 Jeppe Skov
Jeppe Skov Frederik Persson3
Frederik Persson3- 1Department of Endocrinology and Internal Medicine, Aarhus University Hospital, Aarhus, Denmark
- 2Novo Nordisk A/S, Bagsvaerd, Denmark
- 3Steno Diabetes Center, Gentofte, Denmark
- 4Department of Clinical Physiology and Molecular Imaging, Aarhus University Hospital, Aarhus, Denmark
- 5Department of Clinical Medicine, Aarhus University, Aarhus, Denmark
The actions of angiotensin peptides are diverse and locally acting tissue renin–angiotensin systems (RAS) are present in almost all tissues of the body. An activated RAS strongly correlates to metabolic disease (e.g., diabetes) and its complications and blockers of RAS have been demonstrated to prevent diabetes in humans. Hyperglycemia, obesity, hypertension, and cortisol are well-known risk factors of metabolic disease and all stimulate tissue RAS whereas glucagon-like peptide-1, vitamin D, and aerobic exercise are inhibitors of tissue RAS and to some extent can prevent metabolic disease. Furthermore, an activated tissue RAS deteriorates the same risk factors creating a system with several positive feedback pathways. The primary effector hormone of the RAS, angiotensin II, stimulates reactive oxygen species, induces tissue damage, and can be associated to most diabetic complications. Based on these observations, we hypothesize that an activated tissue RAS is the principle cause of metabolic syndrome and type 2 diabetes, and additionally is mediating the majority of the metabolic complications. The involvement of positive feedback pathways may create a self-reinforcing state and explain why metabolic disease initiate and progress. The hypothesis plausibly unifies the major predictors of metabolic disease and places tissue RAS regulation in the center of metabolic control.
Introduction
The renin–angiotensin system (RAS) is classically considered to play key roles in regulating blood pressure as well as water and sodium balance. Liver secreted angiotensinogen (AGT) is enzymatically cleaved to angiotensin (ANG) I by kidney-derived renin. ANG I is, hereafter, cleaved by angiotensin converting enzyme (ACE) to the effector hormone ANG II. This classical theory has, however, been fundamentally revised in recent years. It is now evident that the components of RAS, in addition to the classical pathway, are produced and acting locally in multiple tissues; a concept known as tissue RAS. The local effects are diverse and depend on the specific tissues involved. Additional components (angiotensin peptides, receptors, and enzymes) of the system have been identified but ANG II is still believed to exert the most important actions via the ANG II receptor type 1 (AT1R). Although many details on tissue RAS have been elucidated, the main function of the systems still remains partly unknown. For reviews on tissue RAS see Ref. (1–3).
It is well established that an activated RAS is a major risk factor of both cardiovascular (4) and renal disease (5). Inhibitors of RAS [ACE inhibitors, ANG II receptor blockers (ARB), and renin inhibitors] are therefore widely used in the clinic. The RAS is also closely associated to the metabolic syndrome (6) and recently, inhibitors of RAS have shown to prevent the onset of type 2 diabetes (T2D) in high risk populations (7, 8).
These latter findings suggest an important role of RAS in metabolic disease. However, compared to hormones such as glucagon-like peptide-1 (GLP-1) or cortisol, the RAS may seem only a weak regulator of metabolism. We will challenge this perception and argue that the tissue RAS can be considered as the most central player in metabolic regulation.
This hypothesis originates from reported studies of the tissue RAS regulation. A range of metabolic potent hormones and conditions closely interact with tissue RAS and it is often hard to distinguish direct hormonal actions from actions secondary to tissue RAS stimulation/inhibition. Because of this consistent convergence toward tissue RAS and the well-known potent actions of especially ANG II, we hypothesize that the effects are mediated through regulations of tissue RAS. Implications of this hypothesis may not only change our view on endocrine physiology but also explain both the origin of some metabolic diseases and the accompanying complications.
Tissue RAS Regulation
Below, we will review the interactions between tissue RAS and some of the main players in metabolic disease [hyperglycemia, obesity, hypertension, exercise, GLP-1, cortisol, and vitamin D (VitD)].
Hyperglycemia Stimulates Tissue RAS and Vice Versa
Patients with diabetes are characterized by an impaired ability to secrete insulin and/or a decreased sensitivity to insulin. T2D and metabolic syndrome have consistently been related to an activated RAS, and several in vitro studies find hyperglycemia to stimulate tissue RAS in different tissues (9–14). Renin release after GPR91 receptor activation with succinate may partly be the mechanism of action (15).
A recent 26-weeks randomized controlled trial found that the ARB valsartan improves both beta cell function and insulin sensitivity in subjects with impaired glucose metabolism (16). The large NAVIGATOR study found valsartan treatment to relatively reduce the incidence of diabetes by 14% compared to placebo in patients with impaired glucose tolerance during 5 years follow up (8). The DREAM study did not find ACE inhibition with ramipril for 3 years to significantly reduce the incidence of diabetes but it did increase regression to normoglycemia (17). Several other studies in both animals and humans find that ANG II decreases insulin secretion and sensitivity while these are improved by RAS inhibitors. Conflicting results do exist, however, with regard to insulin sensitivity, as recently reviewed (18, 19).
In addition to acutely diminished insulin release, ANG II decreases beta cell proliferation and stimulates beta cell apoptosis leading to impaired long-term islet function (20).
Obesity Stimulates Tissue RAS and Vice Versa
It is well-known that obesity predisposes to metabolic disease and that many T2D patients are obese. Several studies in humans and animals find obesity associated with enhanced activity of both systemic RAS and adipose tissue RAS (21). Especially, the AGT synthesis is very developed in adipocytes and contributes significantly to the systemic pool (22). The activity of tissue RAS is higher in visceral/central adipose tissue than in subcutaneous tissue (23), which may explain the risks related to upper body visceral fat accumulation (24).
Renin–angiotensin system components are complexly involved in the development of obesity by affections of satiety, energy expenditure, and adipocyte growth and differentiation (21). ARBs reduce body weight in both rodents and patients (25, 26). Surprisingly, chronic ANG II infusion also decreases body weight in rodents (27). However, adipose specific over-activation of AGT expression increases body weight in mice (28), which may be the setting that mimics obesity related RAS activation the best. The apparently contradicting findings probably illustrate the importance of the local nature of RAS in adipose tissue.
