 Etienne Khoury1†
Etienne Khoury1† Stéphane A. Laporte
Stéphane A. Laporte- 1Department of Medicine, McGill University Health Center Research Institute, McGill University, Montreal, QC, Canada
- 2Department of Pharmacology and Therapeutics, McGill University Health Center Research Institute, McGill University, Montreal, QC, Canada
- 3Department of Anatomy and Cell Biology, McGill University Health Center Research Institute, McGill University, Montreal, QC, Canada
G protein-coupled receptors (GPCRs) are seven-transmembrane proteins that participate in many aspects of the endocrine function and are important targets for drug development. They transduce signals mainly, but not exclusively, via hetero-trimeric G proteins, leading to a diversity of intracellular signaling cascades. Ligands binding at the hormone orthosteric sites of receptors have been classified as agonists, antagonists, and/or inverse agonists based on their ability to mainly modulate G protein signaling. Accumulating evidence also indicates that such ligands, alone or in combination with other ones such as those acting outside the orthosteric hormone binding sites (e.g., allosteric modulators), have the ability to selectively engage subsets of signaling responses as compared to the natural endogenous ligands. Such modes of functioning have been variously referred to as “functional selectivity” or “ligand-biased signaling.” In this review, we provide an overview of the current knowledge regarding GPCR-biased signaling and their functional regulation with a focus on the evolving concept that receptor domains can also be targeted to allosterically bias signaling, and discuss the usefulness of such modes of regulation for the design of more efficient therapeutics.
Introduction
G protein-coupled receptors (GPCRs) constitute the largest family of cell-surface receptors and are involved in almost all physiological and hormonal responses. Therefore, it is not surprising that they are the most targeted in drug discovery programs (1). Their activation was first described by a classical two-state model, where receptors exist in equilibrium between active (e.g., G protein-coupled: the “on” state) and inactive states (e.g., G protein-uncoupled: the “off” state), and where extracellular stimuli, such as hormones, neurotransmitters, peptides, and amino acids, shift this equilibrium from one state to the other. Based on this model, the properties of ligands were classified as agonists, antagonists, and inverse agonists, according to their ability to stabilize the “on” state of receptors allowing the full activation of G proteins such as for agonists, reducing the basal spontaneous coupling to G proteins like in the case of the inverse agonists (e.g., maintaining receptor in the “off” state), or inhibiting agonist competitively without changing the equilibrium like for “neutral” antagonists. To explain the biological and physiological responses triggered by these ligands, this binary model also assumes that GPCRs preferentially couple to one G protein subtype, and that either agonists, antagonists, or inverse agonists affect in a similar manner – according to its respective class – the activation of such G protein. Moreover, if more than one G protein subtype binds to its cognate receptor, each class of ligands would also affect it in a similar manner. However, several new lines of evidence now support an alternative multi-state model, where GPCRs can adopt multiple conformations, including active, inactive, and other intermediate ones. In such multi-state model, it is also inferred that ligands have the propensity of stabilizing a unique conformation leading to a specific signaling response, which may or may not always totally mimic the one induced by a natural ligand of reference. These ligands can stabilize a “hybrid” receptor conformation that mimics the “on” conformation with respect to engaging one signaling pathway, while at the same time mimicking the “off” conformation for another signaling pathway that is normally activated by an agonist of reference. Such mode of ligand-mediated differential signaling is commonly referred to as “functional selectivity” or “ligand-biased signaling,” and would in principle allow the activation of specific pathways and cell responses. It is now well-accepted that many orthosteric ligands (OL) have the ability to bias signaling between different G proteins and/or between G proteins and β-arrestins that are involved in the desensitization, internalization, and signaling of GPCRs. This latter mode of biased signaling has already been extensively covered in many recent reviews (2–4), and will not be furthermore expanded here.
