10.1371/journal.pone.0019595
Zhou, G., Kandala, J. C., Tyagi, S. C., Katwa, L. C., and Weber, K. T. (1996). Effects of angiotensin II and aldosterone on collagen gene expression and protein turnover in cardiac fibroblasts. Mol. Cell. Biochem. 154, 171–178.
Keywords: vascular calcification, vascular smooth muscle cells, therapeutic strategies
Citation: Zhu D, Mackenzie NCW, Farquharson C and MacRae VE (2012) Mechanisms and clinical consequences of vascular calcification. Front. Endocrin. 3:95. doi: 10.3389/fendo.2012.00095
Received: 28 April 2012; Accepted: 17 July 2012;
Published online: 06 August 2012.
Edited by:
William Duncan Fraser, University of Liverpool/Royal Liverpool University Hospital, UKReviewed by:
Björn E. Rosengren, Lund University, SwedenBasem M. Abdallah, University of Southern Denmark, Denmark
Copyright: © 2012 Zhu, Mackenzie, Farquharson and MacRae. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Vicky E. MacRae, The Roslin Institute and Royal (Dick) School of Veterinary Studies, University of Edinburgh, Easter Bush, Roslin, Midlothian EH25 9RG, UK. e-mail:dmlja3kubWFjcmFlQHJvc2xpbi5lZC5hYy51aw==