Hypertension Stimulates Tissue RAS and Vice Versa
It is well-known that ANG II induces hypertension through vasoconstriction and sodium retention. Hypertension, however, also activates tissue RAS through mechanical stretch. This has been shown in vitro and in vivo in several studies in cardiac myocytes (29) and mesangial cells (30) as well as in skeletal muscle myoblasts (31). Generally, the mechanical stretch is found to up-regulate tissue RAS synthesis of AGT, ANG II, and AT1Rs.
GLP-1 Inhibits Tissue RAS and Vice Versa
GLP-1 and ANG II have multiple different actions in a variety of tissues that are not included in the classical view of either of the hormones. Interestingly, when the topic is studied in detail, all GLP-1 actions seem to be counteracted by ANG II. We therefore question whether the two systems are independent or if GLP-1 actions partly or totally depend on the more widely distributed tissue RAS. This dependence is supported by several studies.
The circulatory ANG II levels decrease in response to GLP-1 infusion in healthy subjects (32). Exendin-4 (GLP-1R agonist) attenuated the effect of ANG II-induced hypertension in mice (33) and in vitro GLP-1 effectively inhibited ANG II-induced mesangial cell damage (34). GLP-1 and the ARB candesartan additively prevent β-cell apoptosis through the same signaling pathway (20). A dipeptidyl peptidase-4 inhibitor and valsartan additively improve both β-cell structure and function in T2D mice (35). Finally, a recent study provides a novel biochemical pathway on how GLP-1 inhibits ANG II signaling by demonstrating that GLP-1R agonists induce protective actions in glomerular endothelium cells by inhibiting the post-receptor signaling pathway of ANG II at phospho-c-Raf(Ser338) via phospho-c-Raf(Ser259) (12). Thus, it seems likely that GLP-1 from both a functional and biochemical perspective is an inhibitor of ANG II actions. Since it is difficult to identify GLP-1 actions that are not counteracted by ANG II, it is tempting to hypothesize that GLP-1 acts primarily through down-regulation of tissue RAS.
T2D patients have an impaired incretin effect (36), low levels of GLP-1 (37), and an impaired response to GLP-1 (38). In a rat model of metabolic syndrome, ARBs were found to increase fasting plasma levels of GLP-1 by 2.5-fold and increase pancreatic GLP-1R expression (39). This strongly suggests an AT1R-mediated decrease in GLP-1 synthesis and GLP-1R expression.
Cortisol Stimulates Tissue RAS and Vice Versa
Glucocorticoids are steroid hormones with a wide range of important and essential actions. The hormones’ marked impact on metabolism is revealed in Cushing’s syndrome where excess cortisol induces central obesity, hypertension, osteoporosis, and diabetes. These manifestations are similar to those of increased RAS activity.
Glucocorticoids stimulate AGT transcription and secretion in adipocytes (40), kidney tubular cells (41, 42), and cardiac fibroblasts (43). In addition, glucocorticoids up-regulate AT1R expression in vascular smooth muscle cells but do not seem to directly affect post-receptor signaling (44, 45).
Besides ANG II’s well-known actions on adrenal aldosterone secretion, it also stimulates cortisol secretion (46, 47).
The above indicates that the metabolic effects of glucocorticoids may partly be mediated via up-regulation of tissue RAS. The anti-inflammatory properties of glucocorticoids, however, cannot be explained by tissue RAS regulation.
Vitamin D Inhibits Tissue RAS and (maybe) Vice Versa
Vitamin D has become an important topic in metabolic research and VitD deficiency has been linked to insulin resistance and impaired beta cell function (48). The VITAL study found that VitD receptor activation reduced albuminuria in T2D patients in a fashion comparable to RAS-inhibition (49). Several human studies report an inverse relationship between VitD levels and circulating RAS activity (50, 51). In VitD receptor null mice, the renin expression and plasma ANG II concentration are increased several-fold (52). In vitro VitD inhibits pancreatic islet RAS component synthesis in parallel to the improvement in beta cell secretory function (53). The VitD induced suppression of renin gene transcription is believed partly to be the mechanism of action (54). Considerable additional evidence supports the theory of VitD induced inhibition of RAS at both systemic and tissue level; for reviews see (54, 55).
In an interesting recent paper, the close interactions between VitD and the RAS are reviewed from both a functional and evolutionary perspective (56). It is proposed that increased RAS activity negatively regulates the VitD level through inflammatory responses and that this explain the pandemic of VitD deficiency (56). However, the specific mechanisms underlying this effect are not documented.
Regular Exercise Inhibits Tissue RAS
It is well established that moderate aerobic exercise reduces the risk of both metabolic and cardiovascular disease in humans (57). Studies in rodents find chronic exercise to inhibit tissue RAS activation in the brain (58) (reduced local ACE and AT1R), heart (59, 60) (reduced local ACE and ANG II), and kidney (reduced local AT1R) (61). These findings suggest that down-regulation of tissue RAS may partly mediate the beneficial effects related to frequent exercise.
During exercise, however, tissue RAS seem to be acutely activated in the kidneys (increased ANG II, ACE, and AGT), which may function to redistribute blood flow to the muscles (62).
The mechanism behind the different effects of long-term chronic exercise and short-term acute exercise on tissue RAS activity is not fully elucidated but may be related to the differences in sympathetic tone between the two situations. The sympathetic tone increases acutely during exercise whereas regular exercise has shown to decrease an elevated sympathetic tone (63). In the kidneys, at least, RAS activity increases in response to sympathetic stimulation.
Other Hormones and Tissue RAS
Angiotensin II and vasopressin are both vasoconstrictors and antidiuretics. In the kidneys, ANG II and vasopressin stimulate aquaporin-2 insertion in the inner medulla through the AT1R and V2 receptor, respectively. Interestingly, in AT1R knock-out mice the vasopressin-induced signaling and aquaporin-2 insertion are defective (64). This might reflect a dependence of vasopressin on the tissue RAS.
In the kidney, dopamine functions primarily through the D1 receptor, which is a G-protein coupled receptor similar to the GLP-1R. Like GLP-1, and in contrast to ANG II, dopamine inhibits tubular salt reabsorption and seems to prevent kidney damage (65). Interestingly, dopamine is found to inhibit AGT synthesis in proximal tubule cells (65), suppress renin expression (66), and down-regulate AT1Rs (67, 68).
