 Peishen Zhao
Peishen Zhao Matthew Metcalf
Matthew Metcalf Nigel W. Bunnett1,2*
Nigel W. Bunnett1,2*- 1Monash Institute of Pharmaceutical Sciences, Parkville, VIC, Australia
- 2Department of Pharmacology, University of Melbourne, Melbourne, VIC, Australia
In addition to their role in protein degradation and digestion, proteases can also function as hormone-like signaling molecules that regulate vital patho-physiological processes, including inflammation, hemostasis, pain, and repair mechanisms. Certain proteases can signal to cells by cleaving protease-activated receptors (PARs), a family of four G protein-coupled receptors. PARs are expressed by almost all cell types, control important physiological and disease-relevant processes, and are an emerging therapeutic target for major diseases. Most information about PAR activation and function derives from studies of a few proteases, for example thrombin in the case of PAR1, PAR3, and PAR4, and trypsin in the case of PAR2 and PAR4. These proteases cleave PARs at established sites with the extracellular N-terminal domains, and expose tethered ligands that stabilize conformations of the cleaved receptors that activate the canonical pathways of G protein- and/or β-arrestin-dependent signaling. However, a growing number of proteases have been identified that cleave PARs at divergent sites to activate distinct patterns of receptor signaling and trafficking. The capacity of these proteases to trigger distinct signaling pathways is referred to as biased signaling, and can lead to unique patho-physiological outcomes. Given that a different repertoire of proteases are activated in various patho-physiological conditions that may activate PARs by different mechanisms, signaling bias may account for the divergent actions of proteases and PARs. Moreover, therapies that target disease-relevant biased signaling pathways may be more effective and selective approaches for the treatment of protease- and PAR-driven diseases. Thus, rather than mediating the actions of a few proteases, PARs may integrate the biological actions of a wide spectrum of proteases in different patho-physiological conditions.
Introduction
With over 800 members in mammals, G protein-coupled receptors (GPCRs) are the largest family of cell-surface signaling proteins. They are receptors for an extraordinary range of structurally diverse agonists in the extracellular fluid, including endogenous hormones, neurotransmitters, and paracrine regulators, as well as multiple exogenous ligands (1, 2). Due to their critical importance in the control of most patho-physiological processes, GPCRs are the primary target for over 30% of the clinically used drugs (3, 4). The established mechanism of GPCR activation is that agonist binding results in conformational changes in the receptor that activate the Gα subunits of heterotrimeric G proteins, leading to the dissociation of Gβγ dimers from Gα. Activated Gα and Gβγ then initiate downstream signaling processes (5). To control the duration and magnitude of this signaling, activated receptors are phosphorylated by G protein-coupled receptor kinases (GRKs) or other kinases, and then interact with β-arrestins, which mediate receptor desensitization and endocytosis (6). Depending on the receptor and the agonist, internalized receptors are then sorted to lysosomes for degradation, or move to the plasma membrane for another cycle of activation (7, 8). However, a common feature of GPCRs is that a single receptor can interact with multiple endogenous and exogenous ligands, each of which may activate the receptor in different ways. For example, a large number of endogenous opioid neuropeptides as well as many different opiate drugs interact with opioid receptors, and different opioids and opiates result in divergent processes of receptor activation and regulation (9). Thus, the simplistic view of receptor activation and regulation has been revised by the appreciation that different agonists of the same receptor can result in distinct patterns of signaling and regulation.
The early two-state model of receptor function suggested that a receptor adopts active conformation upon ligand binding. This model considered only one active state, leading to a single functional readout. However, increased understanding of receptor signaling has revealed that different ligands can initiate distinct signaling events through the same GPCR. The heterogeneity of signaling events by a single GPCR can include different maximum responses from a single pathway (i.e., full or partial agonism) or activation of distinctly different signaling pathways by different agonists. The capacity of different agonists to initiate signaling of the same GPCR by distinct mechanisms is referred to as biased agonism or signaling (10, 11), and has been described for many GPCRs, including opioid receptors (12), angiotensin receptors (13), and glutamate receptors (14). This phenomenon of signaling bias is not surprising because GPCRs are flexible proteins that interact with multiple ligands and regulatory proteins, all of which may influence the capacity of the receptor to signal by particular mechanisms. Indeed, recent advances in our understanding of the structure of GPCRs in various activation states has revealed that a single GPCR can exist in multiple active conformations that may favor coupling to different signaling pathways (15, 16).
This review focuses on the capacity of different proteases and synthetic ligands to induce biased signaling of protease-activated receptors (PARs). The PARs are a family of four GPCRs (PAR1–4) that belong to group A rhodopsin-like GPCR subfamily. The first family member, PAR1, was identified as a receptor for thrombin, a serine protease coagulation factor (17). PAR2 was subsequently identified as a receptor for the serine protease trypsin (18), followed by PAR3, another thrombin receptor (19), and PAR4, a receptor for both thrombin and trypsin (20). PARs are expressed in many tissues and cell types, where they regulate multiple patho-physiological processes, including hemostasis, inflammation, pain, cellular proliferation, and healing (21–23). However, in addition to thrombin and trypsin, a large number of proteases have been identified that can cleave PARs. In some cases, these proteases cleave at the same sites as thrombin or trypsin and thereby initiate common signaling events. However, in other cases, proteases cleave PARs at distinct sites, and either activate distinct signals (biased agonism), or disarm the receptor by removing or destroying tethered ligand domains (receptor antagonism). We will review mechanisms by which various proteases and synthetic agonists activate PARs, and will discuss the implications of protease-biased signaling of PARs for patho-physiological control and therapeutic targeting.
Mechanisms of Canonical Activation and Signaling of PARs
Unlike other GPCRs, the endogenous ligands for PARs reside within the extracellular N-terminus of the receptors. Receptor cleavage at the defined sites within the N-terminus by proteases such as thrombin and trypsin reveals these tethered ligands that, once exposed, can bind to regions in the second extracellular loops of the cleaved receptors, initiating conformational changes in the receptors that activate downstream signals (23). This is the canonical mechanism of PAR activation (Figure 1A). There are subtle differences in the mechanisms by which different proteases initiate the canonical pathways of receptor activation, which depend on the protease and PAR in question. For example, thrombin first binds to PAR1 and PAR3; this action facilitates receptor cleavage and exposure of the tethered ligand sequence. Mutation of the binding site reduces the efficacy with which thrombin activates these receptors, and mutation of the cleavage site prevents receptor activation (17, 19). On the other hand, trypsin activates PAR2 directly, without first binding to the receptor (18, 24). Accessory proteins can also influence the capacity of proteases to activate PARs. In particular, proteins that anchor proteases to the plasma membrane can enhance proteolytic activation. For example, during tissue damage and inflammation, tissue factor (TF) binds coagulation factors (F) VIIa, which in turn activates FX to FXa. FXa and its co-factor FVa promote conversion of prothrombin to thrombin, and subsequent PAR1 activation (25). Besides promoting thrombin activation, FVIIa and FXa both can signal directly through PAR1 and PAR2, although the efficiency and potency of receptor activation is substantially enhanced when they are coupled with TF (26). Similarly, the proteolytic activity of the anticoagulant activated protein C (APC) toward PARs is largely regulated by its association with the endothelial protein C receptor (EPCR) at the surface of endothelial cells (27, 28).
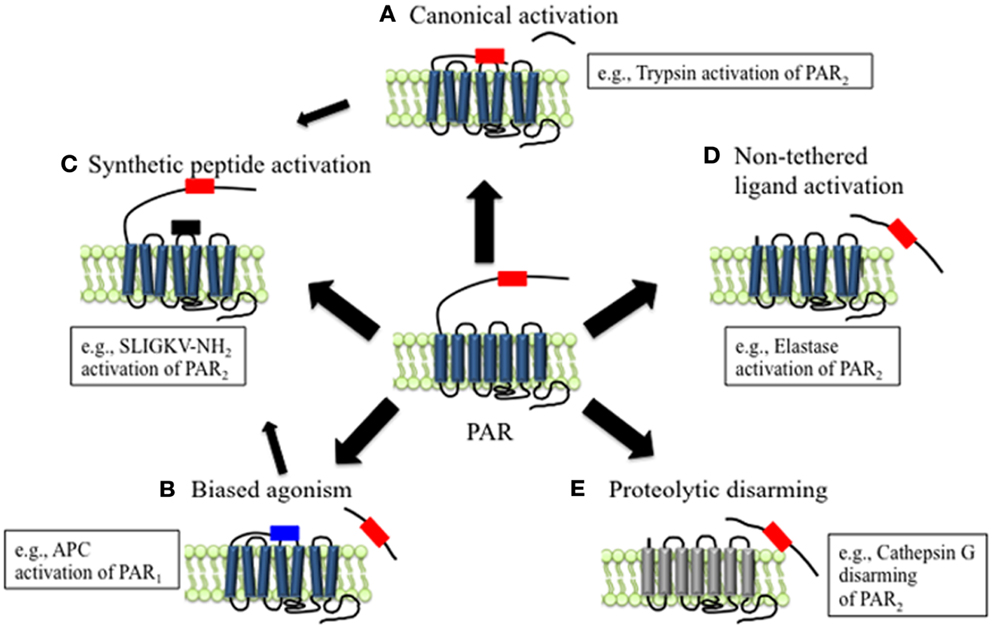
Figure 1. Mechanisms of canonical and biased PAR signaling. (A) Canonical mechanisms of PAR signaling. Proteases such as trypsin and thrombin cleave PARs at canonical cleavage sites, unmasking the tethered ligand domain, which binds to the second extracellular loops of the cleaved receptors. PARs that are activated by such mechanisms often couple to multiple G protein-dependent and β-arrestin-dependent signaling pathways. (B) Biased mechanisms of PAR signaling. Proteases such as elastase, MMP1, and APC cleave PARs at sites distinct from the canonical cleavage site. Cleavage may unmask a new tethered ligand that could interact with domains in the cleaved receptor, leading to the activation of unique and biased signaling pathways. (C) APs are synthetic peptides that mimic the tethered ligands revealed by proteases that cleave at canonical or biased sites. APs can activate the same pathways as proteases, although tethered ligand and soluble peptides may also trigger different signaling pathways and generate biased signal. (D) Some proteases such as elastase that cleave PARs to not appear to reveal tethered ligands, suggesting that proteolysis alone may activate the receptor. (E) Proteolytic disarming of PARs. Proteases such as cathepsin G cleave PARs and remove or destroy tethered ligands, thereby disarming proteolytic activation.
Support for the tethered ligand mechanism of PAR activation is provided by the observation that synthetic peptides, referred to as activating peptides (APs), that mimic the tethered ligand domain can also activate certain PARs directly, without the requirement for proteolysis (Figure 1C). Peptides mimicking the tethered ligands of PAR1, PAR2, and PAR4 can directly activate these receptors, although with a considerably lower potency than the activating proteases, especially in the case of PAR4 (17, 18, 20). The higher EC50 values of APs compare to those of proteases possibly reflect the differences between a tethered ligand and an untethered ligand in solution. PAR3 is not activated by tethered ligand-derived peptides, and appears to be unable to signal directly, but rather to serve as a co-factor for other PARs, such as PAR1 and PAR4 (29, 30).
Activating peptides have been considered to mimic the effects of proteases and have been widely used to probe the functions of PARs without the use of proteases, which can cleave multiple other proteins that may influence outcomes. However, this is not always the case because in some circumstances proteases and APs agonists can exert different effects. For example, in human brain microvascular endothelial cells, thrombin activation of PAR1 triggers endothelial barrier permeability, whereas PAR1-AP (SFLLRN-NH2) has no significant effect (31). In addition, the signaling properties of a PAR2 mutant with substitutions within the trypsin-revealed tethered ligand domain differ from those of APs with the same substitutions, suggesting distinct activation modes by tethered versus soluble peptides (32). The divergent signaling effects of proteases and APs provide evidence for biased signaling of PARs.
Tissue-Specific Complexity and Diversity of PAR Activation and Signaling
In addition to the diversity of signals that can originate from the same receptor after activation by proteases or synthetic agonists (i.e., biased signaling), many other factors also affect patho-physiological outcome of PAR activation. These factors include the availability of activated proteases as well as the existence of regulatory and accessory proteins in different tissues and cell types.
The availability of active, functional proteases is a key requirement of PAR signaling, and the predominant endogenous proteases that activate PARs may vary in different patho-physiological states. For instance, the compliment of available active proteases varies markedly during the course of inflammation and healing, depending on the presence of immune cells, which are the source of many proteases, and on the existence of endogenous inhibitors. The compliment of proteases of mast cells, neutrophils, eosinophil, and macrophages, which participate in different phases of inflammation, varies considerably. For example, as the first responders to microbial infection, neutrophil produce elastase, cathepsin G, and proteinase-3 (33), whereas macrophages, which mediate chronic inflammation, release plasmin, matrix metalloproteinases (MMPs), and cathepsin S (34).
Although PARs can be activated by distinct proteases under different conditions, proteases that cleave PARs at the same sites would be expected to activate the same canonical signaling pathway and to induce common patho-physiological outcomes. However, the consequences of PAR cleavage could vary considerably if the activated proteases cleave PARs at distinct sites and are biased agonists or even antagonists, as discussed below. Indeed, at any one time multiple proteases would likely be activated and capable of cleaving PARs at distinct sites with unique outcomes. Thus, the active conformation of PARs may vary depending on the milieu of available proteases, which may differ between health and disease conditions. For example, during the initial stages of inflammatory processes such as inflammatory bowel diseases or chronic obstructive pulmonary disease, infiltration of neutrophils leads to increased level of elastase (35, 36), a biased agonist of both PAR1, and PAR2 (37, 38) (discussed below). Further complexity is provided by the presence of endogenous protease inhibitors that control the activity of proteases (39).