Functional selectivity is not only limited to OLs, but is also a property that has been described for other allosteric ligands (AL)/effectors. These, which are also known as “allosteric modulators,” include ions, ligands, small and large molecules (e.g., antibodies) and/or protein complexes (e.g., receptor dimers and receptor–effector complexes) that modulate hormone binding and/or the intracellular coupling of receptors to their effectors, and affect responses in different ways: they can have differential cooperative effects – negative or positive ones – on the binding of the OL, the “conduit” of the information of the ligand to the effector through the receptor, and the biasing of receptor signaling. Moreover, an evolving concept also suggests that receptor domains that participate in ligand and/or effector binding, and in the “signaling conformations” of GPCRs, can also be targeted to bias signaling. Here, we will expand on this concept of targeting receptors’ domains to allosterically regulate their signaling for mainly class A and C GPCR, and review some of the potential clinical and pre-clinical uses of biased AL.
Biasing GPCR Signaling by Allosteric Modulators
Allosterism was first described with the hemoglobin, where the binding of oxygen to a specific site increases the affinity of other oxygen molecules to the remaining unoccupied sites (5). Over the years, this concept has proven to be widely spread for various types of proteins, and more recently for transmembrane receptors, such as GPCRs. Interestingly, numerous endogenous allosteric modulators have been identified, and shown to play crucial roles in keeping diverse biological functions mediated by this class of receptors. To date, the best characterized GPCR allosteric modulator is the G protein itself, which binds receptors and stabilizes their active conformation. Moreover, ions, such as Zn2+, Na+, and Ca2+ are other examples of endogenous molecules that have been shown to allosterically modulate GPCRs (6). In addition, other types of endogenous modulators, such as the small tripeptide Pro–Leu–Gly (PLG), also known as the melanocyte-stimulating hormone release inhibiting factor (MIF-1) has also been shown to act as an allosteric modulator on the D2 and the D4 dopamine receptors. PLG increases the affinity of dopamine for its receptors and the agonist-mediated inhibition of adenylyl cyclase (AC) (7). Similarly to OLs that have the capacity to bias GPCR signaling, AL, which bind to topographically distinct sites from the endogenous ligands, can direct receptor signaling (Figure 1). They can either exert positive, negative, or neutral effects on receptor signaling, and these modulatory effects do not always parallel those seen on the binding of OLs. Specifically, AL can influence the binding of the orthosteric hormone to its receptor, which can be independent from its impact on the signaling transduction promoted by the OL itself. Their effects on receptor signaling have been mainly divided into two categories: positive allosteric modulators (PAMs) and negative allosteric modulators (NAMs). Many advantages are known to be associated with the use of AL in terms of fine tuning GPCR responses (8, 9). First, because AL bind sites on GPCRs that are more diverse in nature than the orthosteric ones, greater selectivity can be achieved with such ligands. Indeed, targeting specific receptors belonging to the same family subtype has been often challenging because of their highly conserved sequences and structures within their orthosteric binding sites. For instance, class C GPCRs, group II metabotropic glutamate receptors (mGluRs), including mGluR2 and mGluR3, were selectively targeted by a NAM, which only inhibited the glutamate-induced response of group II receptors without exerting any effect on groups I and III mGluRs (10). Second, the effect of allosteric modulators on receptors is also saturable, since they are not competing with endogenous ligands. In other words, when all allosteric sites are occupied on receptors, no more effects are achieved. Third, most allosteric modulators are known to exert their function only in the presence of the endogenous ligand. Indeed, the AL will modulate the receptors’ conformation and signaling only when the endogenous hormone occupies its orthosteric site. However, some AL have also been described to act as allosteric agonists and promote functional effects in the absence of OLs and those are known as ago-allosteric modulators. Such ligands have also been referred to as “super-agonists,” because they can act synergistically with the natural ligand. An example of ago-AL is the phenylacetamide 1 and 2, which act directly on the free fatty acid receptor 2 (FFA2) (11).
