 Guy Griebel
Guy Griebel Christine Ravinet-Trillou2
Christine Ravinet-Trillou2 Philippe Pichat
Philippe Pichat- 1Exploratory Unit, Sanofi R&D, Chilly-Mazarin, France
- 2Infectious Diseases Therapeutic Strategic Unit, Sanofi R&D, Toulouse, France
Disruption of circadian clock enhances the risk of metabolic syndrome, obesity, and type 2 diabetes. Circadian clocks rely on a highly regulated network of transcriptional and translational loops that drive clock-controlled gene expression. Among these transcribed clock genes are cryptochrome (CRY) family members, which comprise Cry1 and Cry2. While the metabolic effects of deletion of several core components of the clock gene machinery have been well characterized, those of selective inactivation of Cry1 or Cry2 genes have not been described. In this study, we demonstrate that ablation of Cry1, but not Cry2, prevents high-fat diet (HFD)-induced obesity in mice. Despite similar caloric intake, Cry1−/− mice on HFD gained markedly less weight (−18%) at the end of the 16-week experiment and displayed reduced fat accumulation compared to wild-type (WT) littermates (−61%), suggesting increased energy expenditure. Analysis of serum lipid and glucose profiles showed no difference between Cry1−/− and WT mice. Both Cry1−/− and Cry2−/− mice are indistinguishable from WT controls in body weight, fat and protein contents, and food consumption when they are allowed unlimited access to a standard rodent diet. We conclude that although CRY signaling may not be essential for the maintenance of energy homeostasis under steady-state nutritional conditions, Cry1 may play a role in readjusting energy balance under changing nutritional circumstances. These studies reinforce the important role of circadian clock genes in energy homeostasis and suggest that Cry1 is a plausible target for anti-obesity therapy.
Introduction
The biological clock is an extensive molecular network that provides circadian time-keeping, controlling and maintaining daily rhythms of many behavioral and physiological processes. In mammals, it comprises a complex circuitry of transcriptional and translational regulatory feedback loops, including the core transcriptional activators CLOCK and BMAL1, which activate expression of three Period (Per1–3) and two cryptochrome (Cry1 and Cry2) genes (1). These latter products are part of the negative regulatory arm of the circadian clock system as their rhythmical accumulation leads to the formation of a repressor complex that interacts with CLOCK and BMAL1 to inhibit their own transcription. Mammalian clocks not only reside in the suprachiasmatic nucleus (SCN), in which the clock is determined by light signals through the retinohypothalamic tract, but they have been identified in a wide array of peripheral organs including heart, lung, kidney, liver, and pancreas, where their timing is set by metabolic cues (2–4).
Perturbation of endogenous circadian rhythms driven by clock gene mutations or disruptive lifestyles has been shown to give rise to various pathophysiological manifestations (5–7). This association is particularly strong with respect to cancer and metabolic disease. For example, sleep disturbances or long-term shift work has been shown to increase the risk of developing type 2 diabetes (8–11). Moreover, polymorphisms of the core clock genes Clock and Bmal1 are associated with obesity and type 2 diabetes (12, 13). Genetic mouse models also show that mutation in the genes encoding core clock transcription factors resulted in metabolic disturbances [for a review, see Ref. (6)]. This is best illustrated by mice deficient in Clock or Bmal1, which display impaired glucose tolerance and reduced insulin secretion (14). Moreover, disruption of Per2 and Per1–3 in mice results in increased vulnerability to high-fat-induced obesity and/or food intake (15). However, the situation is far from being clear with respect to these repressor clock genes as other studies demonstrated that mutation in Per 1, Per2, and Per1–3 resulted in reduced food intake and/or body weight in mice (16, 17). It was argued that the PER proteins may not be the best representatives of the negative regulatory arm of the molecular clock because they do not directly inhibit the core clock proteins, unlike cryptochromes (CRY) (18). Although there are several studies on the consequences of Cry1, Cry2, and Cry1/2 deletion, mainly on behavioral and molecular rhythmicity, only one has investigated the effects of Cry1−/−Cry2−/− double mutation on metabolism (18). These authors showed that deficiency in CRY resulted in increased susceptibility to diet-induced obesity as a consequence of increased insulin secretion and lipid storage.
To explore further the role of CRY on metabolism, the current study was undertaken to assess the effects of single deletion of either Cry1 or Cry2 on high-fat-induced obesity.