The outcome of PAR activation by the same protease or synthetic agonist can also vary between tissues and cell types. For example, thrombin and PAR1-AP cause relaxation of the intact coronary artery but contraction when the endothelium is removed, indicating distinct outcomes of PAR1 activation in endothelial versus vascular smooth muscle cells, possibility due to formation of different signaling complexes (40).
Mechanisms of Biased Activation and Signaling of PARs
Compared to other GPCRs, the N-terminal domains of PARs are particularly susceptible to proteolysis. Although the reasons for this susceptibility are not fully understood, they probably relate to the presence of protease binding sites on the receptors, the existence of multiple scissile bonds, and the lack of groups that would sterically hinder proteolysis. However, the outcome of PAR activation depends on the site of proteolytic cleavage. Those proteases that cleave PARs at the conserved activating sites reveal tethered ligands that trigger the canonical signaling pathways (Figure 1A). Proteases that cleave PARs at distinct sites can act as biased agonists by triggering signals that are distinct from those activated by the canonical pathways (Figures 1B,D). In some cases, these alternative signaling mechanisms appear to involve exposure of distinct tethered ligands (Figure 1B). However, in other instances receptor cleavage per se may generate a conformational change that is sufficient to activate the receptor (Figure 1D). Alternatively, proteases can destroy or remove tethered ligand domains, forming N-terminally truncated receptors that are unresponsive to further activation by other proteases (Figure 1E).
Activated PARs can couple to multiple G protein-dependent (Gαq, Gα12/13, Gαi, Gαs, and Gβγ) and β-arrestin-dependent pathways. Although in many instances a particular protease or synthetic agonist can activate more than one of these pathways, in some cases proteases and synthetic agonists activate a single pathway. By comparing and categorizing the signaling pathways that are initiated by different proteases and synthetic agonists with the overall outcome of receptor activation, it is possible to identify the primary signaling pathways responsible for PAR-mediated patho-physiological responses (Tables 1–3). Moreover, a comprehensive understanding of the mechanisms and outcomes of PAR signaling by different proteases and synthetic agonists can guide the development of agonists and antagonists that may selectively activate or inhibit disease-relevant pathways. This approach has implications for development of pathway-specific therapies.
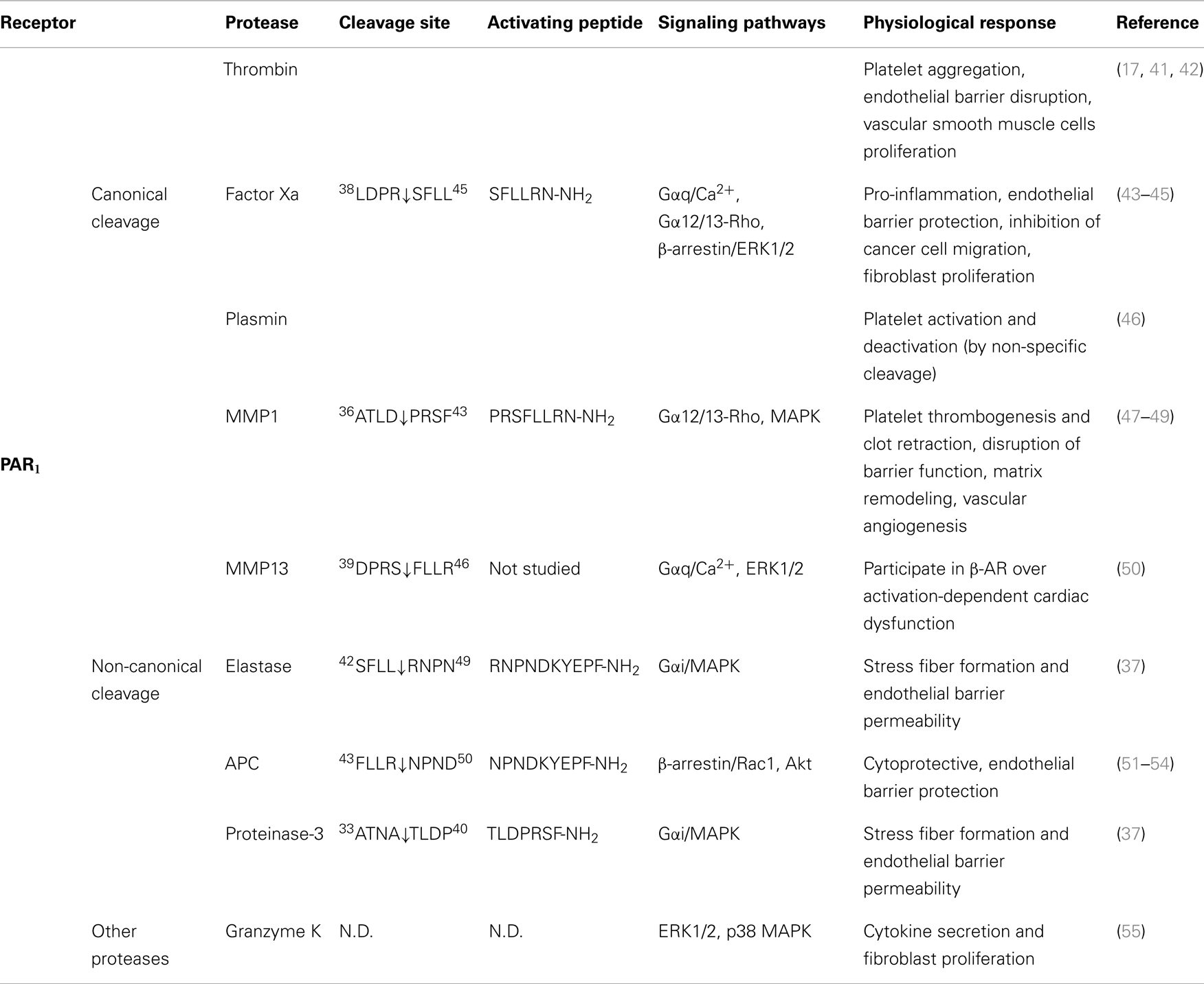
Table 1. Activation of PAR1 by different proteases, their cleavage sites, synthetic activating peptide sequence, signaling pathways, and physiological effects.
PAR1 Activation and Signaling
Canonical Activation of PAR1
As the first identified PAR, the canonical mechanisms of PAR1 activation and signaling have been extensively investigated. An interaction between thrombin’s anion-binding exosite I and a negatively charged region in the extracellular N-terminus of PAR1 (51DKYEPF56) increases the affinity of thrombin for the receptor and facilitates cleavage (17). Binding of thrombin enables the enzyme to cleave the receptor at position R41/S42, which reveals the tethered ligand domain beginning with SFLLRN in human PAR1, and initiates downstream signaling cascades (Figure 2). After cleaving PAR1, thrombin may remain associated with the receptor to facilitate its action on other thrombin receptors, such as PAR4 (83). Thrombin-activated PAR1 can trigger multiple G protein-dependent and -independent signaling pathways, including Gαq, Gαi, and Gα12/13. A region spanning the thrombin cleavage sites act as a “hot spot” for many proteases, including granzyme A, plasmin, and FXa, that cleave at the same site as thrombin and trigger similar cellular responses (Table 1). Proteases that cleave at other sites can induce biased signaling of PAR1.
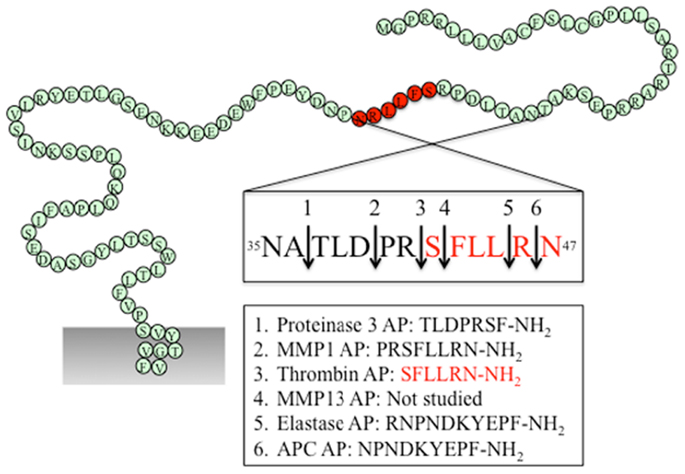
Figure 2. PAR1 N-terminus with major cleavage sites identified. N-terminus of human PAR1 (1–114). The residues in red denote the canonical tethered ligand and a corresponding AP that is revealed by thrombin cleavage. The cleavage sites for different proteases and the corresponding AP for each protease are indicated in the boxes. Gray shading represents membrane.
Biased Activation of PAR1
Several proteases have been identified that cleave PAR1 at sites different from the canonical thrombin site, leading to distinct patho-physiological outcomes.
Activated protein C
Activated protein C is a natural anticoagulant with powerful anti-inflammatory and cytoprotective activities (84). In many cases, APC exerts its protective effect via EPCR and PARs. On the surface of endothelial cells, binding of protein C to EPCR promotes its activation by thrombin, and EPCR-bound APC in turn exerts its cytoprotective effect by cleaving and activating PAR1 (51). Different from thrombin-mediated PAR1 activation, APC activation of PAR1 requires colocalization of PAR1 with EPCR in caveolae microdomains in the form of a signaling complex with caveolin-1 (85, 86). Besides subcellular localization, the differential PAR1-dependent cellular responses induced by thrombin and APC may also be explained by their distinct cleavage sites. APC cleaves PAR1 at the canonical cleavage site R41/S42, as well as at an alternate site R46/N47, with the latter being the primary cleavage site that is responsible for its cytoprotective effect (51, 52) (Figure 2). A synthetic AP corresponding to the tethered ligand that would be revealed by this alternate cleavage (N47PNDKYEPFWEDEEKNESGL66-NH2) mimics the protective effects of APC both in vitro and in vivo. Cleavage of PAR1 at R46/N47 by APC leads to β-arrestin 2-mediated Rac1-activation independent of G protein (53). Both APC and its AP stimulate PAR1-dependent phosphorylation of glycogen synthase kinase 3 β and Akt (51). In contrast to thrombin-activated PAR1, APC-cleaved PAR1 fails to activate extracellular signal-regulated kinase (ERK)1/2. Thus, APC and thrombin cleave PAR1 at different sites leading to the exposure of distinct tethered ligand agonists that activate different signaling pathways.
Matrix metalloproteinases
Matrix metalloproteinases are a family of 28 zinc-dependent proteases that play important roles in regulating platelet and endothelial function (87). Two human MMPs, MMP1 and MMP13, and one murine MMP, MMP1a, exhibit activity toward PAR1. Both MMP1 and MMP13 cleave PAR1 at non-canonical sites (D39/P40 for MMP1, S42/F43 for MMP13, Figure 2), which either generate an extended tethered ligand with two more amino acids or a truncated tethered ligand lacking the first serine residue compared to the tethered ligand exposed by thrombin (Figure 2). Similar to thrombin, MMP1-cleaved PAR1 activates the Gα12/13-Rho-GTPase pathway, and also leads to mitogen-activated protein kinase (MAPK) signaling and platelet shape changes (47). However, MMP1-activated PAR1 is a weak agonist of Ca2+ signaling and platelet aggregation (47, 48). The biased cellular response between thrombin- and MMP1-activated PAR1 has also been studied in vascular smooth muscle cells. Thrombin activation of PAR1 leads to a supercontractile, differentiated phenotype that is pertussis toxin-sensitive, suggesting the involvement of Gαi activation, whereas MMP1 activation of PAR1 results in a dedifferentiated phenotype via a Gαi-independent mechanism (49). These differences in signaling in vascular smooth muscle cells may account for the opposite effects of thrombin and MMP1 on the development of arterial stenosis following arterial injury (49).
Whereas MMP1 is mostly expressed in vascular endothelial cells, platelets, and macrophages, MMP13 is prominently expressed in cardiac fibroblasts and cardiomyocytes. Expression of MMP13 is increased in cardiac fibroblasts after β2-adrenergic receptor activation (50). MMP13 cleaves PAR1 one amino acid downstream from the thrombin site at S42/F43. In ventricular myocytes of neonatal rats, MMP13-activated PAR1 leads to phosphorylation of ERK1/2 and p38 MAPK. However, when compared to thrombin, MMP13 elicits similar levels of ERK1/2 activation but only modestly stimulates inositol phosphate formation (50). Due to the close proximity of the thrombin and MMP13 cleavage sites, it is likely that MMP13 activates PAR1 by a tethered ligand mechanism. Whether this single amino acid difference in the tethered ligands is sufficient to generate biased signaling of PAR1 remains to be determined.
Neutrophil proteases
During acute inflammation, neutrophils are the first cells infiltrate to the inflammatory site and are important mediators of inflammatory response. Elastase and proteinase-3 are stored in large quantities within secretory granules and are activated and released into the extracellular environment during inflammation (88). Recent studies show that both proteases are biased agonists for PAR1 (37). Elastase cleaves PAR1 at L45/R46, and proteinase-3 cleaves PAR1 at A36/T37 (Figure 2). Similar to thrombin, elastase and proteinase-3 activate PAR1 via tethered ligand mechanism. In contrast to thrombin-cleaved PAR1, which activates Gα12/13- as well as Gαq-mediated signaling pathways, elastase, proteinase-3 and their corresponding APs (elastase-AP: RNPNDKYEPF-NH2; proteinase-3-AP: TLDPRSF-NH2) induce Gαi-mediated MAPK activation, regardless of their distinct cleavage positions. Although proteinase-3 cleaves prior to canonical activation site (five amino acids N-terminal to the thrombin cleavage site), proteinase-3 fails to induce Ca2+ signaling, suggesting the possibility that the extra 5 residues (TLDPR) may has an inhibitory role in coupling activated PAR1 to Gαq and Ca2+ mobilization (37).
