

- Department of Neuroscience, University of Wisconsin, Madison, WI, USA
Synaptotagmins (syts) are abundant, evolutionarily conserved integral membrane proteins that play essential roles in regulated exocytosis in nervous and endocrine systems. There are at least 17 syt isoforms in mammals, all with tandem C-terminal C2 domains with highly variable capacities for Ca2+ binding. Many syts play roles in neurotransmitter release or hormone secretion or both, and a growing body of work supports a role for some syts as Ca2+ sensors of exocytosis. Work in many types of endocrine cells has documented the presence of a number of syt isoforms on dense-core vesicles containing various hormones. Syts can influence the kinetics of exocytotic fusion pores and the choice of release mode between kiss-and-run and full-fusion. Vesicles harboring different syt isoforms can preferentially undergo distinct modes of exocytosis with different forms of stimulation. The diverse properties of syt isoforms enable these proteins to shape Ca2+ sensing in endocrine cells, thus contributing to the regulation of hormone release and the organization of complex endocrine functions.
Introduction
Nature employs the same basic molecular machinery for the release of both hormones and neurotransmitters (1–4). Several protein families function broadly in regulated exocytosis, including SNAREs, synaptotagmins (syts), and complexins (5–7). The rich molecular diversity within these families provides a platform for variations in the release process, enabling different cell types to tune and tailor release by blending the different molecular variants of the fusion apparatus. In this way endocrine cells can optimize secretory responses elicited by widely varying signals that are unique to each system. The rates of hormone release from different cell types vary by over two orders of magnitude (8). Endocrine cells secrete an extraordinary variety of hormones by exocytosis from dense-core vesicles (DCVs), and DCVs usually co-package collections of molecules ranging in size from small catecholamines to large peptides (9, 10). The nature of the stimulus can determine which packaged molecules will be released (11, 12), and a single DCV can release both catecholamines and neuropeptides simultaneously (13). Cells also can sort different hormones to different DCVs (14, 15). The diverse forms of storage and release raise questions as to how the exocytotic machinery can be called upon to modulate release rates and enable different types of Ca2+ signals to trigger the release of different substances from the same cell or even the same vesicle. One can hope to gain a better understanding of these problems by studying functional variations within the diverse families of exocytotic proteins.
Exocytosis proceeds through a sequence of distinct steps. Release can start once a fusion pore has formed to create a continuous fluid pathway from the vesicle interior to the extracellular space. The fusion pore is initially very small and can allow only small molecules such as norepinephrine to pass, but after it expands larger molecules such as chromogranins, insulin, and glucagon can escape. It is well established that DCVs of endocrine cells undergo two modes of exocytosis, kiss-and-run, and full-fusion (16–19). In kiss-and-run the pore opens transiently, and closes so that vesicles maintain their integrity as they retreat from the plasma membrane. The fusion pore formed during kiss-and-run can act as a filter to expel small molecules and retain larger molecules. The small molecules can be rapidly restored by vesicular transporters so DCVs can recycle. By contrast, in full-fusion the pore expands and the vesicle membrane collapses into the plasma membrane. After full-fusion a DCV is lost; replacing these DCVs requires the entire production sequence beginning with peptide hormone translation in the endoplasmic reticulum, DCV processing in the trans-Golgi network, and maturation (20). The choice between full-fusion and kiss-and-run thus plays a decisive role in determining the fate of the DCV as well as in selecting what molecules are released.
Ca2+ triggers exocytosis by binding to syt (21–23), but Ca2+ can also influence the kinetics of fusion pores in a variety of ways (24–26), thus raising the possibility that syt isoforms play roles not only in triggering exocytosis, but also in regulating release in subtle ways. Syts regulate the stability of fusion pores (26–30), and fusion pore stability in turn is intimately related to release mode, with the relative frequency of kiss-and-run versus full-fusion described quantitatively in terms of the kinetic rates of fusion pore closure and dilation (26). There are many examples of syts influencing the choice between kiss-and-run and full-fusion (26, 29, 31–33). Syts are conserved proteins with well established functions in membrane trafficking and exocytosis. They contain two C2 domains, and in syt 1, the first isoform to be characterized, each C2 domain binds two or three Ca2+ ions through interactions with key aspartate side chains (34). Syts interact with lipid membranes containing specific phospholipids including phosphatidylserine and phosphatidylinositol-4,5-biphosphate, as well as SNARE proteins. These interactions are regulated by Ca2+, but it remains unclear how binding to these targets enables syts to serve as Ca2+ sensors in exocytosis (21, 22, 30).
The mouse and human genomes encode 17 syt isoforms (35), and the functional significance of syt diversity is a subject of considerable interest (22, 36, 37). Syts vary widely in their Ca2+-dependent lipid binding, with syts 1, 2, and 3 binding rapidly, syts 5, 6, 9, and 10 binding at an intermediate rate, and syt 7 binding slowly (38). The isoforms also vary in their activity in Ca2+-stimulated liposome fusion: Ca2+ concentrations that trigger responses range widely between the isoforms, and many syts completely fail to confer Ca2+ sensitivity on SNARE-mediated liposome fusion in vitro (32, 39, 40). Variations in Ca2+ sensitivity and fusion pore regulation make these proteins ideal candidates for modifying the release apparatus and tuning responses as Ca2+ concentrations rise and fall in distinct spatiotemporal patterns. Here, we survey relevant work on syt functions in hormone release, and where possible draw parallels between syt isoform properties, Ca2+ signals, and forms of secretion.
Syt Function in Endocrine Cells
Synaptotagmins appear broadly throughout the endocrine system, with essentially every cell type examined expressing multiple isoforms (Table 1). Expression varies between cell types and not all reports agree. No effort was made here to distinguish between isoforms untested versus undetected, and the number of isoforms found in endocrine cells will grow as reagents are developed and improved, and as proteomics methods advance. A large body of work supports the expression of syts 1, 4, 7, and 9 in many endocrine systems, and it is remarkable that these four molecules appear in so many different cell types. Syts 1, 4, and 7 are ancient, conserved proteins distributed widely through metazoan genomes (35). These isoforms presumably perform fundamental biological functions, and evidence is accumulating for their roles in a wide range of endocrine and non-endocrine systems.
