

- 1Baker IDI Heart and Diabetes Research Institute, Melbourne, VIC, Australia
- 2Department of Medicine, Monash University, Melbourne, VIC, Australia
A central mechanism driving vascular disease in diabetes is immune cell-mediated inflammation. In diabetes, enhanced oxidation and glycation of macromolecules, such as lipoproteins, insults the endothelium, and activates both innate and adaptive arms of the immune system by generating new antigens for presentation to adaptive immune cells. Chronic inflammation of the endothelium in diabetes leads to continuous infiltration and accumulation of leukocytes at sites of endothelial cell injury. We will describe the central role of the macrophage as a source of signaling molecules and damaging by-products which activate infiltrating lymphocytes in the tissue and contribute to the pro-oxidant and pro-inflammatory microenvironment. An important aspect to be considered is the diabetes-associated defects in the immune system, such as fewer or dysfunctional athero-protective leukocyte subsets in the diabetic lesion compared to non-diabetic lesions. This review will discuss the key pro-inflammatory signaling pathways responsible for leukocyte recruitment and activation in the injured vessel, with particular focus on pro- and anti-inflammatory pathways aberrantly activated or repressed in diabetes. We aim to describe the interaction between advanced glycation end products and their principle receptor RAGE, angiotensin II, and the Ang II type 1 receptor, in addition to reactive oxygen species (ROS) production by NADPH-oxidase enzymes that are relevant to vascular and immune cell function in the context of diabetic vasculopathy. Furthermore, we will touch on recent advances in epigenetic medicine that have revealed high glucose-mediated changes in the transcription of genes with known pro-inflammatory downstream targets. Finally, novel anti-atherosclerosis strategies that target the vascular immune interface will be explored; such as vaccination against modified low-density lipoprotein and pharmacological inhibition of ROS-producing enzymes.
Introduction
A unifying feature of diabetic complications is chronic inflammation of the vasculature. The main vascular diseases that burden diabetic patients include nephropathy, retinopathy, neuropathy, and atherosclerosis. Each of these conditions has a significant immune component. In experimental and human diabetic nephropathy, infiltrating macrophages, and T cells elaborate a host of pro-inflammatory, pro-fibrotic, and pro-angiogenic factors that contribute to disease development and progression in the kidney (Lim and Tesch, 2012). Leukocyte adherence is causally associated with endothelial cell injury and cell death in the diabetic retina (Joussen et al., 2001) while infiltration of post-capillary venules with polymorphonuclear leukocytes is an early feature of proximal diabetic neuropathy (Kelkar et al., 2000). Similar to the microvasculature, immune-mediated inflammation of the macrovasculature plays a central role in the pathogenesis of diabetes-accelerated atherosclerosis (Libby et al., 2002). In this review, we examine the role of the immune response in the pathogenesis of atherosclerosis as a prototype for diabetes-associated vasculopathies.
Triggers of Inflammation Elevated in Diabetes Compared to the Non-Diabetic Disease State
Atherosclerotic lesions represent an excessive inflammatory, fibro-proliferative response against different noxious stimuli (Ross, 1993, 1997). The diabetic milieu comprises of a host of potentially harmful, immunogenic products including modified forms of low-density lipoprotein (LDL), advanced glycation end products (AGEs), reactive oxygen species (ROS) as well as pro-inflammatory chemokines and cytokines. Elevated levels of LDL in patients with diabetes are subject to modification by both oxidation and glycation (Cohen et al., 2004). Furthermore, accelerated generation and vascular deposition of AGEs in addition to AGE interactions with RAGE in diabetes initiate oxidative reactions that promote the formation of oxidized LDL (oxLDL) (Basta et al., 2004). Oxidation of LDL within the sub-endothelial space (intima) enhances the pro-inflammatory properties of the endothelium (Mazière and Mazière, 2009) and activates both innate and adaptive arms of the immune system (Binder et al., 2002; Hansson et al., 2002).
Relationship Between the Endothelium and Leukocytes in Early Atherogenesis
Chronic injury to the endothelium results in endothelial dysfunction, which can be defined as increased permeability, reduced nitric oxide (NO) dependent vasodilatation as well as enhanced pro-thrombotic and pro-inflammatory properties (Hink et al., 2001; Davignon and Ganz, 2004; Hartge et al., 2007). Endothelial dysfunction is a well established precursor of atherosclerosis, particularly in the setting of diabetes (Schalkwijk and Stehouwer, 2005). The diabetes-associated factors that impair normal endothelial function include increased synthesis of vasoconstrictors such as angiotensin II (Ang II) and endothelin-1, uncoupling of endothelial nitric oxide synthase (eNOS; leading to reduced bioavailability of NO), and increased expression and activity of ROS-producing enzymes (such as NADPH oxidases, Nox) which trigger the expression of adhesion and chemotactic molecules that promote the recruitment of inflammatory cells to the arterial wall (Figure 1) (Hink et al., 2001; Guzik et al., 2002; Lüscher et al., 2003; Hartge et al., 2007). Aortic lesions of diabetic ApoE knockout (KO) mice show increased gene expression of pro-inflammatory molecules MCP-1, VCAM-1, and NF-κB subunit p65 associated with increased pro-atherogenic cellularity [macrophages, T cells, and smooth muscle cells (SMCs)] compared to non-diabetic controls (Soro-Paavonen et al., 2008). In line with these in vivo findings, human endothelial cells exposed to high glucose conditions show increased leukocyte binding associated with increased expression of E-selectin, ICAM-1, VCAM-1, and MCP-1 through activation of NF-κB (Kim et al., 1994; Morigi et al., 1998; Piga et al., 2007). In addition, recent studies have revealed a striking association between glucose-induced endothelial expression of adhesion molecules VCAM-1 and P-selectin as well as the chemokines MCP-1 and fractalkine with Nox-derived ROS production (Manduteanu et al., 2010; Gray et al., 2013). These data indicate that diabetes-associated hyperglycemia and oxidative stress promote leukocyte-endothelial cell interactions required for the recruitment of leukocytes to the inflamed vessel.
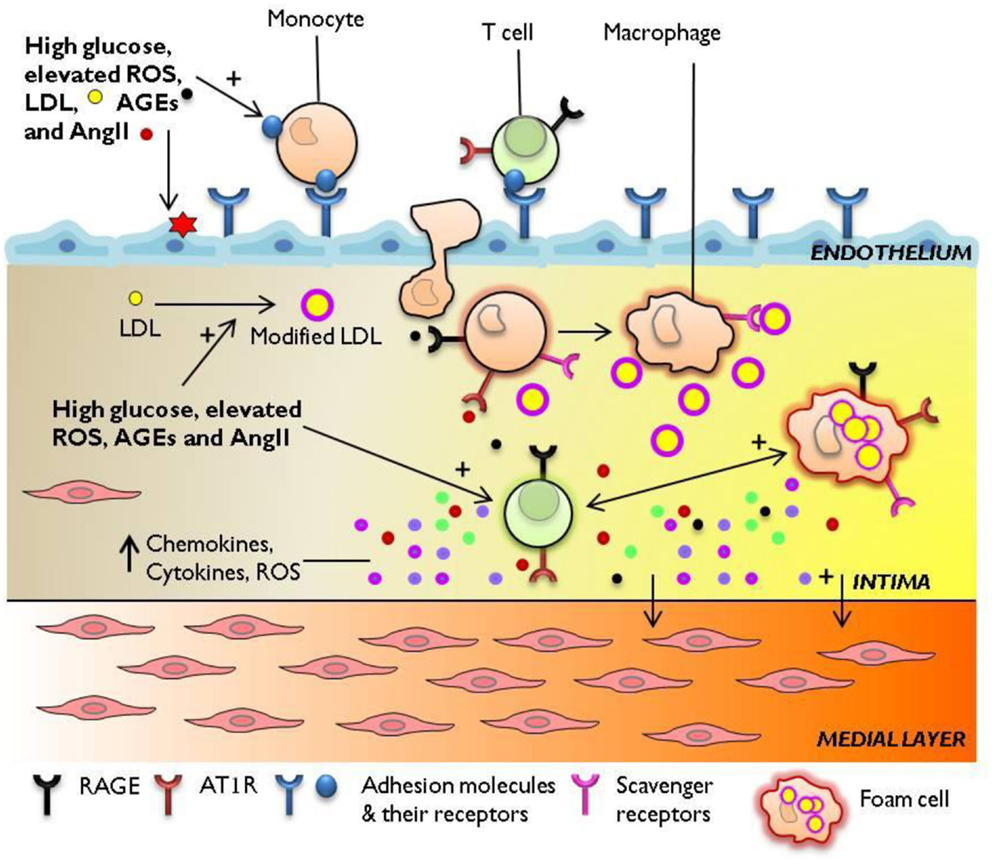
Figure 1. Immune mechanisms engaged in diabetes-accelerated atherosclerosis. Diabetes-associated hyperglycemia, hyperlipidemia, and oxidative stress render the endothelium dysfunctional, leading to the retention and oxidation of LDL molecules in the intimal space. The increased expression of adhesion molecules E-selectin, ICAM-1, VCAM-1 at the endothelial membrane, and upregulation of chemotactic molecules such as MCP-1 facilitate the continuous infiltration of immune cells to the inflamed aorta. Resident and monocyte-derived macrophages engulf LDL to form foam cells which release a host of pro-inflammatory cytokines, protease, and ROS. Activated T cells recruited from the circulation to the lesion also secrete cytokines which amplify pro-inflammatory cellular immune responses in the diabetic plaque. The diabetes-mediated increase in vascular inflammation drives the development and progression of atherosclerosis. AGEs, advanced glycation end-products; AT1R, angiotensin II type 1 receptor; ICAM-1, intercellular adhesion molecule-1; LDL, low-density lipoprotein; MCP-1, monocyte chemotactic protein-1; RAGE, receptor for advanced glycation end products; ROS, reactive oxygen species; VCAM-1, vascular cell adhesion molecule-1.