PAR1 Activation by Synthetic Ligands
Several synthetic APs corresponding to the tethered ligands exposed by proteolytic activation of PAR1 have been evaluated in vitro or in vivo. These include AP corresponding to tethered ligands revealed by thrombin (SFLLRN-NH2), neutrophil elastase (RNPNDKYEPF-NH2), neutrophil proteinase-3 (TLDPRSF-NH2), MMP1 (PRSFLLRN-NH2), and APC (NPNDKYEPF-NH2) (Figure 2). Since these proteases cleave PAR1 between residues 35 and 45, the APs share considerable homology. For example, the thrombin and MMP1 APs differ by only two amino acids, whereas the APC AP is only one amino acid shorter than the elastase-AP. However, regardless of their sequence homology, different APs display considerable signaling bias. For instance, the thrombin AP SFLLRN-NH2 activates Gαq-mediated signaling (89), whereas the MMP1 AP PRSFLLRN-NH2 is a weak agonist of Gαq signaling and preferentially activates the Gα12/13 pathway (47). In addition, elastase-AP RNPNDKYEPF-NH2 stimulates ERK1/2 phosphorylation via Gαi but the APC AP NPNDKYEPF-NH2 activates ERK by a β-arrestin-dependent but G protein-independent mechanism (37, 51).
The differences in AP-induced signals lead to distinct physiological outcomes. For example, in human umbilical vein endothelial cells, elastase-AP and APC-AP suppress thrombin-stimulated increase in endothelial barrier permeability, whereas proteinase-3-AP and MMP1-AP have the opposite effect (37).
Other structurally distinct synthetic peptides can also activate PAR1. YFLLRNP-NH2, a peptide that differs from the thrombin AP by a single amino acid, is able to cause platelet shape changes by a Ca2+-independent mechanism (90) that may involve Gα12/13-dependent activation of Rho kinase (91). TFRRRL-NH2, a peptide derived from the C-terminus of P2Y receptor, can activate PAR1 on human platelets and stimulate Gα12/13- and Gαq-dependent changes in platelet shape and aggregation (92).
In addition to providing evidence for the capacity of PARs to exhibit signaling bias, studies of APs signaling have also provided insights into the molecular mechanisms of PAR1 activation. Thus, the thrombin AP SFLLRN-NH2 is able to activate both Gαq and Gα12/13 pathways, with a preference for Gαq, whereas the MMP1 AP PRSFLLRN and YFLLRNP activate only Gα12/13 signals (47). These observations suggest the importance of the serine residue at position 1 of the peptide for Gαq activation, whereas either replacing it with another residue or extending the peptide with additional N-terminal residues both result in a reduction in Gαq activation. The precise mechanisms of how these peptides interact with cleaved PAR1 to induce these divergent signals remains to be determined.
PAR2 Activation and Signaling
Canonical Activation of PAR2
The canonical mechanism of activation of PAR2 by trypsin involves hydrolysis at position R36/S37, which reveals the tethered ligand SLIGKV (human) (24) or SLIGRL (mouse) (18) (Figure 3). This exposed tethered ligand then interacts with the second extracellular domain of the cleaved receptor and trigger multiple G protein-dependent and -independent signaling pathways (93). Trypsin-activated PAR2 leads to the activation of Gαq-mediated Ca2+ mobilization (94), Gαs-dependent formation of cAMP (95), Gα12/13-mediated increasing in Rho-Kinase activity (95), recruitment of β-arrestin-1 and -2 (96), ERK1/2 phosphorylation (97, 98), and subsequent receptor internalization and degradation (94, 99).
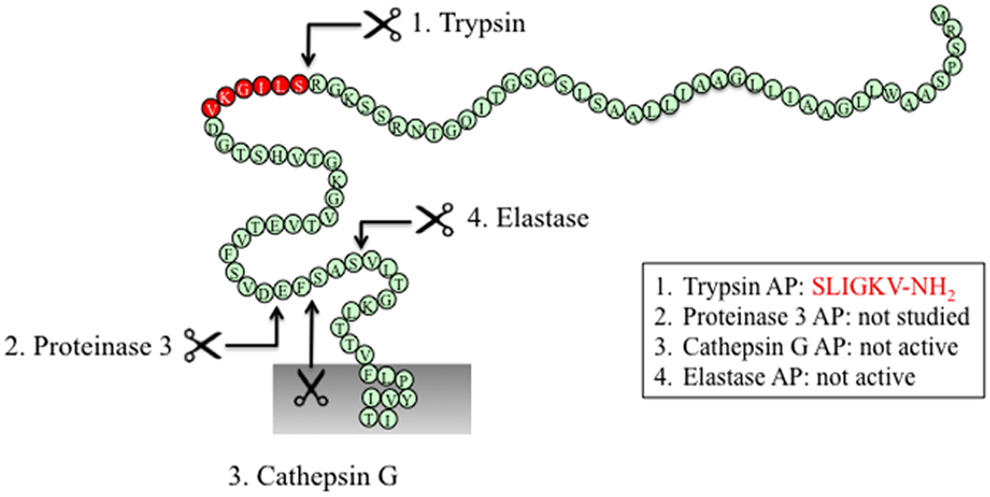
Figure 3. PAR2 N-terminus with major cleavage sites identified. N-terminus of human PAR2 (1–84). The residues in red denote the canonical tethered ligand and a corresponding AP that is revealed by trypsin cleavage. The corresponding APs for each protease are indicated in the boxes. Gray shading represents membrane.
Several other proteases cleave PAR2 at the canonical site (Table 2). Serine proteases that activate canonical PAR2 signaling include trypsin I/II (18, 94), trypsin IV (100, 101), tryptase (59, 102), coagulation factors VIIa and Xa (26), acrosin (103), granzyme A (104), and kallikrein 2, 4, 6, and 14 (63, 64, 105). Proteases that cleave PAR2 at the canonical activation site would be expected to reveal the conserved tethered ligand domain and to activate the same compliment of signaling pathways as trypsin. Despite the fact that these proteases cleave PAR2 at the same site as trypsin, the potency with which they activate PAR2 shows considerable variability. This variability may be due to different rate and efficiency of cleavage between different proteases. Although not all the Kcat values for proteases cleaving PAR2 have been reported, published in vitro peptide proteolytic assays show marked differences in the kinetics of cleavage. Cleavage at sites that disable the receptor may also contribute to the variable potency of proteases-mediated signaling. Compared to trypsin, tryptase is a partial agonist of PAR2-dependent Ca2+ signaling, which may be related to a second cleavage site at R41/S42, which would deactivates the receptor and limit its potential to induce further Ca2+ signals (102). Post-translational modification of PARs may also affect their susceptibility to proteolytic activation. Although mast cell tryptase can activate PAR2, its ability to do so is limited by receptor glycosylation, which presumably sterically hinders hydrolysis at R36/S37 (106). Other proteases such as kallikrein-related peptidase 14 and gingipain-R have been shown to signal by PAR2-dependent mechanism, although the cleavage sites need to be confirmed (70, 71).
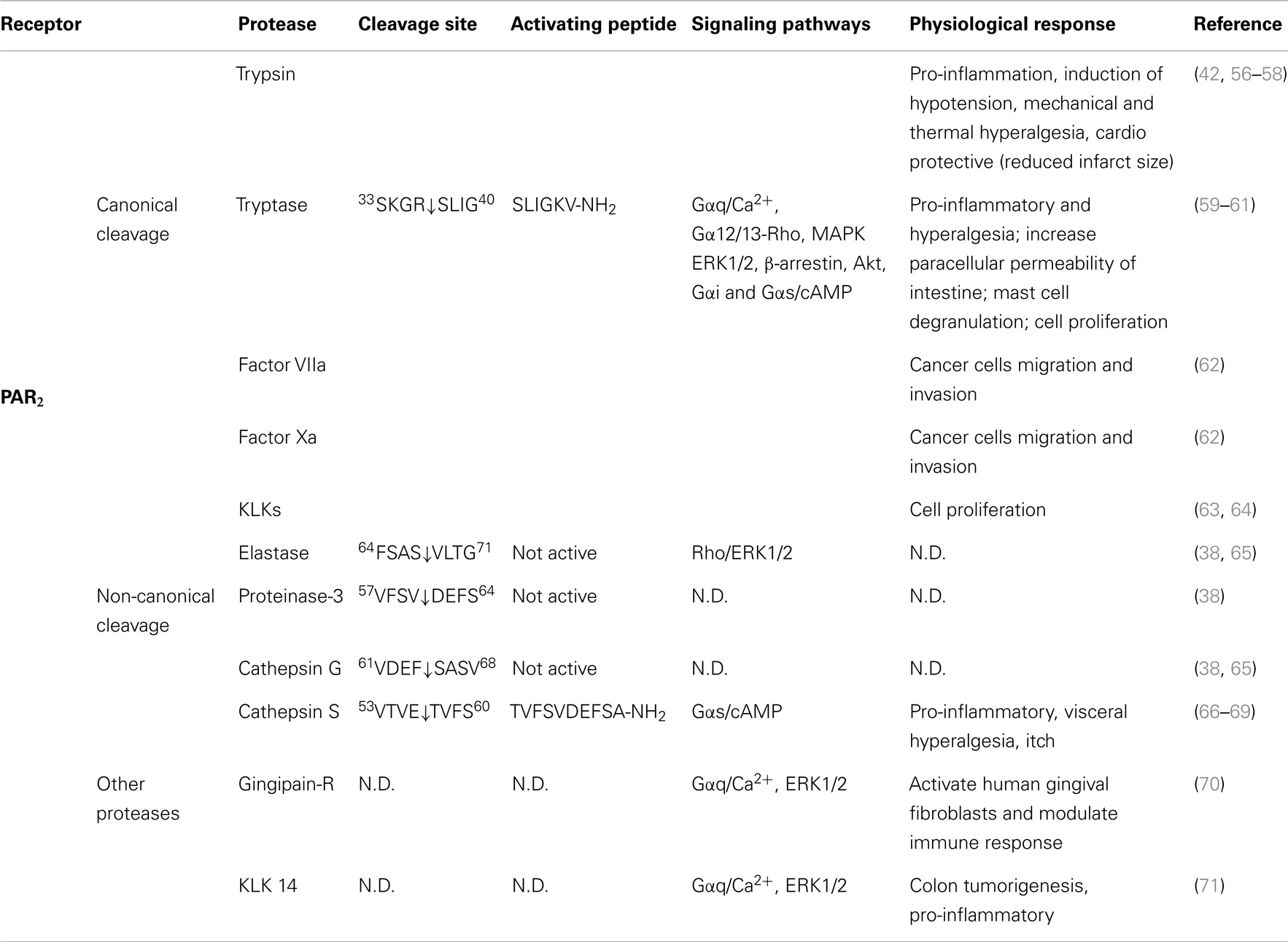
Table 2. Activation of PAR2 by different proteases, their cleavage sites, synthetic activating peptide sequence, signaling pathways, and physiological effects.
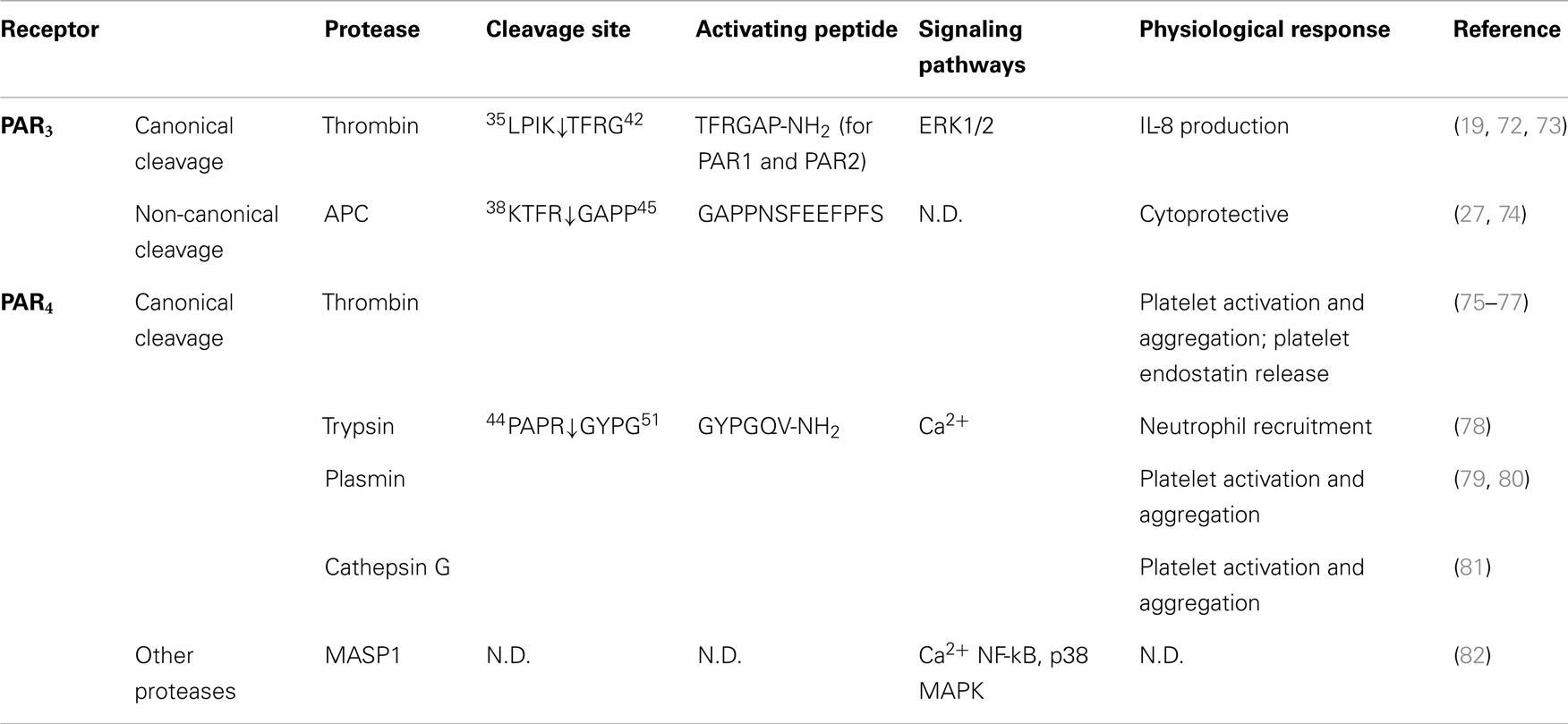
Table 3. Activation of PAR3 and PAR4 by different proteases, their cleavage sites, synthetic activating peptide sequence, signaling pathways, and physiological effects.