Materials and Methods
Animals
In all experiments mice were housed individually at a constant temperature of 21 ± 1°C and humidity (50 ± 10%) on a reverse light–dark cycle under a 12:12 light/dark cycle (light on at 7:00 a.m.). Six- to 10-week-old male Cry1−/−, Cry2−/− mice, and their respective wild-type (WT) counterparts were obtained from The Jackson Laboratories. They were fed ad libitum either under standard-mouse diet (STD) containing 3.7 kcal/g (A04, UAR; 15% fat, 12% protein, 73% carbohydrate) or under a high-fat diet (HFD) of 4.7 kcal/g energy density (D12451, Research Diet; 45% fat, 22% protein, 33% carbohydrate). Genotypes were determined by PCR of tail DNA. Animals were age-matched WT controls (Cry1+/+ or Cry2+/+) and homozygous (Cry1−/− or Cry2−/−) offsprings produced by backcrossing onto C57BL/6J background. All experimental procedures described herein were approved by the Animal Care and Use Committee of Sanofi or the Institutional Animal Care. Our animal facilities and animal care and use programs are in accordance with French legislation which implemented the European directive 86/609/EEC.
Experimental Procedure
In the first experiment, 8- to 10-week-old Cry1−/− mice (n = 9–10) were compared to WT animals (Cry1+/+) in two feeding paradigms for 16 weeks: (a) on a STD laboratory regimen, and (b) on a HFD regimen. In the second experiment, 6- to 9-week-old Cry2−/− mice (n = 12) were compared to their WT counterparts (Cry2+/+) (n = 15) in the two same feeding paradigms for 10 weeks. We have used a shorter time period in the Cry2 study because body weight evolution was not different between groups after 2 months of experiment, regardless the genotype and the regimen.
Body Weight, Body Composition, and Energy Intake
Body weight changes were recorded once a week. Body composition (fat and protein contents) was assessed in Cry1+/+ and Cry1−/− mice fed a HFD at the end of the experiment using the total body electrical conductivity technique (TOBEC system, EMSCAN 3000). This instrument measures total body electrical conductivity of small animals in a non-invasive manner (19). Body composition expressed as percentage protein mass or fat mass was obtained mathematically: protein (a): [0.816662 + 0.002615 × TOBEC + 0.046624 × body weight (BW) − 0.000066 × BW × TOBEC]2; fat (b): (−0.732127 − 0.008899 × TOBEC + 0.198538 × BW)2. Because we observed a different evolution in weight gain in Cry1−/− mice compared to their WT littermates under a HFD, energy intake was assessed weekly between weeks 12 and 16 in these groups.
Serum analysis
Plasma levels of glucose, triglycerides (TGs), free fatty acids (FFAs), and free cholesterol were determined in Cry1+/+ and Cry1−/− mice at the end of the 16-week HFD regimen in fed animals. Animals were sacrificed, blood was collected into EDTA-rinsed capillaries, and plasma was immediately prepared. Free cholesterol, TGs, and glucose were determined using enzymatic kits from Sigma. Plasma FFA levels were measured using the Wako kit (Wako Chemicals, Richmond, VA, USA).
Statistical Analyses
All data are shown as mean ± SEM. ANOVA and post hoc analyses were performed using SAS v.12 software. For body weight changes, statistical analysis was performed using a two-way ANOVA (factor strain × diet, with repeated measures on factor week). For food intake, a two-way ANOVA (factor strain or diet, with repeated measures on factor week) was performed. Finally, for body composition and serum analyses, a one-way ANOVA was used followed by post hoc comparisons with the Dunnett’s test. P-values <0.05 were considered statistically significant.
Results
Susceptibility of Cry1−/− Mice to High-Fat-Induced Obesity
Growth and body temperature
Cry1-deficient mice displayed the same fur quality than WT animals and they grew normally as assessed by naso-anal length, similar in both groups at 26-week of age (Cry1+/+ = 9.7 ± 0.1 cm; Cry1−/− = 9.3 ± 0.2 cm). In addition, there were no significant differences detectable in the body temperature between the two genotypes (Cry1+/+-HFD: 38.3 ± 0.2°C; Cry1−/−-HFD: 38.0 ± 0.2°C).
Body weight
Body weight was not significantly different between mutant mice and WT animals when the feeding trials started (Cry1+/+ = 25.6 ± 0.6 g; Cry1−/− = 24.6 ± 0.4 g). When maintained on a STD for 16 weeks, body weight changes did not differ significantly between Cry1+/+ and Cry1−/− mice (Figure 1A). After 16 weeks on the HFD, Cry1−/− mice showed a significantly lower body weight (37.2 ± 1.2 g, P < 0.001) compared with WT mice (45.3 ± 3.6 g). This effect reached statistical significance from week 8 [three-way ANOVA: F(15, 225) = 3.09, P < 0.001]. The total weight gain during the feeding period (Figure 2A) for Cry1−/− mice on HFD was 12.2 g (+52%), whereas the WT mice on HFD gained 18.7 g (+70%) [three-way ANOVA: F(14, 210) = 2.43, P < 0.01].