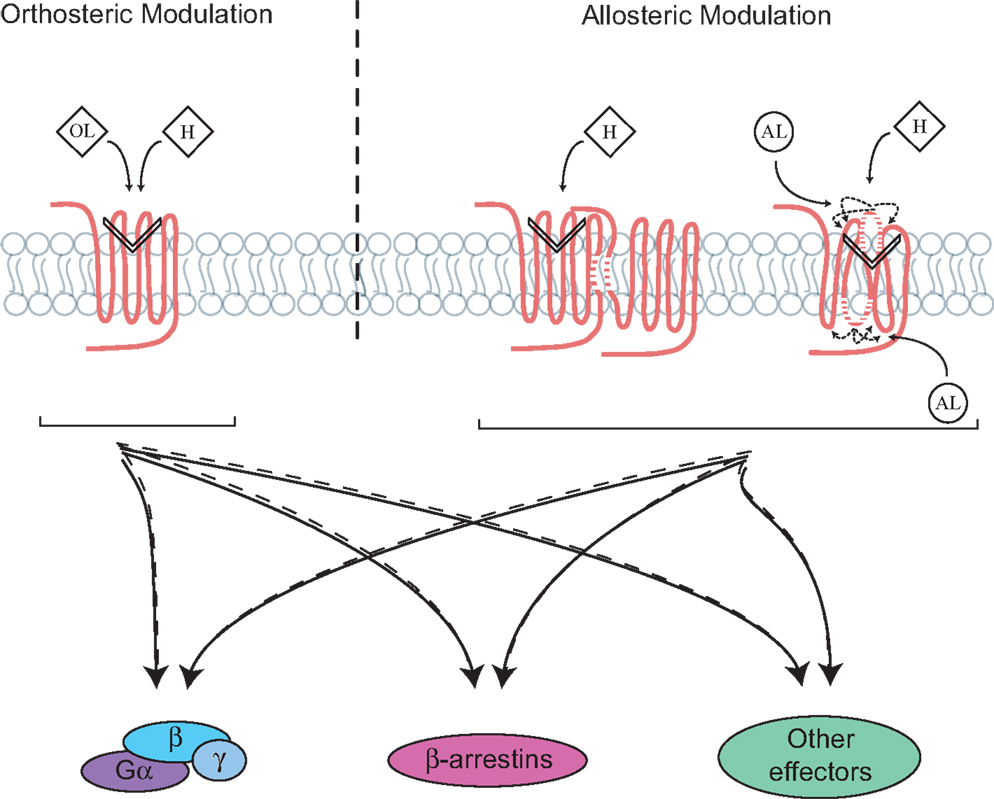
Figure 1. Directing signaling by orthosteric and allosteric GPCR ligands, receptor complexes and domains. Signaling occurs following the binding of endogenous hormones (H) to the orthosteric site on G protein-coupled receptor (GPCR), which involves different domains, mainly the transmembrane and extracellular ones. This leads to the activation of multiple signaling pathways that are balanced between the G proteins, β-arrestins, and/or other signaling effectors. Ligand-directed signaling (i.e., biased signaling) can occur through the binding of either orthosteric (OL) or allosteric (AL) ligands to the receptor, which changes the balance of signaling between the effectors as compared to a ligand of reference. Formation of receptors complexes, such as dimers as well as reorientation of extra/intracellular domains of receptors can also lead to conformational rearrangements or be targeted for biased signaling.
