 Andreas Papadopulos
Andreas Papadopulos Vanesa M. Tomatis
Vanesa M. Tomatis Ravikiran Kasula
Ravikiran Kasula Frederic A. Meunier
Frederic A. Meunier- Queensland Brain Institute, The University of Queensland, St Lucia Campus, Brisbane, QLD, Australia
Dysregulation of regulated exocytosis is linked to an array of pathological conditions, including neurodegenerative disorders, asthma, and diabetes. Understanding the molecular mechanisms underpinning neuroexocytosis including the processes that allow neurosecretory vesicles to access and fuse with the plasma membrane and to recycle post-fusion, is therefore critical to the design of future therapeutic drugs that will efficiently tackle these diseases. Despite considerable efforts to determine the principles of vesicular fusion, the mechanisms controlling the approach of vesicles to the plasma membrane in order to undergo tethering, docking, priming, and fusion remain poorly understood. All these steps involve the cortical actin network, a dense mesh of actin filaments localized beneath the plasma membrane. Recent work overturned the long-held belief that the cortical actin network only plays a passive constraining role in neuroexocytosis functioning as a physical barrier that partly breaks down upon entry of Ca2+ to allow secretory vesicles to reach the plasma membrane. A multitude of new roles for the cortical actin network in regulated exocytosis have now emerged and point to highly dynamic novel functions of key myosin molecular motors. Myosins are not only believed to help bring about dynamic changes in the actin cytoskeleton, tethering and guiding vesicles to their fusion sites, but they also regulate the size and duration of the fusion pore, thereby directly contributing to the release of neurotransmitters and hormones. Here we discuss the functions of the cortical actin network, myosins, and their effectors in controlling the processes that lead to tethering, directed transport, docking, and fusion of exocytotic vesicles in regulated exocytosis.
Introduction
Regulated exocytosis relies on the timely fusion of secretory vesicles or granules (SVs/SGs) with the plasma membrane. For this to occur, SVs need to be mobilized, translocated, docked, and primed at the plasma membrane. Translocation, docking/priming, and fusion of SGs rely on dynamic changes in the cortical actin network, a dense mesh of filamentous actin underneath the plasma membrane (Figures 1A–C) that is controlled by actin effectors and myosin motor proteins. The thick actin ring of the cortical actin network can be visualized in chromaffin cells by staining actin with a variety of methods ranging from classical immunofluorescence to phalloidin (covalently linked to fluorophores), a fungal alkaloid that preferentially binds actin filaments (Figures 1B,C). More recently, the development of lifeact-GFP, a 17-residue peptide from S. cerevisiae that selectively binds to actin without affecting neuroexocytosis (1, 2), has allowed the probing of the dynamic changes occurring during stimulation of exocytosis on the cortical actin network by time-lapse imaging (Figures 1C,D). Following secretagogue stimulation the cortical actin ring fragments, coinciding with a decrease in cortical F-actin labeling (Figure 1B). This process is Ca2+-dependent and involves actin-severing proteins such as scinderin (3–6). Although actin reorganization helps vesicles reach the plasma membrane (7), F-actin also serves as an anchoring point for SGs and provides tracks for their directed motion toward fusion sites (8). Molecular motors associated with F-actin, such as myosins (9), are involved in additional functions (2, 10).
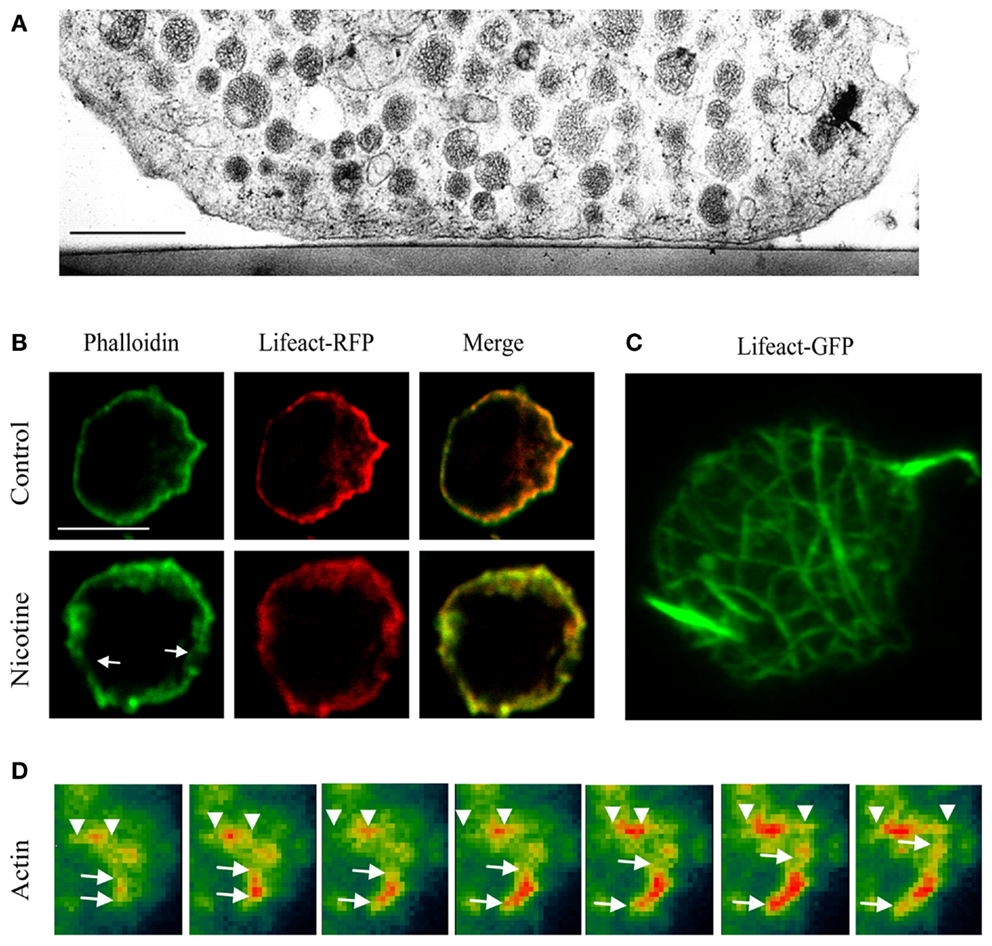
Figure 1. Imaging the actin network in neurosecretory cells. (A) Electron micrograph of a bovine chromaffin cell region attached to the thermanox support. Note the presence of a filamentous cortical region that is devoid of SG. Bar, 1 μm [adapted from Ref. (19)]. (B) Confocal images showing the mid section of bovine chromaffin cells expressing lifeact-RFP and counter stained with FITC-conjugated phalloidin in the presence or absence of nicotine (50 μM). (C) Maximum intensity projection of the footprint of a chromaffin cell. (D) TIRF images showing actin lengthening in a chromaffin cell expressing lifeact-GFP (pseudocolor) after the addition of PI3Kδ inhibitor IC87114. (B–D) Adapted from Ref. (2).
In nerve terminals, actin is a well-known modulator of neurotransmitter release. Actin is involved in synaptic vesicle mobilization as well as axonal vesicle trafficking and synaptic plasticity (11). It is the most abundant cytoskeletal protein in synapses and is highly enriched in dendritic spines, whose formation is initiated by dendritic filopodia formation (12–15), an actin-driven process facilitated by the action of myosin X (16, 17). Neurotransmitter release at central synapses is regulated by actin and depolymerization of F-actin by latrunculin A was found to transiently enhance neurotransmitter release indicating a restraining role of F-actin in active zones (18).