Role of Smooth Muscle Cells in Diabetes-Mediated Vascular Inflammation
Vascular SMCs play a central role in the initiation and progression of atherosclerosis (Doran et al., 2008). The expression of cellular adhesion molecules and pro-inflammatory cytokines by lesional SMCs promote the accumulation and activation of leukocytes in the atherosclerotic lesion (Braun et al., 1999; Raines and Ferri, 2005). Of note, the expression of VCAM-1, ICAM-1, and fractalkine is confined to SMCs within atherosclerotic vessels (or lesion-prone areas) but not in healthy vessels (O’Brien et al., 1993; Endres et al., 1997; Barlic et al., 2007); providing further evidence of the pro-atherogenic role of these molecules. In response to diabetes-associated atherogenic stimuli, such as Ang II (Ruiz-Ortega et al., 2003) and ROS (Su et al., 2001), SMCs elaborate an array of extracellular matrix proteins which facilitate the retention of inflammatory cells and stabilization of the plaque including the formation of a fibrous cap. Diabetes stimulates and sustains the inflammatory “synthetic” phenotype of vascular SMCs. Hyperglycemia enhances the migration and proliferation of resident SMCs within the intimal lesion which contributes to the accelerated formation of advanced atherosclerosis in diabetes (Suzuki et al., 2001). Furthermore, exposure of cultured SMCs to high glucose or AGEs significantly increases the production of ROS by Nox enzymes (Inoguchi et al., 2000; Wautier et al., 2001; Gray et al., 2013). More specifically, recent evidence has identified a central role for the Nox1 isoform in glucose (Gray et al., 2013) and Ang II-induced vascular superoxide production (Lassègue et al., 2001). Therefore, vascular SMCs represent key players in diabetes-mediated pro-inflammatory and pro-oxidant responses that drive atherosclerosis.
Macrophages are a Source of Pro-Oxidant, Pro-Inflammatory Molecules Which Fuel Pro-Atherogenic Processes in the Vessel Wall
The macrophage plays a critical role in the initiation and progression of the atherosclerotic lesion. Circulating monocytes are recruited to inflamed regions of the endothelium where they engulf trapped LDL to form foam cells, histologically visible as the early fatty streak (Ross, 1997). Uptake of glycated LDL by human monocyte-derived macrophages occurs to a greater extent than for native LDL (Klein et al., 1995). Interestingly, glycoxidized LDL increases scavenger receptor expression to a greater extent than glycated LDL or oxLDL alone (Lam et al., 2004). Therefore, hyperglycemia in combination with elevated ROS in diabetes increases macrophage avidity for LDL, and thus foam cell formation, which ultimately activates other macrophages, infiltrating adaptive “effector” cells as well as resident vascular cells leading to accelerated atherosclerosis.
The lipid-laden macrophage acts as a frustrated phagocyte, leaking ROS (refer to Nox section), and metalloproteases (MMPs) into the extracellular space which catalyze the degradation of the extracellular matrix proteins and trigger apoptosis of SMCs that support the plaque’s fibrotic cap, ultimately enhancing the risk of plaque rupture (Shah et al., 1995). Macrophages function in response to environmental cues (Waldo et al., 2008). Monocyte-derived macrophages cultured in high glucose (25 mM) conditions display increased expression and activity of MMP-9 without affecting metalloproteinase inhibitor (TIMP-1) expression (Death et al., 2003). In line with these findings, Devaraj et al. (2006) showed that monocyte superoxide anion release, pro-inflammatory cytokines interleukin (IL) 6, and IL1β were significantly elevated in type 1 diabetic subjects compared with in control subjects. These striking effects of hyperglycemia on macrophage activity may provide some insight into the significantly increased incidence of plaque rupture, thrombosis, and ultimately myocardial infarction in diabetic patients compared to their non-diabetic counterparts (Silva et al., 1995).
Interactions between Leukocytes Within the Lesion Engage Inflammatory Pathways that Drive Atherosclerosis
Monocyte-derived macrophages act as sentinels of pro-inflammatory signals, able to be activated by diverse stimuli and release a range of mediators that contribute to pro- as well as anti-atherogenic processes (Moore and Tabas, 2011). A plethora of cytokines are detected in atherosclerotic vessels, and the reader is directed to excellent reviews by Tedgui and Mallat (2006) and Ait-Oufella et al. (2011). Activated macrophages and foam cells release a number of pro-inflammatory cytokines and chemokines which facilitate the continual recruitment of monocytes and T lymphocytes to the atherosclerotic lesion. Of note, the predominance of IFNγ-producing T lymphocytes in the lesion is likely to contribute significantly to local macrophage activation (de Boer et al., 1999). Further gene expression analysis of advanced human plaques has revealed a cytokine profile indicative of a pro-inflammatory, Th1-type cellular immune response (Frostegård et al., 1999). The increased expression of CD40 and MHC-II molecules on plaque macrophages (and other plaque-associated vascular cells) also suggests an enhanced capacity of these cells to function as antigen-presenting cells to T lymphocytes (Hansson et al., 1991; Li et al., 1993; Mach et al., 1997). The potent immunostimulatory and pro-atherogenic effects of IFNγ are well established (Gupta et al., 1997), and so interactions between macrophages and T lymphocytes that continuously upregulate IFNγ production are expected to contribute to the progression of the lesion.
The Cytokine Profile of Activated Vascular Cells/Leukocytes is Altered in the Diabetic Setting
Macrophages represent a heterogeneous population of distinct subsets capable of differentially affecting Th1- or Th2-type responses (Mills et al., 2000). Lesion macrophages adopt predominantly a pro-inflammatory and pro-atherogenic role indicative of the M1-subtype (Khallou-Laschet et al., 2010; Wilson, 2010; Shalhoub et al., 2011). Furthermore, hyperglycemia interferes with the ability of IL4 to polarize macrophages to the alternatively activated M2 state, characterized by wound repair and anti-inflammatory functions (Auffray et al., 2009; Parathath et al., 2011). A comprehensive review of macrophage differentiation and activation pathways in atherosclerosis can be found by Shalhoub et al. (2011). In a similar manner, hyperglycemia is associated with increased activation of pro-inflammatory human T lymphocytes and decreased suppressor function of T regulatory cell subsets (Marfella et al., 2003; Lindley et al., 2005; Stentz and Kitabchi, 2005). Therefore, not only does the diabetic micro-environment amplify the magnitude and array of new, potentially immunogenic stimuli but it also skews the leukocyte repertoire in favor of adopting a pro-inflammatory functional phenotype. Moreover, high glucose may interfere with the immunomodulatory capacity of T cell and macrophage subsets leading to an unabated prolongation of the inflammatory process.
Patients with diabetes have an increased risk of infection attributable to defects in both innate and adaptive immunity (Geerlings and Hoepelman, 1999). Consistent with these findings in humans, streptozotocin (STZ)-induced diabetes in rodents predisposes to delayed innate (Chin et al., 2012) and adaptive (Vallerskog et al., 2010) responses irrespective of the challenge, leading to increased susceptibility to infection. High glucose conditions have been shown to decrease T and B lymphocyte proliferation, diminish cell viability, and increase apoptosis via mechanisms likely to involve increased oxidative stress (Rubinstein et al., 2008). This seems counter-intuitive in a disease characterized by secondary complications driven by immune cell-mediated inflammation, however the high glucose and high ROS diabetic milieu provides the ideal setting for chronic activation of innate and adaptive cells to non-microbial, modified macromolecules (described in section above). Therefore, it is important to discuss diabetes-induced alterations in the immune system in the context of any insults (infection) or complications (e.g., atherosclerosis) that accentuate as well as catalyze further derangement in both arms of the immune response.