Another property of AL is also their ability to bias receptor signaling. Such allosteric modulation has been mainly studied in class A and C GPCRs. For example, a recent study characterized a new compound, PDC113.824, acting as an allosteric modulator on the class A GPCR, the prostaglandin F2α (FP) receptor. This compound was shown to act as a PAM on the agonist-mediated Gαq signaling pathway, while at the same time acting as a NAM on the Gα12 signaling cascade (12). Another example of G protein-dependent biased signaling is revealed by an autoantibody, which targets a class C GPCR, the calcium-sensing receptor (CaSR) and allosterically modulates the calcium-mediated potentiation of Gαq signaling, while inhibiting the Gαi response (13). Moreover, the gadolinium (Gd3+), a known allosteric modulator of the mGluR1α, was shown to act as a PAM on the agonist-mediated Gαq and a NAM on the Gαs signaling pathway (14). Allosteric-biased modulation on GPCRs is not only limited to G proteins vs. G proteins signaling but can also occur between G proteins and other signaling effectors, such as β-arrestins. For instance, the ATI-2341 compound acting on the C–X–C chemokine receptor type 4 (CXCR4) was recently shown to display differential ago-allosteric properties between G protein and β-arrestins signaling (15). ATI-2341 preferentially promoted the coupling of CXCR4 to Gαi- over Gα13-mediated signaling, but contrarily to the endogenous ligand, did not induce β-arrestin recruitment to the receptor. On the other hand, a study on the cannabinoid receptor 1 (CB1) showed that the ORG27569 compound acted as an NAM on the agonist-mediated Gαi signaling and a PAM on β-arrestin-dependent internalization of CB1; and acted as an ago-allosteric modulator on both receptor internalization and β-arrestin-dependent MAPK signaling (16, 17). These examples highlight the diversity of GPCR allosteric modulators on both receptors and their ensuing signaling (e.g., PAM and NAM effects). However, defining how they differentially direct downstream effectors and responses still remains an empirical endeavor for most receptors. Moreover, because allosteric modulators have the potential to differentially affect distinct effectors and responses, much more work still remains to better survey all the potential signaling pathways that can be affected, even the ones that are not normally suspected to be activated by the receptor studied.
Targeting GPCR Domains for Allosterically Biasing Receptor Signaling
Because different domains of GPCRs [e.g., intracellular loops (ICL) and extracellular loops (ECL) and transmembrane domains (TM)] are known to participate in ligand and/or effector recognition and receptor dimerization, recent attention has also been drawn to understand the role of these domains in receptor conformation and signaling. It is well-recognized that GPCRs can form oligomers within which conformational rearrangements of the receptors can impact signaling (Figure 1) (18, 19). An example is the heterodimerization of the class A GPCRs chemokine receptors, CXCR4 and CXCR7 (19). This study showed that, when receptors are both in complex, agonist-mediated activation of Gαi is impaired, whereas β-arrestin is constitutively recruited to the dimer. Because other reviews have already covered the concept of GPCR oligomers and its ensuing effects on directed signaling, such topic will not be furthermore discussed here (20–22). Of note, however, is a seminal study on the dimerization of the β2-adrenergic receptor (β2AR), which used a peptide consisting of residues 276–296 of the TM6 of the receptor (GIIMGTFTLCWLPFFIVNIVH), to prevent homodimerization, showed that the agonist-mediated cAMP production could be inhibited; thus suggesting that domains of GPCRs could be targeted to regulate receptors signaling in an allosteric fashion (23). In addition, another study has also demonstrated that peptides derived from TM of CXCR4 and CCR5 could be used as specific receptor antagonists (24). Despite that TM are involved in ligand binding and receptor activation, targeting these regions with peptides has proven to be difficult due to their hydrophobic nature.
Receptor ICLs are critical for GPCR signaling and can also be targeted to modulate receptor’s responses (Table 1). For instance, studies using mimics of ICL3 from many different GPCRs, such as the adrenergic receptors α1B and α2A (α1BAR and α2AAR), as well as the muscarinic acetylcholine receptors M1 and M2 (M1AChR and M2AChR), have revealed the importance of this domain in G protein coupling/activation (25). These peptides, which are derived from regions of receptors’ loops were shown to disrupt both Gαq and Gαi coupling to their cognate receptors and to affect downstream signaling. Similar effects were also reported for the angiotensin II type-2 receptor (AT2R) (26). Moreover, because β-arrestins also bind ICLs, peptides derived from the ICL3 and ICL1 of receptors were also shown to block GPCR densensitization (27). For example, a synthetic peptide corresponding to the sequence of the full length of ICL3 of the luteinizing hormone/choriogonadotropin receptor (LH/CGR) was shown to reverse the agonist-mediated desensitization of AC activity when incubated with membranes expressing LH/CGR by preventing the interaction between β-arrestin and the receptor (28). Not only was targeting receptor’s ICL shown to affect G protein and β-arrestin binding, but it was also demonstrated to bias GPCR signaling between these different pathways. Indeed, the ago-allosteric ATI-2341 compound, which belongs to the pepducin family and that is a short lipidated peptide of the ICL1 of the CXCR4, was shown to promote biased signaling between G proteins and β-arrestins (15). Consistent with the idea that intracellular domain of GPCRs can be targeted to allosterically bias receptor signaling, a recent study on the β2AR, using different antibodies directed against its intracellular domains, showed that the recruitment of β-arrestin to the receptor and the activation of G proteins were differentially affected (29). Targeting GPCR’s ICLs with peptide mimics has proven efficient for regulating receptor signaling. However, such approach can suffer from the impediment of having to modifying the peptides (e.g., lipidation) for their cellular delivery. On the other hand, the modification of peptides with lipids would favor reaching higher plasma concentrations of peptide near its target, the receptor, which would presumably increase their activity as compared to a gene delivery approach.