Hypothesis
Tissue RAS is Central to Cell Regulation, and the Effect of Multiple Hormones Depend on the System
Multiple hormones and conditions interact closely with the widely distributed tissue RAS. The local effects of ANG II show great diversity in different tissues and often mimic or oppose the actions of the hormones, which regulate tissue RAS.
We therefore hypothesize that the apparent actions of many hormones are induced through alterations in tissue RAS activity. Thus, it is the cellular pathways of tissue RAS that in the end effect actions. The dependence can be direct and strong (e.g., GLP-1) or partial and less direct (e.g., VitD) mediated via regulations of genes coding for RAS components.
Especially hormones acting through G-Protein coupled receptors seem to be likely candidates for direct tissue RAS dependent hormones. The nature of the receptor (RAS stimulating or inhibiting) along with the distribution will determine the effects of the hormone.
Increased Tissue RAS Activity is Responsible for Metabolic Disease
The observation of a relationship between RAS and metabolic disease is not new (6, 69). An activated RAS promotes all the characteristic properties of metabolic syndrome [hypertension, hyperglycemia, insulin resistance, obesity, and dyslipidemia (6)]. The novelty lies in the explicit coupling between tissue RAS and the major risk factors of metabolic disease. If tissue RAS completely or partly underlies the actions of GLP-1, VitD, and cortisol as hypothesized, then the metabolic power of the system is greater than first anticipated. Therefore, we propose that an activated tissue RAS is mandatory in the pathogenesis of most metabolic diseases, including T2D. The different degrees of tissue RAS activation in different tissues will determine the phenotype of the disease.
The activity of RAS also rises physiologically during pregnancy, which could partly explain the origin of gestational diabetes in predisposed individuals (70). In addition, preeclampsia is potentially the result of extreme RAS activation (70).
Metabolic Disease Initiates Due to the Self-Reinforcing Properties of Tissue RAS
As argued above and illustrated in Figure 1, tissue RAS is regulated by well-known risk factors of diabetes and through several positive feedback mechanisms. ANG II even seems to stimulate its own secretion (71). This makes the basis for an unstable system and we hypothesize that T2D and other metabolic diseases develop when tissue RAS becomes self-reinforcing. This may happen due to one far-off parameter (hyperglycemia in type 1 diabetes (T1D) or high dose glucocorticoid treatment) but is often caused by a sum of risk factors, like the combination: obesity, inactivity, high glycemic index diet, and VitD deficiency.
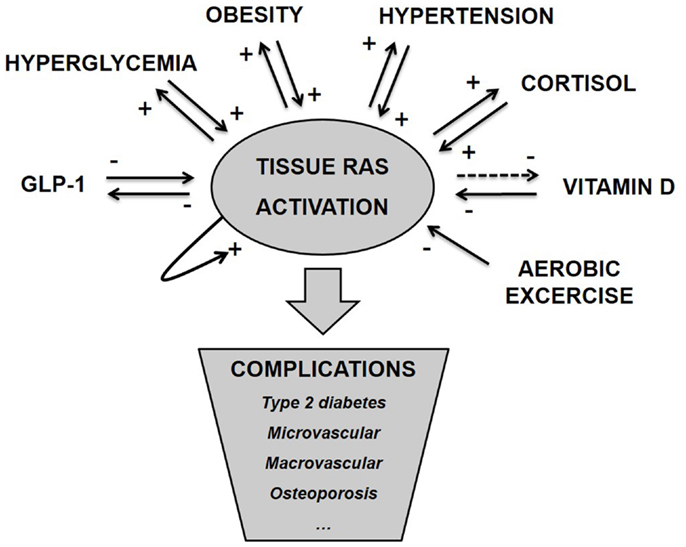
Figure 1. Tissue RAS activity is controlled by several well-known metabolic hormones and conditions. This control often involves positive feedback mechanisms, which makes the basis for an unstable system and progression of disease. We hypothesize that metabolic complications arise as a consequence of pathological activated tissue RAS.
Metabolic Complications are Caused by Increased Tissue RAS Activity
The multiple different effects of ANG II depend on which tissue/cell is involved, the receptor, and the time frame of stimulation. Many of ANG II’s immediate effects (e.g., vasoconstriction) are mediated via the AT1R through classical G-protein-dependent signaling pathways (72).
Stimulation of the AT1R also activates the membrane-bound NAD(P)H oxidase leading to increased generation of reactive oxygen species (ROS). The NAD(P)H formed ROS may activate mitochondrial KATP channels to additionally burst mitochondrial ROS generation (73). Alternatively, the mitochondrial ROS formation may be mediated via local mitochondrial ANG II sensitive receptors (74). After its generation, intracellular ROS can activate many down-stream molecules such as mitogen-activated protein kinases, protein tyrosine phosphatases, protein tyrosine kinases, and transcriptional factors, which if chronically activated will promote inflammation, atherosclerosis, thrombosis, and fibrogenesis (72, 73).
In almost all organ systems, RAS activation has been associated with degeneration, tissue remodeling, and dysfunction that are likely secondary to ROS formation and phenotypic known as the metabolic syndrome. Practically all complications in diabetes are related to the RAS and increased ANG II levels including ischemia, myocardial infarction, stroke, nephropathy, cardiomyopathy, retinopathy (75), polyneuropathy (76), and erectile dysfunction (77).
We therefore hypothesize that the majority of complications seen in metabolic disease are consequences of a pathological-activated tissue RAS. Thus, hyperglycemia may not directly induce complications apart from the ability to stimulate tissue RAS. Although this statement may sound controversial, we do not find it to conflict with the leading theory on the pathogenesis of diabetic complications, The Brownlee hypothesis (78). The Brownlee hypothesis states that all pathological metabolic pathways induced by hyperglycemia are secondary to increased ROS (78). We just add an extra link to the chain so that hyperglycemia stimulates ROS production via stimulation of tissue RAS. By introducing ROS formation secondary to tissue RAS activation as a unifying mediator of complications, we have plausibly linked all the major risk factors in metabolic disease.
Essentially, the full theory simplifies T2D to a complication of a malfunctioning tissue RAS which places it next to other complications. However, the “T2D complication” may be special in the ability to accelerate RAS over-activity (hyperglycemia induced) and thereby other complications. According to the hypothesis, tissue RAS must be pathologically activated prior to the onset of T2D and this may explain why complications often exist at the time of T2D diagnosis. By contrast, the complications seen in T1D will only occur if glucose level is uncontrolled and allowed to secondarily activate tissue RAS.