New Roles for Actin in Exocytosis
The cortical actin network plays an important and well-described role during vesicle exocytosis (5, 7, 9, 10), and in recent years new functions for actin and its associated proteins have emerged (2, 9, 10, 20–24). Ca2+-dependent reorganization and remodeling of the cortical actin network help vesicles move toward the plasma membrane by partial disassembly of the cortical layer (Figure 1B) (3, 6). At the same time, this remodeling provides tracks that extend further toward the center of the cell allowing the mobilization of SGs from the reserve pool (25) to their docking and fusion sites at the plasma membrane (4, 26, 27). Ca2+ regulates the cortical F-actin disassembly in chromaffin cells via two pathways (28, 29). The first involves stimulation-induced influx of extracellular Ca2+ through Ca2+ channels and results in activation of scinderin and ensuing F-actin severing. The second pathway is triggered by Ca2+ release from intracellular stores (30) and can be induced in the absence of secretagogue stimulation, by phorbol esters (3). Here actin disassembly is achieved through protein kinase C (PKC) activation followed by myristoylated alanine-rich C kinase substrate (MARCKS) phosphorylation that inhibits its F-actin-binding and cross-linking properties (28). The cortical actin network provides a layered structure that retains 2–4% of the total vesicles in close proximity to the cell surface that contribute to the burst of catecholamine release at the onset of stimulation (26, 31, 32). Indeed the majority of SGs in the vicinity of the plasma membrane are tethered to the cortical actin network (6), and newly arriving vesicles are also caught in this dense mesh of F-actin (33). Other studies point to the existence of F-actin cages that organize the SNARE proteins SNAP25 and syntaxin-1 as well as L- and P/Q-type calcium channels, creating sites in the cortical actin network where SGs fuse preferentially (34). Consistent with these data, studies using total internal reflection fluorescence (TIRF) microscopy revealed that vesicle motion becomes restricted in the vicinity of the plasma membrane (35, 36). Interestingly, both actin depolymerization (37) and N-WASP- and Cdc42-dependent actin polymerization (Figure 1D) potentiate exocytosis (2, 38). While these results may appear contradictory, such opposing role for actin is not unlikely. Partial actin depolymerization helps SGs to cross the actin layer that acts as barrier, and the remaining (10) as well as newly forming actin fibers provide tracks for vesicles to reach the plasma membrane (2, 38). The balance between actin polymerization and depolymerization is likely regulated by scinderin acting as a molecular switch capable of inducing both actin polymerization and depolymerization (39). An important link connecting membranes and actin during exocytosis is the glycerophospholipid phosphatidylinositol 4,5-bisphosphate (PIP2). Although it is only a minor component of cellular membranes, microdomains, and clusters of PIP2 play a crucial role in exocytosis. PIP2 is known to control actin polymerization by modulating the activity and targeting of actin regulatory proteins (40). PIP2 involvement in SNARE-mediated exocytosis, i.e., its Ca2+-dependent interaction with synaptotagmin-1 and syntaxin, has been described in numerous studies (41–44). Decreased levels of PIP2 in the brain and impairment of its synthesis in nerve terminals lead to early postnatal lethality and synaptic defects in mice, including decreased frequency of miniature currents, enhanced synaptic depression, and a smaller ready release pool of synaptic vesicles, delayed endocytosis, and slower recycling kinetics (45). The formation of PIP2 microdomains at syntaxin-1A clusters with docked SGs seems to be required for Ca2+-dependent exocytosis (46). Both PIP2 and syntaxin-1A have been found in punctate nanoclusters in isolated PC12 cell plasma membrane sheets, and similar PIP2 clusters in PC12 cells have been reported to link synaptotagmin-1 and syntaxin-1A, thus providing a platform for SV recruitment (46, 47). Likewise, the clustering of syntaxin-1A in model membranes has been shown to be modulated by PIP2 (48). PIP2 also plays a role in regulated exocytosis by controlling several proteins involved in modifying the actin cytoskeleton (40), as well as stimulating actin polymerization (49). PIP2 binds scinderin in a Ca2+ and pH-dependent manner (50). PIP2 binding inhibits scinderin-induced actin depolymerization (51, 52), as well as the ADF/cofilin actin-severing activity (53) thereby promoting actin polymerization. A transient increase in PIP2 levels is sufficient to promote the mobilization and recruitment of SVs to the plasma membrane via Cdc42-mediated actin reorganization (2). PIP2 therefore links exocytosis and the actin cytoskeleton by coordinating the actin-based delivery of SVs to the plasma membrane (2). Likewise, decreasing PIP2 levels in neuroendocrine cells by either ATP depletion or sequestering PIP2 rapidly reduces the amount of cortical F-actin (54). In a similar study, nanomolar interaction of HIV-1 transcriptional activator with PIP2 was found to prevent the actin reorganization necessary for bringing SVs to the plasma membrane and severely impaired neurosecretion in PC12 and chromaffin cells (55). Another actin-binding protein that PIP2 has been found to interact with is vilin, with PIP2-vilin association inhibiting actin depolymerization and enhancing actin cross-linking (56). The interplay of Rho GTPases such as Cdc42, RhoA, and Rac with PIP2 and other actin regulatory proteins controls Ca2+-regulated exocytosis in chromaffin cells (9, 22). Other small GTPases implicated in regulated secretion in neurosecretory and endocrine cells are Arf6 (57), Rab27A (58) as well as RalA and Rab3A. RalA has not only been shown to tether insulin granules to R- and L-type calcium channels (59) but also binds to the exocyst complex and regulates filopodia formation linking morphological changes and regulated exocytosis (60). RalA, which is present in GLUT4 vesicles in adipocytes, also interacts with the exocyst complex and its activation is required for insulin-stimulated GLUT4 trafficking. Impairment in the function of RalA in these cells attenuated insulin-stimulated glucose transport. RalA also interacts with Myo1C acting as a cargo receptor for this motor protein (61). In addition RalA has been found to control SG exocytosis in PC12 cells by interacting with phospholipase D1. It is activated during exocytosis and the expression of a constitutively active mutant was found to enhance neuroexocytosis whereas expression of an inactive mutant or silencing resulted in reduced secretion (62). Of the four homologs (A/B/C/D) Rab3A is the best characterized (63). Rab3A is involved in the late steps of exocytosis. Early studies showed that Rab3A is associated with SG in bovine chromaffin cells and rat PC12 cells (64, 65). Overexpression of Rab3A mutant proteins defective in either GTP hydrolysis or in guanine nucleotide-binding inhibited exocytosis (66). Similarly the perfusion of Rab3A and various guanine nucleotides into chromaffin cells resulted in delayed catecholamine secretion suggesting a negative regulatory role in secretion (67). Rab3A plays a role in vesicle priming, where it is involved in Munc13-1 activation and interacts with Munc18-1 to regulate priming and fusion (68). Furthermore, Munc13 and Rab3A localize in the acrosomal region in human sperm, where they stimulate acrosomal exocytosis and play an important role in membrane docking (69). In human spermatozoa Rab3A and Rab27 act in a cascade that regulates dense core granule exocytosis (70). Rab3 interaction with Munc18 has also been shown to regulate SG density at the periphery of PC12 cells (71) and Rab3 guanine cycling is required for Munc18-dependent SG docking (72). However, the high level of redundancy between the four Rab3 isoforms makes it difficult to fully assess their individual contributions and the lack of an obvious exocytic phenotype in double and triple knock-out animals points to a regulatory but not essential role of Rab3A in exocytosis (73).
A number of new functions are now being attributed to the interplay between actin and various myosins. Non-muscle myosin II, and the unconventional myosins 1c/e, Va, and VI are involved in different stages during regulated exocytosis of SGs.
Myosins
Myosins are a 17-member superfamily of actin-based molecular motor proteins (74) that are involved in many aspects of eukaryotic cell functions, including cell movement, establishment of cell shape and polarity (75–80), and vesicular trafficking (61, 81). Myosin function is not limited to that of a molecular motor, as myosins also regulate actin polymerization, serve as molecular anchors (33), and even play a role in signal transduction (82, 83). All myosins contain a heavy chain with a conserved ∼80 kDa N-terminal catalytic domain that includes the ATPase activity and actin-binding regions (Figure 2) (84). This domain is followed by an α-helical neck region containing one or more IQ motifs that allow binding of light chains and calmodulin (CaM). The C-terminal myosin tail contains cargo/membrane-binding domains, kinase activity, and/or mediates heavy chain dimerization depending on the myosin class (Figure 2) (83, 85).
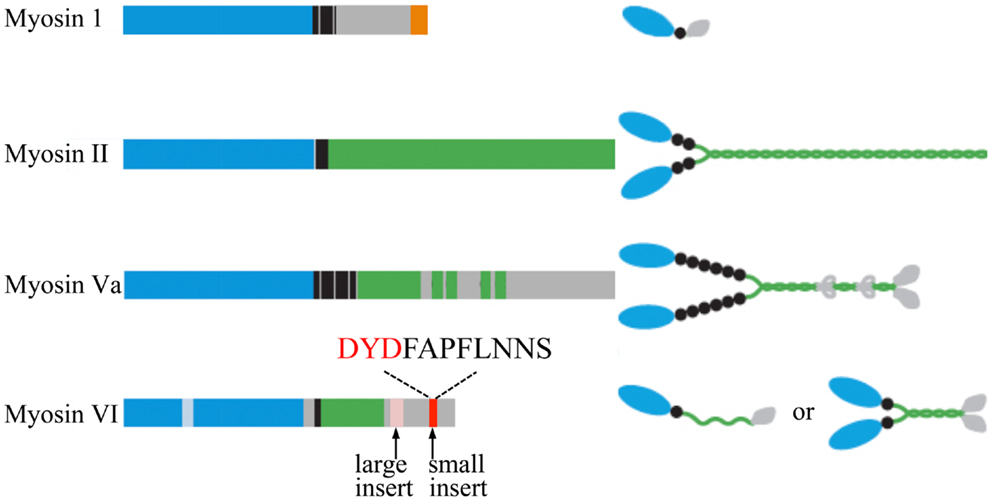
Figure 2. Schematic diagrams of the myosin heavy chains involved in regulated exocytosis. All myosins consist of a head (motor) domain (blue), a neck that contains one or more IQ motifs for light chain and CaM binding (black), and a tail domain with coiled-coil regions (green) and membrane/cargo-binding domains (orange). The small insert of myosin VI, shown to be essential for the tethering of SGs to the cortical actin network, and the DYD-Src phosphorylation motif are highlighted. Cargo-binding induced dimerization of myosin VI is likely to be mediated by the coiled-coil regions and the cargo-binding domains. Adapted from Ref. (83).
Myosin I
Myosin I (Figure 2) is a single-headed membrane-associated protein that is expressed in all eukaryotic cells (84). Although there is currently no evidence for myosin I involvement in neurosecretion two isoforms of the human myosin 1C gene (86), myosin 1C (Myo1C), and myosin 1E (Myo1E) have been implicated in regulated exocytosis. All members of this unconventional myosin family interact with actin through their catalytic head domain (87). Myo1C is also capable of binding phosphoinositides (88) (Figure 3), thereby linking the actin cytoskeleton to the plasma membrane (89). Myo1C is recruited to GLUT4-containing vesicles that undergo regulated exocytosis in 3T3-L1 adipocytes in an insulin-dependent manner, and is involved in their transport to the plasma membrane (Figure 3) (61, 81). In addition, Myo1C also tethers GLUT4-containing vesicles to the cortical actin network (Figure 3) underneath the plasma membrane in response to insulin (90), and promotes GLUT4 insertion to the plasma membrane by fusion (91), thereby regulating glucose uptake in adipose and muscle tissue (92). Myo1C is required for vascular endothelial growth factor receptor-2 (VEGFR2) delivery to the cell surface and for angiogenic signaling (93). VEGF stimulation promotes the recruitment of VEGFR2 to Myo1C and its delivery to the cell surface (93).