Biased Activation of PAR2
As is the case for PAR1, several proteases have been identified that cleave PAR2 at distinct sites, leading to signaling bias.
Neutrophil proteases
Neutrophil elastase, proteinase-3, and cathepsin G can all cleave PAR2. Neutrophil elastase and cathepsin G were first considered to be deactivating proteases due to their ability to cleave downstream from the canonical trypsin site and thereby disarm PAR2 and attenuate trypsin-dependent Ca2+ signals (104). However, a recent study suggests that these cleavage events may also induce PAR2 signaling bias.
Elastase cleaves PAR2 between S68/V69 (38) (Figure 3). Treatment of KNRK-PAR2 cells with elastase does not induce Ca2+ signals but does trigger PAR2-dependent ERK phosphorylation by a pathway that involves Gα12/13-mediated activation of Rho kinase. In contrast to trypsin, elastase does not trigger β-arrestin recruitment or receptor internalization. A synthetic peptide corresponding to a tethered ligand domain that would be revealed by elastase (VLTGKLTTVFL-NH2) fails to mimic the action of elastase and to activate ERK or to stimulate Ca2+ signals in KNRK-PAR2 cells, suggesting that elastase could activate PAR2 by a mechanism that does not require tethered ligand binding to the cleaved receptor. Indeed, the elastase cleavage site is close to the first transmembrane domain of PAR2, which would suggest that a tethered ligand mechanism is unlikely. Presumably, cleavage per se may allow PAR2 to adopt an active conformation that favors activation of certain signaling pathways. Although the functional relevance of elastase activation of PAR2 is uncertain, it may contribute to inflammatory diseases in which this protease-receptor pair is involved. For example, neutrophil elastase activity is elevated in patients with ulcerative colitis as well as dextran sulfate sodium-induced colitis in mice (107), and the elastase inhibitor serpin B1 (107) and PAR2 antagonism and deletion protect against colitis (108). Therefore, it will be important to determine whether elastase triggers PAR2-dependent inflammatory signaling in colonocytes or immune cells that express PAR2.
Similar to elastase, neutrophil cathepsin G and proteinase-3 both cleave PAR2 downstream of the canonical trypsin site (cathepsin G: P65/S66 and proteinase-3 V62/D63) (38) (Figure 3). However, neither cathepsin G nor proteinase-3 stimulate PAR2-dependent Ca2+ signals or activate ERK phosphorylation or receptor internalization (38). Although these proteases can disarm PAR2 by removing the trypsin-exposed tethered ligand, it remains to be determined whether they also induce biased signaling or they act as antagonists for the receptor. The functional relevance of PAR2 cleavage by cathepsin G and proteinase-3 is unknown.
Cathepsin S
Cathepsin S, a lysosome cysteine protease of the papain family, is expressed by antigen-presenting cells, including macrophages, microglial cells, B-lymphocytes, and dendritic cells, and contributes to antigen presentation and adaptive immunity (109). Inflammatory mediators promote cathepsin S secretion from macrophages and microglial cells (66, 110), and there is increased cathepsin S activity in inflamed tissues, including synovial fluid from patients with rheumatoid arthritis (111) and colonic secretions from mice with colitis (67). Cathepsin S is active at both lysosomal acidic pH and extracellular pH (66), and may thus be able to activate PARs. When administered into the colonic lumen of wild-type mice to replicate the increased luminal cathepsin S detected in mice with colitis, cathepsin S causes visceral pain (67). This hyperalgesia is absent from PAR2 knockout mice, suggesting that cathepsin S can activate PAR2. Current studies are investigating whether cathepsin S can activate biased PAR2 signaling and to determine the functional relevance of this process for PAR2-mediated inflammation and pain.
PAR2 Activation by Synthetic Ligands
In addition to proteases, synthetic peptides that mimic the trypsin-exposed tethered ligand can also activate PAR2. The hexapeptides SLIGRL-NH2 and SLIGKV-NH2 corresponding respectively to the mouse and human trypsin-revealed tethered ligands for PAR2 have been extensively used as tools to study PAR2 function despite their relatively low potency. Both peptides have been shown to induce a Gαq-dependent increase in [Ca2+]i (26, 32, 94, 104, 112–114), ERK1/2 activation (62, 98, 115), as well as β-arrestin recruitment and subsequent internalization (98, 112). The effect of SLIGRL-NH2 on cellular cAMP levels is controversial. In rabbit smooth muscle cells, SLIGRL-NH2 decreases forskolin-induced accumulation of cAMP in a pertussis toxin-sensitive manner (114). On the other hand, in HEK293 cells and human keratinocytes, SLIGRL-NH2 increases cAMP formation (95, 116). In keratinocytes, SLIGRL-NH2 stimulates a pertussis toxin-insensitive and cAMP/PKA-independent activation of Rho kinase (95). Thus, activated PAR2 may trigger different G protein pathways depending on the cellular context and the availability of other components of the signaling complex.
Although these APs can cause robust PAR2 signaling, mutagenesis studies have highlighted that different residues within the activating ligand domain may determine the preference of the receptor to activate certain signaling pathways such as Ca2+ versus MAPK. These studies not only support the idea that PAR2 can initiate biased signaling, they also provide important information on the mechanisms of PAR2 activation. Mutagenesis of rat PAR2 has revealed that the first two residues (S37L38) from the AP are critical for PAR2 activation. Both mutated PAR2 receptor with substitution of these two residues to alanine and its corresponding soluble peptide ligand (AAIGRL-NH2) exhibit little or no activity (117). Notably, a subsequent study found that another analog peptide of SLIGRL-NH2, SLAAAA-NH2, also showed minimum Ca2+ signaling activity but was able to induce ERK1/2 phosphorylation via a Rho-kinase-dependent mechanism (32). Although the PAR2 binding pocket for APs has yet to be fully identified, the importance of extracellular loop 2 in PAR2 activation by soluble peptides has been studied, leading to the suggestion that the glutamic acid residues (E232E233 in rat PAR2, and E232Q233 in human PAR2) may interact with the basic arginine residue at position 5 of the AP. This residue is required for activity since mutation of either receptor or the peptide results in loss of Ca2+ signal (118). It will be important to examine whether distinct or overlapping clusters of residues of PAR2 are responsible for binding to different regions of the AP, thereby triggering different signaling events.
PAR2 Activation by Small Molecules
Recent advances in our understanding of the structure-activity relationships of various PAR2 ligands have facilitated the development of small molecule PAR2 agonists, albeit of limited potencies. The potential for these compounds to exhibit signaling bias has not been fully investigated, since most studies have only examined their ability to affect PAR2-dependent Ca2+ signals. AC-98170 is a partial agonist of PAR2-dependent Ca2+ signaling (30% efficacy of SLIGRL-NH2), but with a lower EC50 than AC-55541, another small molecule agonist for PAR2 (119). Whether these compounds are biased agonists of different PAR2 signaling pathways remains to be investigated.
Small molecule PAR2 antagonists have also been developed. One such compound, GB88, is a competitive antagonist for both trypsin- and AP-induced Ca2+ signaling, although GB88 can selectivity activate other PAR2-dependent pathways, including cAMP formation, Rho-kinase stimulation, and ERK1/2 phosphorylation (120). Thus, rather than acting as an antagonist for PAR2, GB88 may act as a biased agonist for this receptor. However, the antagonistic activity of GB88 is sufficient to attenuate PAR2-induced paw edema and acute inflammation, as well as collagen-induced arthritis in rats (121, 122), suggesting that relative contribution of PAR2 in these disease conditions might be primary via Ca2+-dependent pathways.
Like GB88, K-14585 also has a complex pharmacology. In human skin epithelial cells, K-14585 is able to block SLIGKV-NH2-induced inositol phosphate accumulation and p38 MAPK phosphorylation without affecting PAR2-mediated ERK1/2 activation (123). However, although not significant, K-14585 alone does induce a modest IP3 formation. Moreover, at a higher concentration, K-14585 triggers PAR2-dependent p38 MAP kinase phosphorylation (123). Taken together, K-14585 may actually act as a PAR2 agonist with relatively low potency.
PAR3 Activation and Signaling
Although thrombin cleaves PAR3 at K38/T39, there is little evidence that the cleaved receptor is capable of signaling. Thrombin and a synthetic peptide corresponding to the putative tethered ligand failed to generate PAR3-dependent Ca2+ signals (19). However, this PAR3-derived AP is able to activate PAR1 and PAR2 (72). Instead of signaling in its own right, PAR3 appears to be a co-factor for the activation of other PARs, including PAR4 and PAR1 (27, 29, 30). Co-expression of PAR3 with PAR4 in COS-7 cells leads to over 10-fold increase in the efficiency of thrombin cleaving PAR4 compare to PAR4 expressed alone (EC50 from 0.3 to 0.05 nM) (29). However, with the appreciation of PAR biased signaling, further studies are warranted to examine the full repertoire of potential PAR3 signals.
In contrast to other PARs, fewer proteases have been identified that can cleave PAR3. Besides thrombin, APC is the only protease to be identified that exhibits proteolytic activity toward PAR3 (27, 74). In immortalized human and mouse podocytes, which have higher expression of PAR2 and PAR3 than PAR1 and PAR4, the maximum inhibitory effect of APC-dependent podocyte apoptosis requires cleavage of N-terminal domain of PAR3 by APC at the same position as thrombin (27). Cleavage of PAR3 by APC promotes dimerization between PAR2 and PAR3, and this is required for APC-dependent cytoprotective effect. A recent study reported a novel mechanism of APC activation of PAR3 via a cleavage at a non-canonical site (R41/G42 X instead of K38/T39) (74). Peptide hydrolysis experiments revealed a slow kinetics of APC cleavage of PAR3 (50% peptide cleavage reached after ~5 h), suggesting that other modulators may facilitate APC cleaving PAR3 in cells; indeed, the efficiency of this cleavage increased proportional to the expression of EPCR (74). This novel cleavage by APC generates a new tethered ligand domain starting with G42APPNS. Consistent with a tethered ligand activating mechanism, APC-AP (G42APPNSFEEFPFS54-NH2) is able to prevent thrombin-induced endothelial barrier disruption. Interestingly, an extended peptide generated from thrombin cleavage site (40TFGAPPNSFEEFPFS54-NH2) fails to do so. This suggests that APC and thrombin activation of PAR3 mediates different signaling profile, and thus is involved in different cellular response, respectively. However, similar to thrombin activation of PAR3, existence of another receptor, such as PAR1 is necessary for APC/PAR3-mediated signaling (74).
PAR4 Activation and Signaling
PAR4 was identified by a homology search using amino acid query sequence derived from known sequences of PAR1, PAR2, and PAR3 (20). The putative protease cleavage site was identified (R47/G48), and the EC50 of thrombin toward PAR4 was much higher compare to other thrombin sensitive receptors PAR1 and PAR3 (5 nM for PAR4, and 0.2 nM for PAR1 and PAR3) (17, 19, 20). Further investigation suggests that this may be due to the lack of the thrombin binding site within the amino terminus of the receptor. Consistent with this observation, γ-thrombin, another isoform of thrombin that lacks a receptor binding site exhibits similar affinity on PAR4 compare to α-thrombin (20). Different from other PARs that can be cleaved preferentially by trypsin or thrombin, PAR4 exhibits similar sensitivity toward both enzymes. Both thrombin and trypsin activities toward PAR4 can be abolished by R47/A mutation of the receptor, suggesting that both enzymes cleave the receptor at the same site (20).
Compared to PAR1 and PAR2, little is known about biased signaling of PAR4, and to date no additional activating cleavage sites have been identified. However, although proteases such as plasmin can activate PAR4 by cleaving the receptor at the canonical site, thrombin and plasmin cleave PAR4 with different kinetics, possibility due to different mechanisms of action or the affinity of the proteases for the receptor (20). In mouse platelets, PAR3 binds to thrombin and thereby acts as a co-factor to facilitate thrombin cleavage and activation of PAR4 (29). However, in both transfected cell lines as well as platelet, instead of acting as a co-factor, the presence of PAR3 inhibits plasmin-mediated PAR4 activation leading to a decrease in intracellular Ca2+ mobilization and platelet aggregation (79). The mechanism that underlies these findings is not clear. However, since thrombin cleaves both PAR3 and PAR4 whereas plasmin can only cleave PAR4, the conformation of the PAR3–PAR4 receptor pair might be different after plasmin or thrombin cleavage. In addition, difference in binding affinities and kinetics of plasmin and thrombin for PAR4 may also result in distinct receptor-protease complex formation and lead to variation in downstream responses.
Cathepsin G is a neutrophil serine protease that plays an important role in inflammation. Cathepsin G can evoke PAR4-dependent Ca2+ signals in human platelets and in PAR4-transfected fibroblasts (81). Cathepsin G and PAR4 are upregulated in ulcerative colitis patients, and inhibition of cathepsin G and PAR4, but not PAR1 or PAR2, is protective (124). Although no direct evidence suggests that PAR4 is the target for cathepsin G in colitis, this study highlights the possibility of targeting cathepsin G or PAR4 as novel therapeutic approach.
Recently, mannose-binding lectin-associated serine protease-1 has been shown to cleave PAR4 but not PAR1 or PAR2 in endothelial cells, and to induce PAR4-dependent Ca2+ responses and activation of NF-κB and p38 MAPK pathways. However, the exact cleavage site remains to be determined (82).
Signaling by PAR Dimers
Although most studies have examined signaling by monomeric PARs, considerable evidence suggests that PARs may form homo- or hetero-dimers and function as a complex. Dimerized PARs could adopt unique conformations and activate different signaling pathways compare to monomer (125).