Both T1D and T2D are associated with a markedly increased risk of bone fractures despite increased bone mineral density in obese T2D patients (79, 80). Tissue RAS is also present in bones and involved in bone remodeling (81). In an eight-week interventional study, ARBs improved bone mass and strength in osteoporotic rat femurs (82). Consistent with this, an observational study demonstrated that ARB treatment significantly reduced fracture risk (hazard ratio 0.76) in older adults (83). We therefore hypothesize that the osteoporotic complication is caused primarily by an activated bone tissue RAS and that the positive VitD effects on bone metabolism are partly due to RAS-inhibition. Equally, the well-known pro-osteoporotic properties of cortisol as well as the less-known anti-osteoporotic properties of GLP-1 (84, 85) are caused by bone tissue RAS stimulation and inhibition respectively.
Discussion
Tissue RAS is regulated by multiple factors of which we have only mentioned some. We have focused on the interaction between tissue RAS and the major metabolic hormones and conditions, but this is certainly not the complete story. Tissue RAS is present in almost every tissue and exert multiple different actions determined by the involved cells. Thus, we speculate that tissue RAS should be seen as a common effectuating machinery required for multiple different purposes.
From an evolutionary point of view, it is cost-effective to have a common system to potentiate and effect the actions of the regulating hormones. Interestingly, the RAS is also found in primitive animals without a closed circulatory system (86), which indicate that the system is far more than a mediator of vasoconstriction.
The RAS offers multiple targets for regulation as we have seen (AGT, renin, AT1R, ACE, down-stream signaling) and several more which we have excluded (prorenin receptor, ACE2, AT2R, AT4R, neutral endopeptidase, chymase, Mas receptor). We have focused on the ANG II–AT1R interactions since these are the best described and considered the most important. However, the system is complex and several other components probably play significant roles as well. Especially, the AT2Rs have come in focus in recent years and seem to be important counter-regulators of AT1R function (87). This may be of particular importance when the AT1Rs are pharmacologically blocked.
While we have focused only on a few regulators of tissue RAS the same is true regarding the implications of the hypothesis. We will likely identify that many known disorders are caused by a malfunctioning tissue RAS in specific organs and these could likely include inflammatory bowel disease, psoriasis, preeclampsia, Alzheimer’s disease, and depression.
It is essential to emphasize the importance of the local nature of tissue RAS and it is somewhat misleading to talk about a general over-activity. Even the circulating classical RAS should to some degree be considered a local independent system when compared to the tissue level systems. However, since tissue RAS in different tissues have many regulatory hormones and conditions in common, it makes sense to talk about a general direction of activity level. Additionally, although paracrine acting, there seem to be a degree of “spill over” of RAS components from one tissue to another. This is of course very pronounced with the classical circulating RAS but adipocyte AGT synthesis, as an example, also contributes significantly to the total AGT pool.
The local environments with respect to angiotensin concentrations and angiotensin sensitivity are generally hard to assess, especially in humans. In addition, evidence suggests that besides paracrine and autocrine actions, ANG II exerts intracrine effects mediated by intracellular located receptors. ANG II may be internalized or act immediately after intracellular synthesis without ever leaving the cell (2, 88). This may partly explain why the importance of tissue RAS is still broadly unacknowledged and why classical extracellular RAS inhibitors in many ways are insufficient.
If the phenotypes of metabolic diseases and the complications seen in relation hereto are caused by locally activated tissue RAS as hypothesized, diagnosing and treating an activated RAS become the primary target in the clinic. Screening for activated tissue RAS in relevant tissues instead of elevated glycemic level would theoretically serve as an early and more efficient marker of the risk of complications – including T2D. The urinary excretion of AGT has been proposed as a non-invasive marker of renal tissue RAS activity (89). Although this particular method may be imperfect (90), the concept of assessing specific tissue RAS activity may be of great diagnostic value in the future.
Provided that the proposed hypothesis is correct, it should theoretically be possible to prevent T2D by securing that tissue RAS never reaches a high or self-reinforcing level, which emphasizes the importance of prophylaxis and early intervention. Besides lifestyle interventions, drugs aiming to decrease tissue RAS should be preferred and drugs targeting the intracellular part of tissue RAS or some of the newer extracellular components may be the drugs of tomorrow.
A few studies suggest that the ANG II-induced ROS formation is mediated through AT1Rs located directly on the mitochondrial membranes (74), whereas one study cannot confirm this relationship (91). If indeed existing and functional, these mitochondrial AT1Rs seem particular attractive to block selectively to prevent complications.
The hypothesis obviously needs further investigation. While several interactions with tissue RAS are well described, their individual importance is less characterized. In fact, the importance of tissue RAS, in general, is poorly understood and according to this hypothesis greatly underestimated. The local nature of the systems, however, makes the design of appropriate experiments harder. Interesting pathways can be elucidated in vitro but the importance in vivo can be difficult to extrapolate. One possible experimental approach is the use of AT1R knock-out mice. According to the hypothesis, these mice would have defective responses to hormones, which primarily depend on tissue RAS.
In conclusion, we have hypothesized that the tissue RAS is in the very center of metabolic regulation and is the true effector of multiple hormones. Implications of this hypothesis suggest a new unifying understanding of the nature, initiation, and complications of metabolic disease.
Conflict of Interest Statement
Jørgen Frøkiær has no conflicts of interests. Jeppe Skov is employed by Novo Nordisk in a PhD fellowship. Frederik Persson owns stock in Novo Nordisk has received research grants from Novartis, and speaker honoraria from Novartis, Eli Lilly, and Boehringer Ingelheim. Jens Sandahl Christiansen is a recipient of unrestricted research grants from Novo Nordisk, a clinical investigator for Novo Nordisk, receives lecture fees from Novo Nordisk, Eli Lilly, and Pfizer and is a member of Novo Nordisk advisory boards.
Acknowledgments
The work was supported by Novo Nordisk A/S and the Danish Agency for Science Technology and Innovation. Jeppe Skov developed the hypothesis and drafted the manuscript. The content was improved after fruitful discussions with Frederik Persson, Jørgen Frøkiær, and Jens Sandahl Christiansen, who also critically revised the manuscript.