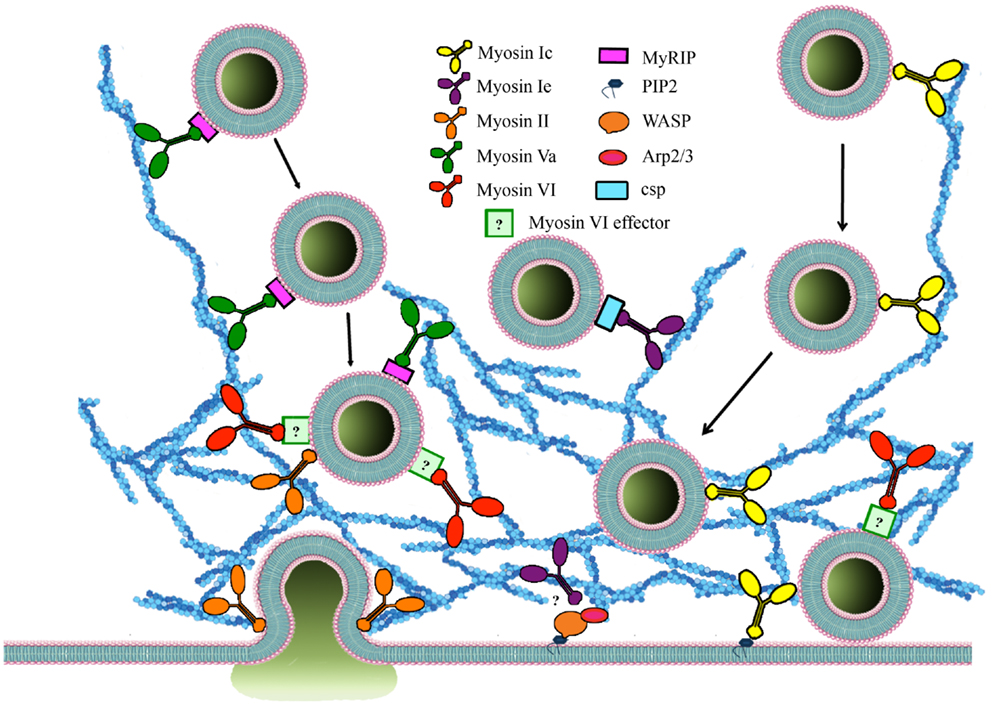
Figure 3. The roles of myosins and accessory proteins involved in regulated exocytosis. Myosins are involved in several steps of regulated exocytosis. Myosin 1C (yellow), myosin 1E (burgundy), myosin II (orange), and myosin Va (green) are involved in secretory vesicle transport. In contrast, myosin VI (red) recruits SGs to the cortical actin network. Myosin 1C interacts with SG through cysteine string proteins and myosin Va binds to MyRIP (purple) on the membrane of SGs. Myosin 1C can be recruited to membranes through PIP2 interaction. The effector that mediates binding between myosin VI and SGs (light green) is currently unknown. Myosin 1E is also involved in regulating actin polymerization through interaction with WASP/Arp 2/3. Cdc42 as well WASP/Arp 2/3 regulate actin polymerization in an activity-dependent manner. Myosin II also regulates size and duration of fusion pore opening.
In Xenopus oocytes Myo1E, the only long-tailed myosin 1 class motor protein has been found to rapidly relocate from the cytosol to cortical SGs upon secretagogue stimulation and to bind to cysteine string proteins, components of cortical SGs that mediate vital steps in regulated exocytosis (94) by interacting with SNAP25 and the calcium sensor synaptotagmin 9 in pancreatic β-cells (95). While cortical granule exocytosis is enhanced by overexpression of Myo1E it is inhibited by injection of Myo1E antibodies (94). Myo1E has also been implicated in the recruitment of several actin-binding proteins leading to N-WASP recruitment and Arp2/3-mediated actin polymerization (Figure 3) (96).
Myosin II
Class II myosins are most abundant in muscle cells where their main function is to generate mechanical force. Non-muscle cells also contain a subset of myosin II molecules with distinct functionality. They all consist of two heavy chains (230 kDa), two regulatory light chains, and two essential light chains (Figure 2). In addition to actin cross-linking, bundling, and contractile properties, myosin II is known to regulate actin polymerization and is therefore linked to a great number of functions in eukaryotic cells including motility, adhesion (97), and regulated exocytosis (24, 98, 99). Non-muscle myosin II has been implicated in vesicle transport through the actin cytoskeleton (Figure 3). Expression of an inactive non-phosphorylatable regulatory light chain mutant myosin II fused to GFP drastically impairs granule mobility and influences actin dynamics, similar to blebbistatin treatment (100).
There is mounting evidence that myosin II is involved in controlling fusion pore dynamics and release kinetics (Figure 3). Expression of non-phosphorylatable regulatory light chain mutant myosin II that produces an inactive protein alters single vesicle fusion kinetics and slows fusion pore expansion (23, 24). Similarly, the release kinetics of fluorescently tagged tissue plasminogen activator and brain-derived neurotrophic (BDNF) factor are prolonged following overexpression of a wild-type form of the myosin II regulatory light chain and shortened by overexpression of a dominant-negative form (101). The use of a green fluorescent pH-sensitive protein (pHluorin) targeted inside the SVs revealed that the altered kinetics of release were caused by changes in the duration of fusion pore opening. Additional evidence indicates that myosin II affects catecholamine release by directly controlling the size of the fusion pore and the duration of its opening (20). Actin cortex disassembly elicited by high frequency stimulation promotes full fusion of SGs – an effect blocked by pharmacological inhibition of myosin II or myosin light chain (MLC) by preventing the fusion pore dilation (102). Inhibition of either actin polymerization with cytochalasin D or myosin II function with blebbistatin also slowed fusion pore expansion and increased its lifetime, suggesting that the interplay between actin and myosin II can accelerate catecholamine release (20). Similar results indicating that myosin II activity maintains an open fusion pore were obtained in exocrine pancreatic cells where myosin II (blebbistatin) and MLC (ML-9) inhibition did not alter the number of fusion events but resulted in a decreased fusion pore lifetime (103).
It has been suggested that myosin II contractility could also help to squeeze secretory cargo out of vesicles surrounded by an actin coat once they are connected to the plasma membrane through a fusion pore (104). Fusion pore opening and closing might not be enough to release large cargo from SVs and myosin II might provide an active extrusion mechanism (104). The direct involvement of MLC and myosin II was also observed in GLUT4-containing vesicle fusion following insulin-stimulated glucose uptake in 3T3-L1 adipocytes. Only active phosphorylated myosin II was recruited to GLUT4 vesicles in an activity-dependent manner. Interestingly, insulin specifically stimulates the myosin IIA isoform via MLC kinase phosphorylation of MLC (105, 106). Myosin II inhibition also increases the distance of SGs from the plasma membrane, and promotes the retraction of the cytoskeleton, suggesting its involvement in the final approach of vesicles toward the plasma membrane (107).
Myosin II involvement in integrin-mediated cell adhesion and exocytosis has been linked to changes in cell adhesion properties (108, 109). Glucose stimulation of pancreatic β-cells promotes the remodeling of integrin focal adhesions and phosphorylation of focal adhesion kinases and myosin II (108, 109). As myosin II is one of the main substrates of Rho-kinase 1/2, which stimulates myosin–actin interactions and induces reorganization of the actin cytoskeleton, this activity could modulate SG translocation and cargo release in response to secretagogue stimulation.
Myosin Va
Myosin Va (Figure 2) has been implicated in exocytosis and vesicle movement to the cell periphery. In melanocytes, in a complex with Rab27a and melanophilin, myosin Va regulates melanosome transport to the plasma membrane (110, 111). In pancreatic β-cells myosin Va also functions in the transport to and retention of insulin granules at the cortical actin network under stimulated conditions as well as their secretion (112– 114). In neurosecretory cells, myosin Va is associated with SGs and plays distinctive roles during SG exocytosis (25, 115). Firstly, it assists the membrane remodeling required for SG maturation by promoting the removal of the transmembrane protein furin from maturing SGs (116). Secondly, in a complex with the SG-associated small GTPase Rab27 and its effector MyRIP, myosin Va regulates the interaction of SGs with the cortical actin network (Figure 3) (58). This complex has been implicated in exocytosis of SGs by modulating the transport of SGs and their retention in the cortical actin network on their way to the plasma membrane (117, 118). The interaction between myosin Va and MyRIP facilitates the dissociation of SGs from microtubules, enhancing their directed motion and the probability of SG docking to the plasma membrane (118). As a conventional processive molecular motor, myosin Va moves selective cargo along actin filaments (117). This feature strongly supports the key role of this protein in the translocation and tethering of SGs to the cell periphery. Blocking myosin Va function reduces the immobilization periods of SGs thereby decreasing the density of docked SGs near the plasma membrane and their exocytosis (117, 119). In resting conditions, myosin Va forms a stable complex with synaptic vesicle membrane proteins, synaptobrevin II, and synaptophysin (120). This complex is rapidly disassembled upon Ca2+ increase in either intact nerve endings or in vitro assays (120). In chromaffin cells, influx of Ca2+ dissociates myosin V from chromaffin vesicles supporting a role for Ca2+ in the regulation of transient interactions between myosin V and its cargo (25). Furthermore, when an antibody against myosin V head was introduced in permeabilized chromaffin cells after a first stimulation of 40 s, the secretory response to a second stimulation several minutes after the first one, was greatly reduced. This points to a role for myosin V in providing SVs for the refilling of the release-ready pool following stimulation (25). The role of Ca2+ as a regulator of the interaction between myosin V and its cargo has also been demonstrated in melanosomes and Xenopus egg extracts (121). Released CaM activates CaM kinase II (CaMK-II), a myosin Va binding partner (122). CaMK-II activation leads to myosin Va phosphorylation and the release of melanosomes from F-actin (121). Similarly, microinjection of CaM antibodies into chromaffin cells resulted in reduced catecholamine output in response to stimulation (123). Ca2+-regulated phosphorylation of myosin Va is believed to represent a universal mechanism that regulates the association between myosin Va and its cargo. These observations suggest that by regulating the interaction between myosin V and SGs, Ca2+ could also control the association between SGs and actin during SG mobilization in the cortical region (124). Importantly, the Ca2+-regulated attachment/release of myosin Va from SGs could be finely coordinated by other molecular motors, such us myosin VI (33). This cooperative model would allow a highly organized and controlled mechanism that regulates SG transport, retention, and anchoring and ultimately SG fusion with the plasma membrane.