Signaling Pathways Important to Hyperglycemia-Induced Vascular Pathophysiology
AGE/RAGE
Advanced glycation end products and their cell surface receptor, RAGE, have been implicated in the amplification and progression of immune-inflammatory responses that underscore diabetic complications (Schmidt et al., 2001; Basta et al., 2004; Jandeleit-Dahm et al., 2008; Bierhaus and Nawroth, 2009). Atherosclerotic lesions in diabetic ApoE KO mice display increased accumulation of AGEs and enhanced expression of RAGE (Kislinger et al., 2001; Wendt et al., 2002; Soro-Paavonen et al., 2008). Ligation of RAGE by AGE stimulates endothelial pro-inflammatory gene expression (VCAM-1, E-selectin) and ROS production in a NADPH-oxidase dependent manner (Schmidt et al., 1995; Wautier et al., 2001; Higai et al., 2006). In macrophages, AGE–RAGE interaction prompts cell chemotaxis and cytokine release via a mechanism involving the activation of NADPH oxidase (Kirstein et al., 1990; Wautier et al., 2001). RAGE also appears to play a role in T lymphocyte activation and differentiation, mediating Th1-type responses such as the synthesis of IFNγ (Chen et al., 2008). Recent work by Akirav et al. (2012) found that RAGE is expressed intracellularly in human T cells following TCR activation but constitutively on T cells from patients with diabetes. The current evidence supports a critical role of AGE/RAGE signaling in the chronic activation of the immune-inflammatory processes that accelerate atherosclerosis in diabetes.
Ang II/AT1R
The renin-angiotensin system (RAS) plays an important role in the regulation of vascular contractility and also represents a primary target for diabetes-induced vascular dysfunction and inflammation. Most of the known pathophysiological effects of Ang II are mediated via the activation of the Ang II type 1 (AT1) receptor. The production of Ang II and the expression of its AT1R are upregulated in the aorta of diabetic mice (Candido et al., 2002, 2004). Signaling via the AT1R has been shown to upregulate the RAGE pathway in diabetic atherosclerosis (Ihara et al., 2007). Furthermore, Ang II induces activation with increased adhesion molecule expression in endothelial cells, monocytes, and T lymphocytes (Hahn, 1994; Tummala et al., 1999; Hoch et al., 2009). Most of the signaling events secondary to Ang II-AT1R binding are redox sensitive, such as NFκB activation, and rely on the production of ROS by Nox enzymes (Pueyo et al., 2000; Alvarez and Sanz, 2001; Liu et al., 2003). Treatment of diabetic mice with an AT1R blocker such as Candesartan attenuated ROS production and Nox activity leading to improvements in endothelial function (Oak and Cai, 2007). Recent work by Valente et al. (2012) revealed a direct physical association between the AT1R and the Nox1-NADPH-oxidase isoform in vascular SMCs, however the potential functional and regulatory implications of this relationship remain to be explored in the context of hyperglycemia. A common feature of AGE and Ang II signaling is the convergence on Nox-derived ROS as secondary mediator molecules required to elicit the effects of RAGE and AT1 receptor binding, respectively.
NOX/ROS
Hyperglycemia induces activation of the Nox family NADPH-oxidase enzymes and the consequent ROS production contributes to the pathophysiological complications of diabetes (Gao and Mann, 2009). Nox enzymes are the major source of ROS in the vessel wall, differentially expressed in vascular and infiltrating inflammatory cells (Griendling and Ushio-Fukai, 1997; Lassègue and Clempus, 2003). In particular, the isoforms Nox1, Nox2, Nox4, and Nox5 play important roles in a wide range of physiological and pathological processes relevant to cardiovascular disease (Sorescu et al., 2002; Bedard and Krause, 2007; Lassègue and Griendling, 2010). Increased ROS production in the diabetic aorta positively correlates with upregulated expression and activity of Nox1, Nox2, and Nox4 (Guzik et al., 2002; Wendt et al., 2005; Hwang et al., 2007). Nox1 KO mice are protected from endothelial dysfunction in diabetes (Youn et al., 2012) while studies in Nox4 KO mice have reported a vasculo-protective role for endogenous Nox4 (Schröder et al., 2012). Furthermore, recent work by Gray et al. (2013) in Nox1 and Nox4/ApoE DKO mice identified a key role for Nox1 but not Nox4-derived ROS in diabetes-accelerated atherosclerosis which is consistent with previous reports suggesting different (patho)physiological functions for Nox isoforms in the vasculature.
Reactive oxygen species are both a product, and modulator, of leukocyte recruitment, activation, and function. Ligation of VCAM by leukocytes activates endothelial NADPH oxidase which results in enhanced ROS production (Deem and Cook-Mills, 2004). In macrophages, Nox2 (gp91phox) is the major source of ROS contributing to the respiratory burst, while Nox4 is inducible by oxLDL (Lee et al., 2010; Tavakoli and Asmis, 2012). T lymphocytes also express a phagocyte-type Nox that is activated after TCR stimulation (Jackson et al., 2004). Further activation of vascular Nox by NFκB-dependent pro-inflammatory cytokines, including TNFα, reaffirms the relationship between oxidative stress and immune-mediated inflammation in diabetes (Park et al., 2006; Gauss et al., 2007; Miller et al., 2007). The potential impact of diabetes-mediated changes in Nox on ROS-sensitive leukocyte functions represents a relatively unexplored area of investigation.
Athero-Protective Mechanisms: Focus on Antioxidants
We have discussed some of the key pro-inflammatory, pro-atherosclerotic effects of Ang II/AT1R and AGE/RAGE signaling pathways that are aberrantly activated in response to hyperglycemia. However, it is important to note that these same pathways can elicit anti-inflammatory effects, and the reader is encouraged to consult Goh and Cooper (2008), Daugherty et al. (2010), and Thomas et al. (2010) for detail on alternative AGE receptors, AT2R, and ACE2. While pro-inflammatory signaling cascades tend to predominate in diabetes, the inactivation of some important athero-protective pathways by the diabetic milieu further exposes the diabetic vasculature to pro-oxidant insults that accelerate atherogenesis. Major anti-oxidant defense systems such as superoxide dismutases (SODs), and glutathione peroxide 1 (Gpx1) are critical in the maintenance of redox balance in the intracellular and extracellular spaces. Diabetic ApoE/GPx1 DKO mice show significantly more aortic atherosclerosis associated with enhanced macrophage recruitment, RAGE, and VCAM-1 expression compared to the diabetic ApoE KO mice (Lewis et al., 2007). Therefore, a decrease in anti-oxidant capacity in diabetes in combination with elevated ROS production from various sources, contributes to the pro-oxidant diabetic milieu (Feillet-Coudray et al., 1999; Vessby et al., 2002).
An Emerging Role for Epigenetic Modifications in Diabetic Complications
The effect of hyperglycemia on the transcription and translation of genes coding proteins involved in the inflammatory response persist beyond the time of exposure. A growing number of studies utilizing the bio-informatic power of epigenetic medicine have already identified chromatic modifications characterized by histone methylation and acetylation sites, and their associated enzymes, involved in glucose-mediated changes in gene expression (Brasacchio et al., 2009; Pirola et al., 2011; Keating and El-Osta, 2012; Miao et al., 2012). El-Osta et al., identified a role for Set7, a H3K4-specific methyltransferase, in transient hyperglycemia-mediated chromatin changes at the promoter of the NF-κB subunit p65 resulting in increased gene expression of p65, MCP-1, and VCAM-1 in vascular endothelial cells that persist during subsequent normoglycemia; a phenomenon termed “glycemic memory” (El-Osta et al., 2008; Okabe et al., 2012). Similarly, ex vivo culture of vascular SMCs from type 2 diabetic db/db mice exhibit a sustained atherogenic and pro-inflammatory phenotype with concomitant depletion of the H3K9-specific methyltransferase, Suv39h1, despite restoration of euglycaemia (Villeneuve et al., 2010). Further investigations suggest that regulation of pro-inflammatory gene expression by Suv39h1 is dependent on glucose-mediated elevations in the microRNA (miR)-125b (Villeneuve et al., 2008).
Recent advances in cancer research have demonstrated cross-talk between miR machinery and DNA methylation (Ting et al., 2008; Yan et al., 2011). While there is growing evidence supporting glucose-induced alterations in chromatin structure and miR expression in the pathogenesis of diabetic complications (Reddy and Natarajan, 2011; Villeneuve et al., 2011), little is known about the interplay of these components in diabetes.
Epigenetic mechanisms play a central role in the pathobiology of endothelial (Piconi et al., 2004; El-Osta et al., 2008) and SMCs (Villeneuve et al., 2008, 2010) in high glucose conditions, yet remain a relatively unexplored area in the context of immune cell-mediated vascular inflammation. Monocytes and lymphocytes, immune cells with well-characterized roles in atherosclerosis (Hansson and Hermansson, 2011), display distinct profiles of histone acetylation and methylation in diabetic patients with corresponding changes in inflammatory gene expression (Miao et al., 2007, 2008). Additionally, Miao et al. (2012) identified marked differences in human leukocyte antigen (HLA)-expression in monocytes from diabetic patients compared to controls which related to differences in the histone acetylation status of the HLA promoter region. In light of these findings, it would be very interesting to explore the epigenetic status of various immune cell populations, especially adaptive immune cells which naturally develop “memory” subsets [present also in complex atherosclerotic lesions (Stemme et al., 1992)] that respond rapidly to repeated challenges of the same antigen. Poorly controlled diabetes whereby blood glucose regularly spikes to pathological levels may be sufficient to reactivate memory subpopulations, leading to rapid and robust inflammatory responses that accelerate atherosclerosis and increase risk of plaque rupture events in diabetes. Future studies examining chromatin modifications in immune cells intimately involved in the pathogenesis of diabetic complications will improve our understanding of the mechanisms underlying glycemic memory and the sustained pro-inflammatory state of the diabetic vasculature.