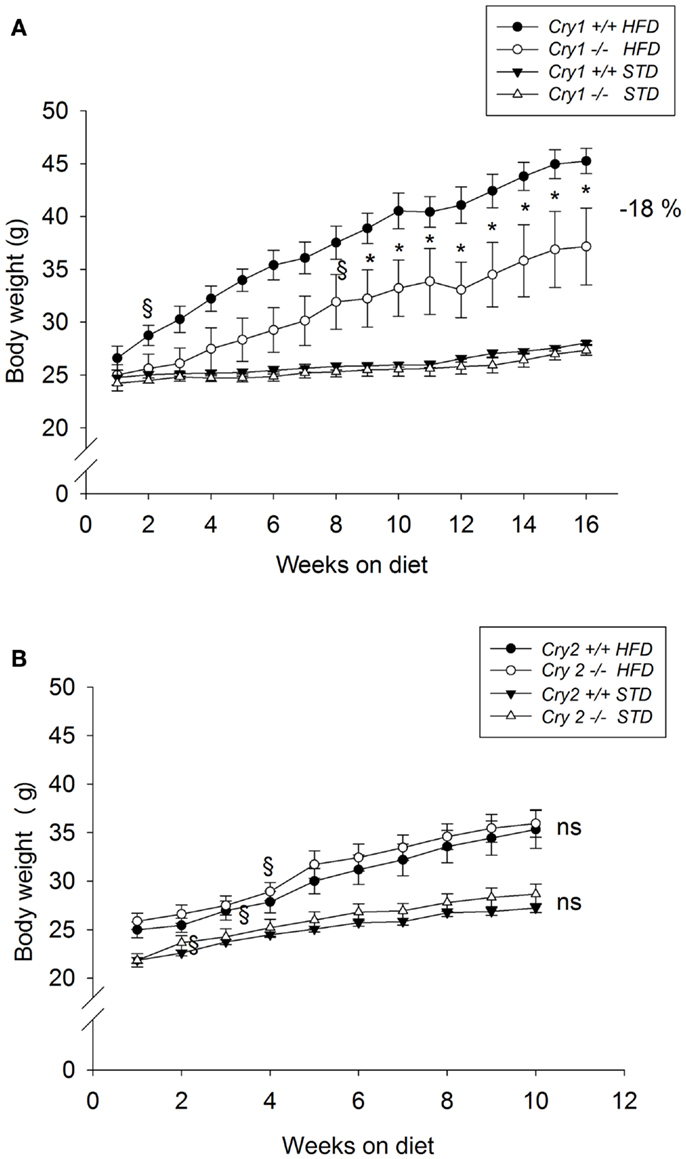
Figure 1. Growth curves of WT control, Cry1-, and Cry2-deficient mice under STD and HFD. Body weight for age-matched controls (A): Cry1+/+ (filled symbols) and Cry1−/− (open symbols) mice, (B): Cry2+/+ (filled symbols) and Cry2−/− mice (open symbols), determined once a week, on STD (triangles) and HFD (circles) regimens. Cry1 group: n = 5 (Cry1+/+-STD) and 5 (Cry1−/−-STD) and n = 4 (Cry1+/+-HFD) and 5 (Cry1−/−-HFD); Cry2 group: n = 6 (Cry2+/+-STD) and n = 7 (Cry2−/−-STD), and n = 8 (Cry2+/+-HFD) and n = 6 (Cry2−/−-STD). *P < 0.05, ns P > 0.05 indicated for genotype comparison, two-way ANOVA for body weight with repeated measures on factor week. §P < 0.05, one-way ANOVA followed by Dunnett’s test for comparison with week 1 with repeated measures for parameter weight. Values shown are mean ± SEM.
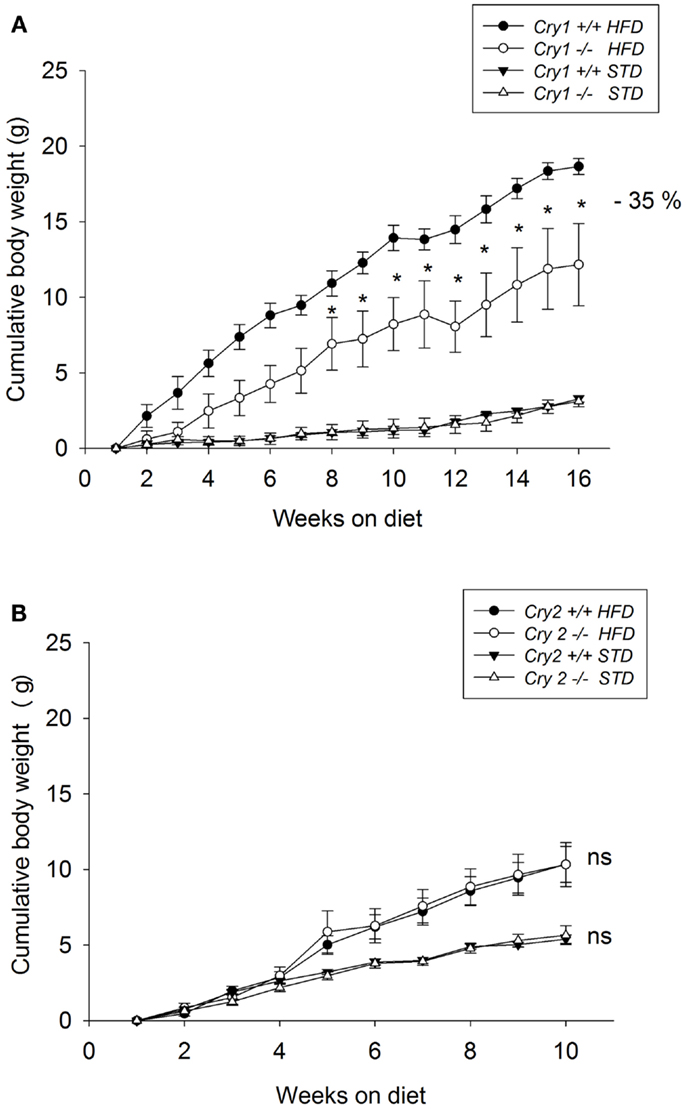
Figure 2. Cumulative weight gain as calculated from mean weights of WT control, Cry1-, and Cry2-deficient mice under STD and HFD. (A) Cry1+/+ (filled symbols) and Cry1−/− (open symbols) mice, (B): Cry2+/+ (filled symbols) and Cry2−/− mice (open symbols), determined once a week, on STD (triangles) and HFD (circles) regimens. Cry1 group: n = 5 (Cry1+/+-STD) and 5 (Cry1−/−-STD) and n = 4 (Cry1+/+-HFD) and 5 (Cry1−/−-HFD); Cry2 group: n = 6 (Cry2+/+-STD) and n = 7 (Cry2−/−-STD), and n = 8 (Cry2+/+-HFD) and n = 6 (Cry2−/−-STD). *P < 0.05, ns P > 0.05 indicated for genotype comparison, two-way ANOVA for body weight with repeated measures on factor week. Values shown are mean ± SEM.
