

 Fausta Alfano
Fausta Alfano
- Department of Clinical Medicine and Surgery, University of Naples “Federico II,”, Naples, Italy
The major product secreted by the thyroid is thyroxine (T4), whereas most of the biologically active triiodothyronine (T3) derives from the peripheral conversion of T4 into T3. The deiodinase enzymes are involved in activation and inactivation of thyroid hormones (THs). Type 1 and type 2 deiodinase (D1 and D2) convert T4 into T3 whereas D3 degrades T4 and T3 into inactive metabolites and is thus the major physiological TH inactivator. The hypothalamic-pituitary-thyroid axis maintains circulating TH levels constant, while the deiodinases tissue-specifically regulate intracellular thyroid status by controlling TH action in a precise spatio-temporal fashion. Here we review the data related to the recent identification of a paraneoplastic syndrome called “consumptive hypothyroidism,” which exemplifies how deiodinases alter substantially the concentration of TH in blood. This syndrome results from the aberrant uncontrolled expression of D3 that can induce a severe form of hypothyroidism by inactivating T4 and T3 in defined tumor tissue. This rare TH insufficiency generally affects patients in the first years of life, and has distinct features in terms of diagnosis, treatment, and prognosis with respect to other forms of hypothyroidism.
Introduction
Thyroid hormones (THs) are circulating hormones widely involved in the development and metabolic homeostasis of virtually all mammalian tissues. Alterations in serum TH levels in the fetus and newborns cause significant deficits in experimental animals and in humans (1). The importance of maintaining the TH-dependent transcriptional signature range is supported by the presence of diverse homeostasis checkpoints. In fact, beyond the hypothalamus-pituitary-thyroid axis, which responds to changes in serum levels of thyroid-stimulating hormone (TSH) (2), there are various ways to affect thyroid status in a specific tissue. First, TH exerts its genomic action only in tissues that express a thyroid hormone receptor (TR), and second, it is possible to locally modulate the amount of TH available for cells at a pre-receptoral level. TH transporters and deiodinases are important in determining the availability of TH in a tissue-specific manner.
Although THs are lipophilic molecules, they require specific plasma membrane transporters to enter target cells. Various transporter families have been identified, but only monocarboxylate transporters 8 and 10 (MCT8 and MCT10) and anion transporting polypeptide 1C1 (OATP1C1) have elevated specificity for THs. These proteins are differentially expressed in several tissues: OATP1C1 is expressed in liver, kidney, and brain, MCT8 is highly expressed in liver and brain, and MCT10 is expressed in intestine, kidney, liver, and placenta. A further level of complexity results from the capacity of these transporters to determine the efflux of THs from the intracellular environment to the external milieu (3). The clinical relevance of these transporters is supported by evidence that mutations in the gene encoding MCT8 are associated with elevated serum triiodothyronine (T3) levels and severe sex-linked psychomotor retardation (Allan–Herndon–Dudley syndrome) (4).
Once inside the nucleus, T3 acts through the binding to ligand-dependent transcription factors namely TRs, which bind, mainly as heterodimers with retinoid-X receptors, to TH response elements in target genes (5). TRs are encoded by two genes, THRA and THRB, which are located on chromosomes 17 and 3, respectively. TRα has one T3-binding splice product, TRα1, predominantly expressed in brain, heart, and skeletal muscle, and two non-T3-binding splice products, TRα2 and TRα3, and several additional truncated forms. TRβ has three major T3-binding splice products: TRβ1 is almost ubiquitously expressed; TRβ2 is expressed primarily in the brain, pituitary, retina, and inner ear; and TRβ3 is expressed in kidney, liver, and lung (6).
Selenodeiodinases catalyze the activation (D1 and D2) and inactivation (D3) of the THs thyroxine (T4) and 3,5,3′-triiodothyronine (T3) by removing distinct iodine moieties. D1 and D2 convert thyroxine into the most active metabolic form of TH, T3, by outer ring deiodination; and D3 converts T4 and T3 into the inactive forms rT3 and 3,3′-diiodiothyronine (T2), respectively, by inner ring deiodination. D1 is expressed mostly in the liver, kidney, thyroid, and pituitary; D2 is expressed primarily in the thyroid, central nervous system, pituitary, developing cochlea, brown adipose tissue, and skeletal muscle; D3 is prevalently expressed in many fetal tissues, through the adult it is expressed in placenta, brain, and skin, and to a lesser extent in the pregnant uterus and pancreatic B-cells. The three deiodinases exert different actions: D1 participates in T3 production within the thyroid gland and controls circulating T3 levels, whereas D2 and D3 are active in local deiodination processes. Recent evidence indicates that D1 and D2 are the major sources of circulating T3 in euthyroid humans, with D1 being the major source of circulating T3 in hyperthyroid patients (7). Conversely, D3 inactivates T3 at tissue and plasma level (8). A dramatic example of the potency of the D3 pathways in TH clearance, at both systemic and tissue level, is the newly identified consumptive hypothyroidism syndrome, which is a rare condition resulting from increased D3 production in neoplastic cells and consequent T4 and T3 catabolism.
In this review, we focus on D3 as the cause of consumptive hypothyroidism, a newly recognized clinical entity which often requires close cooperation between clinical endocrinologists, pediatricians, and oncologists.