Therapeutic Strategies with Clinical Promise
Therapeutic strategies against diabetes-associated atherosclerosis can be targeted at various stages of disease pathogenesis. Interventions that block signaling via RAGE (Soro-Paavonen et al., 2008) or the AT1R (Candido et al., 2004) have proved effective in reducing plaque formation in mouse models with diabetes. There is a growing body of evidence to suggest that a large number of the pro-inflammatory signaling cascades triggered by hyperglycemia converge on Nox-derived ROS making it an excellent target for novel therapies (Sedeek et al., 2012). To date, studies examining pharmacological inhibition of Nox-derived ROS in experimental models of chronic inflammatory diseases including liver fibrosis (Jiang et al., 2012), diabetic nephropathy (Sedeek et al., 2010), and atherosclerosis (Gray et al., 2013) have yielded positive results, and much anticipation surrounds the clinical evaluation of specific Nox inhibitors (Kim et al., 2011). The effects of immune modulation on atherosclerosis and the emerging inflammatory cell/cytokine-directed therapies have been addressed in a recent review by Little et al. (2011). Increasing evidence demonstrates a strong association between circulating oxLDL-immune complexes and cardiovascular risk in diabetic patients (Lopes-Virella et al., 1999, 2011a,b; Orchard et al., 1999). Immunization with oxLDL or other candidate plaque antigens have shown promise in animal models (Palinski et al., 1995; Ameli et al., 1996; George et al., 1998) however evidence demonstrating the pathogenic properties of antibodies to modified LDL (summarized by Lopes-Virella and Virella, 2010) may hamper the clinical utility of this strategy. Cytokine-based therapies that skew macrophage or T cell to anti-inflammatory and regulatory phenotypes have been effective in mouse models of atherosclerosis (Namiki et al., 2004; Sasaki et al., 2009; Cardilo-Reis et al., 2012). The establishment of highly atherosclerosis-specific antigens or cytokines in combination with cell- or tissue-focused delivery systems will be pre-requisites for more fine-tuned immunomodulatory therapies. Overall, significant challenges continue to plague attempts to translate laboratory findings to the clinic and greater emphasis on human studies is required to fully realize the therapeutic potential of targeting immunological mechanisms in disease (Davis, 2008; Libby et al., 2011). A systems approach to immunology, as outlined in a recent review by Brodin et al. (2013), may impart more successful clinical results for novel therapies.
Conclusion
An extensive body of experimental and clinical evidence has improved our understanding of the immune-inflammatory processes involved in vascular disease. However, there are still considerable challenges in alleviating the burden of vascular complications in diabetic patients. In this review we have discussed that, in addition to high glucose, pathological increases in AGEs, Ang II, and ROS induce a chronic state of vascular inflammation. Specifically, we have described how diabetes-mediated aberrations in vascular-leukocyte interactions result in increased accumulation of pro-inflammatory leukocytes to the atherosclerotic vessel wall. Future studies that explore the pro-inflammatory effects of RAGE, AT1R, or Nox-signaling pathways in the diabetic vasculature are likely to offer novel ways of targeting the immune responses inappropriately activated or inactivated in diabetes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ait-Oufella, H., Taleb, S., Mallat, Z., and Tedgui, A. (2011). Recent advances on the role of cytokines in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 31, 969–979. doi:10.1161/ATVBAHA.110.207415
Akirav, E. M., Preston-Hurlburt, P., Garyu, J., Henegariu, O., Clynes, R., Schmidt, A. M., et al. (2012). RAGE expression in human T cells: a link between environmental factors and adaptive immune responses. PLoS ONE 7:e34698. doi:10.1371/journal.pone.0034698
Alvarez, A., and Sanz, M. J. (2001). Reactive oxygen species mediate angiotensin II-induced leukocyte-endothelial cell interactions in vivo. J. Leukoc. Biol. 70, 199–206.
Ameli, S., Hultgårdh-Nilsson, A., Regnström, J., Calara, F., Yano, J., Cercek, B., et al. (1996). Effect of immunization with homologous LDL and oxidized LDL on early atherosclerosis in hypercholesterolemic rabbits. Arterioscler. Thromb. Vasc. Biol. 16, 1074–1079. doi:10.1161/01.ATV.16.8.1074
Auffray, C., Sieweke, M. H., and Geissmann, F. (2009). Blood monocytes: development, heterogeneity, and relationship with dendritic cells. 27, 669–692.
Barlic, J., Zhang, Y., and Murphy, P. M. (2007). Atherogenic lipids induce adhesion of human coronary artery smooth muscle cells to macrophages by up-regulating chemokine CX3CL1 on smooth muscle cells in a TNFα-NFkB-dependent manner. J. Biol. Chem. 282, 19167–19176. doi:10.1074/jbc.M701642200
Basta, G., Schmidt, A. M., and De Caterina, R. (2004). Advanced glycation end products and vascular inflammation: implications for accelerated atherosclerosis in diabetes. Cardiovasc. Res. 63, 582–592. doi:10.1016/j.cardiores.2004.05.001
Bedard, K., and Krause, K. H. (2007). The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 87, 245–313. doi:10.1152/physrev.00044.2005
Bierhaus, A., and Nawroth, P. P. (2009). Multiple levels of regulation determine the role of the receptor for AGE (RAGE) as common soil in inflammation, immune responses and diabetes mellitus and its complications. Diabetologia 52, 2251–2263. doi:10.1007/s00125-009-1458-9
Binder, C. J., Chang, M. K., Shaw, P. X., Miller, Y. I., Hartvigsen, K., Dewan, A., et al. (2002). Innate and acquired immunity in atherogenesis. Nat. Med. 8, 1218–1226. doi:10.1038/nm1102-1218
Brasacchio, D., Okabe, J., Tikellis, C., Balcerczyk, A., George, P., Baker, E. K., et al. (2009). Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes 58, 1229–1236. doi:10.2337/db08-1666
Braun, M., Pietsch, P., Schrör, K., Baumann, G., and Felix, S. B. (1999). Cellular adhesion molecules on vascular smooth muscle cells. Cardiovasc. Res. 41, 395–401. doi:10.1016/S0008-6363(98)00302-2
Brodin, P., Valentini, D., Uhlin, M., Mattsson, J., Zumla, A., and Maeurer, M. J. (2013). Systems level immune response analysis and personalized medicine. Expert Rev. Clin. Immunol. 9, 307–317. doi:10.1586/eci.13.9
Candido, R., Allen, T. J., Lassila, M., Cao, Z., Thallas, V., Cooper, M. E., et al. (2004). Irbesartan but not amlodipine suppresses diabetes-associated atherosclerosis. Circulation 109, 1536–1542. doi:10.1161/01.CIR.0000124061.78478.94
Candido, R., Jandeleit-Dahm, K. A., Cao, Z., Nesteroff, S. P., Burns, W. C., Twigg, S. M., et al. (2002). Prevention of accelerated atherosclerosis by angiotensin-converting enzyme inhibition in diabetic apolipoprotein E-deficient mice. Circulation 106, 246–253. doi:10.1161/01.CIR.0000021122.63813.32
Cardilo-Reis, L., Gruber, S., Schreier, S. M., Drechsler, M., Papac-Milicevic, N., Weber, C., et al. (2012). Interleukin-13 protects from atherosclerosis and modulates plaque composition by skewing the macrophage phenotype. EMBO Mol. Med. 4, 1072–1086. doi:10.1002/emmm.201201374
Chen, Y., Akirav, E. M., Chen, W., Henegariu, O., Moser, B., Desai, D., et al. (2008). RAGE ligation affects T cell activation and controls T cell differentiation. J. Immunol. 181, 4272–4278.