Food intake
Analysis of food intake of animals on HFD shows that both WT and Cry1−/− mice had similar mean energy intake ranging from 13.0 ± 0.4–16.7 ± 0.5 to 11.7 ± 0.7–16.1 ± 0.6 kcal/day (data not shown).
Body composition
Analysis of body content showed that Cry1−/− had reduced fat mass compared to WT animals (−61%) at the end of the 16-week HFD regimen [F(1, 7) = 12.34, P < 0.001], while they had similar lean mass accounting to 17.1 ± 0.2% (WT) and 18.8 ± 1.0% (Cry1−/−) of total body mass (Figure 3).
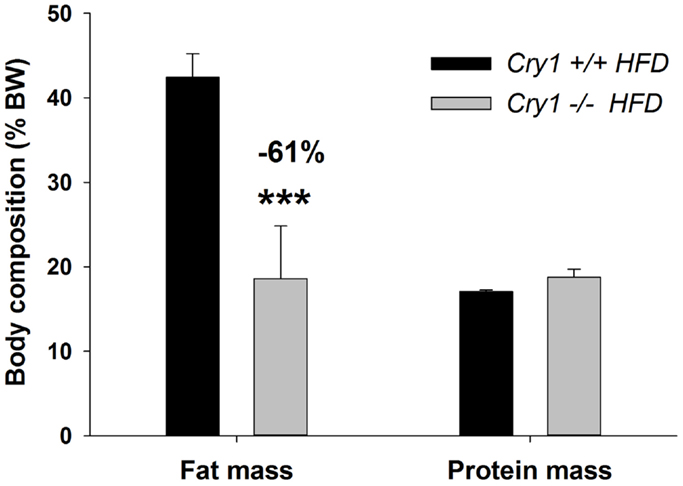
Figure 3. Body composition (% BW) of WT control and Cry1-deficient mice. Fat and protein masses (% body weight) for WT control Cry1+/+ (filled bars) and Cry1−/− (open bars)-deficient mice, determined on a HFD regimen. Cry1 group: n = 4 (Cry1+/+-STD) and 5 (Cry1−/−-STD) and n = 8 (Cry1+/+-HFD) and 6 (Cry1−/−-HFD). One-way ANOVA for body composition. ***P < 0.001 significant difference for body composition of each strain. Values shown are mean ± SEM.
Lipid and glucose profiles
Because changes in weight are often associated with shifts in glucose and lipid metabolism, we surveyed serum levels of glucose, TGs, and FFAs. Tests were performed on mice that were maintained on HFD diet. Results revealed no difference in any of these parameters between WT and Cry1−/− mice (Table 1).
