 Atsufumi Ozoe1Meri Sone1Toshiaki Fukushima1,2Naoyuki Kataoka3Kazuhiro Chida1Tomoichiro Asano2
Atsufumi Ozoe1Meri Sone1Toshiaki Fukushima1,2Naoyuki Kataoka3Kazuhiro Chida1Tomoichiro Asano2 Fumihiko Hakuno1
Fumihiko Hakuno1 Shin-Ichiro Takahashi1*
Shin-Ichiro Takahashi1*- 1Graduate School of Agriculture and Life Sciences, The University of Tokyo, Tokyo, Japan
- 2Institute of Biomedical and Health Sciences, Hiroshima University, Hiroshima, Japan
- 3Laboratory for Malignancy Control Research, Medical Innovation Center, Graduate School of Medicine, Kyoto University, Kyoto, Japan
Insulin receptor substrates (IRSs) are well known to play crucial roles in mediating intracellular signals of insulin-like growth factors (IGFs)/insulin. Previously, we showed that IRS-1 forms high molecular mass complexes containing RNAs. To identify RNAs in IRS-1 complexes, we performed ultraviolet (UV) cross-linking and immunoprecipitation analysis using HEK293 cells expressing FLAG–IRS-1 and FLAG–IRS-2. We detected the radioactive signals in the immunoprecipitates of FLAG–IRS-1 proportional to the UV irradiation, but not in the immunoprecipitates of FLAG–IRS-2, suggesting the direct contact of RNAs with IRS-1. RNAs cross-linked to IRS-1 were then amplified by RT-PCR, followed by sequence analysis. We isolated sequence tags attributed to 25 messenger RNAs and 8 non-coding RNAs, including small nucleolar RNAs (snoRNAs). We focused on the interaction of IRS-1 with U96A snoRNA (U96A) and its host Rack1 (receptor for activated C kinase 1) pre-mRNA. We confirmed the interaction of IRS-1 with U96A, and with RACK1 pre-mRNA by immunoprecipitation with IRS-1 followed by Northern blotting or RT-PCR analyses. Mature U96A in IRS-1−/− mouse embryonic fibroblasts was quantitatively less than WT. We also found that a part of nuclear IRS-1 is localized in the Cajal body, a nuclear subcompartment where snoRNA mature. The unanticipated function of IRS-1 in snoRNA biogenesis highlights the potential of RNA-associated IRS-1 complex to open a new line of investigation to dissect the novel mechanisms regulating IGFs/insulin-mediated biological events.
Introduction
Insulin-like growth factors (IGFs) and insulin display a variety of bioactivities, including embryonic development and growth, postnatal somatic growth, and regulation of glucose, lipid, and protein metabolism (1). These bioactivities have been shown to be accomplished by IGF/insulin signaling pathways composed of many signaling molecules (2–4). Insulin receptor substrates (IRS)-1 and IRS-2 are important substrates of the receptor-intrinsic tyrosine kinases and serve as adaptor proteins transmitting IGF/insulin signaling from the IGF-I/insulin receptor to the downstream effectors. Although the differences in the roles of these IRS isoforms in mediation of the IGF/insulin bioactivities remain largely unknown, it is suggested that IRS-1 plays a predominant role in somatic growth, and IRS-2 in glucose homeostasis, especially in liver (5–7). Following tyrosine phosphorylation of IRSs, Src homology region 2 (SH2) domain-containing proteins bind to IRSs leading to the activation of phosphatidylinositol 3-kinase (PI3K) and mitogen-activated protein kinase (MAPK) pathways. The activated PI3K transmits the signal to up-regulate growth and metabolism through the Akt signaling pathway (8). In particular, the downstream mammalian target of rapamycin (mTOR) is thought to play crucial roles in promoting growth-related intracellular activities such as ribosome biogenesis (9, 10). Since previous reports have shown that aberrant expression of IRS is linked to certain types of cancers (11–13), unraveling the signaling cascade of biological events that is involved in IRS regulation is important to understand the role of IRSs in human pathologies.
The eukaryotic ribosome is a large complex composed of four RNA molecules, including the 5S, 5.8S, 18S, and 28S ribosomal RNAs (rRNAs) (14) and about 80 distinct ribosomal proteins (RPs) (15). Ribosome biogenesis involves several coordinated steps, such as synthesis, post-transcriptional modification and processing of rRNA in the nucleus, synthesis of RPs and their import into the nucleus, the assembly of ribosome subunits, and the transport of the mature 40S and 60S subunits into the cytoplasm (16). The post-transcriptional modifications of rRNA include the two types of chemical modifications, pseudouridine and 2′-O-methylation, and are added by small nucleolar RNP complexes (snoRNPs) (17, 18) consisting of snoRNAs and several proteins. For both types of modification, site specificity is achieved by the snoRNA through base pairing with the target region. In vertebrates, the great majority of snoRNAs is encoded within introns of pre-mRNAs (19, 20) and they are processed from the debranched host introns (21–24). Therefore, the mature intronic snoRNA is produced concomitantly with splicing of the host pre-mRNA (22, 25).
Recently, we found that IRSs form high molecular mass complexes containing RNAs as well as a variety of proteins that modulate or mediate insulin-like bioactivities (29). In addition, we identified components of messenger ribonucleoprotein (mRNP) as IRS-1-associated proteins (30). Here, we present the results of the screening of RNA components of IRS-1 complex and found that IRS-1 can form a complex with snoRNA, U96A, and its host pre-mRNA, Rack1 (receptor for activated C kinase 1) RNA. In this study, we propose the unanticipated role of IRS-1 in the biogenesis of U96A. This may be a novel mechanism to support the induction of IGF/insulin bioactivities.
Materials and Methods
Materials
Anti-IRS-1 antibody was raised in rabbits as described (31). The following peptide and antibodies were purchased: FLAG (F3165, Sigma, St. Louis, MO, USA), anti-FLAG antibody-conjugated agarose beads (A2220, Sigma), and anti-Myc antibody (9E10, Millipore, Billerica, MA, USA). All other chemicals were of reagent grade and were obtained commercially.
Plasmids
To generate FLAG-tagged or Myc-tagged IRS-1 and IRS-2, the open reading frame of rat IRS-1 and human IRS-2 was subcloned into the pCMV–FLAG-2 vector (Sigma) or pCMV–Myc vector in-frame as described previously (32, 33). Full-length rat PABPC1 was subcloned into the pCMV mammalian expression vector containing an N-terminal FLAG tag (FLAG–PABPC1) as follows. An EcoRI restriction site was introduced at the 5′ end of the PABPC1 open reading frame and a SalI site at the 3′- end by PCR using the oligonucleotides 5′-TTAAGAATTCAAGATGAACCCCAGCGCCCCCAGCTA-3′ and 5′-TTAAGTCGACTTAGACAGTTGGAACACCAGTGG-3′, respectively. The PCR product was digested with EcoRI and SalI and subcloned into pCMV–FLAG cut with EcoRI and SalI. Full-length human 15.5K was subcloned into the pCMV mammalian expression vector containing an N-terminal FLAG tag (FLAG-15.5K) or N-terminal GFP tag (GFP-15.5K) as follows. A BglII restriction site was introduced at the 5′ end of the 15.5K open reading frame and a SalI site at the 3′ end by PCR using the oligonucleotides 5′-TTAAGAATTCAAGATGAACCCCAGCGCCCCCAGCTA-3′ and 5′-TTAAGTCGACTTAGACAGTTGGAACACCAGTGG-3′. The PCR product was digested with BglII and SalI and subcloned into pCMV–FLAG and pEGFP-C1 cut with BglII and SalI. Full-length human coilin was subcloned into the pCMV mammalian expression vector containing an N-terminal GFP tag (GFP–coilin) as follows. A KpnI restriction site was introduced at the 5′ end of the coilin open reading frame and a BamHI site at the 3′ end by PCR using the oligonucleotides 5′-TTAAGGTACCATGGCAGCTTCCGAGACGGTTAGGCTACG-3′ and 5′-TTAAGGATCCTCAGGCAGGTTCTGTACTTGATGTGTTACTTGG-3′. The PCR product was digested with KpnI and BamHI and subcloned into pEGFP-C1 cut with KpnI and BamHI.