References
1. Paul M, Poyan Mehr A, Kreutz R. Physiology of local renin-angiotensin systems. Physiol Rev (2006) 86:747–803. doi:10.1152/physrev.00036.2005
2. Ellis B, Li XC, Miguel-Qin E, Gu V, Zhuo JL. Evidence for a functional intracellular angiotensin system in the proximal tubule of the kidney. Am J Physiol Regul Integr Comp Physiol (2012) 302:R494–509. doi:10.1152/ajpregu.00487.2011
3. Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev (2007) 59:251–87. doi:10.1124/pr.59.3.3
4. Unger T. The role of the renin-angiotensin system in the development of cardiovascular disease. Am J Cardiol (2002) 89:3A–9A. doi:10.1016/S0002-9149(01)02321-9
5. Vejakama P, Thakkinstian A, Lertrattananon D, Ingsathit A, Ngarmukos C, Attia J. Reno-protective effects of renin-angiotensin system blockade in type 2 diabetic patients: a systematic review and network meta-analysis. Diabetologia (2012) 55:566–78. doi:10.1007/s00125-011-2398-8
6. Putnam K, Shoemaker R, Yiannikouris F, Cassis LA. The renin-angiotensin system: a target of and contributor to dyslipidemias, altered glucose homeostasis, and hypertension of the metabolic syndrome. Am J Physiol Heart Circ Physiol (2012) 302:H1219–30. doi:10.1152/ajpheart.00796.2011
7. Scheen AJ. Renin-angiotensin system inhibition prevents type 2 diabetes mellitus. Part 1. A meta-analysis of randomised clinical trials. Diabetes Metab (2004) 30:487–96. doi:10.1016/S1262-3636(07)70146-5
8. McMurray JJ, Holman RR, Haffner SM, Bethel MA, Holzhauer B, Hua TA, et al. Effect of valsartan on the incidence of diabetes and cardiovascular events. N Engl J Med (2010) 362:1477–90. doi:10.1056/NEJMoa1001121
9. Singh R, Singh AK, Alavi N, Leehey DJ. Mechanism of increased angiotensin II levels in glomerular mesangial cells cultured in high glucose. J Am Soc Nephrol (2003) 14:873–80. doi:10.1097/01.ASN.0000060804.40201.6E
10. Singh VP, Le B, Bhat VB, Baker KM, Kumar R. High-glucose-induced regulation of intracellular ANG II synthesis and nuclear redistribution in cardiac myocytes. Am J Physiol Heart Circ Physiol (2007) 293:H939–48. doi:10.1152/ajpheart.00391.2007
11. Singh VP, Le B, Khode R, Baker KM, Kumar R. Intracellular angiotensin II production in diabetic rats is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis. Diabetes (2008) 57:3297–306. doi:10.2337/db08-0805
12. Mima A, Hiraoka-Yamomoto J, Li Q, Kitada M, Li C, Geraldes P, et al. Protective effects of GLP-1 on glomerular endothelium and its inhibition by PKCbeta activation in diabetes. Diabetes (2012) 61:2967–79. doi:10.2337/db11-1824
13. Zhang SL, Tang SS, Chen X, Filep JG, Ingelfinger JR, Chan JSD. High levels of glucose stimulate angiotensinogen gene expression via the P38 mitogen-activated protein kinase pathway in rat kidney proximal tubular cells. Endocrinology (2000) 141:4637–46. doi:10.1210/endo.141.12.7844
14. Zhang SL, Filep JG, Hohman TC, Tang SS, Ingelfinger JR, Chan JSD. Molecular mechanisms of glucose action on angiotensinogen gene expression in rat proximal tubular cells. Kidney Int (1999) 55:454–64. doi:10.1046/j.1523-1755.1999.00271.x
15. Peti-Peterdi J. High glucose and renin release: the role of succinate and GPR91. Kidney Int (2010) 78:1214–7. doi:10.1038/ki.2010.333
16. van der Zijl NJ, Moors CC, Goossens GH, Hermans MM, Blaak EE, Diamant M. Valsartan improves {beta}-cell function and insulin sensitivity in subjects with impaired glucose metabolism: a randomized controlled trial. Diabetes Care (2011) 34:845–51. doi:10.2337/dc10-2224
17. Bosch J, Yusuf S, Gerstein HC, Pogue J, Sheridan P, Dagenais G, et al. Effect of ramipril on the incidence of diabetes. N Engl J Med (2006) 355:1551–62. doi:10.1056/NEJMoa065061
18. Goossens GH. The renin-angiotensin system in the pathophysiology of type 2 diabetes. Obes Facts (2012) 5:611–24. doi:10.1159/000342776
19. Henriksen EJ. Improvement of insulin sensitivity by antagonism of the renin-angiotensin system. Am J Physiol Regul Integr Comp Physiol (2007) 293:R974–80. doi:10.1152/ajpregu.00147.2007
20. Wang HW, Mizuta M, Saitoh Y, Noma K, Ueno H, Nakazato M. Glucagon-like peptide-1 and candesartan additively improve glucolipotoxicity in pancreatic beta-cells. Metabolism (2011) 60:1081–9. doi:10.1016/j.metabol.2010.11.004
21. Kalupahana NS, Moustaid-Moussa N. The renin-angiotensin system: a link between obesity, inflammation and insulin resistance. Obes Rev (2012) 13:136–49. doi:10.1111/j.1467-789X.2011.00942.x
22. Lu H, Boustany-Kari CM, Daugherty A, Cassis LA. Angiotensin II increases adipose angiotensinogen expression. Am J Physiol Endocrinol Metab (2007) 292:E1280–7. doi:10.1152/ajpendo.00277.2006
23. Giacchetti G, Faloia E, Mariniello B, Sardu C, Gatti C, Camilloni MA, et al. Overexpression of the renin-angiotensin system in human visceral adipose tissue in normal and overweight subjects. Am J Hypertens (2002) 15:381–8. doi:10.1016/S0895-7061(02)02257-4
24. Bastien M, Poirier P, Lemieux I, Després JP. Overview of epidemiology and contribution of obesity to cardiovascular disease. Prog Cardiovasc Dis (2014) 56:369–81. doi:10.1016/j.pcad.2013.10.016
25. Miesel A, Muller-Fielitz H, Johren O, Vogt FM, Raasch W. Double blockade of angiotensin II (AT(1))-receptors and ACE does not improve weight gain and glucose homeostasis better than single-drug treatments in obese rats. Br J Pharmacol (2012) 165:2721–35. doi:10.1111/j.1476-5381.2011.01726.x
26. Fogari R, Derosa G, Zoppi A, Rinaldi A, Lazzari P, Fogari E, et al. Comparison of the effects of valsartan and felodipine on plasma leptin and insulin sensitivity in hypertensive obese patients. Hypertens Res (2005) 28:209–14. doi:10.1291/hypres.28.209
27. Cassis L, Helton M, English V, Burke G. Angiotensin II regulates oxygen consumption. Am J Physiol Regul Integr Comp Physiol (2002) 282:R445–53. doi:10.1152/ajpregu.00261.2001
28. Massiera F, Bloch-Faure M, Ceiler D, Murakami K, Fukamizu A, Gasc JM, et al. Adipose angiotensinogen is involved in adipose tissue growth and blood pressure regulation. FASEB J (2001) 15:2727–9. doi:10.1096/fj.01-0457fje
29. Baker KM, Chernin MI, Wixson SK, Aceto JF. Renin-angiotensin system involvement in pressure-overload cardiac hypertrophy in rats. Am J Physiol (1990) 259:H324–32.