Type 3 Deiodinase and Inactivation of Thyroid Hormones
As mentioned, type 3 deiodinase is the physiological inactivator of TH: it catalyzes deiodination on the inner rings of T3 and T4 to produce T2 and rT3, respectively. D3 is a selenoenzyme encoded by the DIO3 gene in humans; it is localized on chromosome 14q32 (9). The DIO3 genomic structure contains a single exon and, at the 3′-UTR, a specific RNA structure, named SECIS (selenocysteine insertion element), that is crucial for the insertion of the selenocysteine residue and for maximal enzymatic catalytic efficiency (10). The DIO3 gene is imprinted, with preferential expression of the paternal allele. It belongs to a cluster of imprinted regions, at the DLK1-DIO3 locus (11).
D3 is an integral membrane protein that exerts its role as a homodimer (12). It is recycled through a system of endosomal clathrin-coated vesicles. This suggests a possible mechanism for D3 reactivation, and furthermore the possibility that this enzyme acts on both extracellular and intracellular pools of T3 and T4 (13). Diverse signals are able to regulate D3 expression in vitro and in vivo: retinoic acid, serum growth factors, estrogens and progesterone, TGFβ, Wnt-βcatenin, and Shh/Gli2 increase D3 levels, whereas glucocorticoid and growth hormone reduce D3 levels (14, 15).
Recent data from our laboratory implicates p63, a member of the p53 family (16), in D3 regulation (unpublished data). D3 plays the essential role of protecting tissue from excessive TH levels under normal and disease conditions. Indeed, thyrotoxicosis of any cause induces D3, whereas hypothyroidism suppresses D3 (17). D3 activity is elevated during development, a time when circulating fetal TH levels are much lower than those of the mother (18). D3 is widely expressed in such embryonic tissues as liver, cerebral cortex, cardiovascular apparatus, gonads, gut, skin, and urinary tract (19). The human placenta also expresses elevated D3 levels. Within the placenta, D3 blocks the maternal-to-fetal transfer of T4 thereby protecting the fetus from TH excess (20, 21). During late neonatal and adult life, D3 expression is restricted to a few tissues. It has been detected in skin, the central nervous system, and some endocrine glands (22). However, in adult life, D3 expression is reactivated in several disease conditions, namely inflammation, liver regeneration, cardiac hypertrophy and infarct, and cancer (15, 23–25). Several studies have demonstrated that thyroid status affects tumor formation, growth, and metastasis, which suggests that THs are involved in cell transformation. D3 expression has been widely documented in a variety of malignancies; it is turned-on in some malignant cell lines (26) and in a number of human tumors, i.e., oligodendromas, astrocytomas, gliosarcomas, glioblastoma multiforme, TSH-secreting pituitary adenomas, basal cell carcinomas, colon adenomas, and carcinomas (25). It has been postulated that D3 activity is required to facilitate tumor cell proliferation (27).
Consumptive Hypothyroidism
The paraneoplastic syndrome consumptive hypothyroidism is a rare form of hypothyroidism first identified in newborns with infantile hepatic hemangiomatosis (HHE) (28). The latter is a benign tumor of vascular origin affecting 4–5% of white infants. In most cases, the natural course of HHE is characterized by a rapid proliferation phase during the first 1–2 years of life, and by a slow and steady decline over the next 5–7 years until complete spontaneous involution of the tumor mass. The spectrum of vascular lesions that could be termed “HHE” ranges from benign and self-limiting to aggressive and life-threatening neoplasias. About 10% of cases have aggressive characteristics based on their size, location, and the number of lesions (29). In 2000, Huang et al. reported the first case of severe hypothyroidism (defined as “consumptive hypothyroidism”) in a 6-week-old infant with HHE (28). At the time of diagnosis, TSH level was elevated (156 mUI/ml) and the serum free thyroxine (FT4) concentration was low. Hence, the infant was treated with prednisolone (2 mg/kg) for the hemangioma, and levothyroxine (LT4) replacement (37.5 μg/day) for hypothyroidism, but 16 days later the TSH value was still 256 mUI/ml. At the age of 3 months, the child had intermittent bradycardia and hypothermia, which were probably linked to the hypothyroid status as evidenced by thyroid function tests. In fact, TSH concentration was 177 mUI/ml, serum T4 concentration was 2.5 μg/dl, T3 concentration was low (15 ng/ml), and the level of rT3 was elevated (413 ng/ml). Based on these TH values, replacement hormone therapy was increased to 50 μg/day of LT4, and intravenous administration of 90 μg/day liothyronine was initiated. This therapy resulted in normalization of TSH and T3, but T4 levels remained low. This replacement dose of hormone therapy is very high considering that an athyreotic infant of the same age requires about 7–10 μg/kg/day of LT4 (equivalent to about 3 μg/kg/day of T3), which is about nine times lower than that required by this patient.
After a vertical midline abdominal fasciotomy and embolization of the multiple hemangiomas, the patient died from systemic complications. At that time, the cause of hypothyroidism was not clear, but a markedly elevated serum thyroglobulin concentration argued against a diagnosis of congenital hypothyroidism. Furthermore, biochemical TH parameters suggested that THs were normally synthesized and secreted, but that their rate of degradation was increased. The overexpression of D3 and its activity in the hemangioma tissue were 0.78 pmol of T3 deiodinated/min/mg of protein, a value 7.5 times higher than that normally present in placental tissue, the highest physiological D3-expressing tissue in humans (20).