Chin, C. Y., Monack, D. M., and Nathan, S. (2012). Delayed activation of host innate immune pathways in streptozotocin-induced diabetic hosts leads to more severe disease during infection with Burkholderia pseudomallei. Immunology 135, 312–332. doi:10.1111/j.1365-2567.2011.03544.x
Cohen, M. P., Jin, Y., and Lautenslager, G. T. (2004). Increased plasma glycated low-density lipoprotein concentrations in diabetes: a marker of atherogenic risk. Diabetes Technol. Ther. 6, 348–356. doi:10.1089/152091504774198043
Daugherty, A., Poduri, A., Chen, X., Lu, H., and Cassis, L. A. (2010). Genetic variants of the renin angiotensin system: effects on atherosclerosis in experimental models and humans. Curr. Atheroscler. Rep. 12, 167–173. doi:10.1007/s11883-010-0109-4
Davignon, J., and Ganz, P. (2004). Role of endothelial dysfunction in atherosclerosis. Circulation 109(Suppl. 23), III27–III32. doi:10.1161/01.CIR.0000131515.03336.f8
Davis, M. M. (2008). A prescription for human immunology. Immunity 29, 835–838. doi:10.1016/j.immuni.2008.12.003
de Boer, O. J., van der Wal, A. C., Verhagen, C. E., and Becker, A. E. (1999). Cytokine secretion profiles of cloned T cells from human aortic atherosclerotic plaques. J. Pathol. 188, 174–179. doi:10.1002/(SICI)1096-9896(199906)188:2<174::AID-PATH333>3.0.CO;2-3
Death, A. K., Fisher, E. J., McGrath, K. C., and Yue, D. K. (2003). High glucose alters matrix metalloproteinase expression in two key vascular cells: potential impact on atherosclerosis in diabetes. Atherosclerosis 168, 263–269. doi:10.1016/S0021-9150(03)00140-0
Deem, T. L., and Cook-Mills, J. M. (2004). Vascular cell adhesion molecule 1 (VCAM-1) activation of endothelial cell matrix metalloproteinases: role of reactive oxygen species. Blood 104, 2385–2393. doi:10.1182/blood-2004-02-0665
Devaraj, S., Glaser, N., Griffen, S., Wang-Polagruto, J., Miguelino, E., and Jialal, I. (2006). Increased monocytic activity and biomarkers of inflammation in patients with type 1 diabetes. Diabetes 55, 774–779. doi:10.2337/diabetes.55.03.06.db05-1417
Doran, A. C., Meller, N., and McNamara, C. A. (2008). Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 28, 812–819. doi:10.1161/ATVBAHA.107.159327
El-Osta, A., Brasacchio, D., Yao, D., Pocai, A., Jones, P. L., Roeder, R. G., et al. (2008). Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J. Exp. Med. 205, 2409–2417. doi:10.1084/jem.20081188
Endres, M., Laufs, U., Merz, H., and Kaps, M. (1997). Focal expression of intercellular adhesion molecule-1 in the human carotid bifurcation. Stroke 28, 77–82. doi:10.1161/01.STR.28.1.77
Feillet-Coudray, C., Rock, E., Coudray, C., Grzelkowska, K., Azais-Braesco, V., Dardevet, D., et al. (1999). Lipid peroxidation and antioxidant status in experimental diabetes. Clin. Chim. Acta 284, 31–43. doi:10.1016/S0009-8981(99)00046-7
Frostegård, J., Ulfgren, A. K., Nyberg, P., Hedin, U., Swedenborg, J., Andersson, U., et al. (1999). Cytokine expression in advanced human atherosclerotic plaques: dominance of pro-inflammatory (Th1) and macrophage-stimulating cytokines. Atherosclerosis 145, 33–43. doi:10.1016/S0021-9150(99)00011-8
Gao, L., and Mann, G. E. (2009). Vascular NAD(P)H oxidase activation in diabetes: a double-edged sword in redox signalling. Cardiovasc. Res. 82, 9–20. doi:10.1093/cvr/cvp031
Gauss, K. A., Nelson-Overton, L. K., Siemsen, D. W., Gao, Y., DeLeo, F. R., and Quinn, M. T. (2007). Role of NF-kB in transcriptional regulation of the phagocyte NADPH oxidase by tumor necrosis factor-k. J. Leukoc. Biol. 82, 729–741. doi:10.1189/jlb.1206735
Geerlings, S. E., and Hoepelman, A. I. M. (1999). Immune dysfunction in patients with diabetes mellitus (DM). FEMS Immunol. Med. Microbiol. 26, 259–265. doi:10.1111/j.1574-695X.1999.tb01397.x
George, J., Afek, A., Gilburd, B., Levkovitz, H., Shaish, A., Goldberg, I., et al. (1998). Hyperimmunization of apo-E-deficient mice with homologous malondialdehyde low-density lipoprotein suppresses early atherogenesis. Atherosclerosis 138, 147–152. doi:10.1016/S0021-9150(98)00015-X
Goh, S. Y., and Cooper, M. E. (2008). The role of advanced glycation end products in progression and complications of diabetes. J. Clin. Endocrinol. Metab. 93, 1143–1152. doi:10.1210/jc.2007-1817
Gray, S. P., Di Marco, E., Okabe, J., Szyndralewiez, C., Heitz, F., Montezano, A. C., et al. (2013). NADPH Oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation 127, 1888–1902. doi:10.1161/CIRCULATIONAHA.112.132159
Griendling, K. K., and Ushio-Fukai, M. (1997). NADH/NADPH oxidase and vascular function. Trends Cardiovasc. Med. 7, 301–307. doi:10.1016/S1050-1738(97)00088-1
Gupta, S., Pablo, A. M., Jiang, X. C., Wang, N., Tall, A. R., and Schindler, C. (1997). IFN-γ, potentiates atherosclerosis in ApoE knock-out mice. J. Clin. Invest. 99, 2752–2761. doi:10.1172/JCI119465
Guzik, T. J., Mussa, S., Gastaldi, D., Sadowski, J., Ratnatunga, C., Pillai, R., et al. (2002). Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation 105, 1656–1662. doi:10.1161/01.CIR.0000012748.58444.08
Hahn, A. W. A. (1994). Activation of human peripheral monocytes by angiotensin II. FEBS Lett. 347, 178–180. doi:10.1016/0014-5793(94)00531-1
Hansson, G. K., and Hermansson, A. (2011). The immune system in atherosclerosis. Nat. Immunol. 12, 204–212. doi:10.1038/ni.2001
Hansson, G. K., Libby, P., Schönbeck, U., and Yan, Z. Q. (2002). Innate and adaptive immunity in the pathogenesis of atherosclerosis. Circ. Res. 91, 281–291. doi:10.1161/01.RES.0000029784.15893.10
Hansson, G. K., Seifert, P. S., Olsson, G., and Bondjers, G. (1991). Immunohistochemical detection of macrophages and T lymphocytes in atherosclerotic lesions of cholesterol-fed rabbits. Arterioscler. Thromb. 11, 745–750. doi:10.1161/01.ATV.11.3.745
Hartge, M. M., Unger, T., and Kintscher, U. (2007). The endothelium and vascular inflammation in diabetes. Diabetes Vasc. Dis. Res. 4, 84–88. doi:10.3132/dvdr.2007.025
Higai, K., Shimamura, A., and Matsumoto, K. (2006). Amadori-modified glycated albumin predominantly induces E-selectin expression on human umbilical vein endothelial cells through NADPH oxidase activation. Clin. Chim. Acta 367, 137–143. doi:10.1016/j.cca.2005.12.008
Hink, U., Li, H., Mollnau, H., Oelze, M., Matheis, E., Hartmann, M., et al. (2001). Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ. Res. 88, E14–E22. doi:10.1161/01.RES.88.2.e14
Hoch, N. E., Guzik, T. J., Chen, W., Deans, T., Maalouf, S. A., Gratze, P., et al. (2009). Regulation of T-cell function by endogenously produced angiotensin II. Am. J. Physiol. Regul. Integr. Comp. Physiol. 296, R208–R216. doi:10.1152/ajpregu.90521.2008
Hwang, J., Kleinhenz, D. J., Rupnow, H. L., Campbell, A. G., Thulé, P. M., Sutliff, R. L., et al. (2007). The PPARγ ligand, rosiglitazone, reduces vascular oxidative stress and NADPH oxidase expression in diabetic mice. Vascul. Pharmacol. 46, 456–462. doi:10.1016/j.vph.2007.01.007
Ihara, Y., Egashira, K., Nakano, K., Ohtani, K., Kubo, M., Koga, J., et al. (2007). Upregulation of the ligand-RAGE pathway via the angiotensin II type I receptor is essential in the pathogenesis of diabetic atherosclerosis. J. Mol. Cell. Cardiol. 43, 455–464. doi:10.1016/j.yjmcc.2007.07.044
Inoguchi, T., Li, P., Umeda, F., Yu, H. Y., Kakimoto, M., Imamura, M., et al. (2000). High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 49, 1939–1945. doi:10.2337/diabetes.49.11.1939
Jackson, S. H., Devadas, S., Kwon, J., Pinto, L. A., and Williams, M. S. (2004). T cells express a phagocyte-type NADPH oxidase that is activated after T cell receptor stimulation. Nat. Immunol. 5, 818–827. doi:10.1038/ni1096
Jandeleit-Dahm, K., Watson, A., and Soro-Paavonen, A. (2008). The AGE/RAGE axis in diabetes-accelerated atherosclerosis. Clin. Exp. Pharmacol. Physiol. 35, 329–334. doi:10.1111/j.1440-1681.2007.04875.x
Jiang, J. X., Chen, X., Serizawa, N., Szyndralewiez, C., Page, P., Schröder, K., et al. (2012). Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free Radic. Biol. Med. 53, 289–296. doi:10.1016/j.freeradbiomed.2012.05.007
Joussen, A. M., Murata, T., Tsujikawa, A., Kirchhof, B., Bursell, S. E., and Adamis, A. P. (2001). Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am. J. Pathol. 158, 147–152. doi:10.1016/S0002-9440(10)63952-1
Keating, S. T., and El-Osta, A. (2012). Chromatin modifications associated with diabetes. J. Cardiovasc. Transl. Res. 5, 399–412. doi:10.1007/s12265-012-9380-9
Kelkar, P., Masood, M., and Parry, G. J. (2000). Distinctive pathologic findings in proximal diabetic neuropathy (diabetic amyotrophy). Neurology 55, 83–88. doi:10.1212/WNL.55.1.83
Khallou-Laschet, J., Varthaman, A., Fornasa, G., Compain, C., Gaston, A. T., Clement, M., et al. (2010). Macrophage plasticity in experimental atherosclerosis. PLoS ONE 5:e8852. doi:10.1371/journal.pone.0008852
Kim, J. A., Berliner, J. A., Natarajan, R. D., and Nadler, J. L. (1994). Evidence that glucose increases monocyte binding to human aortic endothelial cells. Diabetes 43, 1103–1107. doi:10.2337/diabetes.43.9.1103
Kim, J. A., Neupane, G. P., Lee, E. S., Jeong, B. S., Park, B. C., and Thapa, P. (2011). NADPH oxidase inhibitors: a patent review. Expert Opin. Ther. Pat. 21, 1147–1158. doi:10.1517/13543776.2011.584870
Kirstein, M., Brett, J., Radoff, S., Ogawa, S., Stern, D., and Vlassara, H. (1990). Advanced protein glycosylation induces transendothelial human monocyte chemotaxis and secretion of platelet-derived growth factor: role in vascular disease of diabetes and aging. Proc. Natl. Acad. Sci. U.S.A. 87, 9010–9014. doi:10.1073/pnas.87.22.9010
Kislinger, T., Tanji, N., Wendt, T., Qu, W., Lu, Y., Ferran, L. J. Jr., et al. (2001). Receptor for advanced glycation end products mediates inflammation and enhanced expression of tissue factor in vasculature of diabetic apolipoprotein E-null mice. Arterioscler. Thromb. Vasc. Biol. 21, 905–910. doi:10.1161/01.ATV.21.6.905
Klein, R. L., Laimins, M., and Lopes-Virella, M. F. (1995). Isolation, characterization, and metabolism of the glycated and nonglycated subfractions of low-density lipoproteins isolated from type I diabetic patients and nondiabetic subjects. Diabetes 44, 1093–1098. doi:10.2337/diabetes.44.9.1093
Lam, M. C. W., Tan, K. C. B., and Lam, K. S. L. (2004). Glycoxidized low-density lipoprotein regulates the expression of scavenger receptors in THP-1 macrophages. Atherosclerosis 177, 313–320. doi:10.1016/j.atherosclerosis.2004.08.003
Lassègue, B., and Clempus, R. E. (2003). Vascular NAD(P)H oxidases: specific features, expression, and regulation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 285, R277–R297.