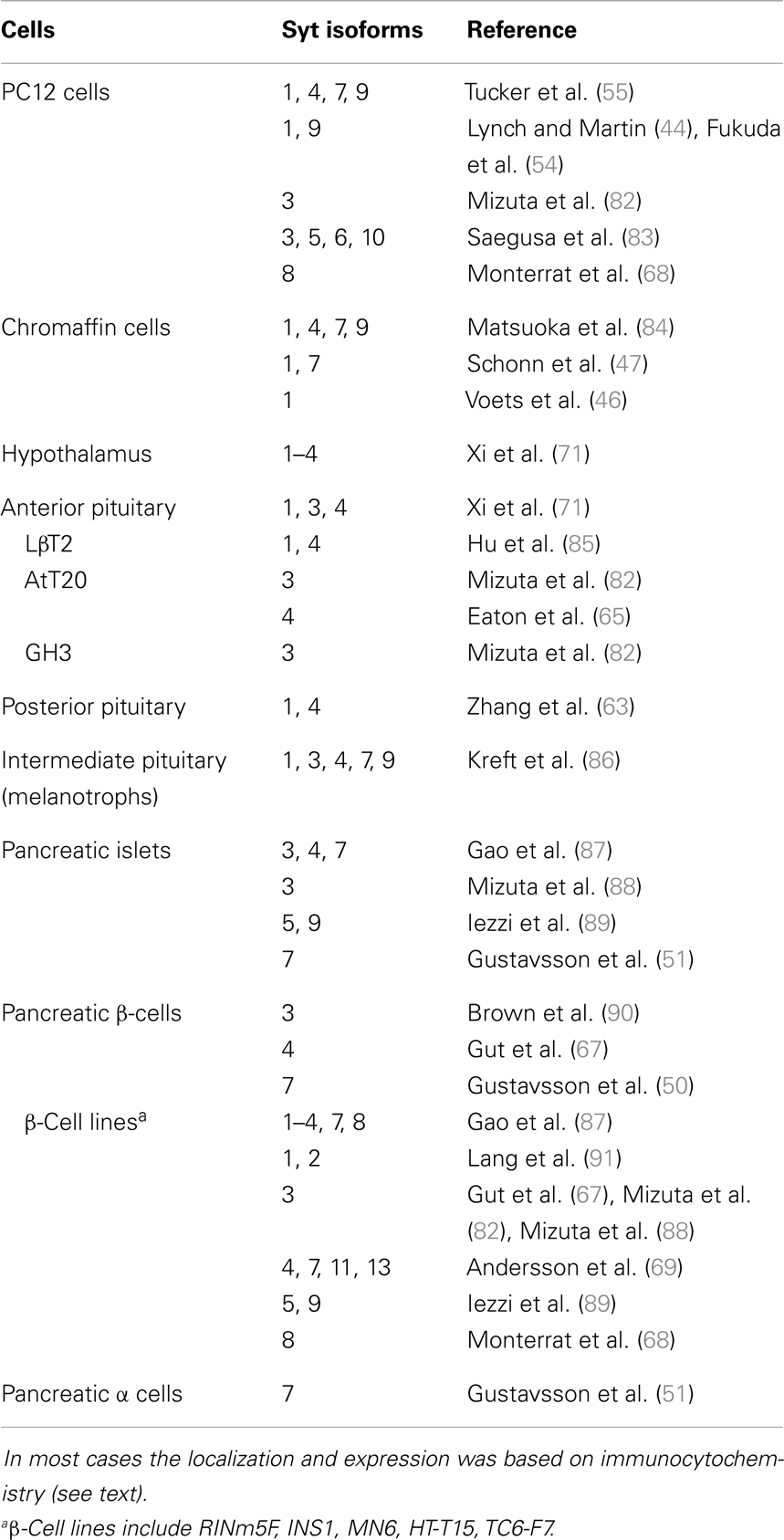
Table 1. Syt isoform expression in various endocrine systems.
Syt 1
Syt 1 is the most widely distributed syt isoform in nervous and endocrine systems. This low-affinity Ca2+ sensor (32, 39, 41, 42) generally triggers rapid exocytosis. The very tight temporal coupling between Ca2+ entry and fusion, within milliseconds, has prompted investigators to use the term “synchronous” to describe this form of release, particularly in the context of synaptic transmission. An early syt 1 knock-down study suggested that PC12 cells can secrete without syt 1 (43), but subsequent work showed that this was due to redundancy with another syt isoform of PC12 cells, syt 9 (33, 44). Overexpression of wild type syt 1 in PC12 cells left the overall time course of secretion unchanged, but overexpression of either wild type syt 1 or a number of syt 1 mutants altered fusion pore kinetics (26–28, 30, 45). Overexpressing syt 1 in PC12 cells also produced more kiss-and-run events than syt 7 and 9 (32).
Deletion of the syt 1 gene in mouse selectively abolished the initial rapid phase of exocytosis in chromaffin cells (33, 46, 47), but had no deleterious effects on slower Ca2+-triggered release. For a given concentration of Ca2+, exocytosis was much slower in chromaffin cells lacking syt 1 than in control cells (46). Mutation of a residue that reduces Ca2+ binding slowed exocytosis in chromaffin cells (48). In PC12 cells syt 1 sorted preferentially to smaller DCVs (32), raising the interesting possibility that hormones packaged in smaller vesicles will be released more rapidly than hormones packaged in larger vesicles.