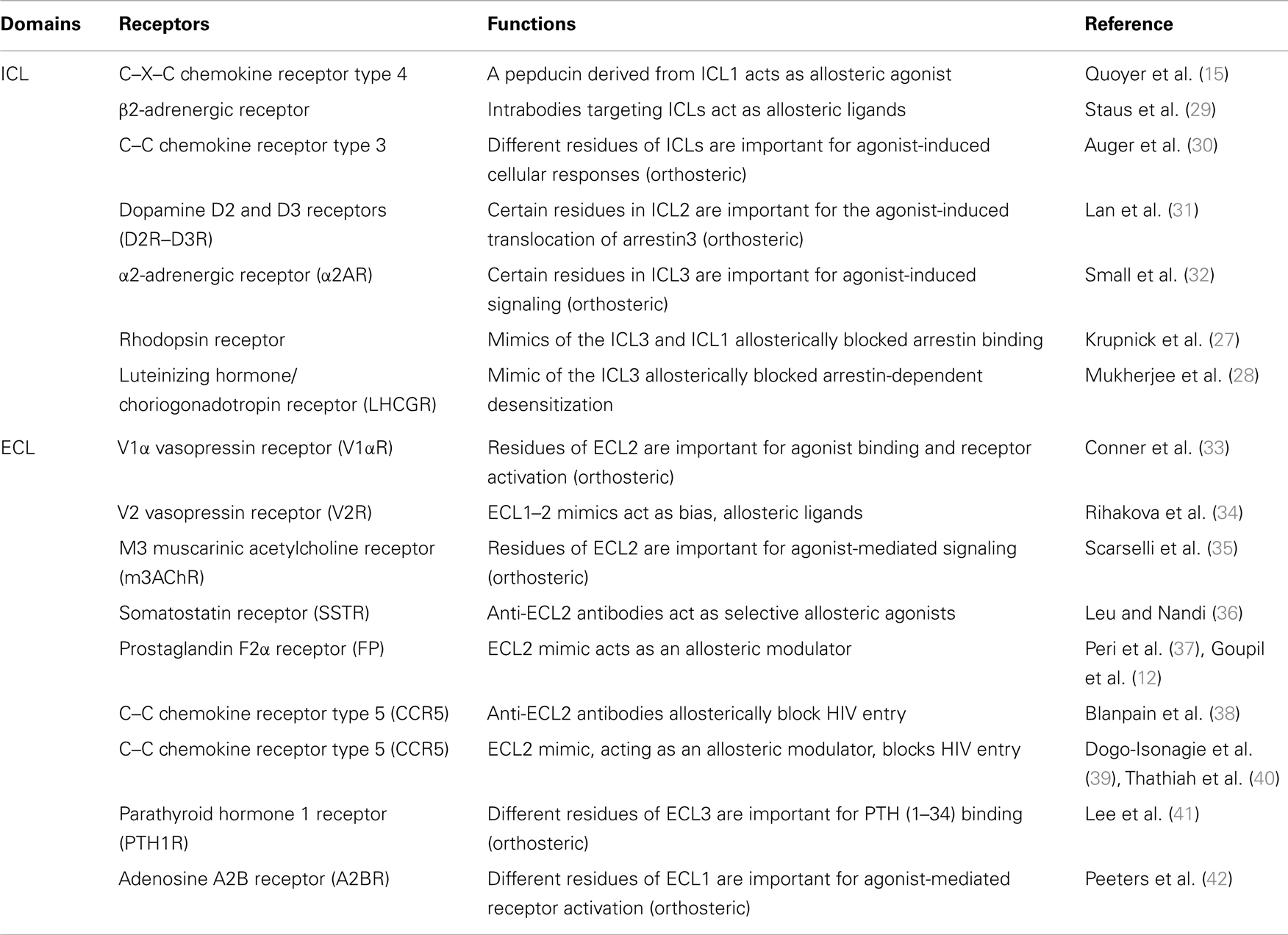
Table 1. Role of GPCRs intracellular (ICL) and extracellular (ECL) domains in receptors function.