Myosin VI
Another member of the myosin family, myosin VI is critical for SV recruitment to the cortical actin network (Figure 2). The cellular functions of myosin VI are attributed to its unique ability to generate movement from the plus to the minus end of actin filaments. Myosin VI has an additional unique 53 aa insert, the “reverse gear,” between the motor domain and the neck region that has been predicted to be responsible for this exceptional inverted movement directionality (Figure 2) (125, 126). Interestingly, this insert binds CaM even though it does not contain a recognizable IQ-CaM motif (127). The tail domain region is the most variable amongst the myosin VI isoforms. Four alternatively spliced isoforms are generated due to the presence of a large insert (21–31 aa), a small insert (9 aa), no insert, or both inserts in this domain (Figure 2) (128, 129).
The function of myosin VI depends on the ability of its cargo-binding domain (CBD) region to interact with different binding partners that target myosin VI to specific cellular compartments (130). Myosin VI undergoes cargo-mediated dimerization a potential regulatory pathway for all myosins (131, 132). Myosin VI has been linked to clathrin- and non-clathrin-mediated endocytosis, as well as maintenance of Golgi organization and cell polarity. The large and no insert isoforms are the main isoforms mediating these functions (128, 133–137). Myosin VI has also been implicated in autophagy (138), stereocilia maintenance (139), spermatid individualization (140–142), nuclear transcription (143), and cell–cell contacts (144, 145). Evidence of a role of myosin VI in secretion were highlighted by Warner et al. (146) using immortalized cells from Snell’s waltzer mice, a strain of myosin VI knock-out mice (146–148). Immortalized fibroblastic cells from these mice have a reduced Golgi complex (∼40% smaller in comparison with that in normal cells) that is accompanied by a similar reduction in constitutive secretion (146). The down-regulation of myosin VI expression using small interfering RNA selectively reduces the secretion of prostate-specific antigen and vascular endothelial growth factor in the prostate cancer cell line LNCaP (149). Myosin VI together with its binding partner optineurin, regulates the final stage of constitutive exocytosis by mechanically controlling the formation of the fusion pore between the SV and the plasma membrane in HeLa cells (150). Less is known about the role of myosin VI in the nervous system (151). Myosin VI is widely and highly expressed in the brain; it is found in synapses and enriched at the postsynaptic density (151). In hippocampal neurons, myosin VI forms a complex with α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR), AP-2 and synaptic-associated protein 97 (SAP-97), and mediates AMPAR clathrin-mediated endocytosis. Importantly, myosin VI function underpins hippocampal neurons synapses and dendritic spines formation (151). Other work supports the role of myosin VI in neurotransmission by demonstrating that myosin VI; together with its binding partner GIPC1 is necessary for BDNF-TrkB-mediated synaptic plasticity (152).
Myosin VI has a very slow rate of release of ADP from its nucleotide-binding pocket, which therefore slows the dissociation of myosin VI from actin (153–155). Studies carried out in Snell’s waltzer mice have shown that myosin VI allows the formation, maturation, and function of sensory hair cells by mediating the attachment of membrane compartments to the F-actin cytoskeleton (148). Together these lines of evidence point toward the possibility that myosin VI could regulate neuroexocytosis by anchoring/recruiting SVs to the actin network before they undergo fusion with the plasma membrane. Although little is known about the precise molecular mechanism(s) underpinning this role, the function of myosin VI in regulated in exocytosis in PC12 cells has been questioned (156). However, Drosophila mutants lacking myosin VI display altered neuromuscular junction morphology and synaptic vesicle localization resulting in impaired synaptic plasticity (157). Myosin VI could therefore mediate the mobilization of synaptic vesicles from different functional pools, by a yet to be elucidated mechanism. We recently described a novel role for the myosin VI small insert isoform (Figure 2) in regulated exocytosis in PC12 cells (33). Using purified SGs in a pull-down approach followed by mass spectrometry, we identified myosin VI as a cytosolic protein that interacts with SGs in a Ca2+-dependent manner. We found that myosin VI maintains an active pool of SGs near the plasma membrane by tethering them to the cortical actin network (Figure 3). This allows the replenishment of the pool of SGs near the plasma membrane and is key to sustaining exocytosis during long periods of stimulation (33). Interestingly, we found that c-Src phosphorylation in a DYD motif located in the CBD of myosin VI small insert is one of the mechanisms controlling its function in regulated neuroexocytosis (33). The mechanisms that target myosin VI to SGs and the regulation of the isoform specific tethering function still need to be elucidated.
There are several other members of the myosin family that could potentially be involved in regulated secretion, including myosin X, a motor protein found predominantly at the tip of filopodia of many cell types including neurons (16, 17). Filopodia are important precursors for dendritic spine and synapse formation but more work is needed to assess whether neurosecretion can occur in these structures.
Conclusion
Understanding the detailed roles of myosins and other accessory proteins in regulated exocytosis is challenging. Although a great deal is known about the involvement of these proteins and their effectors during the different stages of secretion, there is still no comprehensive model of the interplay of the different myosin isoforms, e.g., the transition from myosin Va-mediated directed transport to myosin VI-dependent recruitment to the cortical actin network. Common pathways that are shared by other cellular functions, such as adhesion or migration should also be explored further. Future work should therefore aim at combining in vitro techniques with live cell microscopy experiments in order to explore the complex interplay between the different myosin molecular motors during neuroexocytosis. In particular, it will be necessary to address the nature of the pathways, which coordinate and control myosin functions in order to achieve such precise spatio-temporal trafficking of SVs en route to fusion with the plasma membrane.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Rowan Tweedale for critical reading of the manuscript.
References
1. Jang Y, Soekmadji C, Mitchell JM, Thomas WG, Thorn P. Real-time measurement of F-actin remodelling during exocytosis using lifeact-EGFP transgenic animals. PLoS One (2012) 7:e39815. doi:10.1371/journal.pone.0039815
2. Wen PJ, Osborne SL, Zanin M, Low PC, Wang HT, Schoenwaelder SM, et al. Phosphatidylinositol(4,5)bisphosphate coordinates actin-mediated mobilization and translocation of secretory vesicles to the plasma membrane of chromaffin cells. Nat Commun (2011) 2:491. doi:10.1038/ncomms1500
3. Rose SD, Lejen T, Zhang L, Trifaro JM. Chromaffin cell F-actin disassembly and potentiation of catecholamine release in response to protein kinase C activation by phorbol esters is mediated through myristoylated alanine-rich C kinase substrate phosphorylation. J Biol Chem (2001) 276:36757–63. doi:10.1074/jbc.M006518200
4. Trifaro JM. Scinderin and cortical F-actin are components of the secretory machinery. Can J Physiol Pharmacol (1999) 77:660–71. doi:10.1139/y99-074
5. Trifaro JM, Rodriguez del Castillo A, Vitale ML. Dynamic changes in chromaffin cell cytoskeleton as prelude to exocytosis. Mol Neurobiol (1992) 6:339–58. doi:10.1007/BF02757940
6. Vitale ML, Rodriguez Del Castillo A, Tchakarov L, Trifaro JM. Cortical filamentous actin disassembly and scinderin redistribution during chromaffin cell stimulation precede exocytosis, a phenomenon not exhibited by gelsolin. J Cell Biol (1991) 113:1057–67. doi:10.1083/jcb.113.5.1057
7. Vitale ML, Seward EP, Trifaro JM. Chromaffin cell cortical actin network dynamics control the size of the release-ready vesicle pool and the initial rate of exocytosis. Neuron (1995) 14:353–63. doi:10.1016/0896-6273(95)90291-0
8. Giner D, Neco P, Frances Mdel M, Lopez I, Viniegra S, Gutierrez LM. Real-time dynamics of the F-actin cytoskeleton during secretion from chromaffin cells. J Cell Sci (2005) 118:2871–80. doi:10.1242/jcs.02419
9. Malacombe M, Bader M-F, Gasman SP. Exocytosis in neuroendocrine cells: new tasks for actin. Biochim Biophys Acta (2006) 1763:1175–83. doi:10.1016/j.bbamcr.2006.09.004
10. Trifaro JM, Gasman S, Gutierrez LM. Cytoskeletal control of vesicle transport and exocytosis in chromaffin cells. Acta Physiol (Oxf) (2008) 192:165–72. doi:10.1111/j.1748-1716.2007.01808.x
11. Cingolani LA, Goda Y. Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat Rev Neurosci (2008) 9:344–56. doi:10.1038/nrn2373
12. Fifkova E, Delay RJ. Cytoplasmic actin in neuronal processes as a possible mediator of synaptic plasticity. J Cell Biol (1982) 95:345–50. doi:10.1083/jcb.95.1.345
13. Landis DM, Hall AK, Weinstein LA, Reese TS. The organization of cytoplasm at the presynaptic active zone of a central nervous system synapse. Neuron (1988) 1:201–9. doi:10.1016/0896-6273(88)90140-7
14. Matus A. Actin-based plasticity in dendritic spines. Science (2000) 290:754–8. doi:10.1126/science.290.5492.754
15. Matus A, Ackermann M, Pehling G, Byers HR, Fujiwara K. High actin concentrations in brain dendritic spines and postsynaptic densities. Proc Natl Acad Sci U S A (1982) 79:7590–4. doi:10.1073/pnas.79.23.7590
16. Kerber ML, Cheney RE. Myosin-X: a MyTH-FERM myosin at the tips of filopodia. J Cell Sci (2011) 124:3733–41. doi:10.1242/jcs.023549
17. Plantman S, Zelano J, Novikova LN, Novikov LN, Cullheim S. Neuronal myosin-X is upregulated after peripheral nerve injury and mediates laminin-induced growth of neurites. Mol Cell Neurosci (2013) 56C:96–101. doi:10.1016/j.mcn.2013.04.001
18. Morales M, Colicos MA, Goda Y. Actin-dependent regulation of neurotransmitter release at central synapses. Neuron (2000) 27:539–50. doi:10.1016/S0896-6273(00)00064-7
19. Plattner H, Artalejo AR, Neher E. Ultrastructural organization of bovine chromaffin cell cortex-analysis by cryofixation and morphometry of aspects pertinent to exocytosis. J Cell Biol (1997) 139:1709–17. doi:10.1083/jcb.139.7.1709
20. Berberian K, Torres AJ, Fang Q, Kisler K, Lindau M. F-actin and myosin II accelerate catecholamine release from chromaffin granules. J Neurosci (2009) 29:863–70. doi:10.1523/JNEUROSCI.2818-08.2009
21. Gasman S, Chasserot-Golaz S, Popoff MR, Aunis D, Bader MF. Trimeric G proteins control exocytosis in chromaffin cells. Go regulates the peripheral actin network and catecholamine secretion by a mechanism involving the small GTP-binding protein Rho. J Biol Chem (1997) 272:20564–71. doi:10.1074/jbc.272.33.20564
22. Gasman S, Chasserot-Golaz S, Popoff MR, Aunis D, Bader MF. Involvement of Rho GTPases in calcium-regulated exocytosis from adrenal chromaffin cells. J Cell Sci (1999) 112(Pt 24):4763–71.