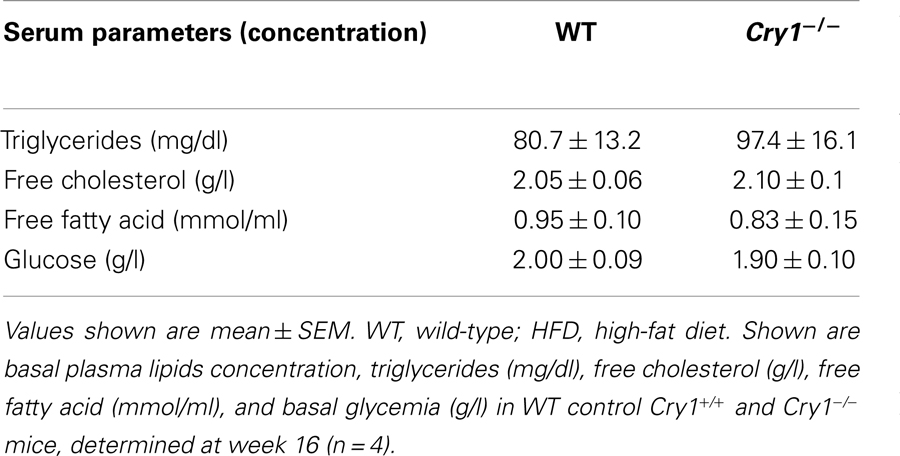
Table 1. Plasma lipids and glycemia in fed WT control and Cry1−/ − mice under HFD.
Susceptibility of Cry2−/− Mice to High-Fat-Induced Obesity
Growth and body temperature
Cry2-deficient mice displayed the same fur quality than WT animals and they grew normally. Naso-anal length was similar in both groups at 15-week of age (Cry2+/+ = 9.1 ± 0.2 cm; Cry2−/− = 9.1 ± 0.1 cm). Neither were significant differences in the body temperature between the two genotypes observed (Cry2+/+-STD: 37.2 ± 0.2°C; Cry2−/−-STD: 37.4 ± 0.2°C).
Body weight
Body weight was not significantly different between mutant mice and WT animals when the feeding trials started (Cry2+/+ = 23.5 ± 0.6 g; Cry2−/− = 23.9 ± 0.8 g). When maintained on a STD for 10 weeks, body weight changes did not differ significantly between Cry2+/+ and Cry2−/− mice (Figure 1B). After 10 weeks on the HFD, Cry2−/− mice showed similar body weight changes (34.9 ± 1.1 g) than WT mice (34.4 ± 1.8 g). The total weight gain during the feeding period for Cry2−/− mice on HFD was 10 g (+39%), whereas the WT mice on HFD gained 10.3 g (+41%) (Figure 2B).
Inasmuch as there were no weight change differences between Cry2+/+ and Cry2−/− under both STD and HFD regimens at week 10, the experiment was discontinued and no additional metabolism parameters were assessed in these mice.
Discussion
Our data provide several new insights into the physiological functions of CRY in mice. Utilizing Cry1−/− and Cry2−/− mice, we demonstrate for the first time that Cry1−/− loss interferes with HFD-induced obesity, and that the physiological basis of this effect is not related to reduced energy intake. Both Cry1- and Cry2-deficient mice show no obvious phenotypical changes when fed a standard high-carbohydrate diet. The animals grow to WT-like size with normal body weights.
Male WT mice that consume a HFD achieve a body weight between 41 (Cry2 group) and 70% (Cry1 group) greater than that of littermates-fed STD and increased adipose tissue mass. HFD are frequently used in rodent models, including C57BL/6 mice (20, 21), to cause obesity associated with metabolic disturbances such as hyperglycemia, hyperinsulinemia, and non-alcoholic fatty liver disease. These phenotypic impairments grossly mimic abdominal obesity phenotypes in humans and are linked to the development of type 2 diabetes (22). In the current study, mice lacking Cry2 did not display any overt phenotypical changes on a HFD. This was in contrast to Cry1−/− mice fed under the same regimen, which showed a markedly reduced weight gain and reduced body fat stores compared with WT counterparts. Significant differences in body weight development generally arise from differences in energy intake and/or energy excretion. However, in the current study, we observed that food intake was not significantly decreased in Cry1−/− mice under HFD conditions, suggesting increased energy expenditure. Body composition analysis revealed that Cry1 deficit resulted in reduced fat content, while that of protein was not affected. Moreover, analysis of serum lipid and glucose profiles showed no difference between Cry1−/− and WT mice at the end of the 16-week HFD regimen.
Deficiency of Cry1 and/or Cry2 has been shown to impair autonomic nervous system activity by increasing sympathetic neural outflow during the light phase, thus activating brown adipose tissue and hypermetabolism (23). It can be hypothesized that the increase in energy expenditure and resistance to HFD observed in the current study in Cry1−/− would be consistent with increased sympathetic activity. Alternatively, it can be argued that the metabolism profile of Cry1−/− mice may be the result of increased locomotor activity in these mice. While this parameter was not assessed here, it is worth mentioning that several previous studies on Cry1−/− mice reported no difference in motor behavior between these mutant animals and their WT counterparts when tested under LD12:12 as was the case in our study (23, 24).