The ECLs of GPCRs are also important in ligand binding and receptor activation (Table 1). Using site-directed mutagenesis, residues in the ECL2 [Phe(189), Trp(206), Phe(209), and Tyr(218)] of the V1α vasopressin receptor, which are highly conserved amongst GPCRs, were shown to be critical for receptor activation (33). Another study focusing on the parathyroid hormone 1 receptor (PTHR) used a similar approach and identified residues in the ECL3 (Trp-437 and Gln-440), which were important for PTH (1–34) binding (41). Mutagenesis studies of the adenosine A2B receptor have also implicated residues of the ECL1 in receptor activation (42). The ECL2 was also shown to be important for agonist binding and the activation of the M3 muscarinic acetylcholine receptor (M3AChR) (35), the C–C chemokine receptor type 5 (CCR5) (43), the dopamine D2 receptor (D2R) (44), and the complement factor 5α receptor (C5αR) (45). Other studies have also explored the contribution of ECL2 in stabilizing receptor conformations. For example, structural studies using NMR spectroscopy reveal that the ECL2 forms a cap that stabilizes the inactive conformation of the receptor and changes its orientation upon receptor activation (46).
The ECL domains of GPCR can also be targeted to allosterically modulate signaling. For instance, a recent study screened potential small molecule for their action on the relaxin/insulin-like family peptide receptor 1 (RXFP1), with the goal of finding agonists. They identified compound 8, which displayed agonistic effects on cellular responses such as cAMP production and cellular impedance. Interestingly, using different RXFP1 constructs and mutagenesis, they showed that the ECL3 of the receptor was required for this effect, suggesting that the compound interacts with an allosteric site within this ECL (47). Conversely, another study showed that the agonist effects of the allosteric modulators (phenylacetamide 1 and 2) on the FFA2 receptor were lost when its ECL2 was replaced with one of the FFA3 subtype receptor (48). Moreover, antibodies against the ECL2 have been shown to modulate receptor activity. In the case of the somatostatin receptors (SSTR), antibodies directed against their ECL2 have agonistic-like properties, while having no effect on agonist binding to receptors. SSTR2, SSTR3, and SSTR5 selective antibodies were also shown to diminish cAMP production and decrease serotonin secretion leading to a suppressed proliferation of neuroendocrine tumor cells (36). Moreover, specific antibodies against the ECL2 of the CCR5 were shown to block HIV entry in cells (38). Similarly, a peptide mimic of the C-terminal portion of CCR5 was shown to act as a NAM for HIV-1 entry (39). Not surprisingly, because ECLs can adopt different conformations in GPCR, either at basal state or upon receptor activation (46, 49), it can also be targeted to allosterically bias receptor signaling. For instance, a study on the vasopressin V2 receptor (V2R) using peptides corresponding to a decapeptide of ECL1 (PPLLARAELA) or an octapeptide of ECL2’s C-terminus (ALCRAVKY) showed physiological functional selectivity on different vasopressin-mediated responses in a non-competitive manner (34). Moreover, a peptide derived from a sequence overlapping the N-terminus of ECL2 and the TM4 of the prostaglandin F2α (FP) receptor, known as THG113 (ILGHRDYK), was able to block PGF2α-mediated contraction of the myometrium in a non-competitive manner (37). Supportive for the allosteric regulation of the FP receptor signaling, is the finding that a peptide mimic of THG113, the PDC113.824 compound, was also able to induce functional selectivity on G protein signaling mediated by PGF2α (12). Also consistent with the idea that the ECL2 can be targeted to direct receptor signaling is the use of an allosteric modulator of the M2 and M4 muscarinic acetylcholine receptors, LY2033298 compound, that was shown to bias downstream signaling (50). Interestingly, a congener of the LY2033298, the LY2119620 compound was later shown, through co-crystallographic studies, to interact with residues of the ECL2, and to allosterically alter M2 receptor active conformation (51). The GPCRs’ ECLs represent promising targets for allosterically modulating receptors, as they are presumably more accessible than ICLs. In particular, ECL2 is a good target, because of its diversity amongst GPCRs and because it is involved in the binding of OLs and in the signaling of many receptors. However, much more needs to be understood about how ECLs regulate ligand binding, receptor conformation, and signaling, and how peptides derived from these regions affect these functions.