23. Neco P, Fernandez-Peruchena C, Navas S, Gutierrez LM, de Toledo GA, Ales E. Myosin II contributes to fusion pore expansion during exocytosis. J Biol Chem (2008) 283:10949–57. doi:10.1074/jbc.M709058200
24. Neco P, Giner D, Viniegra S, Borges R, Villarroel A, Gutierrez LM. New roles of myosin II during vesicle transport and fusion in chromaffin cells. J Biol Chem (2004) 279:27450–7. doi:10.1074/jbc.M311462200
25. Rose SD, Lejen T, Casaletti L, Larson RE, Pene TD, Trifaro JM. Myosins II and V in chromaffin cells: myosin V is a chromaffin vesicle molecular motor involved in secretion. J Neurochem (2003) 85:287–98. doi:10.1046/j.1471-4159.2003.01649.x
26. Neher E, Zucker RS. Multiple calcium-dependent processes related to secretion in bovine chromaffin cells. Neuron (1993) 10:21–30. doi:10.1016/0896-6273(93)90238-M
27. Stevens DR, Schirra C, Becherer U, Rettig J. Vesicle pools: lessons from adrenal chromaffin cells. Front Synaptic Neurosci (2011) 3:2. doi:10.3389/fnsyn.2011.00002
28. Trifaro J, Rose SD, Lejen T, Elzagallaai A. Two pathways control chromaffin cell cortical F-actin dynamics during exocytosis. Biochimie (2000) 82:339–52. doi:10.1016/S0300-9084(00)00193-0
29. Trifaro JM, Lejen T, Rose SD, Pene TD, Barkar ND, Seward EP. Pathways that control cortical F-actin dynamics during secretion. Neurochem Res (2002) 27:1371–85. doi:10.1023/A:1021627800918
30. Zhang L, Rodriguez Del Castillo A, Trifaro JM. Histamine-evoked chromaffin cell scinderin redistribution, F-actin disassembly, and secretion: in the absence of cortical F-actin disassembly, an increase in intracellular Ca2+ fails to trigger exocytosis. J Neurochem (1995) 65:1297–308. doi:10.1046/j.1471-4159.1995.65031297.x
31. Gillis KD, Chow RH. Kinetics of exocytosis in adrenal chromaffin cells. Semin Cell Dev Biol (1997) 8:133–40. doi:10.1006/scdb.1996.0132
32. Parsons TD, Coorssen JR, Horstmann H, Almers W. Docked granules, the exocytic burst, and the need for ATP hydrolysis in endocrine cells. Neuron (1995) 15:1085–96. doi:10.1016/0896-6273(95)90097-7
33. Tomatis VM, Papadopulos A, Malintan NT, Martin S, Wallis T, Gormal RS, et al. Myosin VI small insert isoform maintains exocytosis by tethering secretory granules to the cortical actin. J Cell Biol (2013) 200:301–20. doi:10.1083/jcb.201204092
34. Torregrosa-Hetland CJ, Villanueva J, Lopez-Font I, Garcia-Martinez V, Gil A, Gonzalez-Velez V, et al. Association of SNAREs and calcium channels with the borders of cytoskeletal cages organizes the secretory machinery in chromaffin cells. Cell Mol Neurobiol (2010) 30:1315–9. doi:10.1007/s10571-010-9565-1
35. Johns LM, Levitan ES, Shelden EA, Holz RW, Axelrod D. Restriction of secretory granule motion near the plasma membrane of chromaffin cells. J Cell Biol (2001) 153:177–90. doi:10.1083/jcb.153.1.177
36. Steyer JA, Almers W. Tracking single secretory granules in live chromaffin cells by evanescent-field fluorescence microscopy. Biophys J (1999) 76:2262–71. doi:10.1016/S0006-3495(99)77382-0
37. Cuchillo-Ibanez I, Lejen T, Albillos A, Rose SD, Olivares R, Villarroya M, et al. Mitochondrial calcium sequestration and protein kinase C cooperate in the regulation of cortical F-actin disassembly and secretion in bovine chromaffin cells. J Physiol (2004) 560:63–76. doi:10.1113/jphysiol.2004.064063
38. Gasman S, Chasserot-Golaz S, Malacombe M, Way M, Bader MF. Regulated exocytosis in neuroendocrine cells: a role for subplasmalemmal Cdc42/N-WASP-induced actin filaments. Mol Biol Cell (2004) 15:520–31. doi:10.1091/mbc.E03-06-0402
39. Marcu MG, Zhang L, Elzagallaai A, Trifaro JM. Localization by segmental deletion analysis and functional characterization of a third actin-binding site in domain 5 of scinderin. J Biol Chem (1998) 273:3661–8. doi:10.1074/jbc.273.6.3661
40. Yin HL, Janmey PA. Phosphoinositide regulation of the actin cytoskeleton. Annu Rev Physiol (2003) 65:761–89. doi:10.1146/annurev.physiol.65.092101.142517
41. Bai J, Tucker WC, Chapman ER. PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nat Struct Mol Biol (2004) 11:36–44. doi:10.1038/nsmb709
42. James DJ, Khodthong C, Kowalchyk JA, Martin TF. Phosphatidylinositol 4,5-bisphosphate regulates SNARE-dependent membrane fusion. J Cell Biol (2008) 182:355–66. doi:10.1083/jcb.200801056
43. Kuo W, Herrick DZ, Cafiso DS. Phosphatidylinositol 4,5-bisphosphate alters synaptotagmin 1 membrane docking and drives opposing bilayers closer together. Biochemistry (2011) 50:2633–41. doi:10.1021/bi200049c
44. van den Bogaart G, Meyenberg K, Diederichsen U, Jahn R. Phosphatidylinositol 4,5-bisphosphate increases Ca2+ affinity of synaptotagmin-1 by 40-fold. J Biol Chem (2012) 287:16447–53. doi:10.1074/jbc.M112.343418
45. Di Paolo G, Moskowitz HS, Gipson K, Wenk MR, Voronov S, Obayashi M, et al. Impaired PtdIns(4,5)P2 synthesis in nerve terminals produces defects in synaptic vesicle trafficking. Nature (2004) 431:415–22. doi:10.1038/nature02896
46. Aoyagi K, Sugaya T, Umeda M, Yamamoto S, Terakawa S, Takahashi M. The activation of exocytotic sites by the formation of phosphatidylinositol 4,5-bisphosphate microdomains at syntaxin clusters. J Biol Chem (2005) 280:17346–52. doi:10.1074/jbc.M413307200
47. Honigmann A, van den Bogaart G, Iraheta E, Risselada HJ, Milovanovic D, Mueller V, et al. Phosphatidylinositol 4,5-bisphosphate clusters act as molecular beacons for vesicle recruitment. Nat Struct Mol Biol (2013) 20:679–86. doi:10.1038/nsmb.2570
48. Murray DH, Tamm LK. Clustering of syntaxin-1A in model membranes is modulated by phosphatidylinositol 4,5-bisphosphate and cholesterol. Biochemistry (2009) 48:4617–25. doi:10.1021/bi9003217
49. Lassing I, Lindberg U. Specific interaction between phosphatidylinositol 4,5-bisphosphate and profilactin. Nature (1985) 314:472–4. doi:10.1038/314472a0
50. Rodriguez Del Castillo A, Vitale ML, Trifaro JM. Ca2+ and pH determine the interaction of chromaffin cell scinderin with phosphatidylserine and phosphatidylinositol 4,5,-biphosphate and its cellular distribution during nicotinic-receptor stimulation and protein kinase C activation. J Cell Biol (1992) 119:797–810. doi:10.1083/jcb.119.4.797