To date, more than 20 cases of consumptive hypothyroidism secondary to neoplasias have been reported (Table 1). An evaluation of all these cases strongly suggests that the development of consumptive hypothyroidism is related to the elevated D3 enzyme, which is in turn directly proportional to the size of the tumor mass and its specific activity, and independent of its localization. Furthermore, most cases described so far support the hypothesis advanced by Huang that D3 activity increases rapidly during the proliferative phase of hemangioma (28). It is also well recognized that consumptive hypothyroidism develops when the catabolic D3 activity exceeds the physiological or exogenous replacement of THs (Figure 1). The direct relationship between consumptive hypothyroidism, tumor size, and D3 activity is demonstrated by the spontaneous remission of hypothyroidism and normalization of serum rT3 concentration upon the natural regression of HHE or after surgical resection of the tumor (Table 1).
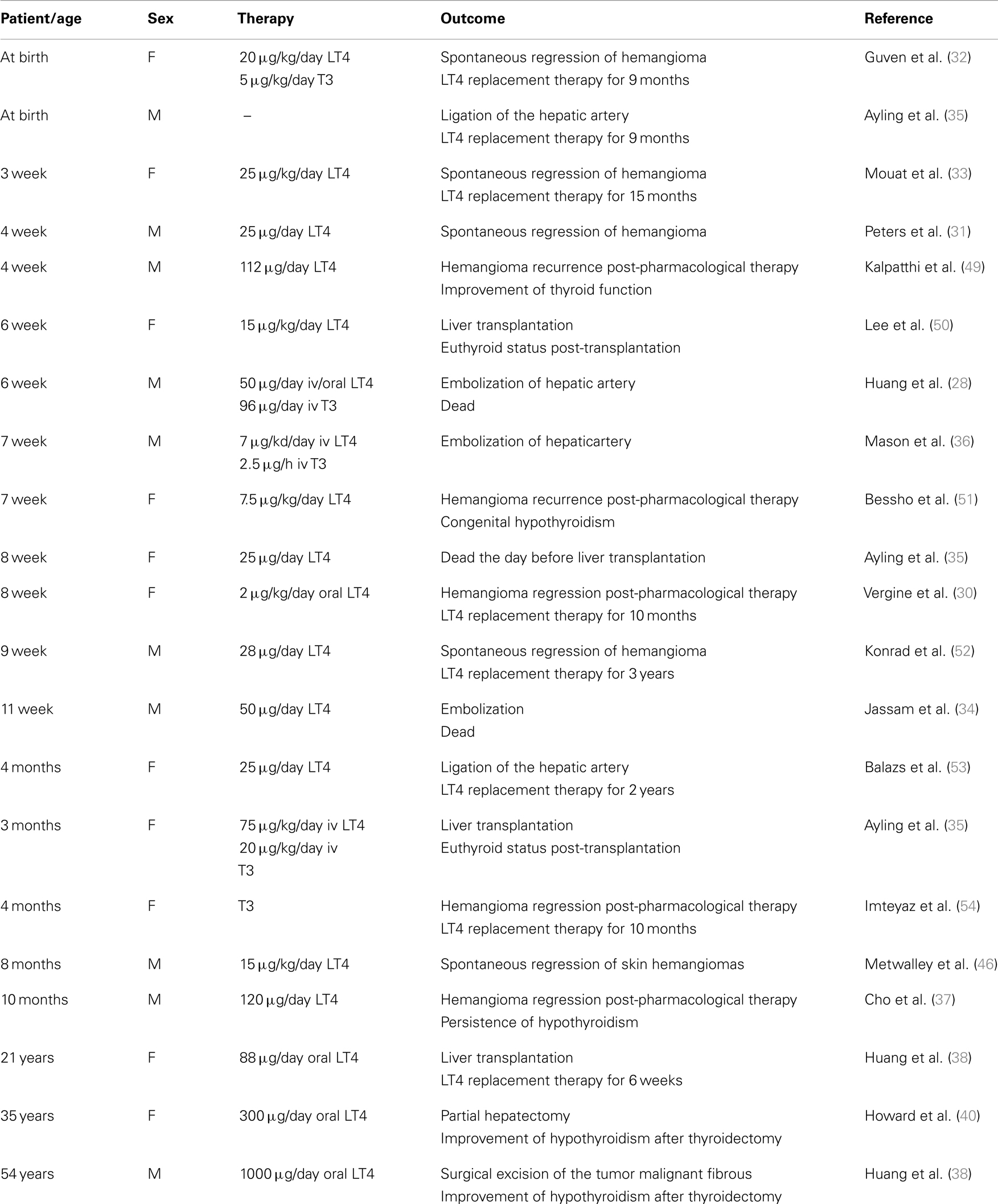
Table 1. Summary of published cases of “consumptive hypothyroidism.”