The metabolism phenotype of Cry1-deficient mice in the current study contrasts with that recently observed with Cry1−/−Cry2−/− double mutant mice (18). In this study, mutant mice were reported to be lean under standard diet and when challenged with HFD, showed reduced food intake. Despite this hypophagia, they rapidly gained weight. The authors of this study suggested that hyperinsulinemia and increased lipid storage might account for the increased vulnerability of this mice to HFD-induced obesity. The discrepancy in metabolism phenotype between single and double mutant Cry mice cannot readily be attributed to methodological differences since the high-fat regimen used in both studies was the same (45% KJ fat). Moreover, background (C57BL/6J) and age range (8- to 10-week-old) of the mice also did not differ between these studies. It cannot completely be excluded that this difference is produced by a multitude of, perhaps small, methodological differences that do not necessarily become clear, even with close scrutiny of published reports. It is striking that in the Barclay et al.’s (18) study, HFD did not induce obesity in WT control mice, which is rather surprising since HFD is frequently used in rodent models, including C57BL/6 mice, to cause obesity associated with metabolic disturbances such as hyperglycemia, hyperinsulinemia, and non-alcoholic fatty liver disease (20, 21).
The apparent resistance of Cry1−/− mice to weight gain with a HFD may seem counterintuitive, since the metabolic consequences of circadian disruption such as those observed in human clinical studies in shift workers or in genetic models involving clock gene ablation are generally associated with increased body weight and onset of metabolic syndrome (25–30). For example, mutant mice of the circadian gene Clock overeat become obese and develop hyperglycemia and dyslipidemia (31). These animals develop the adipocyte hypertrophy and excessive accumulation of fat in the liver that are hallmarks of the metabolic syndrome. Deletion of the three circadian period genes in mice cause increased weight gain on HFD (32). Similarly, Bmal1-deficient mice display increased fat deposition, elevated TGs/FFA levels, and disrupted insulin responsiveness (33–37). However, other studies targeting more selectively the negative arm of the clock have shown that Per1 or Per2 deficiency leads to reduced body weight and fat composition associated with a decrease in plasma levels of TGs and FFAs. These changes could not be attributed to differences in food intake or aberrant circadian regulation, but were suggested to be the result of increased energy expenditure (16, 17). To explain the metabolic effects of Per2 ablation in this study, it was demonstrated that this clock gene modulates peroxisome proliferator-activated receptor γ2 (PPARγ2), a master regulator of adipogenic differentiation and lipid metabolism.
It is unclear whether a comparable scenario (i.e., Cry1 controlling the activity of proteins critical for adipogenesis by operating as their natural modulators) can explain the current observed metabolism profile in Cry1−/− mice. To the best of our knowledge, CRYs have not been described to control lipid metabolism by regulating transcriptional activity of relevant proteins. However, they were shown to participate in the regulation of hepatic gluconeogenesis notably through CRY-mediated inhibition of cAMP-mediated phosphorylation of cAMP response element-binding protein (CREB) (38–40). In particular, it was shown that fasted Cry1 and Cry2 double KO mice displayed increased expression of gluconeogenic genes, and blood glucose level in the morning. Conversely, CRY overexpression resulted in decreased glucose production in db/db mice, a mouse model of obesity and diabetes with excess gluconeogenesis (40). It is difficult to argue that a similar mechanism may be responsible for the phenotype of Cry1−/− mice in the current study as their blood glucose levels were not significantly different from those of WT animals when fed a HFD. Clearly, future studies should provide further insight into the underlying mechanism of the resistance of Cry1−/− mice to weight. It will be important to determine whether the metabolism profile of Cry1 deficiency is the result of disruption of the circadian regulation of metabolism and/or involves transcriptional mechanisms by which Cry regulates metabolic gene expression.
In summary, these results highlight further the importance of CRY as an important regulator of energy homeostasis by showing that ablation of Cry1−/−, but not Cry2−/− is associated with relative resistance to HFD-induced obesity. While our data suggest that Cry1 may be a potential novel therapeutic target to combat diet-induced obesity, future studies will be needed to determine if a pharmacological manipulation of Cry1 also prevents weight gain.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Claire Delgorge for their outstanding technical support.