Syt 7
In contrast to syt 1, syt 7 acts as a high affinity Ca2+ sensor (32, 39, 49). Syt 7 is more abundant on larger DCVs in PC12 cells (32). Syt 7 overexpression in PC12 cells prolonged fusion pore lifetimes more than syt 1 overexpression (30), and favored full-fusion (32). Syt 7 knock-down in zebra fish reduced delayed synaptic release, indicating it is a slow Ca2+ sensor (49). Syt 7 deletion in chromaffin cells reduced Ca2+-triggered release by 50%, also selectively impairing the slow phase of exocytosis, and deletion of both syt 1 and 7 nearly abolished Ca2+-triggered exocytosis. Thus, in chromaffin cells syt 1 and 7 are the primary Ca2+ sensors for the fast and slow kinetic phases of exocytosis, respectively (47). Syt 7 gene ablation also reduced Ca2+-triggered exocytosis of insulin secretion from pancreatic β-cells and of glucagon secretion from pancreatic α-cells (50, 51). It is intriguing that syt 7 also functions in insulin responsive cells (fat and muscle), promoting glucose uptake through Ca2+-stimulated translocation of type-4 glucose transporter to the plasma membrane (52). In syt 7 knock-out α cells, ω-conotoxin further inhibited glucagon secretion to baseline levels, revealing the presence of an N-type Ca2+ channel-dependent component of residual glucagon secretion triggered by another protein (51).
Syt 9
Syt 9 is closely related to syt 1 but exhibits intermediate Ca2+ sensitivity in fusion assays (32, 39). This protein has also been referred to as syt 5 (35, 53); here syt 9 refers to a 386 amino acid isoform. It is abundant on DCVs of PC12 cells (32, 54, 55), and overexpressing it produces fusion pore lifetimes intermediate between those seen with syt 1 and 7 (30). Down-regulating syt 9 alone produced a small, insignificant reduction of fusion rate in PC12 cells, but as noted above, because of the redundancy of syt 1 and 9 as Ca2+ sensors, both must be down-regulated to reduce secretion (33, 44). Silencing of syt 9 strongly inhibited insulin release from islet β-cells and INS-1E cells (56). However, mice with a pancreas-specific knock-out of syt 9 had normal glucose homeostasis and showed no changes in other insulin-dependent functions (57).
Ca2+ Non-Binders
Slightly more than half of the mammalian syts have non-acidic amino acids at some of the positions engaged in Ca2+ binding (35), and these isoforms fail to act as Ca2+ sensors in liposome fusion assays (40). The best characterized of these, syt 4, harbors an evolutionarily conserved serine-for-aspartate substitution at a Ca2+ ligand in the C2A domain (58). Syt 4 is widely expressed in endocrine cells and its expression rises and falls depending on electrical activity (59) and reproductive state (60). Syt 4 negatively regulates both release and Ca2+-dependent liposome fusion (28, 32, 40) and does not bind Ca2+ (61). Syt 4 overexpression reduced exocytosis in PC12 cells (28, 32, 62). Although syt 4 overexpression in PC12 cells shortened the lifetimes of fusion pores capable of dilating to full-fusion (28), another form of release was enhanced in which very small fusion pores could open exclusively as kiss-and-run events with exceptionally long lifetimes. Many but not all of the effects of syt 4 overexpression were mimicked by syt 1 harboring the serine-for-alanine Ca2+ ligand replacement seen in syt 4 (29). Syt 4 overexpression also favored kiss-and-run in MIN6 β-cells (31). Ablation of the syt 4 gene altered exocytosis and fusion pore properties in posterior pituitary nerve terminals. These results suggested that syt 4 reduced exocytosis in response to modest Ca2+ rises but enhanced exocytosis in response to large Ca2+ rises (63). Syt 4 also contributes to the maturation of DCVs (64, 65), and altering syt 4 levels changes DCV size (62, 63, 66).
The Ca2+ non-binding isoforms syt 4, 8, and 13 have been detected in insulin-secreting cells (67–69). Silencing syt 4 and 13 reduced glucose stimulated insulin secretion in INS1 cells (69). Glucose stimulated expression of the syt 8 gene in human islets and syt 8 knock-down impaired both basal and evoked insulin release (70). Since syt 8 fails to stimulate liposome fusion in a Ca2+-dependent manner (40) the precise role of syt 8 in insulin secretion remains unclear. One intriguing possibility is that syt 8 increases the relative proportion of full-fusion events so that fusion pores can grow large enough to allow insulin to escape. It is likely that the Ca2+ non-binders interact with other components of the release machinery in ways that remain to be elucidated. These interactions could allow syts to regulate release in ways that cannot as yet be explained in terms of their biochemical properties.
Reports vary regarding the expression of other syt isoforms in endocrine cells (Table 1) and little is known about their localization and functions in hormone release. Syt 2 has been reported in the hypothalamus (71) but is absent from endocrine cells, and its primary function is likely to be as a synaptic Ca2+ sensor (72, 73).
Specificity of Ca2+ Signaling
The differences in performance of syts as Ca2+ sensors gives a special significance to the spatiotemporal character of Ca2+ signals in different cell types and with different forms of stimuli. Ca2+ can enter cells through a variety of routes so that the dynamics and spatial extent of changes in cytosolic Ca2+ can vary enormously. This creates a scenario in which differences in Ca2+ sensing properties can have a major impact on responses (74). Intracellular Ca2+ rises and falls as Ca2+ enters through Ca2+ channels, diffuses away from these sources into the cytoplasm, binds Ca2+ binding proteins, and is sequestered into stores or pumped out of the cytoplasm (75–77). The opening of one voltage gated Ca2+ channel allows approximately 103 ions to enter per msec. This localized flux sets up a steep gradient to create a domain of high local Ca2+ concentration. In the immediate vicinity of the channel mouth (within tens of nanometer) Ca2+ can rise to >100 μM, which is substantially higher than the average bulk cytoplasmic level, even under conditions of intense stimulation (75, 78). These Ca2+ domains around a Ca2+ channel can form and collapse rapidly (within a few ms) so that the activation of a Ca2+ sensor will depend critically on the kinetics of Ca2+ association and dissociation. A low-affinity sensor can detect this high local Ca2+ as long as it binds with rapid kinetics. A high affinity Ca2+ sensor may fail to respond if it binds too slowly. Syt 1 has properties suited for responding to transient domains with high Ca2+. Syt 7 has properties suited for responding to modest but prolonged rises in bulk Ca2+ concentration (38).