Plasmids expressing Myc–IRS-1 fused with SV40 large T antigen-derived Nuclear Localizing Signal (NLS, PKKKRKV) in its C-terminus was constructed by PCR using pCMV–Myc–IRS-1 as template and the oligonucleotides 5′-AAGGCTGTCCTTGGGGGATCC-3′ and 5′-ATCGGGATCCCTATACCTTTCTCTTCTTTTTTGGTTGACGGTCCTCTGGTTG-3′. The PCR product amplified was digested with BamHI and subcloned into pCMV–Myc–IRS-1 cut with the same restriction enzyme.
For the in vitro RNA synthesis, U96A snoRNA sequence was amplified by PCR using the oligonucleotides 5′-TTAATCTAGACCTGGTGATGACAGATGGCATTGTCAG-3′ and 5′-TTAACTGGAGTTCAGAATTGCAGGACATGTCCTCACTCC-3′. The primers were engineered to contain XbaI and XhoI sites to introduce these restriction sites at the 5′ and 3′ end of the PCR product, respectively. The PCR product was digested with XbaI and XhoI and subcloned into the downstream of T7 promoter of pBS-KS(+) cut with XbaI and XhoI, which is referred to as pBS-KS(+)-U96A snoRNA. U6 small nuclear RNA (snRNA) sequence was also amplified by the PCR using the oligonucleotides 5′-TTAATCTAGAGTGCTCGCTTCGGCAGCACATATACTAAAATTGG-3′ and 5′-TTAACTCGAGAAAATATGGAACGCTTCACGAATTTGCGTGTCATCC-3′, and subcloned into the downstream of T7 promoter of pBS-KS(+) cut with XbaI and XhoI.
Animals
Insulin receptor substrates-1 knockout mice were kindly donated by Dr. Kadowaki and Dr. Ohsugi (Faculty of Medicine, The University of Tokyo). All animal experiments in this study were performed according to procedures approved by the Committee on Laboratory Animal Care, Graduate School of Agriculture and Life Sciences, The University of Tokyo.
Cell Culture and Transfection
Human embryonic kidney 293 (HEK293) cells were kindly provided by Dr. Koichi Suzuki (National Institute of Infectious Diseases, Tokyo, Japan). MCF-7 human breast cancer cells (ATCC No. CRL8305) were a kind gift from Dr. Yoichi Hayakawa (Tokyo University of Science, Tokyo, Japan). HEK293, MCF-7 cells, and HeLa cells were maintained at 37°C in a humidified CO2-controlled atmosphere in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 0.1% NaHCO3, 50 IU/ml penicillin, 50 μg/ml streptomycin, 100 μg/ml kanamycin, and 0.5 μg/ml amphotericin B. For the serum-starvation, cells were starved for 16 h in DMEM supplemented with 0.1% bovine serum albumin (BSA).
Primary mouse embryonic fibroblasts (MEFs) were prepared from littermates according to the previous report (34) and kept in culture for 2–3 weeks, respectively. Briefly, MEFs were prepared from E14.5 embryos. Embryos were dissociated by 0.025% trypsin in 0.2% EDTA at 37°C for 10 min and then treated with DNaseI. After filtering fibroblasts with a 70 μm cell strainer, they were cultured in DMEM supplemented with 10% FBS, 100 μg/ml streptomycin, 100 U/ml penicillin, and 2 mM glutamine. MEFs were cultured at 9% CO2 at 37°C and experiments were performed at first passages (P1–P4).
For the plasmid transfection, the expression vectors were transfected into HEK293 cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA), according to manufacturer’s protocol.
Immunoprecipitation and Immunoblotting
Immunoprecipitation and immunoblotting were performed as described elsewhere (30).
Cross-Linking and Immunoprecipitation
Cross-linking and immunoprecipitation (CLIP) was performed using serum-starved HEK293 cell expressing FLAG–IRS-1 or FLAG–IRS-2 according to Ule et al. (35) with minor modification.
Nucleus and Cytoplasm Fractionation
Nucleus/cytoplasm fractions were prepared according to previous report (36).
RNA Extraction, RT-PCR, and Quantitative RT-PCR
Total RNA was extracted from cells using the TRIzol reagent (Invitrogen) followed by DNase I treatment. For RT-PCR, 1 μg of total RNA was reverse transcribed using random primers (Invitrogen) and SuperScript II reverse transcriptase (Invitrogen). A fraction of the RT reaction products was used in subsequent PCR reactions. Amplification parameters were denaturation at 94°C for 30 s, annealing at 64°C for 30 s, and extension at 72°C for 30 s in a 30-cycle program. Primer sequences were as follows: Rack1 exon1–exon8 (5′-ACTGAGCAGATGACCCTTCGTG 3′ and 5′-GTTGTCCGTGTAGCCAGCAAAC-3′, 912 bp). The simultaneous analysis of serial dilution of the amount of RNA used in the RT-PCR reactions ensured that these reactions were quantitative.
For quantitative RT-PCR, the cDNA prepared was subjected to real-time PCR (ABI StepOne™ Real-Time PCR Systems), using SYBR Green Real-time PCR Master Mix Plus (TOYOBO, Osaka, Japan). Data were expressed as relative mRNA levels normalized to housekeeping gene (Gapdh) expression level in each sample. The primer sequences are as follows: Rack1 exon2–intron2 (5′-GATGGTCAGTTTGCCCTCTC-3′, 5′-CTCAGTTCTGCCCACTTTCC-3′), Rack1 exon3–exon4 (5′-GTCCCGAGACAAGACCATAAAG-3′, 5′-TGATAGGGTTGCTGCTGTTC-3′), Gapdh (5′-GTGTTCCTACCCCCAATGTG-3′, 5′-CCTGCTTCACCACCTTCTTG-3′).
Northern-Blot Analysis
To detect small RNAs, total RNA was separated on a 10% denaturing acrylamide gel and transferred to Hybond-N+ membrane (GE Healthcare Biosciences, Pittsburgh, PA, USA) using a Trans-Blot SD Semi-Dry Transfer Cell (Bio-Rad, Hercules, CA, USA). For the detection of U96A snoRNA, DNA probes were synthesized with a Megaprime DNA labeling Kit (GE healthcare) and [α−32P]dCTP using U96A snoRNA cDNA as a template. The probes were hybridized using prehybridization buffer (50% deionized formamide, 5 × SSPE, 5 × Denhardt, 10% dextran sulfate, 0.1% SDS, 20 μg/ml salmon sperm) at 50°C. Densitometric analysis of RNA bands was performed using ImageJ gel analysis software (http://rsbweb.nih.gov/ij/).
Co-Immunoprecipitation of U96A snoRNA with Proteins
HEK293 cells expressing FLAG-tagged proteins were serum-starved and washed twice with ice-cold PBS and irradiated with 4000 J/m2 ultraviolet (UV) in ice-cold PBS. The cells were collected in 1.5 ml microtubes and pelleted by centrifugation. The cell pellets were resuspended with 1 ml of NET-2 [50 mM Tris–HCl, pH 7.4, 300 mM NaCl, 0.05% NP-40, 500 μM Na3VO4, 10 μg/ml leupeptin, 5 μg/ml pepstatin, 20 μg/ml phenylmethylsulfonyl fluoride (PMSF); 100 KIU/ml aprotinin] containing 100 U of RNase-inhibitor (Ambion, Austin, TX, USA) and were incubated for 10 min on ice. Cells were lysed by 15 (3 × 5) bursts using a Branson Sonifier. Homogenates were centrifuged at 14,000 g for 10 min at 4°C, cleared by incubation with 20 μl Protein G-Sepharose beads (GE Healthcare) for 60 min, and incubated with a specific antibody for 1.5 h at 4°C, after which 30 μl Dynabeads protein G paramagnetic beads (Invitrogen) were added with gentle mixing for another 30 min at 4°C. The beads were washed five times with high-salt wash buffer [50 mM HEPES–KOH, pH 7.5, 500 mM KCl, 0.05% (v/v) NP-40, 0.5 mM DTT] and proteins on beads were eluted by incubation with lysis buffer containing 150 ng/ml FLAG peptide. Nine-tenth of the eluates was treated with Proteinase K, and the eluted RNAs were purified with the TRIzol Reagent and analyzed by Northern blotting. The remainder of the eluates was fractionated by SDS-PAGE and analyzed by immunoblotting.
In vitro RNA Synthesis
pBS-KS(+)-U96A snoRNA 0.5 μg linearized with XhoI and gel purified was used as a template for in vitro transcription with Riboprobe in vitro Transcription kit (Promega, Madison, WI, USA). The RNA products were treated with DNase I and purified with phenol/chloroform/isoamyl alcohol mixture and ethanol precipitation. The amount of transcribed RNA was quantitated by absorbance at 260 nm and the product size and purity were verified by 10% denaturing acrylamide gel.