References
1. Koike N, Yoo SH, Huang HC, Kumar V, Lee C, Kim TK, et al. Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science (2012) 338:349–54. doi: 10.1126/science.1226339
2. Damiola F, Le MN, Preitner N, Kornmann B, Fleury-Olela F, Schibler U. Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Genes Dev (2000) 14:2950–61. doi:10.1101/gad.183500
3. Schibler U, Ripperger J, Brown SA. Peripheral circadian oscillators in mammals: time and food. J Biol Rhythms (2003) 18:250–60. doi:10.1177/0748730403018003007
4. Storch KF, Lipan O, Leykin I, Viswanathan N, Davis FC, Wong WH, et al. Extensive and divergent circadian gene expression in liver and heart. Nature (2002) 417:78–83. doi:10.1038/nature744
5. Bass J, Takahashi JS. Circadian integration of metabolism and energetics. Science (2010) 330:1349–54. doi:10.1126/science.1195027
6. Bechtold DA, Gibbs JE, Loudon AS. Circadian dysfunction in disease. Trends Pharmacol Sci (2010) 31:191–8. doi:10.1016/j.tips.2010.01.002
7. Shi SQ, Ansari TS, McGuinness OP, Wasserman DH, Johnson CH. Circadian disruption leads to insulin resistance and obesity. Curr Biol (2013) 23:372–81. doi:10.1016/j.cub.2013.01.048
8. Kelly MA, Rees SD, Hydrie MZ, Shera AS, Bellary S, O’Hare JP, et al. Circadian gene variants and susceptibility to type 2 diabetes: a pilot study. PLoS One (2012) 7:e32670. doi:10.1371/journal.pone.0032670
9. Pan A, Schernhammer ES, Sun Q, Hu FB. Rotating night shift work and risk of type 2 diabetes: two prospective cohort studies in women. PLoS Med (2011) 8:e1001141. doi:10.1371/journal.pmed.1001141
10. Ruger M, Scheer FA. Effects of circadian disruption on the cardiometabolic system. Rev Endocr Metab Disord (2009) 10:245–60. doi:10.1007/s11154-009-9122-8
11. Spiegel K, Tasali E, Leproult R, Van CE. Effects of poor and short sleep on glucose metabolism and obesity risk. Nat Rev Endocrinol (2009) 5:253–61. doi:10.1038/nrendo.2009.23
12. Scott EM, Carter AM, Grant PJ. Association between polymorphisms in the clock gene, obesity and the metabolic syndrome in man. Int J Obes (Lond) (2008) 32:658–62. doi:10.1038/sj.ijo.0803778
13. Woon PY, Kaisaki PJ, Braganca J, Bihoreau MT, Levy JC, Farrall M, et al. Aryl hydrocarbon receptor nuclear translocator-like (BMAL1) is associated with susceptibility to hypertension and type 2 diabetes. Proc Natl Acad Sci U S A (2007) 104:14412–7. doi:10.1073/pnas.0703247104
14. Marcheva B, Ramsey KM, Buhr ED, Kobayashi Y, Su H, Ko CH, et al. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinemia and diabetes. Nature (2010) 466:627–31. doi:10.1038/nature09253
15. Yang S, Liu A, Weidenhammer A, Cooksey RC, McClain D, Kim MK, et al. The role of mPer2 clock gene in glucocorticoid and feeding rhythms. Endocrinology (2009) 150:2153–60. doi:10.1210/en.2008-0705
16. Dallmann R, Touma C, Palme R, Albrecht U, Steinlechner S. Impaired daily glucocorticoid rhythm in Per1 (Brd) mice. J Comp Physiol A Neuroethol Sens Neural Behav Physiol (2006) 192:769–75. doi:10.1007/s00359-006-0114-9
17. Grimaldi B, Bellet MM, Katada S, Astarita G, Hirayama J, Amin RH, et al. PER2 controls lipid metabolism by direct regulation of PPARgamma. Cell Metab (2010) 12:509–20. doi:10.1016/j.cmet.2010.10.005
18. Barclay JL, Shostak A, Leliavski A, Tsang AH, Johren O, Muller-Fielitz H, et al. High-fat diet-induced hyperinsulinemia and tissue-specific insulin resistance in Cry-deficient mice. Am J Physiol Endocrinol Metab (2013) 304:E1053–63. doi:10.1152/ajpendo.00512.2012
19. Harrison GG, Van Itallie TB. Estimation of body composition: a new approach based on electromagnetic principles. Am J Clin Nutr (1982) 35:1176–9.