The idea of localized Ca2+ signals leads to two types of Ca2+ concentration profile illustrated in Figure 1. When an endocrine cell fires at a moderate rate, Ca2+ domains form and collapse around a few open Ca2+ channels as shown in Figure 1A. The resulting brief period of high Ca2+ concentration will only affect vesicles near the open channels, and will preferentially activate syt 1 over slower isoforms. Basal firing rates corresponding to the resting state of an animal (feed and breed) have been shown to trigger norepinephrine release from chromaffin cells without triggering chromogranin release (11). This implicates exocytosis by kiss-and-run, which is preferentially triggered by syt 1 (32). By contrast, with vigorous electrical activity during stress (fight and flight) the bulk Ca2+ concentration will rise to moderate levels (well under 50 μM) for longer times on the order of seconds (Figure 1B). These signals will activate slow, low-affinity Ca2+ sensors such as syt 7. This form of [Ca2+] signal has been shown to trigger full-fusion (79), with release of both norepinephrine and chromogranin (11). This can be explained by the tendency of syt 7 to trigger full-fusion preferentially (32). Different spatiotemporal patterns of cytosolic Ca2+ can thus target different syt isoforms to modulate fusion kinetics. Furthermore, because syt isoforms favor different modes of exocytosis, by selectively promoting kiss-and-run or full-fusion, Ca2+ signals that activate different syts will determine the relative release of small and large molecules. The sorting of syts to different sized vesicles may also serve as a mechanism for allowing different Ca2+ signals to select different substances for release if content is also found to vary with vesicle size (32).
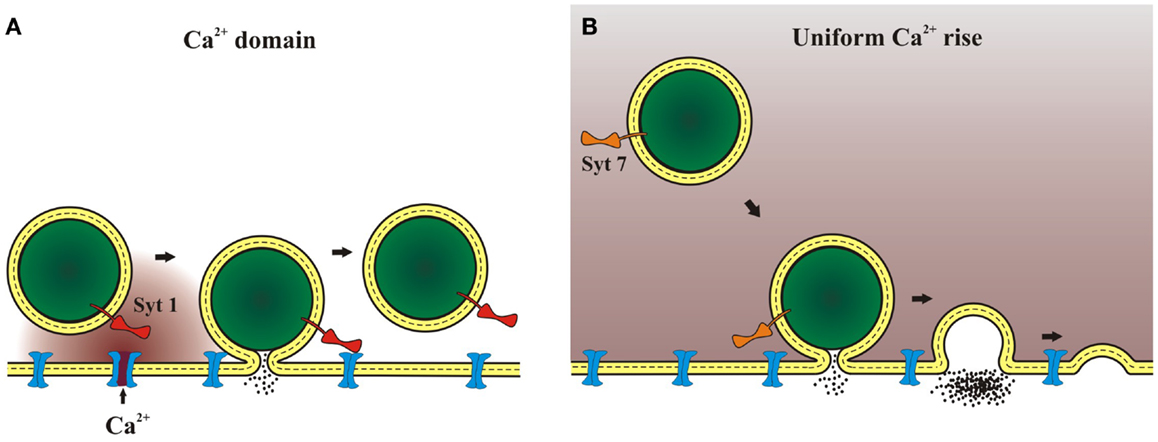
Figure 1. Ca2+ domains and release mechanisms. (A) Weak stimulation opens Ca2+ channels less frequently. One isolated channel can open and as Ca2+ flows a domain of high Ca2+ concentration will form around the channel mouth. This will lead to a highly localized Ca2+ signal that will persist for milliseconds. A Ca2+ sensor such as syt 1, with its rapid kinetics and low affinity, can be activated by a Ca2+ signal of this form. (B) Strong stimulation opens many Ca2+ channels to raise bulk Ca2+. Bulk Ca2+ can also rise as a result of Ca2+ release from internal stores. After the stimulus ends the Ca2+ gradients around individual vesicles will collapse as Ca2+ diffuses through the cytoplasm away from the membrane. This will lead to a more uniform, moderate concentration that can persist for seconds. A Ca2+ sensor such as syt 7, with its slow kinetics and high affinity, can be activated by a Ca2+ signal of this form.
Conclusion
Studies of how different syt isoforms influence the kinetics of exocytosis are starting to resolve functionally relevant distinctions in the mechanisms by which Ca2+ regulates exocytosis. More studies like these will expand our understanding of the diversity of endocrine release mechanisms, but rigorous assessments of syt isoform function in cells face a number of challenges. (1) It is important to address the expression of multiple isoforms, either by ablating endogenous proteins or varying each isoform individually with careful assessment of protein levels. Overexpression of proteins can result in high protein concentrations, possibly leading to mis-targeting and artificial functions. (2) Experiments need to address the different Ca2+ sensitivities of syt isoforms. This requires measurement and control of Ca2+ concentration. (3) Biophysical techniques for measuring exocytosis are very sensitive but often measure surrogates of release and detect events on different time scales. Amperometry measures release and can detect rapid processes but its greatest sensitivity is realized with biogenic amines, and is not nearly as powerful in the measurement of peptides. Furthermore, after content expulsion the closure of a fusion pore can no longer be detected, and the mode of release as kiss-and-run or full-fusion can no longer be ascertained. Capacitance measures membrane area, and single-vesicle capacitance steps provide strong evidence for kiss-and-run. Total internal reflectance microscopy of fluorescent tracers follows the fate of the vesicle content or membrane (80). Like capacitance recording, this method can reveal kiss-and-run but its time resolution is poor. Furthermore, the fate of fluorescent cargo can vary with the design of the fusion construct, raising concerns about experimental artifacts (81). Amperometry, capacitance, and total internal reflectance microscopy are very powerful techniques but they do not always agree and these differences can complicate interpretations and comparisons (62).
Given the functional versatility within the syt protein family, the studies to date probably have only scratched the surface in addressing the roles of syt isoforms in the complex and varied forms of hormone release. By selecting among the different syt isoforms and regulating their expression, and by sorting to different vesicles containing different hormones, cells can regulate the response to diverse forms of Ca2+ signals. The syt isoforms present on a vesicle will determine what form of Ca2+ signal will trigger fusion, how different Ca2+ signals direct the choice between kiss-and-run and full-fusion, and what proportion of small and large molecules are released.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by NIH grants NS044057 and NS072905. We thank Drs. Xuelin Lou and Edwin Chapman for helpful discussions and comments.