51. Marcu MG, Rodriguez del Castillo A, Vitale ML, Trifaro JM. Molecular cloning and functional expression of chromaffin cell scinderin indicates that it belongs to the family of Ca(2+)-dependent F-actin severing proteins. Mol Cell Biochem (1994) 141:153–65. doi:10.1007/BF00926179
52. Zhang L, Marcu MG, Nau-Staudt K, Trifaro JM. Recombinant scinderin enhances exocytosis, an effect blocked by two scinderin-derived actin-binding peptides and PIP2. Neuron (1996) 17:287–96. doi:10.1016/S0896-6273(00)80160-9
53. Ojala PJ, Paavilainen V, Lappalainen P. Identification of yeast cofilin residues specific for actin monomer and PIP2 binding. Biochemistry (2001) 40:15562–9. doi:10.1021/bi0117697
54. Bittner MA, Holz RW. Phosphatidylinositol-4,5-bisphosphate: actin dynamics and the regulation of ATP-dependent and -independent secretion. Mol Pharmacol (2005) 67:1089–98. doi:10.1124/mol.104.008474
55. Tryoen-Toth P, Chasserot-Golaz S, Tu A, Gherib P, Bader MF, Beaumelle B, et al. HIV-1 Tat protein inhibits neurosecretion by binding to phosphatidylinositol 4,5-bisphosphate. J Cell Sci (2013) 126:454–63. doi:10.1242/jcs.111658
56. Kumar N, Zhao P, Tomar A, Galea CA, Khurana S. Association of villin with phosphatidylinositol 4,5-bisphosphate regulates the actin cytoskeleton. J Biol Chem (2004) 279:3096–110. doi:10.1074/jbc.M308878200
57. Galas MC, Helms JB, Vitale N, Thierse D, Aunis D, Bader MF. Regulated exocytosis in chromaffin cells. A potential role for a secretory granule-associated ARF6 protein. J Biol Chem (1997) 272:2788–93. doi:10.1074/jbc.272.5.2788
58. Desnos C, Schonn JS, Huet S, Tran VS, El-Amraoui A, Raposo G, et al. Rab27A and its effector MyRIP link secretory granules to F-actin and control their motion towards release sites. J Cell Biol (2003) 163:559–70. doi:10.1083/jcb.200302157
59. Xie L, Kang Y, Liang T, Dolai S, Xie H, Parsaud L, et al. RalA GTPase tethers insulin granules to L- and R-type calcium channels through binding alpha2 delta-1 subunit. Traffic (2013) 14:428–39. doi:10.1111/tra.12047
60. Sugihara K, Asano S, Tanaka K, Iwamatsu A, Okawa K, Ohta Y. The exocyst complex binds the small GTPase RalA to mediate filopodia formation. Nat Cell Biol (2002) 4:73–8. doi:10.1038/ncb720
61. Chen XW, Leto D, Chiang SH, Wang Q, Saltiel AR. Activation of RalA is required for insulin-stimulated Glut4 trafficking to the plasma membrane via the exocyst and the motor protein Myo1c. Dev Cell (2007) 13:391–404. doi:10.1016/j.devcel.2007.07.007
62. Vitale N, Mawet J, Camonis J, Regazzi R, Bader MF, Chasserot-Golaz S. The small GTPase RalA controls exocytosis of large dense core secretory granules by interacting with ARF6-dependent phospholipase D1. J Biol Chem (2005) 280:29921–8. doi:10.1074/jbc.M413748200
63. Lang T, Jahn R. Core proteins of the secretory machinery. Handb Exp Pharmacol (2008). 184:107–27. doi:10.1007/978-3-540-74805-2_5
64. Darchen F, Senyshyn J, Brondyk WH, Taatjes DJ, Holz RW, Henry JP, et al. The GTPase Rab3a is associated with large dense core vesicles in bovine chromaffin cells and rat PC12 cells. J Cell Sci (1995) 108(Pt 4):1639–49.
65. Darchen F, Zahraoui A, Hammel F, Monteils MP, Tavitian A, Scherman D. Association of the GTP-binding protein Rab3A with bovine adrenal chromaffin granules. Proc Natl Acad Sci U S A (1990) 87: 5692–6. doi:10.1073/pnas.87.15.5692
66. Lin CG, Pan CY, Kao LS. Rab3A delayed catecholamine secretion from bovine adrenal chromaffin cells. Biochem Biophys Res Commun (1996) 221:675–81. doi:10.1006/bbrc.1996.0655
67. Johannes L, Lledo PM, Roa M, Vincent JD, Henry JP, Darchen F. The GTPase Rab3a negatively controls calcium-dependent exocytosis in neuroendocrine cells. EMBO J (1994) 13:2029–37.
68. Huang CC, Yang DM, Lin CC, Kao LS. Involvement of Rab3A in vesicle priming during exocytosis: interaction with Munc13-1 and Munc18-1. Traffic (2011) 12:1356–70. doi:10.1111/j.1600-0854.2011.01237.x
69. Bello OD, Zanetti MN, Mayorga LS, Michaut MA. RIM, Munc13, and Rab3A interplay in acrosomal exocytosis. Exp Cell Res (2012) 318:478–88. doi:10.1016/j.yexcr.2012.01.002
70. Bustos MA, Lucchesi O, Ruete MC, Mayorga LS, Tomes CN. Rab27 and Rab3 sequentially regulate human sperm dense-core granule exocytosis. Proc Natl Acad Sci U S A (2012) 109:E2057–66. doi:10.1073/pnas.1121173109
71. Graham ME, Handley MT, Barclay JW, Ciufo LF, Barrow SL, Morgan A, et al. A gain-of-function mutant of Munc18-1 stimulates secretory granule recruitment and exocytosis and reveals a direct interaction of Munc18-1 with Rab3. Biochem J (2008) 409:407–16. doi:10.1042/BJ20071094
72. van Weering JR, Toonen RF, Verhage M. The role of Rab3a in secretory vesicle docking requires association/dissociation of guanidine phosphates and Munc18-1. PLoS One (2007) 2:e616. doi:10.1371/journal.pone.0000616
73. Schluter OM, Schmitz F, Jahn R, Rosenmund C, Sudhof TC. A complete genetic analysis of neuronal Rab3 function. J Neurosci (2004) 24:6629–37. doi:10.1523/JNEUROSCI.1610-04.2004
74. Berg JS, Powell BC, Cheney RE. A millennial myosin census. Mol Biol Cell (2001) 12:780–94. doi:10.1091/mbc.12.4.780
75. Carlier MF, Valentin-Ranc C, Combeau C, Fievez S, Pantoloni D. Actin polymerization: regulation by divalent metal ion and nucleotide binding, ATP hydrolysis and binding of myosin. Adv Exp Med Biol (1994) 358: 71–81. doi:10.1007/978-1-4615-2578-3_7
76. Lechler T, Shevchenko A, Li R. Direct involvement of yeast type I myosins in Cdc42-dependent actin polymerization. J Cell Biol (2000) 148:363–73. doi:10.1083/jcb.148.2.363
77. van den Boom F, Dussmann H, Uhlenbrock K, Abouhamed M, Bahler M. The myosin IXb motor activity targets the myosin IXb RhoGAP domain as cargo to sites of actin polymerization. Mol Biol Cell (2007) 18:1507–18. doi:10.1091/mbc.E06-08-0771
78. Vicente-Manzanares M, Zareno J, Whitmore L, Choi CK, Horwitz AF. Regulation of protrusion, adhesion dynamics, and polarity by myosins IIA and IIB in migrating cells. J Cell Biol (2007) 176:573–80. doi:10.1083/jcb.20061204320070302c
79. Win TZ, Gachet Y, Mulvihill DP, May KM, Hyams JS. Two type V myosins with non-overlapping functions in the fission yeast Schizosaccharomyces pombe: Myo52 is concerned with growth polarity and cytokinesis, Myo51 is a component of the cytokinetic actin ring. J Cell Sci (2001) 114:69–79.