30. Becker BN, Yasuda T, Kondo S, Vaikunth S, Homma T, Harris RC. Mechanical stretch/relaxation stimulates a cellular renin-angiotensin system in cultured rat mesangial cells. Exp Nephrol (1998) 6:57–66. doi:10.1159/000020505
31. Johnston AP, Baker J, De Lisio M, Parise G. Skeletal muscle myoblasts possess a stretch-responsive local angiotensin signalling system. J Renin Angiotensin Aldosterone Syst (2011) 12:75–84. doi:10.1177/1470320310381795
32. Skov J, Dejgaard A, Frokiaer J, Holst JJ, Jonassen T, Rittig S, et al. Glucagon-like peptide-1 (GLP-1): effect on kidney hemodynamics and renin-angiotensin-aldosterone system in healthy men. J Clin Endocrinol Metab (2013) 98:E664–71. doi:10.1210/jc.2012-3855
33. Hirata K, Kume S, Araki S, Sakaguchi M, Chin-Kanasaki M, Isshiki K, et al. Exendin-4 has an anti-hypertensive effect in salt-sensitive mice model. Biochem Biophys Res Commun (2009) 380:44–9. doi:10.1016/j.bbrc.2009.01.003
34. Ishibashi Y, Matsui T, Ojima A, Nishino Y, Nakashima S, Maeda S, et al. Glucagon-like peptide-1 inhibits angiotensin II-induced mesangial cell damage via protein kinase A. Microvasc Res (2012) 84:395–8. doi:10.1016/j.mvr.2012.06.008
35. Cheng Q, Law PK, de Gasparo M, Leung PS. Combination of the dipeptidyl peptidase IV inhibitor LAF237 [(S)-1-[(3-hydroxy-1-adamantyl)ammo]acetyl-2-cyanopyrrolidine] with the angiotensin II type 1 receptor antagonist valsartan [N-(1-oxopentyl)-N-[[2’-(1H-tetrazol-5-yl)-[1,1’-biphenyl]-4-yl]methyl]-l-valine] enhances pancreatic islet morphology and function in a mouse model of type 2 diabetes. J Pharmacol Exp Ther (2008) 327:683–91. doi:10.1124/jpet.108.142703
36. Bagger JI, Knop FK, Lund A, Vestergaard H, Holst JJ, Vilsboll T. Impaired regulation of the incretin effect in patients with type 2 diabetes. J Clin Endocrinol Metab (2011) 96:737–45. doi:10.1210/jc.2010-2435
37. Toft-Nielsen MB, Damholt MB, Madsbad S, Hilsted LM, Hughes TE, Michelsen BK, et al. Determinants of the impaired secretion of glucagon-like peptide-1 in type 2 diabetic patients. J Clin Endocrinol Metab (2001) 86:3717–23. doi:10.1210/jcem.86.8.7743
38. Kjems LL, Holst JJ, Volund A, Madsbad S. The influence of GLP-1 on glucose-stimulated insulin secretion: effects on beta-cell sensitivity in type 2 and nondiabetic subjects. Diabetes (2003) 52:380–6. doi:10.2337/diabetes.52.2.380
39. Rodriguez R, Viscarra JA, Minas JN, Nakano D, Nishiyama A, Ortiz RM. Angiotensin receptor blockade increases pancreatic insulin secretion and decreases glucose intolerance during glucose supplementation in a model of metabolic syndrome. Endocrinology (2012) 153:1684–95. doi:10.1210/en.2011-1885
40. Aubert J, Darimont C, Safonova I, Ailhaud G, Negrel R. Regulation by glucocorticoids of angiotensinogen gene expression and secretion in adipose cells. Biochem J (1997) 328(Pt 2):701–6.
41. Wang L, Lei C, Zhang SL, Roberts KD, Tang SS, Ingelfinger JR, et al. Synergistic effect of dexamethasone and isoproterenol on the expression of angiotensinogen in immortalized rat proximal tubular cells. Kidney Int (1998) 53:287–95. doi:10.1046/j.1523-1755.1998.00759.x
42. Zhang SL, Chen X, Wei CC, Filep JG, Tang SS, Ingelfinger JR, et al. Insulin inhibits dexamethasone effect on angiotensinogen gene expression and induction of hypertrophy in rat kidney proximal tubular cells in high glucose. Endocrinology (2002) 143:4627–35. doi:10.1210/en.2002-220408
43. Dostal DE, Booz GW, Baker KM. Regulation of angiotensinogen gene expression and protein in neonatal rat cardiac fibroblasts by glucocorticoid and beta-adrenergic stimulation. Basic Res Cardiol (2000) 95:485–90. doi:10.1007/s003950070025
44. Sato A, Suzuki H, Murakami M, Nakazato Y, Iwaita Y, Saruta T. Glucocorticoid increases angiotensin II type 1 receptor and its gene expression. Hypertension (1994) 23:25–30.
45. Sato A, Suzuki H, Nakazato Y, Shibata H, Inagami T, Saruta T. Increased expression of vascular angiotensin II type 1A receptor gene in glucocorticoid-induced hypertension. J Hypertens (1994) 12:511–6.