As a receptor that exhibits little or no activity when expressed alone, PAR3 has been examined as a co-factor for other PARs. PAR3 can modulate the activity of PAR1 by potentiating its response to thrombin, thereby increasing endothelial barrier permeability without altering Ca2+ responses (30). This receptor dimer pair favors coupling to Gα13 over Gαq, whereas both pathways are similarly activated by PAR1 monomer (30). Thus, PAR1 may exhibit distinct signaling profiles in response to the same ligand when coupled to PAR3.
In mouse platelets, dimerization between PAR3 and PAR4 leads to negative regulation of PAR4-mediated Ca2+ mobilization and PKC activation without affecting Gα12/13 and Gαi activation, suggesting that PAR4 signaling is biased away from Gαq activation when coupled to PAR3 (126).
In human podocytes, APC cleavage of PAR3 leads to the formation of PAR2 and PAR3 heterodimers, which is essential for the anti-apoptotic actions of APC (127). Although the signaling pathways that regulate this activity remain to be defined, the observation that both PAR2 and PAR3 activating peptides were able to produce similar effects suggest that the formation of this hetero-dimer may stimulate signaling pathways that are similar to those activated by PAR2 monomers.
Another example of the contribution of receptor dimerization to biased PAR signaling is dimerization between PAR1 and PAR2. PAR1–PAR2 dimerization has been demonstrated in both overexpression system and endogenous expression system (128, 129). When PAR1 forms dimer with PAR2, the thrombin-revealed PAR1 tethered ligand can trans-activate PAR2 and trigger PAR2-dependent Gαi/Rac signaling, while PAR1-mediated Gαq and Gα12/13 signaling is switched off (130). In addition, recruitment of β-arrestin to the PAR1–PAR2 dimer exhibits distinct kinetic compared to each protomer, suggesting a potential alteration in β-arrestin-dependent ERK1/2 signaling (128). During the early development of sepsis, the effect of thrombin is vascular disruption whereas at the later phase of sepsis, with increasing expression of PAR2, thrombin induces a vascular protective effect that is mediated by PAR2/Rac1 activation (130).
Protease-activated receptors may act as co-factors or may dimerize either constitutively or in a ligand-dependent manner. The formation of dimers may play a role in organization of receptors at the cell surface, and may allosterically modulate the activation of either monomer or act as a complex that generates unique signaling outcomes.
Regulation of PAR Signaling by Different G Protein Coupling
Different proteases can lead to biased PARs activation, and PAR signaling can also be modulated by co-factors and receptor dimerization. In addition, the interaction of PARs with different G proteins also has marked influence on the outcome of protease signals. In response to a single protease, PARs are able to couple to multiple G proteins. Although how this occur is not clear, recent studies using bioluminescence resonance energy transfer (BRET) approach suggest dynamic regulation of PAR1 and PAR2 coupling with multiple G proteins. In Cos-7 cells, both receptors spontaneously form pre-assembled complexes with Gαi, whereas they only couple to Gα12 following ligand stimulation, and with slow kinetics (131, 132). Further investigation revealed the existence of two different PAR populations that are responsible for coupling to different Gα protein. The existence of distinct receptor populations may be interpreted as receptor clustering in different membrane microdomains such as membrane raft and caveolin-containing vesicles (131). GPCRs located in lipid raft-enriched domains may assure certain conformations of the receptor that preferentially couple to specific G proteins compared to receptor located in non-raft membrane compartment (133). It would be of great interest to examine whether divergent PARs signaling in various cell types depends on the lipid component of the membrane and whether altering membrane lipid composition leads to a shift in PAR signaling profile, thereby contributing to bias signaling. This mechanism may also account for the tissue specificity of PAR signaling.
Termination of PAR Biased Signaling
A growing number of proteases have been identified that can cleave PARs at distinct sites, leading to diverse signals. One striking observation is the capacity of different proteases to stimulate receptor endocytosis. It is well established that cleavage of PARs at canonical activation site leads to rapid receptor internalization in a β-arrestin-dependent (PAR2) or -independent (PAR1) manner (96, 134). Receptor endocytosis not only contributes to signaling, but is also the first step in the degradation of activated PARs, which irrevocably terminates their ability to signal. Thus, if cleaved PARs remain at the cell surface, how is signaling regulated?
Phosphorylation
Most information about the regulation of PARs has been derived from studies of the canonical mechanisms of PAR activation. After cleavage by thrombin or trypsin, both PAR1 and PAR2 are rapidly phosphorylated by protein kinases, including GRKs, and second messenger kinases such as PKA and PKC (135–137). Phosphorylation within the C-terminal tail of the receptor serves as the primary mechanism to shut down G protein coupling and signaling. PAR1 is phosphorylated by GRK3 in Xenopus laevis oocytes and GRK5 in human endothelial cells (135, 137). Although the specific GRKs that phosphorylate PAR2 in native systems have not been identified, it is clear that PAR2 activation by trypsin recruits multiple GRKs in overexpression system (138). In terms of selectivity of certain GRKs over others, in endothelial cells GRK5 is the critical isoform that mediates thrombin-induced desensitization of PAR1 (137). GRK5 overexpression inhibits thrombin-induced Ca2+ signaling whereas GRK3 and GRK6 have no such effect (137). Whether proteases that cleave PARs at other sites also induce GRK recruitment and differential receptor phosphorylation remains to be determined. However, given the inability of many of these alternatively cleaved receptors to recruit arrestins, alterations in receptor phosphorylation are likely to occur. Studies of other GPCRs, such as β-adrenergic receptors (139) and opioid receptors (140), suggest that receptor phosphorylation occurs in an agonist-selective manner. For example, different biased agonists for β-adrenergic receptor trigger distinct patterns of receptor phosphorylation by different GRKs, which may establish a “barcode” that determines β-arrestin recruitment and functional responses (139). Whether this is also the case for PARs remains to be explored.
Phosphorylation of GPCRs often leads to β-arrestin recruitment and receptor internalization. However, this is not always the case. The morphine-activated μ-opioid receptor is phosphorylated by GRK5 at Ser375, which is sufficient for receptor desensitization but not β-arrestin recruitment or receptor internalization. On the other hand, DAMGO, another agonist for the same receptor, leads to phosphorylation at both Ser375 and Thr370, which leads to both receptor desensitization and internalization (141). Thus, biased proteases signaling might be terminated by phosphorylation without necessary internalization. It will be of interest to determine whether different proteases induce specific patterns of receptor phosphorylation, and to determine the functional relevance of these events.
Intracellular Trafficking and Signaling
β-Arrestins not only act as chaperone proteins that direct receptor trafficking, but also are active participants of signaling by internalized receptors. β-arrestins mediate multiple steps of PAR signaling, including PAR1-mediated Akt activation, and PAR1 and PAR2-dependent ERK1/2 activation (96). After activation by trypsin, PAR2 stably couples to β-arrestin and together they co-translocate to early endosomes where they generate a second wave of intracellular signals (96). As an important scaffolding protein, β-arrestin is essential for the formation of the signaling complex including PAR2-Raf1 and activated ERK. This complex will ensure the appropriate subcellular localization of PAR2-mediated ERK activity. Thus, the stability between activated receptor and β-arrestin is essential for determining the duration of this activation. It has been pointed out that PAR2 may induce distinct cellular response from Gαq pathway via a β-arrestin-mediated mechanism (96, 142, 143). However, proteases such as elastase and cathepsin S are unable to induce β-arrestin recruitment, suggesting their lack of ability to further promote β-arrestin-dependent signals. In contrast to PAR2, where both β-arrestins have similar effects, PAR1-mediated Akt signaling is differentially mediated by different β-arrestins, depending on the mechanism of proteolytic activation. For example, β-arrestin 1 is required for rapid activation of Akt induced by thrombin, whereas APC cleavage leads to β-arrestin 2-dependent Akt activation (53, 134, 144). Although the underlying mechanism is not established, it may relate to different receptor conformations.
Besides desensitization, receptor trafficking to different subcellular compartments also plays an important part in regulation of GPCR signaling. For both PAR1 and PAR2, activation by trypsin or thrombin leads to receptor trafficking to endosomes, followed by lysosome sorting and receptor down-regulation. (145). However, many proteases such as elastase and APC failed to induce PARs endocytosis. The regulatory machinery for biased protease-signaling remains unknown. The potential involvement of compartmentalization and redistribution of the receptors to membrane subdomains seems to be an attractive area to explore. As mentioned earlier, APC-induced PAR1 signals require localization of the signalosome to caveolae, a specific lipid rich plasma membrane microdomain. Caveolae has also been suggested to be involved in TF-mediated PAR2 signaling. In breast carcinoma cells, both TF and PAR2 are observed co-localized in cholesterol-rich caveolae, and depletion or sequestration of plasma membrane cholesterol significantly impaired TF-VII1 induced cell signaling (146). It would be interesting to see if different proteases prefer targeting PARs at certain membrane microdomains or there is PARs redistribution upon activation by different proteases.
Translational Relevance of Protease-Biased Signaling of PARs
The contribution of PARs to important patho-physiological processes, including hemostasis, inflammation, pain, and proliferation, has been extensively investigated through studies of PAR-deficient mice and by use of proteases and synthetic agonists/antagonists of the canonical signaling pathways (21, 22). The capacity of certain proteases to acts as biased PAR agonists or even antagonists adds further complexity to this system, and the relevance of protease-biased signals to complex patho-physiological processes is far from clear. A major difficulty relates to the identities of the proteases that activate PARs under physiological conditions and during disease. Since proteases are regulated through post-translational control of activity (e.g., by zymogen processing and endogenous inhibitors), studies should include assessment of enzymatic activity rather than gene or protein expression. A major advance in this regard is the use of activity-based probes that covalently interact with activated proteases, allowing their localization by whole animal or cellular imaging and identification by proteomic approaches (99). This approach has been used to detect activated cathepsin S in macrophages of tumors and the inflamed colon, as well as in spinal microglial cells during colitis (67, 147). However, the use of probes is likely to reveal that multiple proteases become activated during physiological and pathological events, many of which could activate or disarm PARs. Additional information can be provided by studies of protease knockout mice or through use of selective inhibitors. However, a detailed understanding of the importance of biased signaling of PARs would probably require genetic or pharmacological strategies to selectively disrupt particular biased pathways, and such tools are currently lacking.
Although the patho-physiological importance of biased signaling of PARs is not fully understood, biased agonism could explain certain paradoxes about the patho-physiological contribution of PARs. For example, PARs can have both pro-inflammatory and anti-inflammatory roles, which may depend on the animal models, species, tissues, or the protease that drive the response. In an ovalbumin-induced model of allergic inflammation of the mouse airway, PAR2 deletion is protective, suggesting that PAR2 contributes to the development of immunity and to allergic inflammation of the airway (148). However, in a lipopolysaccharide-induced pulmonary neutrophilia model, PAR2 shows a protective effect (149, 150). The underlying mechanism of these observed differences is unclear. However, differences in the repertoire of proteases that are activated in acute versus chronic inflammation, leading to distinct mechanisms of PAR signaling, could be one explanation. Lipopolysaccharide-induced pulmonary neutrophilia is an acute inflammation characterized by influx of neutrophils and activation of elastase and proteinases 3, biased agonists of PAR1 and PAR2. On the other hand, ovalbumin-induced inflammation is characterized by infiltration of eosinophil and macrophages, leading to activation of distinct proteases (151). Thus, the predominant active proteases for PAR2 may be different in these two models, which could potentially activate different signaling pathways that lead to opposite responses. The contrasting pro-inflammatory and cytoprotective actions of thrombin and APC, respectively, may also be attributed to PAR1 biased signaling. In this instance, the relative concentration of the proteases as well as the occupancy of EPCR by its ligand are critical in determining the PAR1 signaling pathways (28).
Conclusion and Future Directions
Considerable progress has been made in defining the mechanisms by which proteases and synthetic agonists activate PARs. Proteases, peptides, and small molecules have been identified that can activate PARs by distinct mechanisms, leading to the stimulation of divergent pathways of receptor signaling and trafficking. The information derived from these studies has provided insights into the signaling pathways that are responsible for certain patho-physiological processes.
However, there are many unanswered questions about biased signaling of PARs. The ability of a PAR cleaved by different proteases or bound to various synthetic agonists to differentially signal probably arises from distinct receptor conformations. However, the structures of PARs in these different stabilized states remain to be determined. Although some proteases activate PARs by exposure of a tethered ligand, this is not always the case and the mechanism by which proteolysis per se can activate signaling is unknown. There is tantalizing evidence to suggest that biased signaling may underlie contrasting patho-physiological consequences of PAR activation, depending on the available proteases and the nature of PAR signaling. However, the proteases that are responsible for PAR activation in particular cell types in different conditions remain to be identified, and the signaling pathways that give rise to particular patho-physiological outcomes are not fully defined. Finally, very little is known about the mechanisms that regulate protease-biased signaling of PARs, particularly by those proteases that fail to promote the recruitment of β-arrestins and endocytosis of the activated receptors.
Whether protease-biased signaling of PARs can be exploited therapeutically remains an open question. The development of receptor antagonists or agonists that target disease-relevant PAR signaling pathways without affecting beneficial signaling events could provide a route for enhanced selectivity, with fewer on target side-effects. Future challenges will be to identify the primary pathways that mediate PAR-dependent physiological and patho-physiological events, and to develop receptor agonists and antagonists that selectively target these pathways. A deeper understanding of the mechanisms of initiation, regulation, and termination of protease-signaling will have profound implication in developing therapeutics for many critical conditions, including sepsis, thrombosis, inflammation, and pain processes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported by NHMRC 63303, 1049682, 1031886, and Monash University (Nigel W. Bunnett).