References
1. De Camilli P, Jahn R. Pathways to regulated exocytosis in neurons. Annu Rev Physiol (1990) 52:625–45. doi:10.1146/annurev.ph.52.030190.003205
2. Walch-Solimena C, Takei K, Marek KL, Midyett K, Südhof TC, De Camilli P, et al. Synaptotagmin: a membrane constituent of neuropeptide-containing large dense-core vesicles. J Neurosci (1993) 13:3895–903.
3. Brunner Y, Coute Y, Iezzi M, Foti M, Fukuda M, Hochstrasser DF, et al. Proteomics analysis of insulin secretory granules. Mol Cell Proteomics (2007) 6:1007–17. doi:10.1074/mcp.M600443-MCP200
4. Wegrzyn JL, Bark SJ, Funkelstein L, Mosier C, Yap A, Kazemi-Esfarjani P, et al. Proteomics of dense core secretory vesicles reveal distinct protein categories for secretion of neuroeffectors for cell-cell communication. J Proteome Res (2010) 9:5002–24. doi:10.1021/pr1003104
5. Sudhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science (2009) 323:474–7. doi:10.1126/science.1161748
6. Jahn R, Scheller RH. SNAREs – engines for membrane fusion. Nat Rev Mol Cell Biol (2006) 7:631–43. doi:10.1038/nrm2002
7. Jahn R, Fasshauer D. Molecular machines governing exocytosis of synaptic vesicles. Nature (2012) 490:201–7. doi:10.1038/nature11320
8. Martin TF. Tuning exocytosis for speed: fast and slow modes. Biochim Biophys Acta (2003) 1641:157–65. doi:10.1016/S0167-4889(03)00093-4
9. Winkler H, Westhead E. The molecular organization of adrenal chromaffin granules. Neuroscience (1980) 5:1803–23. doi:10.1016/0306-4522(80)90031-7
10. Hokfelt T, Broberger C, Xu ZQ, Sergeyev V, Ubink R, Diez M. Neuropeptides – an overview. Neuropharmacology (2000) 39:1337–56. doi:10.1016/S0028-3908(00)00010-1
11. Fulop T, Radabaugh S, Smith C. Activity-dependent differential transmitter release in mouse adrenal chromaffin cells. J Neurosci (2005) 25:7324–32. doi:10.1523/JNEUROSCI.2042-05.2005
12. MacDonald PE, Braun M, Galvanovskis J, Rorsman P. Release of small transmitters through kiss-and-run fusion pores in rat pancreatic beta cells. Cell Metab (2006) 4:283–90. doi:10.1016/j.cmet.2006.08.011
13. Whim MD. Near simultaneous release of classical and peptide cotransmitters from chromaffin cells. J Neurosci (2006) 26:6637–42. doi:10.1523/JNEUROSCI.5100-05.2006
14. Perello M, Stuart R, Nillni EA. Prothyrotropin-releasing hormone targets its processing products to different vesicles of the secretory pathway. J Biol Chem (2008) 283:19936–47. doi:10.1074/jbc.M800732200
15. Sossin WS, Scheller RH. Biosynthesis and sorting of neuropeptides. Curr Opin Neurobiol (1991) 1:79–83. doi:10.1016/0959-4388(91)90013-W
16. Alvarez de Toledo G, Fernández-Chacón R, Fernández JM. Release of secretory products during transient vesicle fusion. Nature (1993) 363:554–8. doi:10.1038/363554a0
17. Fernandez JM, Neher E, Gomperts BD. Capacitance measurements reveal stepwise fusion events in degranulating mast cells. Nature (1984) 312:453–5. doi:10.1038/312453a0
18. Klyachko VA, Jackson MB. Capacitance steps and fusion pores of small and large-dense-core vesicles in nerve terminals. Nature (2002) 418:89–92. doi:10.1038/nature00852
19. Neher E, Marty A. Discrete changes of cell membrane capacitance observed under conditions of enhanced secretion in bovine adrenal chromaffin cells. Proc Natl Acad Sci U S A (1982) 79:6712–6. doi:10.1073/pnas.79.21.6712
20. Kim T, Gondré-Lewis MC, Arnaoutova I, Loh YP. Dense-core secretory granule biogenesis. Physiology (Bethesda) (2006) 21:124–33. doi:10.1152/physiol.00043.2005
21. Augustine GJ. How does calcium trigger neurotransmitter release? Curr Opin Neurobiol (2001) 11:320–6. doi:10.1016/S0959-4388(00)00214-2
22. Chapman ER. How does synaptotagmin trigger neurotransmitter release? Annu Rev Biochem (2008) 77:615–41. doi:10.1146/annurev.biochem.77.062005.101135
23. Koh TW, Bellen HJ. Synaptotagmin I, a Ca2+ sensor for neurotransmitter release. Trends Neurosci (2003) 26:413–22. doi:10.1016/S0166-2236(03)00195-4
24. Ales E, Tabares L, Poyato JM, Valero V, Lindau M, Alvarez de Toledo G. High calcium concentrations shift the mode of exocytosis to the kiss-and-run mechanism. Nat Cell Biol (1999) 1:40–4. doi:10.1038/9012
25. Fernandez-Chacon R, Alvarez de Toledo G. Cytosolic calcium facilitates release of secretory products after exocytotic vesicle fusion. FEBS Lett (1995) 363:221–5. doi:10.1016/0014-5793(95)00319-5
26. Wang CT, Bai J, Chang PY, Chapman ER, Jackson MB. Synaptotagmin-Ca2+ triggers two sequential steps in regulated exocytosis in rat PC12 cells: fusion pore opening and fusion pore dilation. J Physiol (2006) 570:295–307.