Gel Shift Assay
To obtain FLAG-tagged proteins, whole-cell lysates from HEK293T cells were prepared from transfected cells with lysis buffer (20 mM HEPES, pH7.5, 150 mM NaCl, 1% Triton X-100, 1 mM DTT, 1 mM EDTA, 10% glycerol, 10 μg/ml leupeptin, 5 μg/ml pepstatin, 20 μg/ml PMSF, 100 KIU/ml aprotinin). FLAG-tagged proteins were purified from whole-cell lysates by using anti-FLAG-M2 affinity resin, washed with high-salt buffer (20 mM HEPES, pH7.5, 400 mM NaCl, 1% Triton X-100, 1 mM DTT, 1 mM EDTA, 10% glycerol, 10 μg/ml leupeptin, 5 μg/ml pepstatin, 20 μg/ml PMSF, and 100 KIU/ml aprotinin), and eluted with FLAG peptide. Purity of the eluted proteins was verified by SDS-PAGE, followed by Coomassie Brilliant Blue staining.
Gel shift assay was conducted using ~50,000 cpm in vitro transcribed, [32P]-UTP-labeled U96A snoRNA (~100 nt), U95 snoRNA (~100 nt), or U6 snRNA probe (~120 nt). The probe was heat denatured for 5 min by heating at 65°C and renatured prior to being added to binding reactions containing FLAG–GFP, FLAG–IRS-1, FLAG–15.5K proteins (100 ng). The binding reaction was performed at 4°C for 45 min in 20 μl binding buffer (20 mM HEPES-KOH, pH7.5, 6 mM MgCl2, 60 mM KCl, 7.5% glycerol, 4 mM DTT, 125 ng/μl yeast tRNA, 50 ng/μl BSA, and 5 U RNase-inhibitor) was added. Bound complexes were resolved on native 5% polyacrylamide gels in 0.25 × TAE buffer. Autoradiography was performed using a FLA-5000 imaging system (Fujifilm Life Sciences, Tokyo, Japan).
Measurement of DNA Contents
DNA content in cells was determined using PicoGreen dsDNA Quantitation Reagent (Invitrogen) according to manufacturer’s procedure.
FACS Analysis
For the analysis of cell cycle distribution, the cells were starved for 48 h in DMEM plus 0.1% FBS. MEFs were trypsinized and thoroughly dissociated into single cells in FACS buffer [0.1% FBS, 1mM EDTA, 1 mM NaCl in PBS(−)] and passed through a 40 μm cell strainer (BD Falcon). The collected cells were fixed in 70% ice-cold ethanol overnight at −20°C. Fixed cells were subsequently stained with 20 g/ml Propidium Iodide diluted in PBS containing 0.2 mg/ml RNAse A for 30 min at 37°C. Cells were sorted and counted using BD FACSCalibur (BD Biosciences, San Jose, CA, USA) according to the manufacturer’s instruction. All the analyses were performed at least three times on different genetic backgrounds.
Immunofluorescence
HeLa cells and MCF-7 cells grown on coverslips were serum-starved for 16 h. The cells were fixed in 4% paraformaldehyde/PBS for 15 min at room temperature, permeabilized with PBS containing 0.25% Triton X-100 for 5 min at room temperature, blocked with blocking buffer (3% BSA and 0.025% NaN3 in PBS) for 1 h at room temperature. Then, primary antibody against Myc epitope was added and incubated overnight at 4°C. After the samples were washed with PBS, secondary antibody incubation was done for 1 h at room temperature using anti-mouse Alexa Fluor 594. The slides were mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA, USA). Images were captured using FV500 confocal microscope (Olympus, Tokyo, Japan) and analyzed with Fluoview version 1.4 and Photoshop CS5.
Statistical Analysis
Data are expressed as means ± standard error of mean (SEM). Comparisons between two groups were performed using Student’s t-test.
Results
IRS-1, but Not IRS-2 Forms Complexes with RNAs in Cells
Our previous studies showed that IRS-1 forms high molecular mass complexes with mRNPs (30). Importantly, IRS-1 but not IRS-2 was incorporated into mRNPs (30). Prior to identifying RNAs in IRS-1 complex, we first examined whether IRS-1 interacts with RNAs in close proximity or not. To this end, we performed an UV CLIP analysis (35, 37). HEK293 cells expressing FLAG–IRS-1 or FLAG–IRS-2 were irradiated with UV to cross-link between proteins and RNAs in living cells. Cells were lysed, and RNAs were partially cleaved by the treatment with low-concentrations of RNase, providing short RNA tags covalently cross-linked to target proteins. FLAG–IRSs were then immunoprecipitated and 5′ ends of cleaved RNAs in the precipitates were radiolabeled with polynucleotide kinase (PNK) and [γ−32P]ATP. The immunoprecipitates were then separated by SDS-PAGE, transferred onto nitrocellulose membrane, and detected by autoradiography and immunoblotting (Figures 1A,B). In samples without UV irradiation, we observed radioactive signals at 180 kDa around the band of IRS-1, indicating non-specific phosphorylation of IRS-1 by PNK in vitro. However, UV irradiation greatly increased the radioactive signals at 180 kDa, indicating that IRS-1 covalently interacted with RNAs. In the CLIP analysis, a portion of radioactive signals of the protein–RNA complexes showing slower electrophoretic mobility are often observed because of covalent interaction with longer RNAs (35). Indeed, we actually observed weak but specific radioactive signals with slower electrophoretic mobility than that of IRS-1 itself (Figure 1A). In contrast to IRS-1, UV irradiation did not increase the radioactive signals around the band of IRS-2 (Figure 1A) suggesting that IRS-2 does not interact with RNAs. These results clearly indicated that IRS-1 is in direct contact with RNAs in cells.
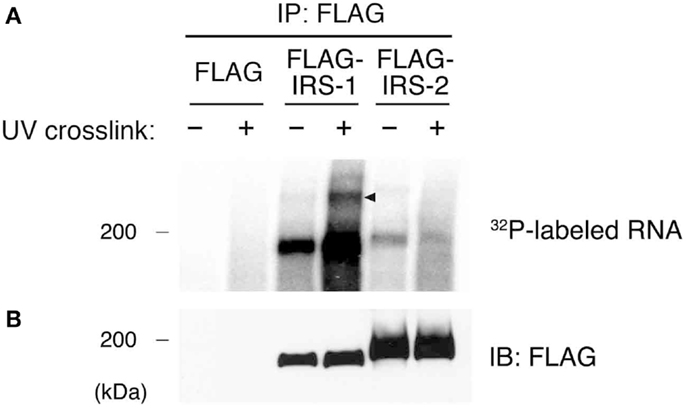
Figure 1. Purification of IRS-1-RNA complexes by CLIP. (A) HEK293 cells expressing FLAG–IRS-1 or IRS-2 were irradiated with or without UV. Protein–RNA complexes were immunoprecipitated with anti-FLAG antibody, and the 5′ ends of immunoprecipitated RNAs were radiolabeled with 32P. Proteins cross-linked with radiolabeled RNAs were separated by SDS-PAGE and visualized by autoradiography. Representative autoradiograms from three experiments are shown. RNA–protein complexes of ~200 kDa (arrowhead) are seen with FLAG–IRS-1 immunoprecipitates (IP) dependent on UV irradiation but not control and FLAG–IRS-2 IP. (B) Immunoblot (IB) analysis of IRS-1 and IRS-2 IP using anti-FLAG antibody.
IRS-1 Complexes Contain U96A snoRNA
We next sought to identify RNAs that are in direct contact with IRS-1 in cells. For this purpose, we extracted RNAs from the radioactive region around IRS-1–RNA complexes (Figure 1, arrowhead) and amplified them by RT-PCR, followed by sequence analysis. We excluded tags with imperfect (<80%) matches to genomic sequences, and accordingly identified 33 IRS-1-bound RNAs from three experiments, which had an average length of 103 nucleotides (Tables A1 and A2 in Appendix). The resulting set of tags included not only tags mapped to 25 messenger-RNA-encoding genes (Table A1 in Appendix) but also tags mapped to 8 non-coding RNAs including snoRNAs and snRNAs (Table A2 in Appendix). RNA sequences that we determined were mapped not only to exons but also to introns or 3′UTRs. As negative control experiments, we repeated CLIP analysis using cells expressing FLAG, but could not amplify the tags.