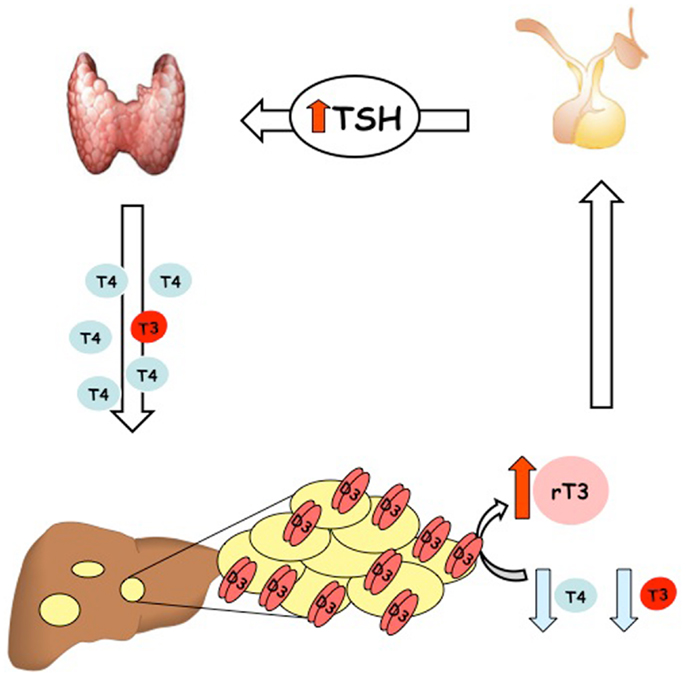
Figure 1. Schematic illustration of the pathogenesis of condition named “consumptive hypothyroidism.”
Recent studies have shed light on the role of D3 in the control of cell proliferation in tumors. In basal cell carcinoma the constitutive activation of the Shh-Gli pathway directly induces D3 mRNA, which in turn reduces TH intracellular activity thereby resulting in increased cyclin D1 expression and keratinocyte proliferation (15). In colon cancer, D3 is a direct target of β-catenin and its activation is a key factor in the regulation of cell proliferation in this tumor (25). Taken together, these findings suggest that local attenuation of TH is an important step in the primary proliferation of some neoplastic cells, which might explain the sustained D3 levels particularly in the initial phase of hemangioma growth.
What induces D3 in hemangioma cells? Data from in vitro studies suggest that D3 overexpression in hemangioma is induced by basic fibroblast growth factor and vascular endothelial growth factor. These angiogenic factors play an important role in the pathogenesis of hemangioma. Indeed, recent evidence suggests that propranolol, an antagonist of β2 adrenergic receptors, is able to block tumor growth by inhibiting the expression of these factors in endothelial tumor cells (30).
In most cases, HHE responds to treatment with steroids and/or propanol. If this first-line therapeutic strategy fails, surgery may be necessary, i.e., tumor resection, liver transplantation, or ligation of the hepatic artery (29). The treatment of HHE-associated hypothyroidism can problematic because exogenous hormones are massively converted to inactive forms. The aim of treatment in such cases is to normalize the T4 level, which is critically important particularly for the developing brain during the neonatal period. Hypothyroidism may be very severe, hence large doses of TH are necessary to normalize the T4 level. Doses of LT4 should be increased gradually until TH levels normalize. Since T4 is rapidly converted to the inactive rT3 form, combined therapy with liothyronine may be necessary. Moreover, in severe cases of HHE, parenteral LT4 administration, with or without liothyronine, may be used to bypass the liver and the hemangioma filter (31). Hypothyroidism usually resolves with involution of the tumor, and LT4 treatment may be gradually reduced as involution progresses (32, 33). Therefore, thyroid functions must be frequently monitored to ensure maintenance of the euthyroid status. Monitoring at 6-week intervals is not sufficient for a rapidly developing condition like consumptive hypothyroidism, and weekly monitoring of T4 and T3 levels should be considered particularly in the initial phase of treatment.
Consumptive hypothyroidism is diagnosed based on the detection of D3 activity in the tumor tissue of a patient with biochemical and clinical signs of hypothyroidism. However, HHE biopsy may be a risky procedure due to the high vascularity of the tumor. Hence, consumptive hypothyroidism should be suspected in each HHE patient whose TH values rapidly change especially during the proliferative phase. Usually, the increased D3 activity is mirrored by an elevation of rT3 level associated with a supraphysiological requirement for exogenous hormone and an high serum thyroglobulin level. In such patients, hypothyroidism usually resolves after medical or surgical treatment of HHE.
The differential diagnosis is between congenital or acquired hypothyroidism, TSH-secreting pituitary adenoma and a hormone resistant state. However, a rapid increase of TSH, low levels of T3 and elevated levels of rT3 associated with rapid proliferation of a vascular tumor is typical of consumptive hypothyroidism. Elevated serum thyroglobulin levels, a normal or increased thyroid uptake or the presence of thyroid gland verified by ultrasound can exclude a diagnosis of congenital hypothyroidism (34). Very rarely, a TSH-like factor may be secreted by HHE, as reported in one infant with hypothyroidism and HHE (35). In such cases, the FT4 index is normal in congenital hypothyroidism and elevated in TSH-secreting pituitary adenomas.
In conclusion, given the long-term negative effects of TH alterations on brain and on overall development, thyroid functions must be frequently monitored in patients with HHE. Conversely, since consumptive hypothyroidism has also been described in two infants with asymptomatic HHE (36, 37), HHE should be suspected in all infants with severe unexplained hypothyroidism.
Consumptive hypothyroidism has been described also in adults. The first reported case was a 21-year-old girl with a large hepatic HHE and hypothyroidism manifested with thyroid enlargement, a serum TSH concentration of 26.2 mU/L and normal serum free T4 in the absence of thyroid antibodies (38). Despite the normal FT4 and total T3 levels, serum rT3 was fivefold higher than normal without significant illness or liver failure. The hypothyroidism and goiter resolved after liver transplantation. Subsequently, Ruppe et al. (39) reported the first case of consumptive hypothyroidism associated with a “non-vascular tumor” in an adult who required an elevated dose of LT4 before resection of a large malignant fibrous tumor. Recently, Howard et al. described a 38-year-old athyreotic woman who presented elevated levels of TSH (37.3 mU/L) after the discovery of a large HHE. The patient also had low T3 and elevated rT3 serum levels (40). An interesting aspect of this case was an increase in TSH after partial hepatic resection despite hormone therapy. This may have been due to the hepatic resection itself. Indeed, regenerative processes in hepatic tissue are associated with rapid induction of D3 in post-hepatectomized mice, so one may speculate that hepatic D3 reactivation occurs in humans after liver resection, and this, in turn, causes an increase in TSH levels (24).