Lassègue, B., and Griendling, K. K. (2010). NADPH oxidases: functions and pathologies in the vasculature. Arterioscler. Thromb. Vasc. Biol. 30, 653–661. doi:10.1161/ATVBAHA.108.181610
Lassègue, B., Sorescu, D., Szöcs, K., Yin, Q., Akers, M., Zhang, Y., et al. (2001). Novel gp91phox homologues in vascular smooth muscle cells: nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ. Res. 88, 888–894. doi:10.1161/hh0901.090299
Lee, C. F., Qiao, M., Schröder, K., Zhao, Q., and Asmis, R. (2010). Nox4 is a novel inducible source of reactive oxygen species in monocytes and macrophages and mediates oxidized low density lipoprotein-induced macrophage death. Circ. Res. 106, 1489–1497. doi:10.1161/CIRCRESAHA.109.215392
Lewis, P., Stefanovic, N., Pete, J., Calkin, A. C., Giunti, S., Thallas-Bonke, V., et al. (2007). Lack of the antioxidant enzyme glutathione peroxidase-1 accelerates atherosclerosis in diabetic apolipoprotein E-deficient mice. Circulation 115, 2178–2187. doi:10.1161/CIRCULATIONAHA.106.664250
Li, H., Cybulsky, M. I., Gimbrone, M. A. Jr., and Libby, P. (1993). An atherogenic diet rapidly induces VCAM-1, a cytokine-regulatable mononuclear leukocyte adhesion molecule, in rabbit aortic endothelium. Arterioscler. Thromb. 13, 197–204. doi:10.1161/01.ATV.13.2.197
Libby, P., Ridker, P. M., and Maseri, A. (2002). Inflammation and atherosclerosis. Circulation 105, 1135–1143. doi:10.1161/hc0902.104353
Libby, P., Ridker, P. M., and Hansson, G. K. (2011). Progress and challenges in translating the biology of atherosclerosis. Nature 473, 317–325. doi:10.1038/nature10146
Lim, A. K. H., and Tesch, G. H. (2012). Inflammation in diabetic nephropathy. Mediators Inflamm. 2012, doi:10.1155/2012/146154
Lindley, S., Dayan, C. M., Bishop, A., Roep, B. O., Peakman, M., and Tree, T. I. (2005). Defective suppressor function in CD4+CD25+ T-cells from patients with type 1 diabetes. Diabetes 54, 92–99. doi:10.2337/diabetes.54.1.92
Little, P. J., Chait, A., and Bobik, A. (2011). Cellular and cytokine-based inflammatory processes as novel therapeutic targets for the prevention and treatment of atherosclerosis. Pharmacol. Ther. 131, 255–268. doi:10.1016/j.pharmthera.2011.04.001
Liu, J., Yang, F., Yang, X. P., Jankowski, M., and Pagano, P. J. (2003). NAD(P)H oxidase mediates angiotensin II-induced vascular macrophage infiltration and medial hypertrophy. Arterioscler. Thromb. Vasc. Biol. 23, 776–782. doi:10.1161/01.ATV.0000066684.37829.16
Lopes-Virella, M. F., Hunt, K. J., Baker, N. L., Lachin, J., Nathan, D. M., Virella, G., et al. (2011a). Levels of oxidized LDL and advanced glycation end products – modified LDL in circulating immune complexes are strongly associated with increased levels of carotid intima-media thickness and its progression in type 1 diabetes. Diabetes 60, 582–589. doi:10.2337/db10-0915
Lopes-Virella, M. F., Baker, N. L., Hunt, K. J., Lachin, J., Nathan, D., Virella, G., et al. (2011b). Oxidized LDL immune complexes and coronary artery calcification in type 1 diabetes. Atherosclerosis 214, 462–467. doi:10.1016/j.atherosclerosis.2010.11.012
Lopes-Virella, M. F., and Virella, G. (2010). Clinical significance of the humoral immune response to modified LDL. Clin. Immunol. 134, 55–65. doi:10.1016/j.clim.2009.04.001
Lopes-Virella, M. F., Virella, G., Orchard, T. J., Koskinen, S., Evans, R. W., Becker, D. J., et al. (1999). Antibodies to oxidized LDL and LDL-containing immune complexes as risk factors for coronary artery disease in diabetes mellitus. Clin. Immunol. 90, 165–172. doi:10.1006/clim.1998.4631
Lüscher, T. F., Creager, M. A., Beckman, J. A., and Cosentino, F. (2003). Diabetes and vascular disease. Pathophysiology, clinical consequences, and medical therapy: part II. Circulation 108, 1655–1661. doi:10.1161/01.CIR.0000089189.70578.E2
Mach, F., Schönbeck, U., Sukhova, G. K., Bourcier, T., Bonnefoy, J. Y., Pober, J. S., et al. (1997). Functional CD40 ligand is expressed on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for CD40-CD40 ligand signaling in atherosclerosis. Proc. Natl. Acad. Sci. U.S.A. 94, 1931–1936. doi:10.1073/pnas.94.5.1931
Manduteanu, I., Pirvulescu, M., Gan, A. M., Stan, D., Simion, V., Dragomir, E., et al. (2010). Similar effects of resistin and high glucose on P-selectin and fractalkine expression and monocyte adhesion in human endothelial cells. Biochem. Biophys. Res. Commun. 391, 1443–1448. doi:10.1016/j.bbrc.2009.12.089
Marfella, R., Siniscalchi, M., Esposito, K., Sellitto, A., De Fanis, U., Romano, C., et al. (2003). Effects of stress hyperglycemia on acute myocardial infarction: role of inflammatory immune process in functional cardiac outcome. Diabetes Care 26, 3129–3135. doi:10.2337/diacare.26.11.3129
Mazière, C., and Mazière, J. C. (2009). Activation of transcription factors and gene expression by oxidized low-density lipoprotein. Free Radic. Biol. Med. 46, 127–137. doi:10.1016/j.freeradbiomed.2008.10.024
Miao, F., Chen, Z., Zhang, L., Liu, Z., Wu, X., Yuan, Y. C., et al. (2012). Profiles of epigenetic histone post-translational modifications at type 1 diabetes susceptible genes. J. Biol. Chem. 287, 16335–16345. doi:10.1074/jbc.M111.330373
Miao, F., Smith, D. D., Zhang, L., Min, A., Feng, W., and Natarajan, R. (2008). Lymphocytes from patients with type 1 diabetes display a distinct profile of chromatin histone H3 lysine 9 dimethylation an epigenetic study in diabetes. Diabetes 57, 3189–3198. doi:10.2337/db08-0645
Miao, F., Wu, X., Zhang, L., Yuan, Y. C., Riggs, A. D., and Natarajan, R. (2007). Genome-wide analysis of histone lysine methylation variations caused by diabetic conditions in human monocytes. J. Biol. Chem. 282, 13854–13863. doi:10.1074/jbc.M609446200
Miller, F. J. Jr., Filali, M., Huss, G. J., Stanic, B., Chamseddine, A., Barna, T. J., et al. (2007). Cytokine activation of nuclear factor kB in vascular smooth muscle cells requires signaling endosomes containing Nox1 and ClC-3. Circ. Res. 101, 663–671. doi:10.1161/CIRCRESAHA.107.151076
Mills, C. D., Kincaid, K., Alt, J. M., Heilman, M. J., and Hill, A. M. (2000). M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 164, 6166–6173.