Abbreviations
AP, activating peptide; APC, activated protein C; EPCR, endothelial protein C receptor; ERK, extracellular signal-regulated kinase; F, coagulation factor; GPCR, G protein-coupled receptor; GRK, G protein-coupled receptor kinase; MAPK, mitogen-activated protein kinase; MMP, matrix metalloproteinase; PAR, protease-activated receptor; TF, tissue factor.
References
1. Wittinghofer A, Vetter IR. Structure-function relationships of the G domain, a canonical switch motif. Biochemistry (2011) 80:943–71. doi: 10.1146/annurev-biochem-062708-134043
2. Zhao P, Cladman W, Van Tol HHM, Chidiac P. Fine-tuning of GPCR signals by intracellular G protein modulators. Prog Mol Biol Transl Sci (2013) 115:421–53. doi:10.1016/B978-0-12-394587-7.00010-5
3. Jacoby E, Bouhelal R, Gerspacher M, Seuwen K. The 7 TM G-protein-coupled receptor target family. ChemMedChem (2006) 1(8):760–82. doi:10.1002/cmdc.200600134
4. McNeely PM, Naranjo AN, Robinson AS. Structure-function studies with G protein-coupled receptors as a paradigm for improving drug discovery and development of therapeutics. Biotechnol J (2012) 7(12):1451–61. doi:10.1002/biot.201200076
5. Neves SR, Ram PT, Iyengar R. G protein pathways. Science (2002) 296(5573):1636–9. doi:10.1126/science.1071550
6. Moore CAC, Milano SK, Benovic JL. Regulation of receptor trafficking by GRKs and arrestins. Annu Rev Physiol (2007) 69:451–82. doi:10.1146/annurev.physiol.69.022405.154712
7. Marchese A, Paing MM, Temple BRS, Trejo J. G protein-coupled receptor sorting to endosomes and lysosomes. Annu Rev Pharmacol Toxicol (2008) 48:601–29. doi:10.1146/annurev.pharmtox.48.113006.094646
8. Ferguson SS, Zhang J, Barak LS, Caron MG. Molecular mechanisms of G protein-coupled receptor desensitization and resensitization. Life Sci (1998) 62(17–18):1561–5. doi:10.1016/S0024-3205(98)00107-6
9. Pradhan AA, Smith ML, Kieffer BL, Evans CJ. Ligand-directed signalling within the opioid receptor family. Br J Pharmacol (2012) 167(5):960–9. doi:10.1111/j.1476-5381.2012.02075.x
10. Kenakin T. Functional selectivity and biased receptor signaling. J Pharmacol Exp Ther (2011) 336(2):296–302. doi:10.1124/jpet.110.173948
11. Wisler JW, Xiao K, Thomsen AR, Lefkowitz RJ. Recent developments in biased agonism. Curr Opin Cell Biol (2014) 27C:18–24. doi:10.1016/j.ceb.2013.10.008
12. Pradhan AAA, Walwyn W, Nozaki C, Filliol D, Erbs E, Matifas A, et al. Ligand-directed trafficking of the δ-opioid receptor in vivo: two paths toward analgesic tolerance. J Neurosci (2010) 30(49):16459–68. doi:10.1523/JNEUROSCI.3748-10.2010
13. Godin CM, Ferguson SSG. Biased agonism of the angiotensin II type 1 receptor. Mini Rev Med Chem (2012) 12(9):812–6. doi:10.2174/138955712800959134
14. Emery AC, DiRaddo JO, Miller E, Hathaway HA, Pshenichkin S, Takoudjou GR, et al. Ligand bias at metabotropic glutamate 1a receptors: molecular determinants that distinguish β-arrestin-mediated from G protein-mediated signaling. Mol Pharmacol (2012) 82(2):291–301. doi:10.1124/mol.112.078444
15. Kobilka BK, Deupi X. Conformational complexity of G-protein-coupled receptors. Trends Pharmacol Sci (2007) 28(8):397–406. doi:10.1016/j.tips.2007.06.003
16. Rosenbaum DM, Rasmussen SGF, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature (2009) 459(7245):356–63. doi:10.1038/nature08144
17. Vu TK, Hung DT, Wheaton VI, Coughlin SR. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell (1991) 64(6):1057–68. doi:10.1016/0092-8674(91)90261-V
18. Nystedt S, Emilsson K, Wahlestedt C, Sundelin J. Molecular cloning of a potential proteinase activated receptor. Proc Natl Acad Sci U S A (1994) 91(20):9208–12. doi:10.1073/pnas.91.20.9208
19. Ishihara H, Connolly AJ, Zeng D, Kahn ML, Zheng YW, Timmons C, et al. Protease-activated receptor 3 is a second thrombin receptor in humans. Nature (1997) 386(6624):502–6. doi:10.1038/386502a0
20. Xu WF, Andersen H, Whitmore TE, Presnell SR, Yee DP, Ching A, et al. Cloning and characterization of human protease-activated receptor 4. Proc Natl Acad Sci U S A (1998) 95(12):6642–6. doi:10.1073/pnas.95.12.6642
21. Adams MN, Ramachandran R, Yau M-K, Suen JY, Fairlie DP, Hollenberg MD, et al. Structure, function and pathophysiology of protease activated receptors. Pharmacol Ther (2011) 130(3):248–82. doi:10.1016/j.pharmthera.2011.01.003
22. Ossovskaya VSV, Bunnett NWN. Protease-activated receptors: contribution to physiology and disease. Physiol Rev (2004) 84(2):579–621. doi:10.1152/physrev.00028.2003
23. Soh UJK, Dores MR, Chen B, Trejo J. Signal transduction by protease-activated receptors. Br J Pharmacol (2010) 160(2):191–203. doi:10.1111/j.1476-5381.2010.00705.x
24. Nystedt S, Emilsson K, Larsson AK, Strömbeck B, Sundelin J. Molecular cloning and functional expression of the gene encoding the human proteinase-activated receptor 2. Eur J Biochem (1995) 232(1):84–9. doi:10.1111/j.1432-1033.1995.tb20784.x
25. Riewald M, Ruf W. Science review: role of coagulation protease cascades in sepsis. Crit Care (2003) 7(2):123–9. doi:10.1186/cc2012
26. Camerer E, Huang W, Coughlin SR. Tissue factor- and factor X-dependent activation of protease-activated receptor 2 by factor VIIa. Proc Natl Acad Sci U S A (2000) 97(10):5255–60. doi:10.1073/pnas.97.10.5255
27. Madhusudhan T, Wang H, Straub BK, Gröne E, Zhou Q, Shahzad K, et al. Cytoprotective signaling by activated protein C requires protease-activated receptor-3 in podocytes. Blood (2012) 119(3):874–83. doi:10.1182/blood-2011-07-365973
28. Rezaie AR. The occupancy of endothelial protein C receptor by its ligand modulates the par-1 dependent signaling specificity of coagulation proteases. IUBMB Life (2011) 63(6):390–6. doi:10.1002/iub.447
29. Nakanishi-Matsui M, Zheng YW, Sulciner DJ, Weiss EJ, Ludeman MJ, Coughlin SR. PAR3 is a cofactor for PAR4 activation by thrombin. Nature (2000) 404(6778):609–13. doi:10.1038/35007085
30. McLaughlin JN, Patterson MM, Malik AB. Protease-activated receptor-3 (PAR3) regulates PAR1 signaling by receptor dimerization. Proc Natl Acad Sci U S A (2007) 104(13):5662–7. doi:10.1073/pnas.0700763104
31. Kim YV, Di Cello F, Hillaire CS, Kim KS. Differential Ca2+ signaling by thrombin and protease-activated receptor-1-activating peptide in human brain microvascular endothelial cells. Am J Physiol Cell Physiol (2004) 286(1):C31–42. doi:10.1152/ajpcell.00157.2003
32. Ramachandran R, Mihara K, Mathur M, Rochdi MD, Bouvier M, DeFea K, et al. Agonist-biased signaling via proteinase activated receptor-2: differential activation of calcium and mitogen-activated protein kinase pathways. Mol Pharmacol (2009) 76(4):791–801. doi:10.1124/mol.109.055509
33. Pham CT. Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol (2006) 6(7):541–50. doi:10.1038/nri1841
34. Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis (2010) 30(3):245–57. doi:10.1055/s-0030-1255354
35. Chin AC, Lee WY, Nusrat A, Vergnolle N, Parkos CA. Neutrophil-mediated activation of epithelial protease-activated receptors-1 and -2 regulates barrier function and transepithelial migration. J Immunol (2008) 181(8):5702–10. doi:10.4049/jimmunol.181.8.5702
36. Pettersen CA, Adler KB. Airways inflammation and COPD: epithelial-neutrophil interactions. Chest (2002) 121(5 Suppl):142S–50S. doi:10.1378/chest.121.5_suppl.142S
37. Mihara K, Ramachandran R, Renaux B, Saifeddine M, Hollenberg MD. Neutrophil elastase and proteinase-3 trigger G protein-biased signaling through proteinase-activated receptor-1 (PAR1). J Biol Chem (2013) 288(46):32979–90. doi:10.1074/jbc.M113.483123
38. Ramachandran R, Mihara K, Chung H, Renaux B, Lau CS, Muruve DA, et al. Neutrophil elastase acts as a biased agonist for proteinase-activated receptor-2 (PAR2). J Biol Chem (2011) 286(28):24638–48. doi:10.1074/jbc.M110.201988
39. Hiemstra PS. Novel roles of protease inhibitors in infection and inflammation. Biochem Soc Trans (2002) 30(2):116–20. doi:10.1042/BST0300116
40. Ku DD, Dai J. Expression of thrombin receptors in human atherosclerotic coronary arteries leads to an exaggerated vasoconstrictory response in vitro. J Cardiovasc Pharmacol (1997) 30(5):649–57. doi:10.1097/00005344-199711000-00016
41. Dorsam RT, Kim S, Jin J, Kunapuli SP. Coordinated signaling through both G12/13 and G(i) pathways is sufficient to activate GPIIb/IIIa in human platelets. J Biol Chem (2002) 277(49):47588–95. doi:10.1074/jbc.M208778200
42. van Nieuw Amerongen GP, van Delft S, Vermeer MA, Collard JG, van Hinsbergh VW. Activation of RhoA by thrombin in endothelial hyperpermeability: role of Rho kinase and protein tyrosine kinases. Circ Res (2000) 87(4):335–40. doi:10.1161/01.RES.87.4.335
43. Blanc-Brude OP, Archer F, Leoni P, Derian C, Bolsover S, Laurent GJ, et al. Factor Xa stimulates fibroblast procollagen production, proliferation, and calcium signaling via PAR1 activation. Exp Cell Res (2005) 304(1):16–27. doi:10.1016/j.yexcr.2004.10.021
44. Feistritzer C, Lenta R, Riewald M. Protease-activated receptors-1 and -2 can mediate endothelial barrier protection: role in factor Xa signaling. J Thromb Haemost (2005) 3(12):2798–805. doi:10.1111/j.1538-7836.2005.01610.x
45. Schuepbach RA, Riewald M. Coagulation factor Xa cleaves protease-activated receptor-1 and mediates signaling dependent on binding to the endothelial protein C receptor. J Thromb Haemost (2010) 8(2):379–88. doi:10.1111/j.1538-7836.2009.03682.x
46. Kuliopulos A, Covic L, Seeley SK, Sheridan PJ, Helin J, Costello CE. Plasmin desensitization of the PAR1 thrombin receptor: kinetics, sites of truncation, and implications for thrombolytic therapy. Biochemistry (1999) 38(14):4572–85. doi:10.1021/bi9824792
47. Trivedi V, Boire A, Tchernychev B, Kaneider NC, Leger AJ, Callaghan K, et al. Platelet matrix metalloprotease-1 mediates thrombogenesis by activating PAR1 at a cryptic ligand site. Cell (2009) 137(2):332–43. doi:10.1016/j.cell.2009.02.018
48. Tressel SL, Kaneider NC, Kasuda S, Foley C, Koukos G, Austin K, et al. A matrix metalloprotease-PAR1 system regulates vascular integrity, systemic inflammation and death in sepsis. EMBO Mol Med (2011) 3(7):370–84. doi:10.1002/emmm.201100145
49. Austin KM, Nguyen N, Javid G, Covic L, Kuliopulos A. Noncanonical matrix metalloprotease-1-protease-activated receptor-1 signaling triggers vascular smooth muscle cell dedifferentiation and arterial stenosis. J Biol Chem (2013) 288(32):23105–15. doi:10.1074/jbc.M113.467019
50. Jaffré F, Friedman AE, Hu Z, Mackman N, Blaxall BC. β-adrenergic receptor stimulation transactivates protease-activated receptor 1 via matrix metalloproteinase 13 in cardiac cells. Circulation (2012) 125(24):2993–3003. doi:10.1161/CIRCULATIONAHA.111.066787
51. Mosnier LOL, Sinha RKR, Burnier LL, Bouwens EAE, Griffin JHJ. Biased agonism of protease-activated receptor 1 by activated protein C caused by noncanonical cleavage at Arg46. Blood (2012) 120(26):5237–46. doi:10.1182/blood-2012-08-452169
52. Schuepbach RA, Madon J, Ender M, Galli P, Riewald M. Protease-activated receptor-1 cleaved at R46 mediates cytoprotective effects. J Thromb Haemost (2012) 10(8):1675–84. doi:10.1111/j.1538-7836.2012.04825.x
53. Soh UJK, Trejo J. Activated protein C promotes protease-activated receptor-1 cytoprotective signaling through β-arrestin and dishevelled-2 scaffolds. Proc Natl Acad Sci U S A (2011) 108(50):E1372–80. doi:10.1073/pnas.1112482108
54. Schuepbach RA, Feistritzer C, Fernandez JA, Griffin JH, Riewald M. Protection of vascular barrier integrity by activated protein C in murine models depends on protease-activated receptor-1. Thromb Haemost (2009) 101(4):724–33. doi:10.1160/TH08-10-0632
55. Cooper DM, Pechkovsky DV, Hackett TL, Knight DA, Granville DJ. Granzyme K activates protease-activated receptor-1. PLoS One (2011) 6(6):e21484. doi:10.1371/journal.pone.0021484
56. Meyer-Hoffert U, Rogalski C, Seifert S, Schmeling G, Wingertszahn J, Proksch E, et al. Trypsin induces epidermal proliferation and inflammation in murine skin. Exp Dermatol (2004) 13(4):234–41. doi:10.1111/j.0906-6705.2004.00159.x
57. Miike S, McWilliam AS, Kita H. Trypsin induces activation and inflammatory mediator release from human eosinophils through protease-activated receptor-2. J Immunol (2001) 167(11):6615–22. doi:10.4049/jimmunol.167.11.6615
58. Cicala C, Pinto A, Bucci M, Sorrentino R, Walker B, Harriot P, et al. Protease-activated receptor-2 involvement in hypotension in normal and endotoxemic rats in vivo. Circulation (1999) 99(19):2590–7. doi:10.1161/01.CIR.99.19.2590
59. Corvera CU, Dery O, McConalogue K, Bohm SK, Khitin LM, Caughey GH, et al. Mast cell tryptase regulates rat colonic myocytes through proteinase-activated receptor 2. J Clin Invest (1997) 100(6):1383–93. doi:10.1172/JCI119658
60. Berger P, Perng DW, Thabrew H, Compton SJ, Cairns JA, McEuen AR, et al. Tryptase and agonists of PAR-2 induce the proliferation of human airway smooth muscle cells. J Appl Physiol (2001) 91(3):1372–9.