Among IRS-1 bound RNAs, we focused on U96A snoRNA (U96A), a member of the Box C/D type snoRNAs, which are well established to function in post-transcriptional chemical modification of rRNA (19). U96A is encoded in the second intron of the Rack1 gene (Figure 2A) (38). We first examined the expression of U96A in MCF-7 cells by Northern-blot analysis and the result showed that U96A was expressed and localized in the nucleus (Figure 2B), which is the same subcellular distribution as canonical box C/D snoRNAs. The interaction of IRS-1 with U96A was confirmed by co-immunoprecipitation assay, followed by Northern-blot analysis using labeled U96A as a probe (Figure 2C). We neither detected U96A in the immunoprecipitates of GFP, IRS-2 nor poly-A binding protein (PABP). In a positive control experiment, U96A was detected in the RNAs co-purified with 15.5K, which is known to primarily interact with Box C/D type snoRNAs as a component of snoRNP (19, 22, 39). Interestingly, U95 snoRNA, which is encoded in the first intron of Rack1 pre-mRNA was not co-immunoprecipitated with IRS-1. The direct interaction between IRS-1 and U96A was also confirmed by in vitro binding assay (Figure 2D).
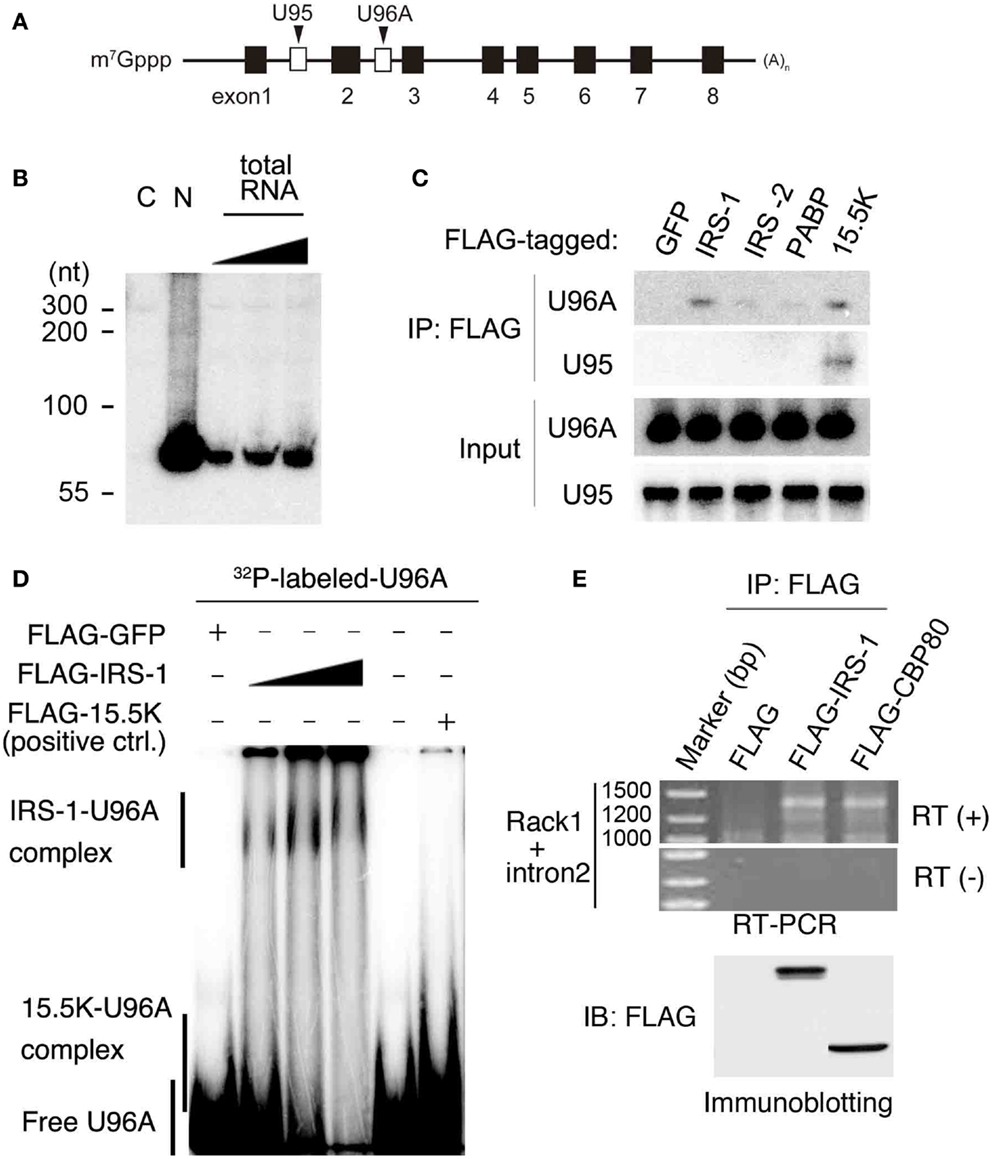
Figure 2. Complex formation of IRS-1 with U96A snoRNA. (A) U96A is located in the second intron of human Rack1 gene. Human Rack1 gene contains 8 exons (black boxes) and 2 snoRNAs (white boxes). Rack1 produces mature Rack1 mRNA and two non-coding RNAs, U95 and U96A. (B) MCF-7 cells were separated into cytosolic (C) and nuclear (N) fractions. RNA was prepared from both fractions and 10 μg RNAs from each fraction were analyzed for U96A snoRNA abundance by Northern-blot analysis. 100, 33, and 11% of 10 μg total RNAs were also loaded. Representative results from at least four independent experiments are shown. (C) Lysates from HEK 293 cells overexpressing FLAG–IRS-1, IRS-2, PABPC1 (negative control) or 15.5 K (positive control) were subjected to immunoprecipitation with anti-FLAG antibody. RNA–protein complexes were eluted with 3 × FLAG peptides, and RNAs bound to the proteins were extracted with phenol/chloroform/isoamyl alcohol mixture and ethanol precipitation. RNAs were subjected to Northern blotting with the indicated probes. Representative results from three independent experiments are shown. (D) Gel shift assay was conducted using in vitro transcribed, [32P]-UTP-labeled U96A probe. The U96A probe was added to binding reactions containing FLAG–IRS-1 proteins (50, 100, 200 ng), FLAG-tagged GFP (negative control) or FLAG-15.5K (positive control). Representative results from three independent experiments are shown. (E) Lysates from HEK 293 cells overexpressing FLAG, FLAG–IRS-1, FLAG–CBP80 were subjected to immunoprecipitation with anti-FLAG antibody. RNA–protein complexes were eluted with 3 × FLAG peptides, and RNA bound to the proteins was extracted with phenol/chloroform/isoamyl alcohol mixture and ethanol precipitation. RNAs were subjected to RT-PCR using the primers specific for Rack1, which are designed within the first and last exon. Representative results from three independent experiments are shown.
Because snoRNAs were generally excised from an intron of protein-coding mRNAs coupled with splicing (22, 25), we examined the possibility that IRS-1 also interacts with Rack1 pre-mRNA. RNAs co-immunoprecipitated IRS-1 was subjected to RT-PCR using Rack1 specific primers designed within the first and last exon. As a result, we detected single band with a size around 1400 bases (Figure 2E). By sequencing analysis, The PCR product was proved to be Rack1 pre-mRNA that retains only intron 2. A similar size DNA was amplified using RNAs co-immunoprecipitated with CBP80, a protein that can bind pre-mRNAs (40, 41).
IRS-1 Positively Regulates U96A snoRNA Biogenesis
To gain insight into the biological significance of the association of IRS-1 with Rack1 pre-mRNA and U96A, we examined the amount of U96A in embryonic fibroblasts derived from IRS-1-deficient mice (IRS-1−/− MEFs) and wild-type MEFs (IRS-1+/+ MEFs). Embryonic fibroblasts prepared from littermate IRS-1+/+ MEFs and IRS-1−/− MEFs were cultured under serum-free conditions and total RNAs were normalized to DNA content since cell cycle distribution was the same in IRS-1+/+ and IRS-1−/− MEFs (Figure 3A). Northern-blot analysis showed that the U96A levels in IRS-1−/− MEFs were less than those in WT MEFs (Figure 3B). These results demonstrated that IRS-1 positively regulates the abundance of U96A. It is also important to note that the amounts of both Rack1 mature mRNA and Rack1 pre-mRNA retaining only intron 2 were unchanged, suggesting that the effect of IRS-1 depletion on the reduction of U96A in IRS-1−/− MEFs is not due to altered Rack1 transcription and splicing (Figures 3C,D).