80. Yin H, Pruyne D, Huffaker TC, Bretscher A. Myosin V orientates the mitotic spindle in yeast. Nature (2000) 406:1013–5. doi:10.1038/35023024
81. Bose A, Guilherme A, Robida SI, Nicoloro SM, Zhou QL, Jiang ZY, et al. Glucose transporter recycling in response to insulin is facilitated by myosin Myo1c. Nature (2002) 420:821–4. doi:10.1038/nature01246
82. Bahler M. Myosins on the move to signal transduction. Curr Opin Cell Biol (1996) 8:18–22. doi:10.1016/S0955-0674(96)80043-3
83. Mermall V, Post PL, Mooseker MS. Unconventional myosins in cell movement, membrane traffic, and signal transduction. Science (1998) 279:527–33. doi:10.1126/science.279.5350.527
84. Sellers JR. Myosins: a diverse superfamily. Biochim Biophys Acta (2000) 1496:3–22. doi:10.1016/S0167-4889(00)00005-7
85. Krendel M, Mooseker MS. Myosins: tails (and heads) of functional diversity. Physiology (Bethesda) (2005) 20:239–51. doi:10.1152/physiol.00014.2005
86. Gillespie PG, Cyr JL. Myosin-1c, the hair cell’s adaptation motor. Annu Rev Physiol (2004) 66:521–45. doi:10.1146/annurev.physiol.66.032102.112842
87. Barylko B, Binns DD, Albanesi JP. Regulation of the enzymatic and motor activities of myosin I. Biochim Biophys Acta (2000) 1496:23–35. doi:10.1016/S0167-4889(00)00006-9
88. Hokanson DE, Laakso JM, Lin T, Sept D, Ostap EM. Myo1c binds phosphoinositides through a putative pleckstrin homology domain. Mol Biol Cell (2006) 17:4856–65. doi:10.1091/mbc.E06-05-0449
89. Tang N, Lin T, Yang J, Foskett JK, Ostap EM. CIB1 and CaBP1 bind to the myo1c regulatory domain. J Muscle Res Cell Motil (2007) 28:285–91. doi:10.1007/s10974-007-9124-7
90. Boguslavsky S, Chiu T, Foley KP, Osorio-Fuentealba C, Antonescu CN, Bayer KU, et al. Myo1c binding to submembrane actin mediates insulin-induced tethering of GLUT4 vesicles. Mol Biol Cell (2012) 23:4065–78. doi:10.1091/mbc.E12-04-0263
91. Bose A, Robida S, Furcinitti PS, Chawla A, Fogarty K, Corvera S, et al. Unconventional myosin Myo1c promotes membrane fusion in a regulated exocytic pathway. Mol Cell Biol (2004) 24:5447–58. doi:10.1128/MCB.24.12.5447-5458.2004
92. Toyoda T, An D, Witczak CA, Koh HJ, Hirshman MF, Fujii N, et al. Myo1c regulates glucose uptake in mouse skeletal muscle. J Biol Chem (2011) 286:4133–40. doi:10.1074/jbc.M110.174938
93. Tiwari A, Jung JJ, Inamdar SM, Nihalani D, Choudhury A. The myosin motor Myo1c is required for VEGFR2 delivery to the cell surface and for angiogenic signaling. Am J Physiol Heart Circ Physiol (2013) 304:H687–96. doi:10.1152/ajpheart.00744.2012
94. Schietroma C, Yu HY, Wagner MC, Umbach JA, Bement WM, Gundersen CB. A role for myosin 1e in cortical granule exocytosis in Xenopus oocytes. J Biol Chem (2007) 282:29504–13. doi:10.1074/jbc.M705825200
95. Boal F, Laguerre M, Milochau A, Lang J, Scotti PA. A charged prominence in the linker domain of the cysteine-string protein Cspalpha mediates its regulated interaction with the calcium sensor synaptotagmin 9 during exocytosis. FASEB J (2011) 25:132–43. doi:10.1096/fj.09-152033
96. Cheng J, Grassart A, Drubin DG. Myosin 1E coordinates actin assembly and cargo trafficking during clathrin-mediated endocytosis. Mol Biol Cell (2012) 23:2891–904. doi:10.1091/mbc.E11-04-0383
97. Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol (2009) 10:778–90. doi:10.1038/nrm2786
98. Bond LM, Brandstaetter H, Sellers JR, Kendrick-Jones J, Buss F. Myosin motor proteins are involved in the final stages of the secretory pathways. Biochem Soc Trans (2011) 39:1115–9. doi:10.1042/BST0391115
99. DePina AS, Wollert T, Langford GM. Membrane associated nonmuscle myosin II functions as a motor for actin-based vesicle transport in clam oocyte extracts. Cell Motil Cytoskeleton (2007) 64:739–55. doi:10.1002/cm.20219
100. Neco P, Gil A, Del Mar Frances M, Viniegra S, Gutierrez LM. The role of myosin in vesicle transport during bovine chromaffin cell secretion. Biochem J (2002) 368:405–13. doi:10.1042/BJ20021090
101. Aoki R, Kitaguchi T, Oya M, Yanagihara Y, Sato M, Miyawaki A, et al. Duration of fusion pore opening and the amount of hormone released are regulated by myosin II during kiss-and-run exocytosis. Biochem J (2010) 429:497–504. doi:10.1042/BJ20091839
102. Doreian BW, Fulop TG, Smith CB. Myosin II activation and actin reorganization regulate the mode of quantal exocytosis in mouse adrenal chromaffin cells. J Neurosci (2008) 28:4470–8. doi:10.1523/JNEUROSCI.0008-08.2008
103. Bhat P, Thorn P. Myosin 2 maintains an open exocytic fusion pore in secretory epithelial cells. Mol Biol Cell (2009) 20:1795–803. doi:10.1091/mbc.E08-10-1048
104. Miklavc P, Hecht E, Hobi N, Wittekindt OH, Dietl P, Kranz C, et al. Actin coating and compression of fused secretory vesicles are essential for surfactant secretion – a role for Rho, formins and myosin II. J Cell Sci (2012) 125:2765–74. doi:10.1242/jcs.105262
105. Choi YO, Ryu HJ, Kim HR, Song YS, Kim C, Lee W, et al. Implication of phosphorylation of the myosin II regulatory light chain in insulin-stimulated GLUT4 translocation in 3T3-F442A adipocytes. Exp Mol Med (2006) 38:180–9. doi:10.1038/emm.2006.22
106. Fulcher FK, Smith BT, Russ M, Patel YM. Dual role for myosin II in GLUT4-mediated glucose uptake in 3T3-L1 adipocytes. Exp Cell Res (2008) 314:3264–74. doi:10.1016/j.yexcr.2008.08.007
107. Villanueva J, Torres V, Torregrosa-Hetland CJ, Garcia-Martinez V, Lopez-Font I, Viniegra S, et al. F-actin-myosin II inhibitors affect chromaffin granule plasma membrane distance and fusion kinetics by retraction of the cytoskeletal cortex. J Mol Neurosci (2012) 48:328–38. doi:10.1007/s12031-012-9800-y
108. Arous C, Rondas D, Halban PA. Non-muscle myosin IIA is involved in focal adhesion and actin remodelling controlling glucose-stimulated insulin secretion. Diabetologia (2013) 56:792–802. doi:10.1007/s00125-012-2800-1
109. Rondas D, Tomas A, Halban PA. Focal adhesion remodeling is crucial for glucose-stimulated insulin secretion and involves activation of focal adhesion kinase and paxillin. Diabetes (2011) 60:1146–57. doi:10.2337/db10-0946
110. Deacon SW, Gelfand VI. Of yeast, mice, and men. Rab proteins and organelle transport. J Cell Biol (2001) 152:F21–4. doi:10.1083/jcb.152.4.F21
111. Matesic LE, Yip R, Reuss AE, Swing DA, O’Sullivan TN, Fletcher CF, et al. Mutations in Mlph, encoding a member of the Rab effector family, cause the melanosome transport defects observed in leaden mice. Proc Natl Acad Sci U S A (2001) 98:10238–43. doi:10.1073/pnas.181336698
112. Bizario JC, Castro FA, Sousa JF, Fernandes RN, Damiao AD, Oliveira MK, et al. Myosin-V colocalizes with MHC class II in blood mononuclear cells and is up-regulated by T-lymphocyte activation. J Leukoc Biol (2002) 71:195–204.