46. Breidert M, Bornstein SR, Ehrhart-Bornstein M, Scherbaum WA, Holst JJ. Angiotensin II regulates both adrenocortical and adrenomedullary function in isolated perfused pig adrenals. Peptides (1996) 17:287–92. doi:10.1016/0196-9781(95)02106-X
47. Romero DG, Welsh BL, Gomez-Sanchez EP, Yanes LL, Rilli S, Gomez-Sanchez CE. Angiotensin II-mediated protein kinase D activation stimulates aldosterone and cortisol secretion in H295R human adrenocortical cells. Endocrinology (2006) 147:6046–55. doi:10.1210/en.2006-0794
48. Chiu KC, Chu A, Go VLW, Saad MF. Hypovitaminosis D is associated with insulin resistance and beta cell dysfunction. Am J Clin Nutr (2004) 79:820–5.
49. de Zeeuw D, Agarwal R, Amdahl M, Audhya P, Coyne D, Garimella T, et al. Selective vitamin D receptor activation with paricalcitol for reduction of albuminuria in patients with type 2 diabetes (VITAL study): a randomised controlled trial. Lancet (2010) 376:1543–51. doi:10.1016/S0140-6736(10)61032-X
50. Tomaschitz A, Pilz S, Ritz E, Grammer T, Drechsler C, Boehm BO, et al. Independent association between 1,25-dihydroxyvitamin D, 25-hydroxyvitamin D and the renin-angiotensin system: the Ludwigshafen Risk and Cardiovascular Health (LURIC) study. Clin Chim Acta (2010) 411:1354–60. doi:10.1016/j.cca.2010.05.037
51. Forman JP, Williams JS, Fisher ND. Plasma 25-hydroxyvitamin D and regulation of the renin-angiotensin system in humans. Hypertension (2010) 55:1283–8. doi:10.1161/HYPERTENSIONAHA.109.148619
52. Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest (2002) 110:229–38. doi:10.1172/JCI15219
53. Cheng Q, Li YC, Boucher BJ, Leung PS. A novel role for vitamin D: modulation of expression and function of the local renin-angiotensin system in mouse pancreatic islets. Diabetologia (2011) 54:2077–81. doi:10.1007/s00125-011-2100-1
54. Li YC. Chapter 40 – vitamin d and the renin-angiotensin system. Third ed. In: David F, Pike JW, Adams JohnS, Feldman David, John SA editors. Vitamin D. San Diego: Academic Press (2011). p. 707–23. doi:10.1016/B978-0-12-381978-9.10040-X
55. Vaidya A, Williams JS. The relationship between vitamin D and the renin-angiotensin system in the pathophysiology of hypertension, kidney disease, and diabetes. Metabolism (2012) 61:450–8. doi:10.1016/j.metabol.2011.09.007
56. Ferder M, Inserra F, Manucha W, Ferder L. The world pandemic of vitamin D deficiency could possibly be explained by cellular inflammatory response activity induced by the renin-angiotensin system. Am J Physiol Cell Physiol (2013) 304:C1027–39. doi:10.1152/ajpcell.00403.2011
57. Pan XR, Li GW, Hu YH, Wang JX, Yang WY, An ZX, et al. Effects of diet and exercise in preventing NIDDM in people with impaired glucose tolerance. The Da Qing IGT and Diabetes Study. Diabetes Care (1997) 20:537–44. doi:10.2337/diacare.20.4.537
58. Agarwal D, Welsch MA, Keller JN, Francis J. Chronic exercise modulates RAS components and improves balance between pro- and anti-inflammatory cytokines in the brain of SHR. Basic Res Cardiol (2011) 106:1069–85. doi:10.1007/s00395-011-0231-7
59. Barretti DL, Magalhaes Fde C, Fernandes T, do Carmo EC, Rosa KT, Irigoyen MC, et al. Effects of aerobic exercise training on cardiac renin-angiotensin system in an obese zucker rat strain. PLoS One (2012) 7:e46114. doi:10.1371/journal.pone.0046114
60. Pereira MG, Ferreira JC, Bueno CR Jr, Mattos KC, Rosa KT, Irigoyen MC, et al. Exercise training reduces cardiac angiotensin II levels and prevents cardiac dysfunction in a genetic model of sympathetic hyperactivity-induced heart failure in mice. Eur J Appl Physiol (2009) 105:843–50. doi:10.1007/s00421-008-0967-4
61. Ciampone S, Borges R, de Lima IP, Mesquita FF, Cambiucci EC, Gontijo JA. Long-term exercise attenuates blood pressure responsiveness and modulates kidney angiotensin II signalling and urinary sodium excretion in SHR. J Renin Angiotensin Aldosterone Syst (2011) 12:394–403. doi:10.1177/1470320311408750
62. Maeda S, Iemitsu M, Jesmin S, Miyauchi T. Acute exercise causes an enhancement of tissue renin-angiotensin system in the kidney in rats. Acta Physiol Scand (2005) 185:79–86. doi:10.1111/j.1365-201X.2005.01459.x
63. Mueller PJ. Exercise training and sympathetic nervous system activity: evidence for physical activity dependent neural plasticity. Clin Exp Pharmacol Physiol (2007) 34:377–84. doi:10.1111/j.1440-1681.2007.04590.x
64. Schrier RW. Interactions between angiotensin II and arginine vasopressin in water homeostasis. Kidney Int (0000) 76:137–9. doi:10.1038/ki.2009.103
65. Yang S, Yao B, Zhou Y, Yin H, Zhang MZ, Harris RC. Intrarenal dopamine modulates progressive angiotensin II-mediated renal injury. Am J Physiol Renal Physiol (2012) 302:F742–9. doi:10.1152/ajprenal.00583.2011
66. Zhang MZ, Yao B, Fang X, Wang S, Smith JP, Harris RC. Intrarenal dopaminergic system regulates renin expression. Hypertension (2009) 53:564–70. doi:10.1161/HYPERTENSIONAHA.108.127035
67. Li H, Armando I, Yu P, Escano C, Mueller SC, Asico L, et al. Dopamine 5 receptor mediates Ang II type 1 receptor degradation via a ubiquitin-proteasome pathway in mice and human cells. J Clin Invest (2008) 118:2180–9. doi:10.1172/JCI33637