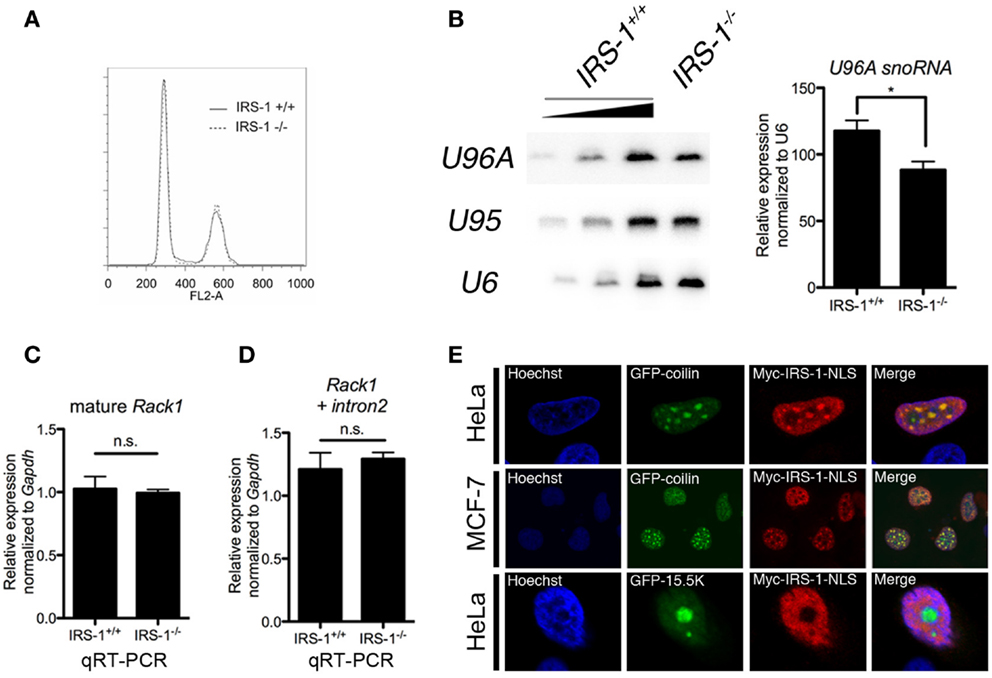
Figure 3. Effects of IRS-1 on expression of U96A snoRNA. Embryonic fibroblasts prepared from IRS-1+/+ and IRS-1−/− mice were serum-starved for 48 h. Cells were harvested and split into three portions. One portion was subjected to DNA extraction, which was used to normalize the amount of RNA. Another portion was used as a source of RNA, which was analyzed by Northern blotting (B), and by RT-PCR (C,D). The remaining portion was used for cell cycle analysis (A). (A) FACS analysis was performed to analyze the cell cycle distribution (n = 3). (B) Relative expression of U96A in MEFs from IRS-1+/+ and IRS-1−/− animals. 100, 33, and 11% of IRS-1+/+ samples were loaded. n = 3, bars indicate SEM. *P < 0.05 (Student’s t-test). (C,D) Relative expression of mature Rack1 mRNA and Rack1 pre-mRNA retaining intron2 in MEFs from IRS-1+/+ and IRS-1−/− animals. n = 5, bars indicate SEM. n.s., non-significant (Student’s t-test). (E) HeLa cells and MDF-7 cells were co-transfected with the plasmids expressing Myc–IRS-1 and either GFP–coilin (Cajal body marker) or GFP-15.5K (nucleolus marker). Following serum-starvation, cells were fixed and stained with anti-Myc antibody and Hoechist33342. A merge of the two images is shown with yellow indicating areas of colocalization. Representative stainings from at least three experiments using each cell are shown.
Small nucleolar RNAs are generally produced concomitantly with transcription/splicing of pre-mRNA in nucleoplasm and translocated to the nucleolus, which is the site of rRNA transcription and processing. Several snoRNAs are also reported to be detected in Cajal bodies, where they mature (20, 42). On the other hand, a part of IRS-1 is reported to be localized in nucleus (43–49). To investigate the possibility that nuclear IRS-1 is linked to snoRNA biogenesis, we performed immunofluorescent staining of NLS-fused Myc–IRS-1 (Myc–IRS-1–NLS) using HeLa cells and MCF-7 cells expressing either GFP–coilin or GFP-15.5K, a molecular component of Cajal bodies and nucleolar. As shown in Figure 3E, Myc–IRS-1–NLS certainly localizes to nucleus and exhibits a punctate staining pattern as well as general staining of the nucleoplasm. We found that Myc–IRS-1–NLS significantly colocalizes with GFP–coilin, suggesting that a function of IRS-1 in the nucleus might be related to snoRNA biogenesis.
Discussion
We had shown that IRS-1 and RNAs co-exist in high molecular weight complexes in cells. In this study, we identified several RNAs that physically interact with IRS-1. They included some mRNA species and non-coding RNAs. Among them, we focused on U96A snoRNA, and our results suggest a novel role of IRS-1 in U96A biogenesis.
Using CLIP analysis, we demonstrated that IRS-1 is in close proximity to RNAs including some mRNA species and non-coding RNAs. In vitro binding assay using purified IRS-1 and U96A, one of RNAs associating with IRS-1, also suggest that they interact with each other directly, although it cannot be ruled out that another RNA-binding protein(s) is co-purified with IRS-1 and mediates the interaction of IRS-1 with U96A. Because IRS-1 does not possess a known RNA-binding domain as far as we searched, it is important to determine the region in IRS-1 responsible for interaction with RNAs.
Since snoRNAs are encoded in protein-coding genes (host genes), the expression level/efficiency of snoRNA is dependent on the transcription of their host genes and splicing (21, 22). We observed that the U96A levels were reduced in IRS-1−/− MEF (Figure 3A), while the amounts of mature and intron2-retained pre-mRNA of Rack1 were not affected (Figure 3C), indicating that IRS-1 positively regulates the U96A levels after the step in which U96A was removed from Rack1 pre-mRNA by splicing. These steps include assembly of snoRNPs, debranching of removed lariat intron, or translocation of snoRNP into nucleolus.
We found that Rack1 pre-mRNA that retains the only intron 2 was accumulated in the IRS-1-immunoprecipitates. We found that the second intron of Rack1 where U96A is encoded possesses an atypical 5′ splice site (5′ss) with a GC dinucleotide in the first two intron positions, whose splicing-promoting activity is relatively weak. Thus, the retention of only the second intron may be due to this unique 5′ss sequence rather than the binding of IRS-1 to the intron. The mechanism by which this intrinsically weak 5′ss is efficiently selected by splicing factors and the GC 5′ ss-containing intron removal is promoted are poorly understood (50, 51). It is possible that the retention of the intron 2 is the rate-limiting step and plays important roles in expression of U96A. Given that our result shows IRS-1 forms a complex with U96A but not U95 (Figure 2C), which is encoded within the first intron of Rack1, splicing retention of intron 2 may provide the temporal opportunity for IRS-1 to access to the site of encoding U96A.
It is well established that snoRNAs form snoRNP complexes, and function to guide modification enzymes to newly synthesized rRNAs in the nucleolus (52, 53). Thus, snoRNAs are essential for the post-transcriptional chemical modification of rRNA, which is required for ribosome maturation and functions such as rRNA processing, ribosome structure, and IRES-mediated translation (53–56). Although the loss of all of snoRNAs individually in yeast causes no obvious effect on cell growth (57), depletion of the modifications in multiple sites resulted in severe growth retardation (53, 58). Furthermore, recent studies have shown that dysfunction of snoRNAs and their host pre-mRNA may have a role in the human cancers (26), implying their contribution to the cancer development caused by the alterations of ribosome biogenesis (27, 28). On the other hand, although IRS-1 is predominantly a cytoplasmic protein, earlier studies demonstrated that endogenous IRS-1 is also detectable in the nucleus (43–49), where its precise functions and biological relevance remain largely unknown. So far, in the nucleus, it has been reported that IRS-1 regulates transcription of rRNA and several genes implicated in cell proliferation and cancer progression (47, 48). Our findings that repression of U96A in IRS-1 knockout cells and the presence of nucleus-targeted IRS-1 in Cajal bodies suggest that IRS-1 functions in ribosome biogenesis by affecting the quality control of rRNA as well as rRNA abundance, which may contribute to the efficient cell growth mediated by IRS-1 and cancer development as well.