61. Vergnolle N, Bunnett NW, Sharkey KA, Brussee V, Compton SJ, Grady EF, et al. Proteinase-activated receptor-2 and hyperalgesia: a novel pain pathway. Nat Med (2001) 7(7):821–6. doi:10.1038/89945
62. Morris DR, Ding Y, Ricks TK, Gullapalli A, Wolfe BL, Trejo J. Protease-activated receptor-2 is essential for factor VIIa and Xa-induced signaling, migration, and invasion of breast cancer cells. Cancer Res (2006) 66(1):307–14. doi:10.1158/0008-5472.CAN-05-1735
63. Ramsay AJ, Dong Y, Hunt ML, Linn M, Samaratunga H, Clements JA, et al. Kallikrein-related peptidase 4 (KLK4) initiates intracellular signaling via protease-activated receptors (PARs). KLK4 and PAR-2 are co-expressed during prostate cancer progression. J Biol Chem (2008) 283(18):12293–304. doi:10.1074/jbc.M709493200
64. Stefansson K, Brattsand M, Roosterman D, Kempkes C, Bocheva G, Steinhoff M, et al. Activation of proteinase-activated receptor-2 by human kallikrein-related peptidases. J Invest Dermatol (2008) 128(1):18–25. doi:10.1038/sj.jid.5700965
65. Dulon S, Candé C, Bunnett NW, Hollenberg MD, Chignard M, Pidard D. Proteinase-activated receptor-2 and human lung epithelial cells: disarming by neutrophil serine proteinases. Am J Respir Cell Mol Biol (2003) 28(3):339–46. doi:10.1165/rcmb.4908
66. Liuzzo JP, Petanceska SS, Moscatelli D, Devi LA. Inflammatory mediators regulate cathepsin S in macrophages and microglia: a role in attenuating heparan sulfate interactions. Mol Med (1999) 5(5):320–33.
67. Cattaruzza F, Lyo V, Jones E, Pham D, Hawkins J, Kirkwood K, et al. Cathepsin S is activated during colitis and causes visceral hyperalgesia by a PAR2-dependent mechanism in mice. Gastroenterology (2011) 141(5):e1–3. doi:10.1053/j.gastro.2011.07.035
68. Clark AK, Grist J, Al-Kashi A, Perretti M, Malcangio M. Spinal cathepsin S and fractalkine contribute to chronic pain in the collagen-induced arthritis model. Arthritis Rheum (2012) 64(6):2038–47. doi:10.1002/art.34351
69. Reddy VB, Shimada SG, Sikand P, Lamotte RH, Lerner EA. Cathepsin S elicits itch and signals via protease-activated receptors. J Invest Dermatol (2010) 130(5):1468–70. doi:10.1038/jid.2009.430
70. Lourbakos A, Chinni C, Thompson P, Potempa J, Travis J, Mackie EJ, et al. Cleavage and activation of proteinase-activated receptor-2 on human neutrophils by gingipain-R from Porphyromonas gingivalis. FEBS Lett (1998) 435(1):45–8. doi:10.1016/S0014-5793(98)01036-9
71. Gratio V, Loriot C, Virca GD, Oikonomopoulou K, Walker F, Diamandis EP, et al. Kallikrein-related peptidase 14 acts on proteinase-activated receptor 2 to induce signaling pathway in colon cancer cells. Am J Pathol (2011) 179(5):2625–36. doi:10.1016/j.ajpath.2011.07.016
72. Hansen KK, Saifeddine M, Hollenberg MD. Tethered ligand-derived peptides of proteinase-activated receptor 3 (PAR3) activate PAR1 and PAR2 in Jurkat T cells. Immunology (2004) 112(2):183–90. doi:10.1111/j.1365-2567.2004.01870.x
73. Ostrowska E, Reiser G. The protease-activated receptor-3 (PAR-3) can signal autonomously to induce interleukin-8 release. Cell Mol Life Sci (2008) 65(6):970–81. doi:10.1007/s00018-008-7555-y
74. Burnier L, Mosnier LO. Novel mechanisms for activated protein C cytoprotective activities involving noncanonical activation of protease-activated receptor 3. Blood (2013) 122(5):807–16. doi:10.1182/blood-2013-03-488957
75. Faruqi TR, Weiss EJ, Shapiro MJ, Huang W, Coughlin SR. Structure-function analysis of protease-activated receptor 4 tethered ligand peptides. Determinants of specificity and utility in assays of receptor function. J Biol Chem (2000) 275(26):19728–34. doi:10.1074/jbc.M909960199
76. Ma L, Hollenberg MD, Wallace JL. Thrombin-induced platelet endostatin release is blocked by a proteinase activated receptor-4 (PAR4) antagonist. Br J Pharmacol (2001) 134(4):701–4. doi:10.1038/sj.bjp.0704312
77. Hollenberg MD, Saifeddine M. Proteinase-activated receptor 4 (PAR4): activation and inhibition of rat platelet aggregation by PAR4-derived peptides. Can J Physiol Pharmacol (2001) 79(5):439–42. doi:10.1139/y01-013
78. Gomides LF, Duarte ID, Ferreira RG, Perez AC, Francischi JN, Klein A. Proteinase-activated receptor-4 plays a major role in the recruitment of neutrophils induced by trypsin or carrageenan during pleurisy in mice. Pharmacology (2012) 89(5–6):275–82. doi:10.1159/000337378
79. Mao Y, Jin J, Daniel JL, Kunapuli SP. Regulation of plasmin-induced protease-activated receptor 4 activation in platelets. Platelets (2009) 20(3):191–8. doi:10.1080/09537100902803635
80. Quinton TM, Kim S, Derian CK, Jin J, Kunapuli SP. Plasmin-mediated activation of platelets occurs by cleavage of protease-activated receptor 4. J Biol Chem (2004) 279(18):18434–9. doi:10.1074/jbc.M401431200
81. Sambrano GR, Huang W, Faruqi T, Mahrus S, Craik C, Coughlin SR. Cathepsin G activates protease-activated receptor-4 in human platelets. J Biol Chem (2000) 275(10):6819–23. doi:10.1074/jbc.275.10.6819
82. Megyeri M, Makó V, Beinrohr L, Doleschall Z, Prohászka Z, Cervenak L, et al. Complement protease MASP-1 activates human endothelial cells: PAR4 activation is a link between complement and endothelial function. Mol Immunol (2009) 46(14):2. doi:10.4049/jimmunol.0900879
83. Kahn ML, Zheng YW, Huang W, Bigornia V, Zeng D, Moff S, et al. A dual thrombin receptor system for platelet activation. Nature (1998) 394(6694):690–4. doi:10.1038/29325
84. Neyrinck AP, Liu KD, Howard JP, Matthay MA. Protective mechanisms of activated protein C in severe inflammatory disorders. Br J Pharmacol (2009) 158(4):1034–47. doi:10.1111/j.1476-5381.2009.00251.x
85. Bae JS, Yang L, Rezaie AR. Receptors of the protein C activation and activated protein C signaling pathways are colocalized in lipid rafts of endothelial cells. Proc Natl Acad Sci U S A (2007) 104(8):2867–72. doi:10.1073/pnas.0611493104
86. Russo A, Soh UJK, Trejo J. Proteases display biased agonism at protease-activated receptors: location matters! Mol Interv (2009) 9(2):87–96. doi:10.1124/mi.9.2.8
87. Siefert SA, Sarkar R. Matrix metalloproteinases in vascular physiology and disease. Vascular (2012) 20(4):210–6. doi:10.1258/vasc.2011.201202
88. Korkmaz B, Horwitz MS, Jenne DE, Gauthier F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol Rev (2010) 62(4):726–59. doi:10.1124/pr.110.002733
89. McLaughlin JN, Shen L, Holinstat M, Brooks JD, DiBenedetto E, Hamm HE. Functional selectivity of G protein signaling by agonist peptides and thrombin for the protease-activated receptor-1. J Biol Chem (2005) 280(26):25048–59. doi:10.1074/jbc.M414090200
90. Rasmussen UB, Gachet C, Schlesinger Y, Hanau D, Ohlmann P, Van Obberghen-Schilling E, et al. A peptide ligand of the human thrombin receptor antagonizes alpha-thrombin and partially activates platelets. J Biol Chem (1993) 268(19):14322–8.
91. Bauer M, Retzer M, Wilde JI, Maschberger P, Essler M, Aepfelbacher M, et al. Dichotomous regulation of myosin phosphorylation and shape change by Rho-kinase and calcium in intact human platelets. Blood (1999) 94(5):1665–72.