Moore, K. J., and Tabas, I. (2011). Macrophages in the pathogenesis of atherosclerosis. Cell 145, 341–355. doi:10.1016/j.cell.2011.04.005
Morigi, M., Angioletti, S., Imberti, B., Donadelli, R., Micheletti, G., Figliuzzi, M., et al. (1998). Leukocyte-endothelial interaction is augmented by high glucose concentrations and hyperglycemia in a NF-kB-dependent fashion. J. Clin. Invest. 101, 1905–1915. doi:10.1172/JCI656
Namiki, M., Kawashima, S., Yamashita, T., Ozaki, M., Sakoda, T., Inoue, N., et al. (2004). Intramuscular gene transfer of interleukin-10 cDNA reduces atherosclerosis in apolipoprotein E-knockout mice. Atherosclerosis 172, 21–29. doi:10.1016/j.atherosclerosis.2003.08.032
Oak, J. H., and Cai, H. (2007). Attenuation of angiotensin II signaling recouples eNOS and inhibits nonendothelial NOX activity in diabetic mice. Diabetes 56, 118–126. doi:10.2337/db06-0288
O’Brien, K. D., Allen, M. D., McDonald, T. O., Chait, A., Harlan, J. M., Fishbein, D., et al. (1993). Vascular cell adhesion molecule-1 is expressed in human coronary atherosclerotic plaques: implications for the mode of progression of advanced coronary atherosclerosis. J. Clin. Invest. 92, 945–951. doi:10.1172/JCI116670
Okabe, J., Orlowski, C., Balcerczyk, A., Tikellis, C., Thomas, M. C., Cooper, M. E., et al. (2012). Distinguishing hyperglycemic changes by set7 in vascular endothelial cells. Circ. Res. 110, 1067–1076. doi:10.1161/CIRCRESAHA.112.266171
Orchard, T. J., Virella, G., Forrest, K. Y., Evans, R. W., Becker, D. J., and Lopes-Virella, M. F. (1999). Antibodies to oxidized LDL predict coronary artery disease in type 1 diabetes: a nested case-control study from the Pittsburgh epidemiology of diabetes complications study. Diabetes 48, 1454–1458. doi:10.2337/diabetes.48.7.1454
Palinski, W., Miller, E., and Witztum, J. L. (1995). Immunization of low density lipoprotein (LDL) receptor-deficient rabbits with homologous malondialdehyde-modified LDL reduces atherogenesis. Proc. Natl. Acad. Sci. U.S.A. 92, 821–825. doi:10.1073/pnas.92.3.821
Parathath, S., Grauer, L., Huang, L. S., Sanson, M., Distel, E., Goldberg, I. J., et al. (2011). Diabetes adversely affects macrophages during atherosclerotic plaque regression in mice. Diabetes 60, 1759–1769. doi:10.2337/db10-0778
Park, H. S., Chun, J. N., Jung, H. Y., Choi, C., and Bae, Y. S. (2006). Role of NADPH oxidase 4 in lipopolysaccharide-induced proinflammatory responses by human aortic endothelial cells. Cardiovasc. Res. 72, 447–455. doi:10.1016/j.cardiores.2006.09.012
Piconi, L., Quagliaro, L., Da Ros, R., Assaloni, R., Giugliano, D., Esposito, K., et al. (2004). Intermittent high glucose enhances ICAM-1, VCAM-1, E-selectin and interleukin-6 expression in human umbilical endothelial cells in culture: the role of poly(ADP-ribose) polymerase. J. Thromb. Haemost. 2, 1453–1459. doi:10.1111/j.1538-7836.2004.00835.x
Piga, R., Naito, Y., Kokura, S., Handa, O., and Yoshikawa, T. (2007). Short-term high glucose exposure induces monocyte-endothelial cells adhesion and transmigration by increasing VCAM-1 and MCP-1 expression in human aortic endothelial cells. Atherosclerosis 193, 328–334. doi:10.1016/j.atherosclerosis.2006.09.016
Pirola, L., Balcerczyk, A., Tothill, R. W., Haviv, I., Kaspi, A., Lunke, S., et al. (2011). Genome-wide analysis distinguishes hyperglycemia regulated epigenetic signatures of primary vascular cells. Genome Res. 21, 1601–1615. doi:10.1101/gr.116095.110
Pueyo, M. E., Gonzalez, W., Nicoletti, A., Savoie, F., Arnal, J. F., and Michel, J. B. (2000). Angiotensin II stimulates endothelial vascular cell adhesion molecule-1 via nuclear factor-kB activation induced by intracellular oxidative stress. Arterioscler. Thromb. Vasc. Biol. 20, 645–651. doi:10.1161/01.ATV.20.3.645
Raines, E. W., and Ferri, N. (2005). Cytokines affecting endothelial and smooth muscle cells in vascular disease. J. Lipid Res. 46, 1081–1092. doi:10.1194/jlr.R500004-JLR200
Reddy, M. A., and Natarajan, R. (2011). Epigenetic mechanisms in diabetic vascular complications. Cardiovasc. Res. 90, 421–429. doi:10.1093/cvr/cvr024
Ross, R. (1993). The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature 362, 801–809. doi:10.1038/362801a0
Ross, R. (1997). Cellular and molecular studies of atherogenesis. Atherosclerosis 131(Suppl.), S3–S4. doi:10.1016/S0021-9150(97)06114-5
Rubinstein, R., Genaro, A. M., Motta, A., Cremaschi, G., and Wald, M. R. (2008). Impaired immune responses in streptozotocin-induced type I diabetes in mice. Involvement of high glucose. Clin. Exp. Immunol. 154, 235–246. doi:10.1111/j.1365-2249.2008.03742.x
Ruiz-Ortega, M., Ruperez, M., Esteban, V., and Egido, J. (2003). Molecular mechanisms of angiotensin II-induced vascular injury. Curr. Hypertens. Rep. 5, 73–79. doi:10.1007/s11906-003-0014-0
Sasaki, N., Yamashita, T., Takeda, M., Shinohara, M., Nakajima, K., Tawa, H., et al. (2009). Oral anti-CD3 antibody treatment induces regulatory t cells and inhibits the development of atherosclerosis in mice. Circulation 120, 1996–2005. doi:10.1161/CIRCULATIONAHA.109.863431
Schalkwijk, C. G., and Stehouwer, C. D. A. (2005). Vascular complications in diabetes mellitus: the role of endothelial dysfunction. Clin. Sci. 109, 143–159. doi:10.1042/CS20050025
Schmidt, A. M., Hori, O., Chen, J. X., Li, J. F., Crandall, J., Zhang, J., et al. (1995). Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule-1 (VCAM-1) in cultured human endothelial cells and in mice: a potential mechanism for the accelerated vasculopathy of diabetes. J. Clin. Invest. 96, 1395–1403. doi:10.1172/JCI118175
Schmidt, A. M., Yan, S. D., Yan, S. F., and Stern, D. M. (2001). The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J. Clin. Invest. 108, 949–955. doi:10.1172/JCI14002
Schröder, K., Zhang, M., Benkhoff, S., Mieth, A., Pliquett, R., Kosowski, J., et al. (2012). Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ. Res. 110, 1217–1225. doi:10.1161/CIRCRESAHA.112.267054
Sedeek, M., Callera, G., Montezano, A., Gutsol, A., Heitz, F., Szyndralewiez, C., et al. (2010). Critical role of Nox4-based NADPH oxidase in glucose-induced oxidative stress in the kidney: implications in type 2 diabetic nephropathy. Am. J. Physiol. Renal Physiol. 299, F1348–F1358. doi:10.1152/ajprenal.00028.2010
Sedeek, M., Montezano, A. C., Hebert, R. L., Gray, S. P., Di, M. arcoE., Jha, J. C., and Etalname, A. (2012). Oxidative stress, Nox isoforms and complications of diabetes-potential targets for novel therapies. J. Cardiovasc. Transl. Res. 5, 509–518. doi:10.1007/s12265-012-9387-2
Shah, P. K., Falk, E., Badimon, J. J., Fernandez-Ortiz, A., Mailhac, A., Villareal-Levy, G., et al. (1995). Human monocyte-derived macrophages induce collagen breakdown in fibrous caps of atherosclerotic plaques: potential role of matrix-degrading metalloproteinases and implications for plaque rupture. Circulation 92, 1565–1569.