As explained in the introduction, IGF/insulin signals through IRSs induce translation of mRNA by activating the PI3K and MAPK pathways. In this study, we propose a novel mechanism by which IRSs could potentiate protein synthesis through the interaction of IRS-1 with snoRNA (Figure 4).
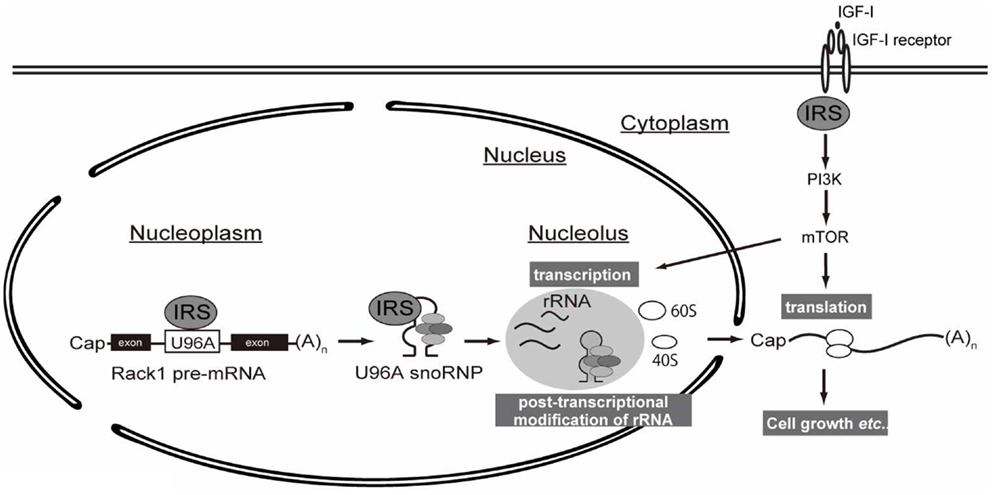
Figure 4. A working hypothesis of IRS-mediated control of protein synthesis. Following IGF stimulation, intracellular signaling through IRS activates downstream PI3K–mTOR signaling pathway, which is well known to promote translation of mRNAs. Up-regulation of protein synthesis is supported by ribosome maturation, which is accomplished by multiple coordinated processes, including rRNA transcription and ribosome activation (e.g., ribosomal protein S6 phosphorylation). The transcribed rRNAs in nucleus undergo chemical modification by snoRNP complexes whose biogenesis is controlled by nuclear IRS-1.
Author Contributions
Atsufumi Ozoe, Toshiaki Fukushima, Naoyuki Kataoka, Kazuhiro Chida, Tomoichiro Asano, Fumihiko Hakuno, and Shin-Ichiro Takahashi conceived and designed the experiments. Atsufumi Ozoe, Meri Sone, and Toshiaki Fukushima performed the experiments. Atsufumi Ozoe, Meri Sone, Toshiaki Fukushima, Naoyuki Kataoka, Kazuhiro Chida, Fumihiko Hakuno, and Shin-Ichiro Takahashi analyzed the data. Atsufumi Ozoe, Meri Sone, Toshiaki Fukushima, Naoyuki Kataoka, Fumihiko Hakuno, and Shin-Ichiro Takahashi contributed reagents/materials/analysis tools. Atsufumi Ozoe, Naoyuki Kataoka, Fumihiko Hakuno, and Shin-Ichiro Takahashi wrote the paper. Shin-Ichiro Takahashi took the primary responsibility for final content. Atsufumi Ozoe, Meri Sone, Toshiaki Fukushima, Naoyuki Kataoka, Kazuhiro Chida, Tomoichiro Asano, Fumihiko Hakuno, and Shin-Ichiro Takahashi read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge the help of Dr. Susan Hall (The University of North Carolina at Chapel Hill) in writing the manuscript. We thank Astellas Pharma Inc. (Tokyo, Japan) for donating recombinant human IGF-I. Funding: this work was partially supported by Grant-in-Aid [(A)(2)#16208028, (A) #22248030 and (S)#25221204], Challenging Exploratory Research (#20658065), Core-to-core program from the Japan Society for the Promotion of Science (JSPS) and Program for Promotion of Basic and Applied Researches for Innovations in Bio-oriented Industry to Shin-Ichiro Takahashi. This work was also partially supported by Research Activity Start-up (#20658065) to Toshiaki Fukushima from JSPS. Atsufumi Ozoe is a recipient of a JSPS Research Fellowship for young Scientists. The funders had no role in study design, data collection, and analysis, decision to publish, or preparation of the manuscript.
Abbreviations
CLIP, UV cross-linking and immunoprecipitation; DMEM, Dulbecco’s modified Eagle’s medium; FBS, fetal bovine serum; HEK293, human embryonic kidney 293; IGFs, insulin-like growth factors; IRSs, insulin receptor substrates; MAPK, mitogen-activated protein kinase; mRNA, messenger RNA; mRNP, messenger ribonucleoprotein; mTOR, mammalian target of rapamycin; PI3K, phosphatidylinositol 3-kinase; RNase A, ribonuclease A; snoRNA, small nucleolar RNA.
References
1. Clayton PE, Banerjee I, Murray PG, Renehan AG. Growth hormone, the insulin-like growth factor axis, insulin and cancer risk. Nat Rev Endocrinol (2010) 7(1):11–24. doi:10.1038/nrendo.2010.171
2. Copps KD, White MF. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia (2012) 55(10):2565–82. doi:10.1007/s00125-012-2644-8
3. Sun XJ, Miralpeix M, Myers MG Jr, Glasheen EM, Backer JM, Kahn CR, et al. Expression and function of IRS-1 in insulin signal transmission. J Biol Chem (1992) 267(31):22662–72.
4. Sun XJ, Wang LM, Zhang Y, Yenush L, Myers MG Jr, Glasheen E, et al. Role of IRS-2 in insulin and cytokine signalling. Nature (1995) 377(6545):173–7. doi:10.1038/377173a0
5. Withers DJ, Gutierrez JS, Towery H, Burks DJ, Ren JM, Previs S, et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature (1998) 391(6670):900–4. doi:10.1038/36116
6. Araki E, Lipes MA, Patti ME, Bruning JC, Haag B III, Johnson RS, et al. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature (1994) 372(6502):186–90. doi:10.1038/372186a0
7. Tamemoto H, Kadowaki T, Tobe K, Yagi T, Sakura H, Hayakawa T, et al. Insulin resistance and growth retardation in mice lacking insulin receptor substrate-1. Nature (1994) 372(6502):182–6. doi:10.1038/372182a0
8. Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet (2006) 7(8):606–19. doi:10.1038/nrg1879
9. Mayer C, Grummt I. Ribosome biogenesis and cell growth: mTOR coordinates transcription by all three classes of nuclear RNA polymerases. Oncogene (2006) 25(48):6384–91. doi:10.1038/sj.onc.1209883
10. Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell (2006) 124(3):471–84. doi:10.1016/j.cell.2006.01.016
11. Tanaka S, Wands JR. A carboxy-terminal truncated insulin receptor substrate-1 dominant negative protein reverses the human hepatocellular carcinoma malignant phenotype. J Clin Invest (1996) 98(9):2100–8. doi:10.1172/JCI119016
12. Bergmann U, Funatomi H, Kornmann M, Beger HG, Korc M. Increased expression of insulin receptor substrate-1 in human pancreatic cancer. Biochem Biophys Res Commun (1996) 220(3):886–90. doi:10.1006/bbrc.1996.0500
13. Chang Q, Li Y, White MF, Fletcher JA, Xiao S. Constitutive activation of insulin receptor substrate 1 is a frequent event in human tumors: therapeutic implications. Cancer Res (2002) 62(21):6035–8.