68. Cheng HF, Becker BN, Harris RC. Dopamine decreases expression of type-1 angiotensin II receptors in renal proximal tubule. J Clin Invest (1996) 97:2745–52. doi:10.1172/JCI118729
69. de Kloet AD, Krause EG, Woods SC. The renin angiotensin system and the metabolic syndrome. Physiol Behav (2010) 100:525–34. doi:10.1016/j.physbeh.2010.03.018
70. Irani RA, Xia Y. The functional role of the renin-angiotensin system in pregnancy and preeclampsia. Placenta (2008) 29:763–71. doi:10.1016/j.placenta.2008.06.011
71. Shao W, Seth DM, Navar LG. Augmentation of endogenous intrarenal angiotensin II levels in Val5-ANG II-infused rats. Am J Physiol Renal Physiol (2009) 296:F1067–71. doi:10.1152/ajprenal.90596.2008
72. Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol (2007) 292:C82–97. doi:10.1152/ajpcell.00287.2006
73. Zhang GX, Lu XM, Kimura S, Nishiyama A. Role of mitochondria in angiotensin II-induced reactive oxygen species and mitogen-activated protein kinase activation. Cardiovasc Res (2007) 76:204–12. doi:10.1016/j.cardiores.2007.07.014
74. Erdmann B, Fuxe K, Ganten D. Subcellular localization of angiotensin II immunoreactivity in the rat cerebellar cortex. Hypertension (1996) 28:818–24. doi:10.1161/01.HYP.28.5.818
75. Wilkinson-Berka JL, Agrotis A, Deliyanti D. The retinal renin-angiotensin system: roles of angiotensin II and aldosterone. Peptides (2012) 36:142–50. doi:10.1016/j.peptides.2012.04.008
76. Kasselman LJ, Rutkove SB. Application of angiotensin II to healthy rat sciatic nerve can produce neuropathy without associated vasculopathy. Muscle Nerve (2010) 42:959–65. doi:10.1002/mus.21767
77. Jin LM. Angiotensin II signaling and its implication in erectile dysfunction. J Sex Med (2009) 3(6 Suppl):302–10. doi:10.1111/j.1743-6109.2008.01188.x
78. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature (2001) 414:813–20. doi:10.1038/414813a
79. Janghorbani M, Van Dam RM, Willett WC, Hu FB. Systematic review of type 1 and type 2 diabetes mellitus and risk of fracture. Am J Epidemiol (2007) 166:495–505. doi:10.1093/aje/kwm106
80. Vestergaard P. Discrepancies in bone mineral density and fracture risk in patients with type 1 and type 2 diabetes – a meta-analysis. Osteoporos Int (2007) 18:427–44. doi:10.1007/s00198-006-0253-4
81. Gebru Y, Diao TY, Pan H, Mukwaya E, Zhang Y. Potential of RAS inhibition to improve metabolic bone disorders. Biomed Res Int (2013) 2013:932691. doi:10.1155/2013/932691
82. Donmez BO, Ozdemir S, Sarikanat M, Yaras N, Koc P, Demir N, et al. Effect of angiotensin II type 1 receptor blocker on osteoporotic rat femurs. Pharmacol Rep (2012) 64:878–88.
83. Solomon DH, Mogun H, Garneau K, Fischer MA. Risk of fractures in older adults using antihypertensive medications. J Bone Miner Res (2011) 26:1561–7. doi:10.1002/jbmr.356
84. Nuche-Berenguer B, Moreno P, Portal-Nunez S, Dapia S, Esbrit P, Villanueva-Penacarrillo ML. Exendin-4 exerts osteogenic actions in insulin-resistant and type 2 diabetic states. Regul Pept (2010) 159:61–6. doi:10.1016/j.regpep.2009.06.010
85. Nuche-Berenguer B, Moreno P, Esbrit P, Dapia S, Caeiro JR, Cancelas J, et al. Effect of GLP-1 treatment on bone turnover in normal, type 2 diabetic, and insulin-resistant states. Calcif Tissue Int (2009) 84:453–61. doi:10.1007/s00223-009-9220-3
86. Nishimura H. Angiotensin receptors – evolutionary overview and perspectives. Comp Biochem Physiol A Mol Integr Physiol (2001) 128:11–30. doi:10.1016/S1095-6433(00)00294-4
87. Jones ES, Vinh A, McCarthy CA, Gaspari TA, Widdop RE. AT2 receptors: functional relevance in cardiovascular disease. Pharmacol Ther (2008) 120:292–316. doi:10.1016/j.pharmthera.2008.08.009
88. Kumar R, Thomas CM, Yong QC, Chen W, Baker KM. The intracrine renin-angiotensin system. Clin Sci (1979) 123(2012):273–84. doi:10.1042/CS20120089
89. Kobori H, Navar LG. Urinary angiotensinogen as a novel biomarker of intrarenal renin-angiotensin system in chronic kidney disease. Int Rev Thromb (2011) 6:108–16.
90. Persson F, Lu X, Rossing P, Garrelds IM, Danser AH, Parving HH. Urinary renin and angiotensinogen in type 2 diabetes: added value beyond urinary albumin? J Hypertens (2013) 31:1646–52. doi:10.1097/HJH.0b013e328362217c
Keywords: renin–angiotensin system, diabetes, metabolic syndrome, obesity, GLP-1, cortisol, vitamin D, osteoporosis
Citation: Skov J, Persson F, Frøkiær J and Christiansen JS (2014) Tissue renin–angiotensin systems: a unifying hypothesis of metabolic disease. Front. Endocrinol. 5:23. doi: 10.3389/fendo.2014.00023
Received: 02 December 2013; Paper pending published: 10 January 2014;
Accepted: 13 February 2014; Published online: 28 February 2014.
Edited by:
Anca Dana Dobrian, Eastern Virginia Medical School, USAReviewed by:
Brian M. Shewchuk, Brody School of Medicine at East Carolina University, USAKay Waud, Jones Institute for Reproductive Medicine, USA
Copyright: © 2014 Skov, Persson, Frøkiær and Christiansen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeppe Skov, Department of Endocrinology and Internal Medicine, Aarhus University Hospital, Norrebrogade 44, Aarhus DK-8000, Denmark e-mail:anNrQGRhZGxuZXQuZGs=