Can Pregnancy be Considered a Mild Form of “Consumptive Hypothyroidism”?
Pregnancy is a physiological condition characterized by changes in thyroidal economy due to a combination of pregnancy-related factors that determine the need for increased TH production. During pregnancy, thyroid function adapts to the increased concentration of T4-binding globulin, the effect of chorionic gonadotropin on maternal thyroid function, the increased requirement of iodine, and the effect of placental deiodinases on TH concentration. Fetal thyroid physiology differs greatly from maternal thyroid physiology, but the two systems interact through the placenta and amniotic fluid. These systems control the transfer of iodine and TH from the mother to the fetus (41). It is important that an appropriate amount of TH be transferred from the mother to the fetus early in gestation because high TH levels are deleterious for developing fetal tissues (42). Therefore, the transplacental supply of TH to the fetus must be finely modulated to ensure appropriate hormone levels. The deiodinases and plasma membrane transporters, which regulate the passage of TH in and out of cells, are major control mechanisms for transplacental TH passage (20).
Fetal thyroid activity begins in mid gestation, at about 18–20 weeks. TSH level peaks at 24–28 weeks, with consequent elevated T4 levels at 35–40 weeks (43). T3 levels remain low during gestation whereas D3 causes an increase in rT3 levels. High D3 expression has been found in human placental syncytiotrophoblasts and cytotrophoblasts, maternal deciduas, and endothelium of fetal vessels (28). During pregnancy, estrogens are potent inducers of D3 (14). Elevation of D3 during pregnancy is necessary to “burn off” the elevated amount of T4 in maternal circulation and acts as a barrier to maternal-fetal exchanges. The injection of 700 μg of T4 into amniotic fluid of pregnant women at term caused an increase of about 13-fold in amniotic fluid T3 concentration, and of about 30-fold in rT3 concentration measured 24 h later (44). Thus, syncytiotrophoblasts, which express high D3 levels, can control the amount and forms of THs taken up by human placenta.
Further evidence that D3 plays a relevant role in pregnancy is the finding that at 6–12 weeks of gestation there is a gradient of T4 from the mother to fetus, which is in the opposite direction of the rT3 gradient. In celomic and amniotic fluid, the concentration of rT3 is 3.8 and 15 times maternal levels, thereby indicating elevated D3 activity (45). The D3 activity in the maternal-fetal unit locally converts most of the T4 derived from the mother into rT3. Low T4 and T3 levels and a high rT3 level is typical of consumptive hypothyroidism. Thus, pregnancy could be considered a physiological form of compensated “consumptive hypothyroidism,” in which the “physiologically increased” expression of D3 increases the inactivation rate of maternal T4 thereby enhancing the need for TH.
Other Conditions Associated with Elevated D3 Levels
Consumptive hypothyroidism has also been associated with extra-hepatic HHE. In fact, two cases of consumptive hypothyroidism associated with cutaneous hemangioma have been reported in infants (40). Moreover, a case of neonatal hemangioma in the skin, associated with consumptive hypothyroidism, has been reported in an 8-month-old infant (46). The first case of consumptive hypothyroidism secondary to a large parotid hemangioma was reported in 2012 in a child with congenital hypothyroidism and hypoplastic thyroid gland (47). The first case of consumptive hypothyroidism from a non-vascular tumor was reported in 2005 in a patient with a large malignant solitary fibrous tumor, expressing elevated D3 mRNA, and proteins (39). At diagnosis, the patient was administered a supraphysiologic dose of levothyroxine prior to the discovery of the tumor, whose identification suggested that the cause of hypothyroidism could have been the paraneoplastic syndrome.
Notably, D3 is frequently overexpressed in malignant cells, which raises the question “why are circulating TH levels not always perturbed by neoplastic cells?” It is likely that, in most conditions, due to the low D3 expression in tumor cells, and more importantly, to the efficient homeostatic hypothalamic-pituitary-thyroid axis, paraneoplastic D3 is unable to perturb significantly the circulating level of TH.
Finally, tyrosine kinase inhibitors have been recently proposed as therapeutic options for patients with recurrent non-resectable radio-resistant thyroid cancer. In some of these patients undergoing LT4 replacement therapy, treatment with these drugs has been associated with an increase in TSH level. This increase has been associated with the induction (probably at hepatic level) of D3 and, the consequent need to increase the amount of T4 in replacement therapy (48).
Conclusion
In conclusion, increased catabolism of TH by increased levels of D3 can cause a rare form of acquired hypothyroidism. While this rare syndrome was initially described in association with massive hemangiomas in infants, it was later recognized also in adults, and, in some cases, as a consequence of non-vascular tumors. Although this condition is usually reversible, it requires prompt recognition and immediate TH replacement therapy usually at higher doses than required in other types of hypothyroidism. Furthermore, given that D3 reactivation in tumors is a rather common event in the neoplastic context in vitro, and that D3 increase may be a side effect of new pharmacological treatments, it is conceivable that consumptive hypothyroidism may be more frequent than expected. For the clinician, this form of hypothyroidism requires particular attention given its refractory nature toward replacement therapy and the risk of mental and growth retardation in neonatal patients.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Jean Ann Gilder (Scientific Communication Srl., Naples, Italy) for editing the text.