113. Ivarsson R, Jing X, Waselle L, Regazzi R, Renstrom E. Myosin 5a controls insulin granule recruitment during late-phase secretion. Traffic (2005) 6:1027–35. doi:10.1111/j.1600-0854.2005.00342.x
114. Wu X, Bowers B, Rao K, Wei Q, Hammer JA III. Visualization of melanosome dynamics within wild-type and dilute melanocytes suggests a paradigm for myosin V function in vivo. J Cell Biol (1998) 143:1899–918. doi:10.1083/jcb.143.7.1899
115. Rudolf R, Kogel T, Kuznetsov SA, Salm T, Schlicker O, Hellwig A, et al. Myosin Va facilitates the distribution of secretory granules in the F-actin rich cortex of PC12 cells. J Cell Sci (2003) 116:1339–48. doi:10.1242/jcs.00317
116. Kogel T, Rudolf R, Hodneland E, Hellwig A, Kuznetsov SA, Seiler F, et al. Distinct roles of myosin Va in membrane remodeling and exocytosis of secretory granules. Traffic (2010) 11:637–50. doi:10.1111/j.1600-0854.2010.01048.x
117. Desnos C, Huet S, Fanget I, Chapuis C, Bottiger C, Racine V, et al. Myosin va mediates docking of secretory granules at the plasma membrane. J Neurosci (2007) 27:10636–45. doi:10.1523/JNEUROSCI.1228-07.2007
118. Huet S, Fanget I, Jouannot O, Meireles P, Zeiske T, Larochette N, et al. Myrip couples the capture of secretory granules by the actin-rich cell cortex and their attachment to the plasma membrane. J Neurosci (2012) 32:2564–77. doi:10.1523/JNEUROSCI.2724-11.2012
119. Varadi A, Tsuboi T, Rutter GA. Myosin Va transports dense core secretory vesicles in pancreatic MIN6 beta-cells. Mol Biol Cell (2005) 16:2670–80. doi:10.1091/mbc.E04-11-1001
120. Prekeris R, Terrian DM. Brain myosin V is a synaptic vesicle-associated motor protein: evidence for a Ca2+-dependent interaction with the synaptobrevin-synaptophysin complex. J Cell Biol (1997) 137:1589–601. doi:10.1083/jcb.137.7.1589
121. Karcher RL, Roland JT, Zappacosta F, Huddleston MJ, Annan RS, Carr SA, et al. Cell cycle regulation of myosin-V by calcium/calmodulin-dependent protein kinase II. Science (2001) 293:1317–20. doi:10.1126/science.1061086
122. Costa MC, Mani F, Santoro W Jr, Espreafico EM, Larson RE. Brain myosin-V, a calmodulin-carrying myosin, binds to calmodulin-dependent protein kinase II and activates its kinase activity. J Biol Chem (1999) 274:15811–9. doi:10.1074/jbc.274.22.15811
123. Kenigsberg RL, Trifaro JM. Microinjection of calmodulin antibodies into cultured chromaffin cells blocks catecholamine release in response to stimulation. Neuroscience (1985) 14:335–47. doi:10.1016/0306-4522(85)90183-6
124. Rudolf R, Bittins CM, Gerdes HH. The role of myosin V in exocytosis and synaptic plasticity. J Neurochem (2011) 116:177–91. doi:10.1111/j.1471-4159.2010.07110.x
125. Bryant Z, Altman D, Spudich JA. The power stroke of myosin VI and the basis of reverse directionality. Proc Natl Acad Sci U S A (2007) 104:772–7. doi:10.1073/pnas.0610144104
126. Wells AL, Lin AW, Chen LQ, Safer D, Cain SM, Hasson T, et al. Myosin VI is an actin-based motor that moves backwards. Nature (1999) 401:505–8. doi:10.1038/46835
127. Bahloul A, Chevreux G, Wells AL, Martin D, Nolt J, Yang Z, et al. The unique insert in myosin VI is a structural calcium-calmodulin binding site. Proc Natl Acad Sci U S A (2004) 101:4787–92. doi:10.1073/pnas.0306892101
128. Au JS, Puri C, Ihrke G, Kendrick-Jones J, Buss F. Myosin VI is required for sorting of AP-1B-dependent cargo to the basolateral domain in polarized MDCK cells. J Cell Biol (2007) 177:103–14. doi:10.1083/jcb.200608126
129. Buss F, Arden SD, Lindsay M, Luzio JP, Kendrick-Jones J. Myosin VI isoform localized to clathrin-coated vesicles with a role in clathrin-mediated endocytosis. EMBO J (2001) 20:3676–84. doi:10.1093/emboj/20.14.3676
130. Buss F, Kendrick-Jones J. Multifunctional myosin VI has a multitude of cargoes. Proc Natl Acad Sci U S A (2011) 108:5927–8. doi:10.1073/pnas.1103086108
131. Phichith D, Travaglia M, Yang Z, Liu X, Zong AB, Safer D, et al. Cargo binding induces dimerization of myosin VI. Proc Natl Acad Sci U S A (2009) 106:17320–4. doi:10.1073/pnas.0909748106
132. Yu C, Feng W, Wei Z, Miyanoiri Y, Wen W, Zhao Y, et al. Myosin VI undergoes cargo-mediated dimerization. Cell (2009) 138:537–48. doi:10.1016/j.cell.2009.05.030
133. Aschenbrenner L, Lee T, Hasson T. Myo6 facilitates the translocation of endocytic vesicles from cell peripheries. Mol Biol Cell (2003) 14:2728–43. doi:10.1091/mbc.E02-11-0767
134. Buss F, Luzio JP, Kendrick-Jones J. Myosin VI, a new force in clathrin mediated endocytosis. FEBS Lett (2001) 508:295–9. doi:10.1016/S0014-5793(01)03065-4
135. Hasson T. Myosin VI: two distinct roles in endocytosis. J Cell Sci (2003) 116:3453–61. doi:10.1242/jcs.00669
136. Puri C. Loss of myosin VI no insert isoform (NoI) induces a defect in clathrin-mediated endocytosis and leads to caveolar endocytosis of transferrin receptor. J Biol Chem (2009) 284:34998–5014. doi:10.1074/jbc.M109.012328
137. Roberts R, Lister I, Schmitz S, Walker M, Veigel C, Trinick J, et al. Myosin VI: cellular functions and motor properties. Philos Trans R Soc Lond B Biol Sci (2004) 359:1931–44. doi:10.1098/rstb.2004.1563
138. Tumbarello DA, Waxse BJ, Arden SD, Bright NA, Kendrick-Jones J, Buss F. Autophagy receptors link myosin VI to autophagosomes to mediate Tom1-dependent autophagosome maturation and fusion with the lysosome. Nat Cell Biol (2012) 14:1024–35. doi:10.1038/ncb2589
139. Hertzano R, Shalit E, Rzadzinska AK, Dror AA, Song L, Ron U, et al. A Myo6 mutation destroys coordination between the myosin heads, revealing new functions of myosin VI in the stereocilia of mammalian inner ear hair cells. PLoS Genet (2008) 4:e1000207. doi:10.1371/journal.pgen.1000207
140. Hicks JL, Deng WM, Rogat AD, Miller KG, Bownes M. Class VI unconventional myosin is required for spermatogenesis in Drosophila. Mol Biol Cell (1999) 10:4341–53. doi:10.1091/mbc.10.12.4341
141. Noguchi T, Frank DJ, Isaji M, Miller KG. Coiled-coil-mediated dimerization is not required for myosin VI to stabilize actin during spermatid individualization in Drosophila melanogaster. Mol Biol Cell (2009) 20:358–67. doi:10.1091/mbc.E08-07-0776
142. Noguchi T, Lenartowska M, Miller KG. Myosin VI stabilizes an actin network during Drosophila spermatid individualization. Mol Biol Cell (2006) 17:2559–71. doi:10.1091/mbc.E06-01-0031
143. Vreugde S, Ferrai C, Miluzio A, Hauben E, Marchisio PC, Crippa MP, et al. Nuclear myosin VI enhances RNA polymerase II-dependent transcription. Mol Cell (2006) 23:749–55. doi:10.1016/j.molcel.2006.07.005
144. Maddugoda MP, Crampton MS, Shewan AM, Yap AS. Myosin VI and vinculin cooperate during the morphogenesis of cadherin cell cell contacts in mammalian epithelial cells. J Cell Biol (2007) 178:529–40. doi:10.1083/jcb.200612042
145. Millo H, Leaper K, Lazou V, Bownes M. Myosin VI plays a role in cell-cell adhesion during epithelial morphogenesis. Mech Dev (2004) 121:1335–51. doi:10.1016/j.mod.2004.06.007
146. Warner CL, Stewart A, Luzio JP, Steel KP, Libby RT, Kendrick-Jones J, et al. Loss of myosin VI reduces secretion and the size of the Golgi in fibroblasts from Snell’s waltzer mice. EMBO J (2003) 22:569–79. doi:10.1093/emboj/cdg055
147. Avraham KB, Hasson T, Steel KP, Kingsley DM, Russell LB, Mooseker MS, et al. The mouse Snell’s waltzer deafness gene encodes an unconventional myosin required for structural integrity of inner ear hair cells. Nat Genet (1995) 11:369–75. doi:10.1038/ng1295-369
148. Self T, Sobe T, Copeland NG, Jenkins NA, Avraham KB, Steel KP. Role of myosin VI in the differentiation of cochlear hair cells. Dev Biol (1999) 214:331–41. doi:10.1006/dbio.1999.9424
149. Puri C, Chibalina MV, Arden SD, Kruppa AJ, Kendrick-Jones J, Buss F. Overexpression of myosin VI in prostate cancer cells enhances PSA and VEGF secretion, but has no effect on endocytosis. Oncogene (2010) 29:188–200. doi:10.1038/onc.2009.328
150. Bond LM, Peden AA, Kendrick-Jones J, Sellers JR, Buss F. Myosin VI and its binding partner optineurin are involved in secretory vesicle fusion at the plasma membrane. Mol Biol Cell (2011) 22:54–65. doi:10.1091/mbc.E10-06-0553
151. Osterweil E, Wells DG, Mooseker MS. A role for myosin VI in postsynaptic structure and glutamate receptor endocytosis. J Cell Biol (2005) 168:329–38. doi:10.1083/jcb.200410091
152. Yano H, Ninan I, Zhang H, Milner TA, Arancio O, Chao MV. BDNF-mediated neurotransmission relies upon a myosin VI motor complex. Nat Neurosci (2006) 9:1009–18. doi:10.1038/nn1730
153. Iwaki M, Tanaka H, Iwane AH, Katayama E, Ikebe M, Yanagida T. Cargo-binding makes a wild-type single-headed myosin-VI move processively. Biophys J (2006) 90:3643–52. doi:10.1529/biophysj.105.075721
154. Naccache SN, Hasson T. Myosin VI altered at threonine 406 stabilizes actin filaments in vivo. Cell Motil Cytoskeleton (2006) 63:633–45. doi:10.1002/cm.20150
155. Rock RS, Rice SE, Wells AL, Purcell TJ, Spudich JA, Sweeney HL. Myosin VI is a processive motor with a large step size. Proc Natl Acad Sci U S A (2001) 98:13655–9. doi:10.1073/pnas.191512398
156. Majewski L, Sobczak M, Redowicz MJ. Myosin VI is associated with secretory granules and is present in the nucleus in adrenal medulla chromaffin cells. Acta Biochim Pol (2010) 57:109–14.
Keywords: cortical actin, myosin, regulated exocytosis, cdc42, phosphoinositides, secretory vesicles
Citation: Papadopulos A, Tomatis VM, Kasula R and Meunier FA (2013) The cortical acto-myosin network: from diffusion barrier to functional gateway in the transport of neurosecretory vesicles to the plasma membrane. Front. Endocrinol. 4:153. doi: 10.3389/fendo.2013.00153
Received: 15 August 2013; Accepted: 05 October 2013;
Published online: 21 October 2013.
Edited by:
Stephane Gasman, Centre National de la Recherche Scientifique, FranceReviewed by:
Nicolas Vitale, Centre National de la Recherche Scientifique, FranceJose-Maria Trifaro, University of Ottawa, Canada
Copyright: © 2013 Papadopulos, Tomatis, Kasula and Meunier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Frederic A. Meunier, Queensland Brain Institute, The University of Queensland, St Lucia Campus, QBI Building #79, St Lucia, QLD 4072, Australia e-mail:Zi5tZXVuaWVyQHVxLmVkdS5hdQ==