20. Surwit RS, Kuhn CM, Cochrane C, McCubbin JA, Feinglos MN. Diet-induced type II diabetes in C57BL/6J mice. Diabetes (1988) 37:1163–7. doi:10.2337/diab.37.9.1163
21. Wallis K, Walters JR, Gabe S. Short bowel syndrome: the role of GLP-2 on improving outcome. Curr Opin Clin Nutr Metab Care (2009) 12:526–32. doi:10.1097/MCO.0b013e32832d23cd
22. Collins S, Martin TL, Surwit RS, Robidoux J. Genetic vulnerability to diet-induced obesity in the C57BL/6J mouse: physiological and molecular characteristics. Physiol Behav (2004) 81:243–8. doi:10.1016/j.physbeh.2004.02.006
23. Ikeda H, Yong Q, Kurose T, Todo T, Mizunoya W, Fushiki T, et al. Clock gene defect disrupts light-dependency of autonomic nerve activity. Biochem Biophys Res Commun (2007) 364:457–63. doi:10.1016/j.bbrc.2007.10.058
24. Vitaterna MH, Selby CP, Todo T, Niwa H, Thompson C, Fruechte EM, et al. Differential regulation of mammalian period genes and circadian rhythmicity by cryptochromes 1 and 2. Proc Natl Acad Sci U S A (1999) 96:12114–9. doi:10.1073/pnas.96.21.12114
25. Fonken LK, Workman JL, Walton JC, Weil ZM, Morris JS, Haim A, et al. Light at night increases body mass by shifting the time of food intake. Proc Natl Acad Sci U S A (2010) 107:18664–9. doi:10.1073/pnas.1008734107
26. Gimble JM, Sutton GM, Bunnell BA, Ptitsyn AA, Floyd ZE. Prospective influences of circadian clocks in adipose tissue and metabolism. Nat Rev Endocrinol (2011) 7:98–107. doi:10.1038/nrendo.2010.214
27. Gimble JM, Sutton GM, Ptitsyn AA, Floyd ZE, Bunnell BA. Circadian rhythms in adipose tissue: an update. Curr Opin Clin Nutr Metab Care (2011) 14:554–61. doi:10.1097/MCO.0b013e32834ad94b
28. Green CB, Takahashi JS, Bass J. The meter of metabolism. Cell (2008) 134:728–42. doi:10.1016/j.cell.2008.08.022
29. Karatsoreos IN, Bhagat S, Bloss EB, Morrison JH, McEwen BS. Disruption of circadian clocks has ramifications for metabolism, brain, and behavior. Proc Natl Acad Sci U S A (2011) 108:1657–62. doi:10.1073/pnas.1018375108
30. Scheer FA, Hilton MF, Mantzoros CS, Shea SA. Adverse metabolic and cardiovascular consequences of circadian misalignment. Proc Natl Acad Sci U S A (2009) 106:4453–8. doi:10.1073/pnas.0808180106
31. Turek FW, Joshu C, Kohsaka A, Lin E, Ivanova G, McDearmon E, et al. Obesity and metabolic syndrome in circadian clock mutant mice. Science (2005) 308:1043–5. doi:10.1126/science.1108750
32. Dallmann R, Weaver DR. Altered body mass regulation in male mPeriod mutant mice on high-fat diet. Chronobiol Int (2010) 27:1317–28. doi:10.3109/07420528.2010.489166
33. Lamia KA, Storch KF, Weitz CJ. Physiological significance of a peripheral tissue circadian clock. Proc Natl Acad Sci U S A (2008) 105:15172–7. doi:10.1073/pnas.0806717105
34. Rudic RD, McNamara P, Curtis AM, Boston RC, Panda S, Hogenesch JB, et al. BMAL1 and CLOCK, two essential components of the circadian clock, are involved in glucose homeostasis. PLoS Biol (2004) 2:e377. doi:10.1371/journal.pbio.0020377
35. Sadacca LA, Lamia KA, deLemos AS, Blum B, Weitz CJ. An intrinsic circadian clock of the pancreas is required for normal insulin release and glucose homeostasis in mice. Diabetologia (2011) 54:120–4. doi:10.1007/s00125-010-1920-8
36. Shi S, Hida A, McGuinness OP, Wasserman DH, Yamazaki S, Johnson CH. Circadian clock gene Bmal1 is not essential; functional replacement with its paralog, Bmal2. Curr Biol (2010) 20:316–21. doi:10.1016/j.cub.2009.12.034
37. Shimba S, Ogawa T, Hitosugi S, Ichihashi Y, Nakadaira Y, Kobayashi M, et al. Deficient of a clock gene, brain and muscle Arnt-like protein-1 (BMAL1), induces dyslipidemia and ectopic fat formation. PLoS One (2011) 6:e25231. doi:10.1371/journal.pone.0025231
38. Hirota T, Lee JW, St John PC, Sawa M, Iwaisako K, Noguchi T, et al. Identification of small molecule activators of cryptochrome. Science (2012) 337:1094–7. doi:10.1126/science.1223710
39. Lamia KA, Papp SJ, Yu RT, Barish GD, Uhlenhaut NH, Jonker JW, et al. Cryptochromes mediate rhythmic repression of the glucocorticoid receptor. Nature (2011) 480:552–6. doi:10.1038/nature10700
Keywords: clock genes, cryptochromes, obesity, diet-induced obesity, knockout mice, mice, triglycerides
Citation: Griebel G, Ravinet-Trillou C, Beeské S, Avenet P and Pichat P (2014) Mice deficient in cryptochrome 1 (Cry1−/−) exhibit resistance to obesity induced by a high-fat diet. Front. Endocrinol. 5:49. doi: 10.3389/fendo.2014.00049
Received: 24 February 2014; Accepted: 26 March 2014;
Published online: 09 April 2014.
Edited by:
Romesh Khardori, Eastern Virginia Medical School, USAReviewed by:
Romesh Khardori, Eastern Virginia Medical School, USAFrancis Kim, University of Washington, USA
Copyright: © 2014 Griebel, Ravinet-Trillou, Beeské, Avenet and Pichat. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guy Griebel, Exploratory Unit, Sanofi R&D, 1 Avenue Pierre Brossolette, Chilly-Mazarin 91385, France e-mail:Z3V5LmdyaWViZWxAc2Fub2ZpLmNvbQ==