27. Bai J, Wang CT, Richards DA, Jackson MB, Chapman ER. Fusion pore dynamics are regulated by synaptotagmin*t-SNARE interactions. Neuron (2004) 41:929–42. doi:10.1016/S0896-6273(04)00117-5
28. Wang CT, Grishanin R, Earles CA, Chang PY, Martin TF, Chapman ER, et al. Synaptotagmin modulation of fusion pore kinetics in regulated exocytosis of dense-core vesicles. Science (2001) 294:1111–5. doi:10.1126/science.1064002
29. Wang CT, Lu JC, Bai J, Chang PY, Martin TF, Chapman ER, et al. Different domains of synaptotagmin control the choice between kiss-and-run and full fusion. Nature (2003) 424:943–7. doi:10.1038/nature01857
30. Zhang Z, Hui E, Chapman ER, Jackson MB. Regulation of exocytosis and fusion pores by synaptotagmin-effector interactions. Mol Biol Cell (2010) 21:2821–31. doi:10.1091/mbc.E10-04-0285
31. Tsuboi T, Rutter GA. Multiple forms of “kiss-and-run” exocytosis revealed by evanescent wave microscopy. Curr Biol (2003) 13:563–7. doi:10.1016/S0960-9822(03)00176-3
32. Zhang Z, Wu Y, Wang Z, Dunning FM, Rehfuss J, Ramanan D, et al. Release mode of large and small dense-core vesicles specified by different synaptotagmin isoforms in PC12 cells. Mol Biol Cell (2011) 22:2324–36. doi:10.1091/mbc.E11-02-0159
33. Zhu D, Zhou W, Liang T, Yang F, Zhang RY, Wu ZX, et al. Synaptotagmin I and IX function redundantly in controlling fusion pore of large dense core vesicles. Biochem Biophys Res Commun (2007) 361:922–7. doi:10.1016/j.bbrc.2007.07.083
34. Ubach J, Zhang X, Shao X, Sudhof TC, Rizo J. Ca2+ binding to synaptotagmin: how many Ca2+ ions bind to the tip of a C2-domain? EMBO J (1998) 17:3921–30. doi:10.1093/emboj/17.14.3921
35. Craxton M. A manual collection of Syt, Esyt, Rph3a, Rph3al, Doc2, and Dblc2 genes from 46 metazoan genomes – an open access resource for neuroscience and evolutionary biology. BMC Genomics (2010) 11:37. doi:10.1186/1471-2164-11-37
36. Marqueze B, Berton F, Seagar M. Synaptotagmins in membrane traffic: which vesicles do the tagmins tag? Biochimie (2000) 82:409–20. doi:10.1016/S0300-9084(00)00220-0
37. Sudhof TC. Synaptotagmins: why so many? J Biol Chem (2002) 277:7629–32. doi:10.1074/jbc.R100052200
38. Hui E, Bai J, Wang P, Sugimori M, Llinas RR, Chapman ER. Three distinct kinetic groupings of the synaptotagmin family: candidate sensors for rapid and delayed exocytosis. Proc Natl Acad Sci U S A (2005) 102:5210–4. doi:10.1073/pnas.0500941102
39. Bhalla A, Tucker WC, Chapman ER. Synaptotagmin isoforms couple distinct ranges of Ca2+, Ba2+, and Sr2+ concentration to SNARE-mediated membrane fusion. Mol Biol Cell (2005) 16:4755–64. doi:10.1091/mbc.E05-04-0277
40. Bhalla A, Chicka MC, Chapman ER. Analysis of the synaptotagmin family during reconstituted membrane fusion. Uncovering a class of inhibitory isoforms. J Biol Chem (2008) 283:21799–807. doi:10.1074/jbc.M709628200
41. Li C, Ullrich B, Zhang JZ, Anderson RG, Brose N, Südhof TC. Ca(2+)-dependent and -independent activities of neural and non-neural synaptotagmins. Nature (1995) 375:594–9. doi:10.1038/375594a0
42. Sugita S, Shin OH, Han W, Lao Y, Sudhof TC. Synaptotagmins form a hierarchy of exocytotic Ca(2+) sensors with distinct Ca(2+) affinities. EMBO J (2002) 21:270–80. doi:10.1093/emboj/21.3.270
43. Shoji-Kasai Y, Yoshida A, Sato K, Hoshino T, Ogura A, Kondo S, et al. Neurotransmitter release from synaptotagmin-deficient clonal variants of PC12 cells. Science (1992) 256:1821–3.
44. Lynch KL, Martin TF. Synaptotagmins I and IX function redundantly in regulated exocytosis but not endocytosis in PC12 cells. J Cell Sci (2007) 120:617–27. doi:10.1242/jcs.03375
45. Lynch KL, Gerona RR, Kielar DM, Martens S, McMahon HT, Martin TF. Synaptotagmin-1 utilizes membrane bending and SNARE binding to drive fusion pore expansion. Mol Biol Cell (2008) 19:5093–103. doi:10.1091/mbc.E08-03-0235
46. Voets T, Moser T, Lund PE, Chow RH, Geppert M, Sudhof TC, et al. Intracellular calcium dependence of large dense-core vesicle exocytosis in the absence of synaptotagmin I. Proc Natl Acad Sci U S A (2001) 98:11680–5. doi:10.1073/pnas.201398798
47. Schonn JS, Maximov A, Lao Y, Sudhof TC, Sorensen JB. Synaptotagmin-1 and -7 are functionally overlapping Ca2+ sensors for exocytosis in adrenal chromaffin cells. Proc Natl Acad Sci U S A (2008) 105:3998–4003. doi:10.1073/pnas.0712373105
48. Sorensen JB, Fernandez-Chacon R, Sudhof TC, Neher E. Examining synaptotagmin 1 function in dense core vesicle exocytosis under direct control of Ca2+. J Gen Physiol (2003) 122:265–76. doi:10.1085/jgp.200308855
49. Wen H, Linhoff MW, McGinley MJ, Li GL, Corson GM, Mandel G, et al. Distinct roles for two synaptotagmin isoforms in synchronous and asynchronous transmitter release at zebrafish neuromuscular junction. Proc Natl Acad Sci U S A (2010) 107:13906–11. doi:10.1073/pnas.1008598107
50. Gustavsson N, Lao Y, Maximov A, Chuang JC, Kostromina E, Repa JJ, et al. Impaired insulin secretion and glucose intolerance in synaptotagmin-7 null mutant mice. Proc Natl Acad Sci U S A (2008) 105:3992–7. doi:10.1073/pnas.0711700105
51. Gustavsson N, Wei SH, Hoang DN, Lao Y, Zhang Q, Radda GK, et al. Synaptotagmin-7 is a principal Ca2+ sensor for Ca2+-induced glucagon exocytosis in pancreas. J Physiol (2009) 587:1169–78. doi:10.1113/jphysiol.2008.168005
52. Li Y, Wang P, Xu J, Gorelick F, Yamazaki H, Andrews N, et al. Regulation of insulin secretion and GLUT4 trafficking by the calcium sensor synaptotagmin VII. Biochem Biophys Res Commun (2007) 362:658–64. doi:10.1016/j.bbrc.2007.08.023
53. Fukuda M, Sagi-Eisenberg R. Confusion in the nomenclature of synaptotagmins V and IX. Calcium Bind Proteins (2008) 3:1–4.