References
1. Fisher DA. Clinical review 19: management of congenital hypothyroidism. J Clin Endocrinol Metab (1991) 72(3):523–9. doi:10.1210/jcem-72-3-523
2. Alkemade A, Friesema EC, Unmehopa UA, Fabriek BO, Kuiper GG, Leonard JL, et al. Neuroanatomical pathways for thyroid hormone feedback in the human hypothalamus. J Clin Endocrinol Metab (2005) 90(7):4322–34. doi:10.1210/jc.2004-2567
3. Visser WE, Friesema EC, Jansen J, Visser TJ. Thyroid hormone transport in and out of cells. Trends Endocrinol Metab (2008) 19(2):50–6. doi:10.1016/j.tem.2007.11.003
4. Heuer H, Maier MK, Iden S, Mittag J, Friesema EC, Visser TJ, et al. The monocarboxylate transporter 8 linked to human psychomotor retardation is highly expressed in thyroid hormone-sensitive neuron populations. Endocrinology (2005) 146(4):1701–6. doi:10.1210/en.2004-1179
5. Cheng SY, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocr Rev (2010) 31(2):139–70. doi:10.1210/er.2009-0007
6. Flamant F, Gauthier K. Thyroid hormone receptors: the challenge of elucidating isotype-specific functions and cell-specific response. Biochim Biophys Acta (2013) 1830(7):3900–7. doi:10.1016/j.bbagen.2012.06.003
7. Marsili A, Zavacki AM, Harney JW, Larsen PR. Physiological role and regulation of iodothyronine deiodinases: a 2011 update. J Endocrinol Invest (2011) 34(5):395–407. doi:10.3275/7615
8. Dentice M, Marsili A, Zavacki A, Larsen PR, Salvatore D. The deiodinases and the control of intracellular thyroid hormone signaling during cellular differentiation. Biochim Biophys Acta (2013) 1830(7):3937–45. doi:10.1016/j.bbagen.2012.05.007
9. Salvatore D, Low SC, Berry M, Maia AL, Harney JW, Croteau W, et al. Type 3 lodothyronine deiodinase: cloning, in vitro expression, and functional analysis of the placental selenoenzyme. J Clin Invest (1995) 96(5):2421–30. doi:10.1172/JCI118299
10. Kuiper GG, Wassen F, Klootwijk W, Van Toor H, Kaptein E, Visser TJ. Molecular basis for the substrate selectivity of cat type I iodothyronine deiodinase. Endocrinology (2003) 144(12):5411–21. doi:10.1210/en.2003-0728
11. Charalambous M, Hernandez A. Genomic imprinting of the type 3 thyroid hormone deiodinase gene: regulation and developmental implications. Biochim Biophys Acta (2013) 1830(7):3946–55. doi:10.1016/j.bbagen.2012.03.015
12. Curcio-Morelli C, Gereben B, Zavacki AM, Kim BW, Huang S, Harney JW, et al. In vivo dimerization of types 1, 2, and 3 iodothyronine selenodeiodinases. Endocrinology (2003) 144(3):937–46. doi:10.1210/en.2002-220960
13. Baqui M, Botero D, Gereben B, Curcio C, Harney JW, Salvatore D, et al. Human type 3 iodothyronine selenodeiodinase is located in the plasma membrane and undergoes rapid internalization to endosomes. J Biol Chem (2003) 278(2):1206–11. doi:10.1074/jbc.M210266200
14. Wasco EC, Martinez E, Grant KS, St Germain EA, St Germain DL, Galton VA. Determinants of iodothyronine deiodinase activities in rodent uterus. Endocrinology (2003) 144(10):4253–61. doi:10.1210/en.2003-0490
15. Dentice M, Luongo C, Huang S, Ambrosio R, Elefante A, Mirebeau-Prunier D, et al. Sonic hedgehog-induced type 3 deiodinase blocks thyroid hormone action enhancing proliferation of normal and malignant keratinocytes. Proc Natl Acad Sci U S A (2007) 104(36):14466–71. doi:10.1073/pnas.0706754104
16. Ferone G, Mollo MR, Thomason HA, Antonini D, Zhou H, Ambrosio R, et al. p63 Control of desmosome gene expression and adhesion is compromised in AEC syndrome. Hum Mol Genet (2013) 22(3):531–43. doi:10.1093/hmg/dds464
17. Escobar-Morreale HF, Obregón MJ, Hernandez A, Escobar del Rey F, Morreale de Escobar G. Regulation of iodothyronine deiodinase activity as studied in thyroidectomized rats infused with thyroxine or triiodothyronine. Endocrinology (1997) 138(6):2559–68. doi:10.1210/en.138.6.2559
18. Bianco AC, Salvatore D, Gereben B, Berry MJ, Larsen PR. Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr Rev (2002) 23(1):38–89. doi:10.1210/er.23.1.38
19. Hernandez A, Fiering S, Martinez E, Galton VA, St Germain D. The gene locus encoding iodothyronine deiodinase type 3 (Dio3) is imprinted in the fetus and expresses antisense transcripts. Endocrinology (2002) 143(11):4483–6. doi:10.1210/en.2002-220800