92. Mao Y, Jin J, Kunapuli SP. Characterization of a new peptide agonist of the protease-activated receptor-1. Biochem Pharmacol (2008) 75(2):438–47. doi:10.1016/j.bcp.2007.09.002
93. Al-Ani B, Saifeddine M, Kawabata A, Hollenberg MD. Proteinase activated receptor 2: role of extracellular loop 2 for ligand-mediated activation. Br J Pharmacol (1999) 128(5):1105–13. doi:10.1038/sj.bjp.0702834
94. Böhm SKS, Khitin LML, Grady EFE, Aponte GG, Payan DGD, Bunnett NWN. Mechanisms of desensitization and resensitization of proteinase-activated receptor-2. J Biol Chem (1996) 271(36):22003–16. doi:10.1074/jbc.271.36.22003
95. Scott G, Leopardi S, Parker L, Babiarz L, Seiberg M, Han R. The proteinase-activated receptor-2 mediates phagocytosis in a Rho-dependent manner in human keratinocytes. J Invest Dermatol (2003) 121(3):529–41. doi:10.1046/j.1523-1747.2003.12427.x
96. DeFea KA, Zalevsky J, Thoma MS, Déry O, Mullins RD, Bunnett NW. beta-arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J Cell Biol (2000) 148(6):1267–81. doi:10.1083/jcb.148.6.1267
97. Kanke T, Macfarlane SR, Seatter MJ, Davenport E, Paul A, McKenzie RC, et al. Proteinase-activated receptor-2-mediated activation of stress-activated protein kinases and inhibitory kappa B kinases in NCTC 2544 keratinocytes. J Biol Chem (2001) 276(34):31657–66. doi:10.1074/jbc.M100377200
98. Stalheim L, Ding Y, Gullapalli A, Paing MM, Wolfe BL, Morris DR, et al. Multiple independent functions of arrestins in the regulation of protease-activated receptor-2 signaling and trafficking. Mol Pharmacol (2005) 67(1):78–87. doi:10.1124/mol.104.006072
99. Sadaghiani AM, Verhelst SH, Bogyo M. Tagging and detection strategies for activity-based proteomics. Curr Opin Chem Biol (2007) 11(1):20–8. doi:10.1016/j.cbpa.2006.11.030
100. Cottrell GS, Amadesi S, Grady EF, Bunnett NW. Trypsin IV, a novel agonist of protease-activated receptors 2 and 4. J Biol Chem (2004) 279(14):13532–9. doi:10.1074/jbc.M312090200
101. Knecht W, Cottrell GS, Amadesi S, Mohlin J, Skaregarde A, Gedda K, et al. Trypsin IV or mesotrypsin and p23 cleave protease-activated receptors 1 and 2 to induce inflammation and hyperalgesia. J Biol Chem (2007) 282(36):26089–100. doi:10.1074/jbc.M703840200
102. Molino M, Barnathan ES, Numerof R, Clark J, Dreyer M, Cumashi A, et al. Interactions of mast cell tryptase with thrombin receptors and PAR-2. J Biol Chem (1997) 272(7):4043–9. doi:10.1074/jbc.272.7.4043
103. Smith R, Jenkins A, Lourbakos A, Thompson P, Ramakrishnan V, Tomlinson J, et al. Evidence for the activation of PAR-2 by the sperm protease, acrosin: expression of the receptor on oocytes. FEBS Lett (2000) 484(3):285–90. doi:10.1016/S0014-5793(00)02146-3
104. Dulon S, Leduc D, Cottrell GS, D’Alayer J, Hansen KK, Bunnett NW, et al. Pseudomonas aeruginosa elastase disables proteinase-activated receptor 2 in respiratory epithelial cells. Am J Respir Cell Mol Biol (2005) 32(5):411–9. doi:10.1165/rcmb.2004-0274OC
105. Oikonomopoulou K, Hansen KK, Saifeddine M, Vergnolle N, Tea I, Blaber M, et al. Kallikrein-mediated cell signalling: targeting proteinase-activated receptors (PARs). Biol Chem (2006) 387(6):817–24. doi:10.1515/BC.2006.104
106. Compton SJ, Renaux B, Wijesuriya SJ, Hollenberg MD. Glycosylation and the activation of proteinase-activated receptor 2 (PAR(2)) by human mast cell tryptase. Br J Pharmacol (2001) 134(4):705–18. doi:10.1038/sj.bjp.0704303
107. Morohoshi Y, Matsuoka K, Chinen H, Kamada N, Sato T, Hisamatsu T, et al. Inhibition of neutrophil elastase prevents the development of murine dextran sulfate sodium-induced colitis. J Gastroenterol (2006) 41(4):318–24. doi:10.1007/s00535-005-1768-8
108. Lohman RJ, Cotterell AJ, Suen J, Liu L, Do AT, Vesey DA, et al. Antagonism of protease-activated receptor 2 protects against experimental colitis. J Pharmacol Exp Ther (2012) 340(2):256–65. doi:10.1124/jpet.111.187062
109. Clark AK, Malcangio M. Microglial signalling mechanisms: Cathepsin S and Fractalkine. Exp Neurol (2012) 234(2):283–92. doi:10.1016/j.expneurol.2011.09.012
110. Clark AK, Wodarski R, Guida F, Sasso O, Malcangio M. Cathepsin S release from primary cultured microglia is regulated by the P2X7 receptor. Glia (2010) 58(14):1710–26. doi:10.1002/glia.21042
111. Pozgan U, Caglic D, Rozman B, Nagase H, Turk V, Turk B. Expression and activity profiling of selected cysteine cathepsins and matrix metalloproteinases in synovial fluids from patients with rheumatoid arthritis and osteoarthritis. Biol Chem (2010) 391(5):571–9. doi:10.1515/BC.2010.035
112. Gardell LR, Ma J-N, Seitzberg JG, Knapp AE, Schiffer HH, Tabatabaei A, et al. Identification and characterization of novel small-molecule protease-activated receptor 2 agonists. J Pharmacol Exp Ther (2008) 327(3):799–808. doi:10.1124/jpet.108.142570
113. McGuire JJ, Saifeddine M, Triggle CR, Sun K, Hollenberg MD. 2-furoyl-LIGRLO-amide: a potent and selective proteinase-activated receptor 2 agonist. J Pharmacol Exp Ther (2004) 309(3):1124–31. doi:10.1124/jpet.103.064584
114. Sriwai W, Mahavadi S, Al-Shboul O, Grider JR, Murthy KS. Distinctive G protein-dependent signaling by protease-activated receptor 2 (PAR2) in smooth muscle: feedback inhibition of RhoA by cAMP-independent PKA. PLoS One (2013) 8(6):e66743. doi:10.1371/journal.pone.0066743
115. Tanaka Y, Sekiguchi F, Hong H, Kawabata A. PAR2 triggers IL-8 release via MEK/ERK and PI3-kinase/Akt pathways in GI epithelial cells. Biochem Biophys Res Commun (2008) 377(2):622–6. doi:10.1016/j.bbrc.2008.10.018
116. Amadesi S, Cottrell GS, Divino L, Chapman K, Grady EF, Bautista F, et al. Protease-activated receptor 2 sensitizes TRPV1 by protein kinase Cepsilon- and A-dependent mechanisms in rats and mice. J Physiol (2006) 575(Pt 2):555–71. doi:10.1113/jphysiol.2006.111534
117. Al-Ani B, Hansen KK, Hollenberg MD. Proteinase-activated receptor-2: key role of amino-terminal dipeptide residues of the tethered ligand for receptor activation. Mol Pharmacol (2004) 65(1):149–56. doi:10.1124/mol.65.1.149
118. Al-Ani B, Wijesuriya SJ, Hollenberg MD. Proteinase-activated receptor 2: differential activation of the receptor by tethered ligand and soluble peptide analogs. J Pharmacol Exp Ther (2002) 302(3):1046–54. doi:10.1124/jpet.302.3.1046
119. Seitzberg JG, Knapp AE, Lund BW, Mandrup Bertozzi S, Currier EA, Ma J-N, et al. Discovery of potent and selective small-molecule PAR-2 agonists. J Med Chem (2008) 51(18):5490–3. doi:10.1021/jm800754r
120. Yau MK, Liu L, Fairlie DP. Toward drugs for protease-activated receptor 2 (PAR2). J Med Chem (2013) 56(19):7477–97. doi:10.1021/jm400638v
121. Lohman RJ, Cotterell AJ, Barry GD, Liu L, Suen JY, Vesey DA, et al. An antagonist of human protease activated receptor-2 attenuates PAR2 signaling, macrophage activation, mast cell degranulation, and collagen-induced arthritis in rats. FASEB J (2012) 26(7):2877–87. doi:10.1096/fj.11-201004
122. Suen JY, Barry GD, Lohman RJ, Halili MA, Cotterell AJ, Le GT, et al. Modulating human proteinase activated receptor 2 with a novel antagonist (GB88) and agonist (GB110). Br J Pharmacol (2012) 165(5):1413–23. doi:10.1111/j.1476-5381.2011.01610.x
123. Goh FG, Ng PY, Nilsson M, Kanke T, Plevin R. Dual effect of the novel peptide antagonist K-14585 on proteinase-activated receptor-2-mediated signalling. Br J Pharmacol (2009) 158(7):1695–704. doi:10.1111/j.1476-5381.2009.00415.x
124. Dabek M, Ferrier L, Roka R, Gecse K, Annahazi A, Moreau J, et al. Luminal cathepsin g and protease-activated receptor 4: a duet involved in alterations of the colonic epithelial barrier in ulcerative colitis. Am J Pathol (2009) 175(1):207–14. doi:10.2353/ajpath.2009.080986
125. Lin H, Liu AP, Smith TH, Trejo J. Cofactoring and dimerization of proteinase-activated receptors. Pharmacol Rev (2013) 65(4):1198–213. doi:10.1124/pr.111.004747
126. Arachiche A, de la Fuente M, Nieman MT. Calcium mobilization and protein kinase C activation downstream of protease activated receptor 4 (PAR4) is negatively regulated by PAR3 in mouse platelets. PLoS One (2013) 8(2):e55740. doi:10.1371/journal.pone.0055740
127. Madhusudhan T, Wang H, Straub BK, Grone E, Zhou Q, Shahzad K, et al. Cytoprotective signaling by activated protein C requires protease-activated receptor-3 in podocytes. Blood (2012) 119(3):874–83. doi:10.1182/blood-2011-07-365973
128. Lin H, Trejo J. Transactivation of the PAR1-PAR2 heterodimer by thrombin elicits β-arrestin-mediated endosomal signaling. J Biol Chem (2013) 288(16):11203–15. doi:10.1074/jbc.M112.439950
129. Sevigny LM, Zhang P, Bohm A, Lazarides K, Perides G, Covic L, et al. Interdicting protease-activated receptor-2-driven inflammation with cell-penetrating pepducins. Proc Natl Acad Sci U S A (2011) 108(20):8491–6. doi:10.1073/pnas.1017091108
130. Kaneider NC, Leger AJ, Agarwal A, Nguyen N, Perides G, Derian C, et al. ‘Role reversal’ for the receptor PAR1 in sepsis-induced vascular damage. Nat Immunol (2007) 8(12):1303–12. doi:10.1038/ni1525
131. Ayoub MA, Trinquet E, Pfleger KD, Pin JP. Differential association modes of the thrombin receptor PAR1 with Galphai1, Galpha12, and beta-arrestin 1. FASEB J (2010) 24(9):3522–35. doi:10.1096/fj.10-154997
132. Ayoub MA, Pin JP. Interaction of protease-activated receptor 2 with G proteins and beta-arrestin 1 studied by bioluminescence resonance energy transfer. Front Endocrinol (2013) 4:196. doi:10.3389/fendo.2013.00196
133. Patel HH, Murray F, Insel PA. G-protein-coupled receptor-signaling components in membrane raft and caveolae microdomains. Handb Exp Pharmacol (2008) 186:167–84. doi:10.1007/978-3-540-72843-6_7
134. Paing MM, Stutts AB, Kohout TA, Lefkowitz RJ, Trejo J. beta -Arrestins regulate protease-activated receptor-1 desensitization but not internalization or Down-regulation. J Biol Chem (2002) 277(2):1292–300. doi:10.1074/jbc.M109160200
135. Ishii K, Chen J, Ishii M, Koch WJ, Freedman NJ, Lefkowitz RJ, et al. Inhibition of thrombin receptor signaling by a G-protein coupled receptor kinase. Functional specificity among G-protein coupled receptor kinases. J Biol Chem (1994) 269(2):1125–30.
136. Ricks TK, Trejo J. Phosphorylation of protease-activated receptor-2 differentially regulates desensitization and internalization. J Biol Chem (2009) 284(49):34444–57. doi:10.1074/jbc.M109.048942
137. Tiruppathi C, Yan W, Sandoval R, Naqvi T, Pronin AN, Benovic JL, et al. G protein-coupled receptor kinase-5 regulates thrombin-activated signaling in endothelial cells. Proc Natl Acad Sci U S A (2000) 97(13):7440–5. doi:10.1073/pnas.97.13.7440
138. Jensen DD, Godfrey CB, Niklas C, Canals M, Kocan M, Poole DP, et al. The bile acid receptor TGR5 does not interact with β-arrestins or traffic to endosomes but transmits sustained signals from plasma membrane rafts. J Biol Chem (2013) 288(32):22942–60. doi:10.1074/jbc.M113.455774
139. Nobles KN, Xiao K, Ahn S, Shukla AK, Lam CM, Rajagopal S, et al. Distinct phosphorylation sites on the beta(2)-adrenergic receptor establish a barcode that encodes differential functions of beta-arrestin. Sci Signal (2011) 4(185):ra51. doi:10.1126/scisignal.2001707
140. Johnson EA, Oldfield S, Braksator E, Gonzalez-Cuello A, Couch D, Hall KJ, et al. Agonist-selective mechanisms of mu-opioid receptor desensitization in human embryonic kidney 293 cells. Mol Pharmacol (2006) 70(2):676–85. doi:10.1124/mol.106.022376
141. Doll C, Poll F, Peuker K, Loktev A, Gluck L, Schulz S. Deciphering micro-opioid receptor phosphorylation and dephosphorylation in HEK293 cells. Br J Pharmacol (2012) 167(6):1259–70. doi:10.1111/j.1476-5381.2012.02080.x
142. Ge L, Ly Y, Hollenberg M, DeFea K. A beta-arrestin-dependent scaffold is associated with prolonged MAPK activation in pseudopodia during protease-activated receptor-2-induced chemotaxis. J Biol Chem (2003) 278(36):34418–26. doi:10.1074/jbc.M300573200
143. Zoudilova M, Kumar P, Ge L, Wang P, Bokoch GM, DeFea KA. Beta-arrestin-dependent regulation of the cofilin pathway downstream of protease-activated receptor-2. J Biol Chem (2007) 282(28):20634–46. doi:10.1074/jbc.M701391200
144. Goel R, Baldassare JJ. beta-Arrestin 1 couples thrombin to the rapid activation of the Akt pathway. Ann N Y Acad Sci (2002) 973:138–41. doi:10.1111/j.1749-6632.2002.tb04622.x
145. Trejo J. Protease-activated receptors: new concepts in regulation of G protein-coupled receptor signaling and trafficking. J Pharmacol Exp Ther (2003) 307(2):437–42. doi:10.1124/jpet.103.052100
146. Awasthi V, Mandal SK, Papanna V, Rao LV, Pendurthi UR. Modulation of tissue factor-factor VIIa signaling by lipid rafts and caveolae. Arterioscler Thromb Vasc Biol (2007) 27(6):1447–55. doi:10.1161/ATVBAHA.107.143438
147. Verdoes M, Edgington LE, Scheeren FA, Leyva M, Blum G, Weiskopf K, et al. A nonpeptidic cathepsin S activity-based probe for noninvasive optical imaging of tumor-associated macrophages. Chem Biol (2012) 19(5):619–28. doi:10.1016/j.chembiol.2012.03.012
148. Schmidlin F, Amadesi S, Dabbagh K, Lewis DE, Knott P, Bunnett NW, et al. Protease-activated receptor 2 mediates eosinophil infiltration and hyperreactivity in allergic inflammation of the airway. J Immunol (2002) 169(9):5315–21. doi:10.4049/jimmunol.169.9.5315
149. Knight DA, Lim S, Scaffidi AK, Roche N, Chung KF, Stewart GA, et al. Protease-activated receptors in human airways: upregulation of PAR-2 in respiratory epithelium from patients with asthma. J Allergy Clin Immunol (2001) 108(5):797–803. doi:10.1067/mai.2001.119025
150. Moffatt JD, Jeffrey KL, Cocks TM. Protease-activated receptor-2 activating peptide SLIGRL inhibits bacterial lipopolysaccharide-induced recruitment of polymorphonuclear leukocytes into the airways of mice. Am J Respir Cell Mol Biol (2002) 26(6):680–4. doi:10.1165/ajrcmb.26.6.4693
Keywords: PARs, proteases, biased signaling, G proteins, β-arrestins, signal transduction
Citation: Zhao P, Metcalf M and Bunnett NW (2014) Biased signaling of protease-activated receptors. Front. Endocrinol. 5:67. doi: 10.3389/fendo.2014.00067
Received: 17 March 2014; Paper pending published: 07 April 2014;
Accepted: 22 April 2014; Published online: 13 May 2014.
Edited by:
Stuart Maudsley, Vlaams Instituut voor Biotechnologie, BelgiumReviewed by:
Mohammed Akli Ayoub, King Saud University, Saudi ArabiaAnne-Françoise Burnol, Cochin Institute, France
Copyright: © 2014 Zhao, Metcalf and Bunnett. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nigel W. Bunnett, Monash Institute of Pharmaceutical Sciences, 381 Royal Parade, Parkville, VIC 3052, Australia e-mail:bmlnZWwuYnVubmV0dEBtb25hc2guZWR1