54. Fukuda M, Kowalchyk JA, Zhang X, Martin TF, Mikoshiba K. Synaptotagmin IX regulates Ca2+-dependent secretion in PC12 cells. J Biol Chem (2002) 277:4601–4. doi:10.1074/jbc.C100588200
55. Tucker WC, Edwardson JM, Bai J, Kim HJ, Martin TF, Chapman ER. Identification of synaptotagmin effectors via acute inhibition of secretion from cracked PC12 cells. J Cell Biol (2003) 162:199–209. doi:10.1083/jcb.200302060
56. Iezzi M, Eliasson L, Fukuda M, Wollheim CB. Adenovirus-mediated silencing of synaptotagmin 9 inhibits Ca2+-dependent insulin secretion in islets. FEBS Lett (2005) 579:5241–6. doi:10.1016/j.febslet.2005.08.047
57. Gustavsson N, Wang X, Wang Y, Seah T, Xu J, Radda GK, et al. Neuronal calcium sensor synaptotagmin-9 is not involved in the regulation of glucose homeostasis or insulin secretion. PLoS One (2010) 5:e15414. doi:10.1371/journal.pone.0015414
58. von Poser C, Ichtchenko K, Shao X, Rizo J, Sudhof TC. The evolutionary pressure to inactivate. A subclass of synaptotagmins with an amino acid substitution that abolishes Ca2+ binding. J Biol Chem (1997) 272:14314–9. doi:10.1074/jbc.272.22.14314
59. Vician L, Lim IK, Ferguson G, Tocco G, Baudry M, Herschman HR. Synaptotagmin IV is an immediate early gene induced by depolarization in PC12 cells and in brain. Proc Natl Acad Sci U S A (1995) 92:2164–8. doi:10.1073/pnas.92.6.2164
60. Poopatanapong A, Teramitsu I, Byun JS, Vician LJ, Herschman HR, White SA. Singing, but not seizure, induces synaptotagmin IV in zebra finch song circuit nuclei. J Neurobiol (2006) 66:1613–29. doi:10.1002/neu.20329
61. Dai H, Shin OH, Machius M, Tomchick DR, Sudhof TC, Rizo J. Structural basis for the evolutionary inactivation of Ca2+ binding to synaptotagmin 4. Nat Struct Mol Biol (2004) 11:844–9. doi:10.1038/nsmb817
62. Zhang Z, Zhang Z, Jackson MB. Synaptotagmin IV modulation of vesicle size and fusion pores in PC12 cells. Biophys J (2010) 98:968–78. doi:10.1016/j.bpj.2009.11.024
63. Zhang Z, Bhalla A, Dean C, Chapman ER, Jackson MB. Synaptotagmin IV: a multifunctional regulator of peptidergic nerve terminals. Nat Neurosci (2009) 12:163–71. doi:10.1038/nn.2252
64. Ahras M, Otto GP, Tooze SA. Synaptotagmin IV is necessary for the maturation of secretory granules in PC12 cells. J Cell Biol (2006) 173(2):242–51. doi:10.1083/jcb.200506163
65. Eaton BA, Haugwitz M, Lau D, Moore HP. Biogenesis of regulated exocytotic carriers in neuroendocrine cells. J Neurosci (2000) 20:7334–44.
66. Wang P, Wang CT, Bai J, Jackson MB, Chapman ER. Mutations in the effector binding loops in the C2A and C2B domains of synaptotagmin I disrupt exocytosis in a nonadditive manner. J Biol Chem (2003) 278:47030–7. doi:10.1074/jbc.M306728200
67. Gut A, Kiraly CE, Fukuda M, Mikoshiba K, Wollheim CB, Lang J. Expression and localisation of synaptotagmin isoforms in endocrine beta-cells: their function in insulin exocytosis. J Cell Sci (2001) 114:1709–16.