20. Kurlak LO, Mistry HD, Kaptein E, Visser TJ, Broughton Pipkin F. Thyroid hormones and their placental deiodination in normal and pre-eclamptic pregnancy. Placenta (2013) 34(5):395–400. doi:10.1016/j.placenta.2013.02.009
21. Huang SA, Dorfman DM, Genest DR, Salvatore D, Larsen PR. Type 3 iodothyronine deiodinase is highly expressed in the human uteroplacental unit and in fetal epithelium. J Clin Endocrinol Metab (2003) 88(3):1384–8. doi:10.1210/jc.2002-021291
22. Dentice M, Salvatore D. Deiodinases: the balance of thyroid hormone: local impact of thyroid hormone inactivation. J Endocrinol (2011) 209(3):273–82. doi:10.1530/JOE-11-0002
23. Boelen A, Kwakkel J, Alkemade A, Renckens R, Kaptein E, Kuiper G, et al. Induction of type 3 deiodinase activity in inflammatory cells of mice with chronic local inflammation. Endocrinology (2005) 146(12):5128–34. doi:10.1210/en.2005-0608
24. Kester MH, Toussaint MJ, Punt CA, Matondo R, Aarnio AM, Darras VM, et al. Large induction of type III deiodinase expression after partial hepatectomy in the regenerating mouse and rat liver. Endocrinology (2009) 150(1):540–5. doi:10.1210/en.2008-0344
25. Dentice M, Luongo C, Ambrosio R, Sibilio A, Casillo A, Iaccarino A, et al. Beta-catenin regulates deiodinase levels and thyroid hormone signaling in colon cancer cells. Gastroenterology (2012) 143(4):1037–47. doi:10.1053/j.gastro.2012.06.042
26. Kester MH, Kuiper GG, Versteeg R, Visser TJ. Regulation of type III iodothyronine deiodinase expression in human cell lines. Endocrinology (2006) 147(12):5845–54. doi:10.1210/en.2006-0590
27. Kress E, Samarut J, Plateroti M. Thyroid hormones and the control of cell proliferation or cell differentiation: paradox or duality? Mol Cell Endocrinol (2009) 313(1–2):36–49. doi:10.1016/j.mce.2009.08.028
28. Huang SA, Tu HM, Harney JW, Venihaki M, Butte AJ, Kozakewich HP, et al. Severe hypothyroidism caused by type 3 iodothyronine deiodinase in infantile hemangiomas. N Engl J Med (2000) 343(3):185–9. doi:10.1056/NEJM200007203430305
29. Christison-Lagay ER, Burrows PE, Alomari A, Dubois J, Kozakewich HP, Lane TS, et al. Hepatic hemangiomas: subtype classification and development of a clinical practice algorithm and registry. J Pediatr Surg (2007) 42(1):62–7. doi:10.1016/j.jpedsurg.2006.09.041 discussion 67–8,
30. Vergine G, Marsciani A, Pedini A, Brocchi S, Marsciani M, Desiderio E, et al. Efficacy of propranolol treatment in thyroid dysfunction associated with severe infantile hepatic hemangioma. Horm Res Paediatr (2012) 78(4):256–60. doi:10.1159/000337253
31. Peters C, Langham S, Mullis PE, Dattani MT. Use of combined liothyronine and thyroxine therapy for consumptive hypothyroidism associated with hepatic haemangiomas in infancy. Horm Res Paediatr (2010) 74(2):149–52. doi:10.1159/000281884
32. Güven A, Aygun C, Ince H, Aydin M, Pinarli FG, Baysal K, et al. Severe hypothyroidism caused by hepatic hemangioendothelioma in an infant of a diabetic mother. Horm Res (2005) 63(2):86–9. doi:10.1159/000083879
33. Mouat F, Evans HM, Cutfield WS, Hofman PL, Jefferies C. Massive hepatic hemangioendothelioma and consumptive hypothyroidism. J Pediatr Endocrinol Metab (2008) 21(7):701–3. doi:10.1515/JPEM.2008.21.7.701
34. Jassam N, Visser TJ, Brisco T, Bathia D, McClean P, Barth JH. Consumptive hypothyroidism: a case report and review of the literature. Ann Clin Biochem (2011) 48(Pt 2):186–9. doi:10.1258/acb.2010.010170
35. Ayling RM, Davenport M, Hadzic N, Metcalfe R, Buchanan CR, Howard ER, et al. Hepatic hemangioendothelioma associated with production of humoral thyrotropin-like factor. J Pediatr (2001) 138(6):932–5. doi:10.1067/mpd.2001.113104
36. Mason KP, Koka BV, Eldredge EA, Fishman SJ, Burrows PE. Perioperative considerations in a hypothyroid infant with hepatic haemangioma. Paediatr Anaesth (2001) 11(2):228–32. doi:10.1046/j.1460-9592.2001.00624.x
37. Cho YH, Taplin C, Mansour A, Howman-Giles R, Hardwick R, Lord D, et al. Case report: consumptive hypothyroidism consequent to multiple infantile hepatic haemangiomas. Curr Opin Pediatr (2008) 20(2):213–5. doi:10.1097/MOP.0b013e3282f409c3
38. Huang SA, Fish SA, Dorfman DM, Salvatore D, Kozakewich HP, Mandel SJ, et al. A 21-year-old woman with consumptive hypothyroidism due to a vascular tumor expressing type 3 iodothyronine deiodinase. J Clin Endocrinol Metab (2002) 87(10):4457–61. doi:10.1210/jc.2002-020627