14. van Riggelen J, Yetil A, Felsher DW. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat Rev Cancer (2010) 10(4):301–9. doi:10.1038/nrc2819
15. Doudna JA, Rath VL. Structure and function of the eukaryotic ribosome: the next frontier. Cell (2002) 109(2):153–6. doi:10.1016/S0092-8674(02)00725-0
16. Rodnina MV, Wintermeyer W. Recent mechanistic insights into eukaryotic ribosomes. Curr Opin Cell Biol (2009) 21(3):435–43. doi:10.1016/j.ceb.2009.01.023
17. Kiss T. Small nucleolar RNAs: an abundant group of noncoding RNAs with diverse cellular functions. Cell (2002) 109(2):145–8. doi:10.1016/S0092-8674(02)00718-3
18. Tran E, Brown J, Maxwell ES. Evolutionary origins of the RNA-guided nucleotide-modification complexes: from the primitive translation apparatus? Trends Biochem Sci (2004) 29(7):343–50. doi:10.1016/j.tibs.2004.05.001
19. Kiss T, Fayet E, Jady BE, Richard P, Weber M. Biogenesis and intranuclear trafficking of human box C/D and H/ACA RNPs. Cold Spring Harb Symp Quant Biol (2006) 71:407–17. doi:10.1101/sqb.2006.71.025
20. Weinstein LB, Steitz JA. Guided tours: from precursor snoRNA to functional snoRNP. Curr Opin Cell Biol (1999) 11(3):378–84. doi:10.1016/S0955-0674(99)80053-2
21. Hirose T, Ideue T, Nagai M, Hagiwara M, Shu MD, Steitz JA. A spliceosomal intron binding protein, IBP160, links position-dependent assembly of intron-encoded box C/D snoRNP to pre-mRNA splicing. Mol Cell (2006) 23(5):673–84. doi:10.1016/j.molcel.2006.07.011
22. Hirose T, Shu MD, Steitz JA. Splicing-dependent and -independent modes of assembly for intron-encoded box C/D snoRNPs in mammalian cells. Mol Cell (2003) 12(1):113–23. doi:10.1016/S1097-2765(03)00267-3
23. Tycowski KT, Shu MD, Steitz JA. A small nucleolar RNA is processed from an intron of the human gene encoding ribosomal protein S3. Genes Dev (1993) 7(7A):1176–90. doi:10.1101/gad.7.7a.1176
24. Cavaille J, Bachellerie JP. Processing of fibrillarin-associated snoRNAs from pre-mRNA introns: an exonucleolytic process exclusively directed by the common stem-box terminal structure. Biochimie (1996) 78(6):443–56. doi:10.1016/0300-9084(96)84751-1
25. Ooi SL, Samarsky DA, Fournier MJ, Boeke JD. Intronic snoRNA biosynthesis in Saccharomyces cerevisiae depends on the lariat-debranching enzyme: intron length effects and activity of a precursor snoRNA. RNA (1998) 4(9):1096–110. doi:10.1017/S1355838298980785
26. Williams GT, Farzaneh F. Are snoRNAs and snoRNA host genes new players in cancer? Nat Rev Cancer (2012) 12(2):84–8. doi:10.1038/nrc3195
27. Montanaro L, Trere D, Derenzini M. Nucleolus, ribosomes, and cancer. Am J Pathol (2008) 173(2):301–10. doi:10.2353/ajpath.2008.070752
28. Shiue CN, Berkson RG, Wright AP. c-Myc induces changes in higher order rDNA structure on stimulation of quiescent cells. Oncogene (2009) 28(16):1833–42. doi:10.1038/onc.2009.21
29. Fukushima T, Arai T, Ariga-Nedachi M, Okajima H, Ooi Y, Iijima Y, et al. Insulin receptor substrates form high-molecular-mass complexes that modulate their availability to insulin/insulin-like growth factor-I receptor tyrosine kinases. Biochem Biophys Res Commun (2010) 404(3):767–73. doi:10.1016/j.bbrc.2010.12.045
30. Ozoe A, Sone M, Fukushima T, Kataoka N, Arai T, Chida K, et al. Insulin receptor substrate-1 (IRS-1) forms a ribonucleoprotein complex associated with polysomes. FEBS Lett (2013) 587(15):2319–24. doi:10.1016/j.febslet.2013.05.066
31. Ariga M, Nedachi T, Akahori M, Sakamoto H, Ito Y, Hakuno F, et al. Signalling pathways of insulin-like growth factor-I that are augmented by cAMP in FRTL-5 cells. Biochem J (2000) 348(Pt 2):409–16. doi:10.1042/0264-6021:3480409
32. Hakuno F, Kurihara S, Watson RT, Pessin JE, Takahashi S. 53BP2S, interacting with insulin receptor substrates, modulates insulin signaling. J Biol Chem (2007) 282(52):37747–58. doi:10.1074/jbc.M702472200
33. Kabuta T, Hakuno F, Asano T, Takahashi S. Insulin receptor substrate-3 functions as transcriptional activator in the nucleus. J Biol Chem (2002) 277(9):6846–51. doi:10.1074/jbc.M107058200
34. Robertson EJ. Teratocarcinomas and embryonic stem cells: a practical approach edited by E.J. Robertson. IRL Press, Oxford, 71–112 (1987).
35. Ule J, Jensen K, Mele A, Darnell RB. CLIP: a method for identifying protein-RNA interaction sites in living cells. Methods (2005) 37(4):376–86. doi:10.1016/j.ymeth.2005.07.018
36. Ninomiya K, Kataoka N, Hagiwara M. Stress-responsive maturation of Clk1/4 pre-mRNAs promotes phosphorylation of SR splicing factor. J Cell Biol (2011) 195(1):27–40. doi:10.1083/jcb.201107093
37. Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, Darnell RB. CLIP identifies Nova-regulated RNA networks in the brain. Science (2003) 302(5648):1212–5. doi:10.1126/science.1090095
38. Vitali P, Royo H, Seitz H, Bachellerie JP, Huttenhofer A, Cavaille J. Identification of 13 novel human modification guide RNAs. Nucleic Acids Res (2003) 31(22):6543–51. doi:10.1093/nar/gkg849
39. Watkins NJ, Dickmanns A, Luhrmann R. Conserved stem II of the box C/D motif is essential for nucleolar localization and is required, along with the 15.5K protein, for the hierarchical assembly of the box C/D snoRNP. Mol Cell Biol (2002) 22(23):8342–52. doi:10.1128/MCB.22.23.8342-8352.2002
40. Izaurralde E, Lewis J, McGuigan C, Jankowska M, Darzynkiewicz E, Mattaj IW. A nuclear cap binding protein complex involved in pre-mRNA splicing. Cell (1994) 78(4):657–68. doi:10.1016/0092-8674(94)90530-4
41. Kataoka N, Ohno M, Kangawa K, Tokoro Y, Shimura Y. Cloning of a complementary DNA encoding an 80 kilodalton nuclear cap binding protein. Nucleic Acids Res (1994) 22(19):3861–5. doi:10.1093/nar/22.19.3861
42. Shaw PJ, Beven AF, Leader DJ, Brown JW. Localization and processing from a polycistronic precursor of novel snoRNAs in maize. J Cell Sci (1998) 111(Pt 15):2121–8.