68. Monterrat C, Boal F, Grise F, Hemar A, Lang J. Synaptotagmin 8 is expressed both as a calcium-insensitive soluble and membrane protein in neurons, neuroendocrine and endocrine cells. Biochim Biophys Acta (2006) 1763:73–81. doi:10.1016/j.bbamcr.2005.11.008
69. Andersson SA, Olsson AH, Esguerra JL, Heimann E, Ladenvall C, Edlund A, et al. Reduced insulin secretion correlates with decreased expression of exocytotic genes in pancreatic islets from patients with type 2 diabetes. Mol Cell Endocrinol (2012) 364:36–45. doi:10.1016/j.mce.2012.08.009
70. Xu Z, Wei G, Chepelev I, Zhao K, Felsenfeld G. Mapping of INS promoter interactions reveals its role in long-range regulation of SYT8 transcription. Nat Struct Mol Biol (2011) 18:372–8. doi:10.1038/nsmb.1993
71. Xi D, Chin H, Gainer H. Analysis of synaptotagmin I-IV messenger RNA expression and developmental regulation in the rat hypothalamus and pituitary. Neuroscience (1999) 88:425–35. doi:10.1016/S0306-4522(98)00234-6
72. Kochubey O, Schneggenburger R. Synaptotagmin increases the dynamic range of synapses by driving Ca(2)+-evoked release and by clamping a near-linear remaining Ca(2)+ sensor. Neuron (2011) 69:736–48. doi:10.1016/j.neuron.2011.01.013
73. Sun J, Pang ZP, Qin D, Fahim AT, Adachi R, Südhof TC. A dual-Ca2+-sensor model for neurotransmitter release in a central synapse. Nature (2007) 450:676–82. doi:10.1038/nature06308
74. Kasai H. Comparative biology of Ca2+-dependent exocytosis: implications of kinetic diversity for secretory function. Trends Neurosci (1999) 22:88–93. doi:10.1016/S0166-2236(98)01293-4
75. Neher E. Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron (1998) 20:389–99. doi:10.1016/S0896-6273(00)80983-6
76. Neher E. Usefulness and limitations of linear approximations to the understanding of Ca++ signals. Cell Calcium (1998) 24:345–57. doi:10.1016/S0143-4160(98)90058-6
77. Augustine GJ, Santamaria F, Tanaka K. Local calcium signaling in neurons. Neuron (2003) 40:331–46. doi:10.1016/S0896-6273(03)00639-1
78. Yamada WM, Zucker RS. Time course of transmitter release calculated from simulations of a calcium diffusion model. Biophys J (1992) 61:671–82. doi:10.1016/S0006-3495(92)81872-6
79. Elhamdani A, Azizi F, Artalejo CR. Double patch clamp reveals that transient fusion (kiss-and-run) is a major mechanism of secretion in calf adrenal chromaffin cells: high calcium shifts the mechanism from kiss-and-run to complete fusion. J Neurosci (2006) 26:3030–6. doi:10.1523/JNEUROSCI.5275-05.2006
80. Michael DJ, Cai H, Xiong W, Ouyang J, Chow RH. Mechanisms of peptide hormone secretion. Trends Endocrinol Metab (2006) 17:408–15. doi:10.1016/j.tem.2006.10.011
81. Michael DJ, Geng X, Cawley NX, Loh YP, Rhodes CJ, Drain P, et al. Fluorescent cargo proteins in pancreatic beta-cells: design determines secretion kinetics at exocytosis. Biophys J (2004) 87:L03–5. doi:10.1529/biophysj.104.052175
82. Mizuta M, Inagaki N, Nemoto Y, Matsukura S, Takahashi M, Seino S. Synaptotagmin III is a novel isoform of rat synaptotagmin expressed in endocrine and neuronal cells. J Biol Chem (1994) 269:11675–8.
83. Saegusa C, Fukuda M, Mikoshiba K. Synaptotagmin V is targeted to dense-core vesicles that undergo calcium-dependent exocytosis in PC12 cells. J Biol Chem (2002) 277:24499–505. doi:10.1074/jbc.M202767200
84. Matsuoka H, Harada K, Nakamura J, Fukuda M, Inoue M. Differential distribution of synaptotagmin-1, -4, -7, and -9 in rat adrenal chromaffin cells. Cell Tissue Res (2011) 344:41–50. doi:10.1007/s00441-011-1131-8
85. Hu ZT, Chen MR, Ping Z, Dong YM, Zhang RY, Xu T, et al. Synaptotagmin IV regulates dense core vesicle (DCV) release in LbetaT2 cells. Biochem Biophys Res Commun (2008) 371:781–6. doi:10.1016/j.bbrc.2008.04.174
86. Kreft M, Kuster V, Grilc S, Rupnik M, Milisav I, Zorec R. Synaptotagmin I increases the probability of vesicle fusion at low [Ca2+] in pituitary cells. Am J Physiol Cell Physiol (2003) 284:C547–54. doi:10.1152/ajpcell.00333.2002
87. Gao Z, Reavey-Cantwell J, Young RA, Jegier P, Wolf BA. Synaptotagmin III/VII isoforms mediate Ca2+-induced insulin secretion in pancreatic islet beta-cells. J Biol Chem (2000) 275:36079–85. doi:10.1074/jbc.M004284200
88. Mizuta M, Kurose T, Miki T, Shoji-Kasai Y, Takahashi M, Seino S, et al. Localization and functional role of synaptotagmin III in insulin secretory vesicles in pancreatic beta-cells. Diabetes (1997) 46:2002–6. doi:10.2337/diabetes.46.12.2002
89. Iezzi M, Kouri G, Fukuda M, Wollheim CB. Synaptotagmin V and IX isoforms control Ca2+-dependent insulin exocytosis. J Cell Sci (2004) 117:3119–27. doi:10.1242/jcs.01179
90. Brown H, Meister B, Deeney J, Corkey BE, Yang SN, Larsson O, et al. Synaptotagmin III isoform is compartmentalized in pancreatic beta-cells and has a functional role in exocytosis. Diabetes (2000) 49:383–91. doi:10.2337/diabetes.49.3.383
Keywords: exocytosis, neuropeptides, dense-core vesicle, norepinephrine, insulin, calcium, fusion pores, kiss-and-run
Citation: Moghadam PK and Jackson MB (2013) The functional significance of synaptotagmin diversity in neuroendocrine secretion. Front. Endocrinol. 4:124. doi: 10.3389/fendo.2013.00124
Received: 23 June 2013; Accepted: 31 August 2013;
Published online: 18 September 2013.
Edited by:
Stephane Gasman, Centre national de la recherche scientifique, FranceReviewed by:
Ling-Gang Wu, National Institute of Neurological Disorders and Stroke (NINDS), USARalf Schneggenburger, Ecole Polytechnique Fédérale de Lausanne, Switzerland
Copyright: © 2013 Moghadam and Jackson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Meyer B. Jackson, Department of Neuroscience, University of Wisconsin, 1300 University Avenue, Madison, WI 53706-1510, USA e-mail:bWJqYWNrc29Ad2lzYy5lZHU=