39. Ruppe MD, Huang SA, Jan de Beur SM. Consumptive hypothyroidism caused by paraneoplastic production of type 3 iodothyronine deiodinase. Thyroid (2005) 15(12):1369–72. doi:10.1089/thy.2005.15.1369
40. Howard D, La Rosa FG, Huang S, Salvatore D, Mulcahey M, Sang-Lee J, et al. Consumptive hypothyroidism resulting from hepatic vascular tumors in an athyreotic adult. J Clin Endocrinol Metab (2011) 96(7):1966–70. doi:10.1210/jc.2010-2104
41. Mandel SJ, Larsen PR, Seely EW, Brent GA. Increased need for thyroxine during pregnancy in women with primary hypothyroidism. N Engl J Med (1990) 323(2):91–6. doi:10.1056/NEJM199007123230204
42. Parkes IL, Schenker JG, Shufaro Y. Thyroid disorders during pregnancy. Gynecol Endocrinol (2012) 28(12):993–8. doi:10.3109/09513590.2012.692001
43. Thorpebeeston JG, Nicolaides KH, Mcgregor AM. Fetal thyroid-function. Thyroid (1992) 2(3):207–17. doi:10.1089/thy.1992.2.207
44. Klein AH, Hobel CJ, Sack J, Fisher DA. Effect of intraamniotic fluid thyroxine injection on fetal serum and amniotic fluid iodothyronine concentrations. J Clin Endocrinol Metab (1978) 47(5):1034–7. doi:10.1210/jcem-47-5-1034
45. Contempré B, Jauniaux E, Calvo R, Jurkovic D, Campbell S, de Escobar GM. Detection of thyroid hormones in human embryonic cavities during the first trimester of pregnancy. J Clin Endocrinol Metab (1993) 77(6):1719–22. doi:10.1210/jc.77.6.1719
46. Metwalley KA, Farghaly HS. Consumptive hypothyroidism in an Egyptian baby with benign neonatal hemangiomatosis: a case report. J Med Case Rep (2013) 7(1):48. doi:10.1186/1752-1947-7-48
47. Vigone MC, Cortinovis F, Rabbiosi S, Di Frenna M, Passoni A, Persani L, et al. Difficult treatment of consumptive hypothyroidism in a child with massive parotid hemangioma. J Pediatr Endocrinol Metab (2012) 25(1-2):153–5. doi:10.1515/jpem.2011.438
48. Kappers MH, van Esch JH, Smedts FM, de Krijger RR, Eechoute K, Mathijssen RH, et al. Sunitinib-induced hypothyroidism is due to induction of type 3 deiodinase activity and thyroidal capillary regression. J Clin Endocrinol Metab (2011) 96(10):3087–94. doi:10.1210/jc.2011-1172
49. Kalpatthi R, Germak J, Mizelle K, Yeager N. Thyroid abnormalities in infantile hepatic hemangioendothelioma. Pediatr Blood Cancer (2007) 49(7):1021–4. doi:10.1002/pbc.20801
50. Lee TC, Barshes NR, Agee EE, O’Mahony CA, Karpen SJ, Carter BA, et al. Resolution of medically resistant hypothyroidism after liver transplantation for hepatic hemangioendothelioma. J Pediatr Surg (2006) 41(10):1783–5. doi:10.1016/j.jpedsurg.2006.05.037
51. Bessho K, Etani Y, Ichimori H, Miyoshi Y, Namba N, Yoneda A, et al. Increased type 3 iodothyronine deiodinase activity in a regrown hepatic hemangioma with consumptive hypothyroidism. Eur J Pediatr (2010) 169(2):215–21. doi:10.1007/s00431-009-1009-x
52. Konrad D, Ellis G, Perlman K. Spontaneous regression of severe acquired infantile hypothyroidism associated with multiple liver hemangiomas. Pediatrics (2003) 112(Pt 1):1424–6. doi:10.1542/peds.112.6.1424
53. Balazs AE, Athanassaki I, Gunn SK, Tatevian N, Huang SA, Haymond MW, et al. Rapid resolution of consumptive hypothyroidism in a child with hepatic hemangioendothelioma following liver transplantation. Ann Clin Lab Sci (2007) 37(3):280–4.
Keywords: deiodinase, thyroid hormones, hypothyroidism, thyroid gland, thyroid neoplasms
Citation: Luongo C, Trivisano L, Alfano F and Salvatore D (2013) Type 3 deiodinase and consumptive hypothyroidism: a common mechanism for a rare disease. Front. Endocrinol. 4:115. doi: 10.3389/fendo.2013.00115
Received: 21 July 2013; Accepted: 18 August 2013;
Published online: 04 September 2013.
Edited by:
P. Reed Larsen, Harvard Medical School, USAReviewed by:
Salvatore Benvenga, Sezione di Endocrinologia, ItalyStephen Albert Huang, Boston Children’s Hospital, USA
Copyright: © 2013 Luongo, Trivisano, Alfano and Salvatore. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Domenico Salvatore, Department of Clinical Medicine and Surgery, University of Naples “Federico II,” Via S. Pansini 5, 80131 Naples, Italy e-mail:ZG9tc2FsdmFAdW5pbmEuaXQ=