43. Chen J, Wu A, Sun H, Drakas R, Garofalo C, Cascio S, et al. Functional significance of type 1 insulin-like growth factor-mediated nuclear translocation of the insulin receptor substrate-1 and beta-catenin. J Biol Chem (2005) 280(33):29912–20. doi:10.1074/jbc.M504516200
44. Lassak A, Del Valle L, Peruzzi F, Wang JY, Enam S, Croul S, et al. Insulin receptor substrate 1 translocation to the nucleus by the human JC virus T-antigen. J Biol Chem (2002) 277(19):17231–8. doi:10.1074/jbc.M110885200
45. Morelli C, Garofalo C, Sisci D, del Rincon S, Cascio S, Tu X, et al. Nuclear insulin receptor substrate 1 interacts with estrogen receptor alpha at ERE promoters. Oncogene (2004) 23(45):7517–26. doi:10.1038/sj.onc.1208014
46. Prisco M, Santini F, Baffa R, Liu M, Drakas R, Wu A, et al. Nuclear translocation of insulin receptor substrate-1 by the simian virus 40 T antigen and the activated type 1 insulin-like growth factor receptor. J Biol Chem (2002) 277(35):32078–85. doi:10.1074/jbc.M204658200
47. Sun H, Tu X, Prisco M, Wu A, Casiburi I, Baserga R. Insulin-like growth factor I receptor signaling and nuclear translocation of insulin receptor substrates 1 and 2. Mol Endocrinol (2003) 17(3):472–86. doi:10.1210/me.2002-0276
48. Tu X, Batta P, Innocent N, Prisco M, Casaburi I, Belletti B, et al. Nuclear translocation of insulin receptor substrate-1 by oncogenes and IGF-I. Effect on ribosomal RNA synthesis. J Biol Chem (2002) 277(46):44357–65. doi:10.1074/jbc.M208001200
49. Yoon JC, Ng A, Kim BH, Bianco A, Xavier RJ, Elledge SJ. Wnt signaling regulates mitochondrial physiology and insulin sensitivity. Genes Dev (2010) 24(14):1507–18. doi:10.1101/gad.1924910
50. Kralovicova J, Hwang G, Asplund AC, Churbanov A, Smith CI, Vorechovsky I. Compensatory signals associated with the activation of human GC 5’ splice sites. Nucleic Acids Res (2011) 39(16):7077–91. doi:10.1093/nar/gkr306
51. Thanaraj TA, Clark F. Human GC-AG alternative intron isoforms with weak donor sites show enhanced consensus at acceptor exon positions. Nucleic Acids Res (2001) 29(12):2581–93. doi:10.1093/nar/29.12.2581
52. Omer AD, Ziesche S, Ebhardt H, Dennis PP. In vitro reconstitution and activity of a C/D box methylation guide ribonucleoprotein complex. Proc Natl Acad Sci U S A (2002) 99(8):5289–94. doi:10.1073/pnas.082101999
53. Tollervey D, Lehtonen H, Jansen R, Kern H, Hurt EC. Temperature-sensitive mutations demonstrate roles for yeast fibrillarin in pre-rRNA processing, pre-rRNA methylation, and ribosome assembly. Cell (1993) 72(3):443–57. doi:10.1016/0092-8674(93)90120-F
54. Jack K, Bellodi C, Landry DM, Niederer RO, Meskauskas A, Musalgaonkar S, et al. rRNA pseudouridylation defects affect ribosomal ligand binding and translational fidelity from yeast to human cells. Mol Cell (2011) 44(4):660–6. doi:10.1016/j.molcel.2011.09.017
55. Liang XH, Liu Q, Fournier MJ. rRNA modifications in an intersubunit bridge of the ribosome strongly affect both ribosome biogenesis and activity. Mol Cell (2007) 28(6):965–77. doi:10.1016/j.molcel.2007.10.012
56. Yoon A, Peng G, Brandenburger Y, Zollo O, Xu W, Rego E, et al. Impaired control of IRES-mediated translation in X-linked dyskeratosis congenita. Science (2006) 312(5775):902–6. doi:10.1126/science.1123835
57. Piekna-Przybylska D, Decatur WA, Fournier MJ. New bioinformatic tools for analysis of nucleotide modifications in eukaryotic rRNA. RNA (2007) 13(3):305–12. doi:10.1261/rna.373107
58. Zebarjadian Y, King T, Fournier MJ, Clarke L, Carbon J. Point mutations in yeast CBF5 can abolish in vivo pseudouridylation of rRNA. Mol Cell Biol (1999) 19(11):7461–72.
Appendix
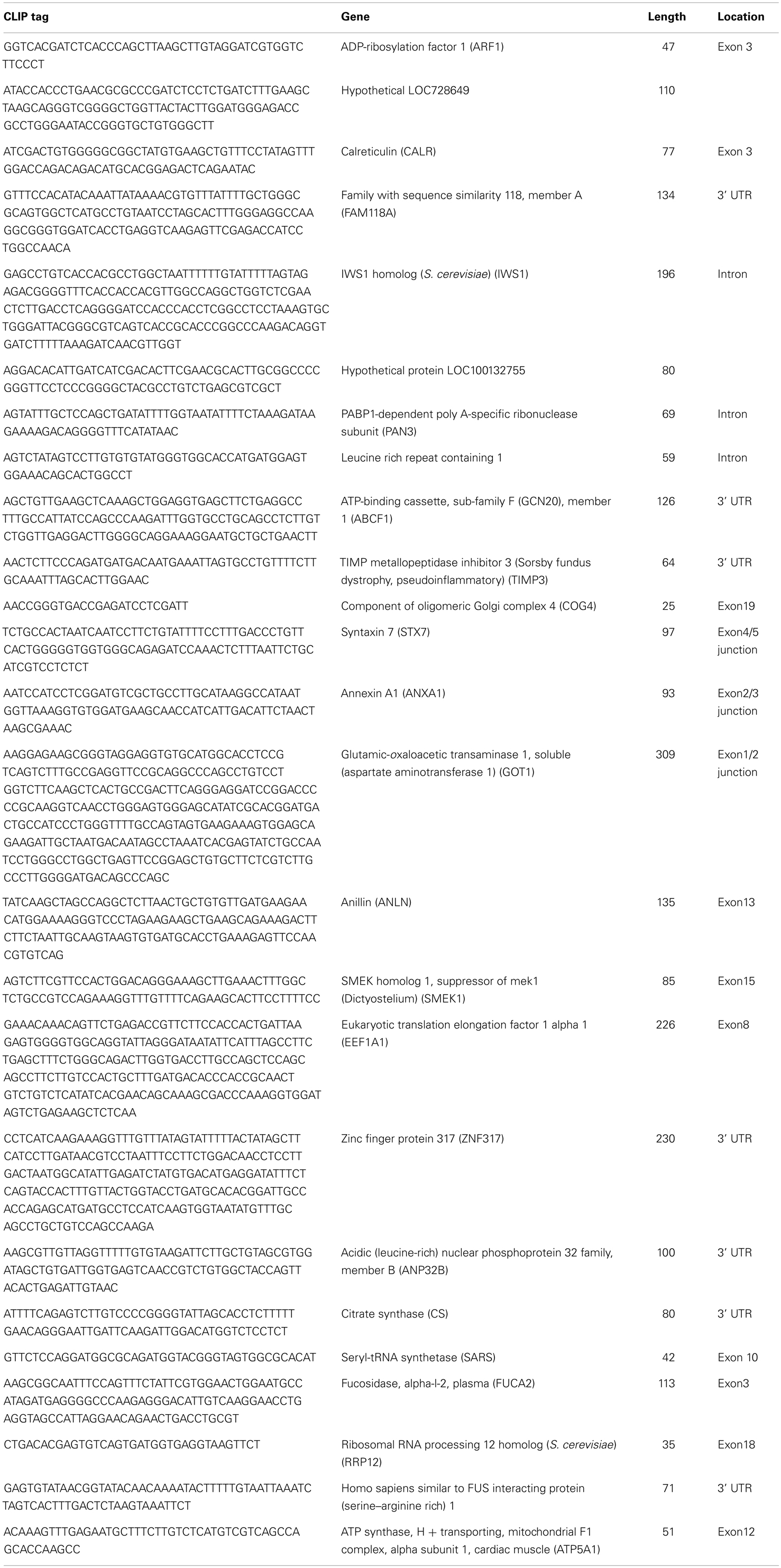
Table A1. Annotated list of IRS-1 CLIP tags (mRNA).

Table A2. Annotated list of IRS-1 CLIP tags (non-coding RNA).
Keywords: insulin-like growth factor, insulin receptor substrate, snoRNA, U96A, RACK1
Citation: Ozoe A, Sone M, Fukushima T, Kataoka N, Chida K, Asano T, Hakuno F and Takahashi S-I (2014) Insulin receptor substrate-1 associates with small nucleolar RNA which contributes to ribosome biogenesis. Front. Endocrinol. 5:24. doi: 10.3389/fendo.2014.00024
Received: 11 January 2014; Paper pending published: 24 January 2014;
Accepted: 14 February 2014; Published online: 04 March 2014.
Edited by:
Haim Werner, Tel Aviv University, IsraelReviewed by:
Patricia Alejandra Pennisi, Consejo Nacional de Investigaciones Científicas y Tecnológicas, ArgentinaMichael Danilenko, Ben-Gurion University of the Negev, Israel
Copyright: © 2014 Ozoe, Sone, Fukushima, Kataoka, Chida, Asano, Hakuno and Takahashi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shin-Ichiro Takahashi, Laboratory of Cell Regulation, Departments of Animal Sciences and Applied Biological Chemistry, Graduate School of Agriculture and Life Science, The University of Tokyo, 1-1-1 Yayoi, Bunkyo-ku, Tokyo 113-8657, Japan e-mail:YXRrc2hpbkBtYWlsLmVjYy51LXRva3lvLmFjLmpw