Shalhoub, J., Falck-Hansen, M. A., Davies, A. H., and Monaco, C. (2011). Innate immunity and monocyte-macrophage activation in atherosclerosis. J. Inflamm. (Lond.) 8, 9. doi:10.1186/1476-9255-8-9
Silva, J. A., Escobar, A., Collins, T. J., Ramee, S. R., and White, C. J. (1995). Unstable angina: a comparison of angioscopic findings between diabetic and nondiabetic patients. Circulation 92, 1731–1736. doi:10.1161/01.CIR.92.7.1731
Sorescu, D., Weiss, D., Lassègue, B., Clempus, R. E., Szöcs, K., Sorescu, G. P., et al. (2002). Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation 105, 1429–1435. doi:10.1161/01.CIR.0000012917.74432.66
Soro-Paavonen, A., Watson, A. M., Li, J., Paavonen, K., Koitka, A., Calkin, A. C., et al. (2008). Receptor for advanced glycation end products (RAGE) deficiency attenuates the development of atherosclerosis in diabetes. Diabetes 57, 2461–2469. doi:10.2337/db07-1808
Stemme, S., Holm, J., and Hansson, G. K. (1992). T lymphocytes in human atherosclerotic plaques are memory cells expressing CD45RO and the integrin VLA-1. Arterioscler. Thromb. 12, 206–211. doi:10.1161/01.ATV.12.2.206
Stentz, F. B., and Kitabchi, A. E. (2005). Hyperglycemia-induced activation of human T-lymphocytes with de novo emergence of insulin receptors and generation of reactive oxygen species. Biochem. Biophys. Res. Commun. 335, 491–495. doi:10.1016/j.bbrc.2005.07.109
Su, B., Mitra, S., Gregg, H., Flavahan, S., Chotani, M. A., Clark, K. R., et al. (2001). Redox regulation of vascular smooth muscle cell differentiation. Circ. Res. 89, 39–46. doi:10.1161/hh1301.093615
Suzuki, L. A., Poot, M., Gerrity, R. G., and Bornfeldt, K. E. (2001). Diabetes accelerates smooth muscle accumulation in lesions of atherosclerosis: lack of direct growth-promoting effects of high glucose levels. Diabetes 50, 851–860. doi:10.2337/diabetes.50.4.851
Tavakoli, S., and Asmis, R. (2012). Reactive oxygen species and thiol redox signaling in the macrophage biology of atherosclerosis. Antioxid. Redox Signal. 17, 1785–1795. doi:10.1089/ars.2012.4638
Tedgui, A., and Mallat, Z. (2006). Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiol. Rev. 86, 515–581. doi:10.1152/physrev.00024.2005
Thomas, M. C., Pickering, R. J., Tsorotes, D., Koitka, A., Sheehy, K., Bernardi, S., et al. (2010). Genetic Ace2 deficiency accentuates vascular inflammation and atherosclerosis in the ApoE knockout mouse. Circ. Res. 107, 888–897. doi:10.1161/CIRCRESAHA.110.219279
Ting, A. H., Suzuki, H., Cope, L., Schuebel, K. E., Lee, B. H., Toyota, M., et al. (2008). A requirement for DICER to maintain full promoter CpG island hypermethylation in human cancer cells. Cancer Res. 68, 2570–2575. doi:10.1158/0008-5472.CAN-07-6405
Tummala, P. E., Chen, X. L., Sundell, C. L., Laursen, J. B., Hammes, C. P., Alexander, R. W., et al. (1999). Angiotensin II induces vascular cell adhesion molecule-1 expression in rat vasculature: a potential link between the renin-angiotensin system and atherosclerosis. Circulation 100, 1223–1229. doi:10.1161/01.CIR.100.11.1223
Valente, A. J., Yoshida, T., Murthy, S. N., Sakamuri, S. S., Katsuyama, M., Clark, R. A., et al. (2012). Angiotensin II enhances AT 1-Nox1 binding and stimulates arterial smooth muscle cell migration and proliferation through AT 1, Nox1, and interleukin-18. Am. J. Physiol. Heart Circ. Physiol. 303, 282–296. doi:10.1152/ajpheart.00231.2012
Vallerskog, T., Martens, G. W., and Kornfeld, H. (2010). Diabetic mice display a delayed adaptive immune response to Mycobacterium tuberculosis. J. Immunol. 184, 6275–6282. doi:10.4049/jimmunol.1000304
Vessby, J., Basu, S., Mohsen, R., Berne, C., and Vessby, B. (2002). Oxidative stress and antioxidant status in type 1 diabetes mellitus. J. Intern. Med. 251, 69–76. doi:10.1046/j.1365-2796.2002.00927.x
Villeneuve, L. M., Kato, M., Reddy, M. A., Wang, M., Lanting, L., and Natarajan, R. (2010). Enhanced levels of microRNA-125b in vascular smooth muscle cells of diabetic db/db mice lead to increased inflammatory gene expression by targeting the histone methyltransferase Suv39h1. Diabetes 59, 2904–2915. doi:10.2337/db10-0208
Villeneuve, L. M., Reddy, M. A., Lanting, L. L., Wang, M., Meng, L., and Natarajan, R. (2008). Epigenetic histone H3 lysine 9 methylation in metabolic memory and inflammatory phenotype of vascular smooth muscle cells in diabetes. Proc. Natl. Acad. Sci. U.S.A. 105, 9047–9052. doi:10.1073/pnas.0803623105
Villeneuve, L. M., Reddy, M. A., and Natarajan, R. (2011). Epigenetics: deciphering its role in diabetes and its chronic complications. Clin. Exp. Pharmacol. Physiol. 38, 401–409. doi:10.1111/j.1440-1681.2011.05497.x
Waldo, S. W., Li, Y., Buono, C., Zhao, B., Billings, E. M., Chang, J., et al. (2008). Heterogeneity of human macrophages in culture and in atherosclerotic plaques. Am. J. Pathol. 172, 1112–1126. doi:10.2353/ajpath.2008.070513
Wautier, M. P., Chappey, O., Corda, S., Stern, D. M., Schmidt, A. M., and Wautier, J. L. (2001). Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am. J. Physiol. Endocrinol. Metab. 280, E685–E694.
Wendt, M. C., Daiber, A., Kleschyov, A. L., Mülsch, A., Sydow, K., Schulz, E., et al. (2005). Differential effects of diabetes on the expression of the gp91 phox homologues nox1 and nox4. Free Radic. Biol. Med. 39, 381–391. doi:10.1016/j.freeradbiomed.2005.03.020
Wendt, T., Bucciarelli, L., Qu, W., Lu, Y., Yan, S. F., Stern, D. M., et al. (2002). Receptor for advanced glycation endproducts (RAGE) and vascular inflammation: insights into the pathogenesis of macrovascular complications in diabetes. Curr. Atheroscler. Rep. 4, 228–237. doi:10.1007/s11883-002-0024-4
Wilson, H. M. (2010). Macrophages heterogeneity in atherosclerosis – implications for therapy. J. Cell. Mol. Med. 14, 2055–2065. doi:10.1111/j.1582-4934.2010.01121.x
Yan, H., Choi, A. J., Lee, B. H., and Ting, A. H. (2011). Identification and functional analysis of epigenetically silenced micrornas in colorectal cancer cells. PLoS ONE 6:e20628. doi:10.1371/journal.pone.0020628
Youn, J. Y., Gao, L., and Cai, H. (2012). The p47 phox- and NADPH oxidase organiser 1 (NOXO1)-dependent activation of NADPH oxidase 1 (NOX1) mediates endothelial nitric oxide synthase (eNOS) uncoupling and endothelial dysfunction in a streptozotocin-induced murine model of diabetes. Diabetologia 55, 2069–2079. doi:10.1007/s00125-012-2557-6
Keywords: Nox, diabetes complications, atherosclerosis, immune cells, inflammation
Citation: Di Marco E, Gray SP and Jandeleit-Dahm K (2013) Diabetes alters activation and repression of pro- and anti-inflammatory signaling pathways in the vasculature. Front. Endocrinol. 4:68. doi: 10.3389/fendo.2013.00068
Received: 01 February 2013; Accepted: 21 May 2013;
Published online: 05 June 2013.
Edited by:
Gabriel Virella, Universidade de Lisboa, PortugalReviewed by:
Gabriel Virella, Universidade de Lisboa, PortugalIleana Manduteanu, Institute of Cellular Biology and Pathology Nicolae Simionescu, Romania
Copyright: © 2013 Di Marco, Gray and Jandeleit-Dahm. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Karin Jandeleit-Dahm, Head, Diabetes Complications Division, Baker IDI Heart and Diabetes Research Institute, PO Box 6492, St Kilda Road Central, Melbourne, VIC 8008, Australia e-mail:a2FyaW4uamFuZGVsZWl0LWRhaG1AYmFrZXJpZGkuZWR1LmF1