Therapeutic Potentials of Allosteric and Biased Signaling
Recently, great attention has been devoted to functional selectivity as a new paradigm applicable for the development of better therapeutic drugs with potentially fewer off-target and/or side effects. An example is highlighted with the use of the biased agonist pilocarpine, which selectively acts on the M1 muscarinic acetylcholine receptor and shows positive therapeutic effects in different Alzheimer’s disease models. Specifically, pilocarpine biased Gαq-mediated phospholipase C activation over the Gαs-mediated AC stimulation, whereas the non-selective muscarinic agonist carbachol equally stimulated responses mediated by Gαs and Gαq (52, 53). Moreover, biasing β-arrestin-dependent signaling has also been shown to be potentially beneficial in heart diseases. For example, TRV120027 (Sar–Arg–Val–Tyr–Ile–His–Pro-d-Ala-OH), a peptide antagonist of the AT1R-dependent Gαq pathway was recently shown to selectively induce β-arrestin signaling. TRV120027 increases cardiomyocyte contractility in vitro, and cardiac performance both in rats and dogs (54, 55), suggesting that this orthosteric-biased ligand could be beneficial in acute heart failure treatment (54). Another example is the biased ligand TRV130, which acts on the μ-opioid receptor and induces cAMP inhibition through a Gαi-dependent mechanism without inducing either β-arrestin recruitment or receptor internalization. This biased agonist, which has similar potency and efficacy on Gαi signaling as morphine, showed higher analgesic efficacy, lower respiratory suppression, and less gastrointestinal dysfunction when compared to morphine (56).
Because AL can bias GPCR signaling, such modulators also represent interesting opportunities for drug discovery (Table 2). Moreover, greater subtype selectivity amongst receptors can be achieved using AL, which may also improve therapeutic benefits. For instance, the ADX10059 compound, a selective NAM for the mGluR5, was shown to improve symptoms in patients suffering from gastro-esophageal reflux disease (GERD) (57, 58). On the other hand, Reparixin (formerly Repertaxin), which acts as a NAM on both chemokine receptors, CXCR1 and CXCR2, shows promising therapeutic effects on the prevention of delayed graft dysfunction after kidney transplantation and early stages of breast cancer (59, 60). Research efforts focusing on targeting the ECL2 of GPCRs to allosterically modulate signaling, have also led to the development of new drugs. Maraviroc, a peptide that was originally developed from a mimic of the ECL2 of CCR5 (discussed in previous section), decreases the viral load in HIV-1 patients (61). Because of the putative role of CCR5 in alloreactivity, Maraviroc is also in phase II clinical trial for acute graft-versus-host disease (GVHD). Moreover, the ECL2 mimic of the FP receptor, PDC31 (derived from the THG113 peptide), which was shown to inhibit preterm labor in different animal models, is now being evaluated in a clinical phase II trial for primary dysmenorrhea (37, 62). Other examples of allosteric and biased drugs are currently under pre-clinical investigation or in clinical trials (Table 2).
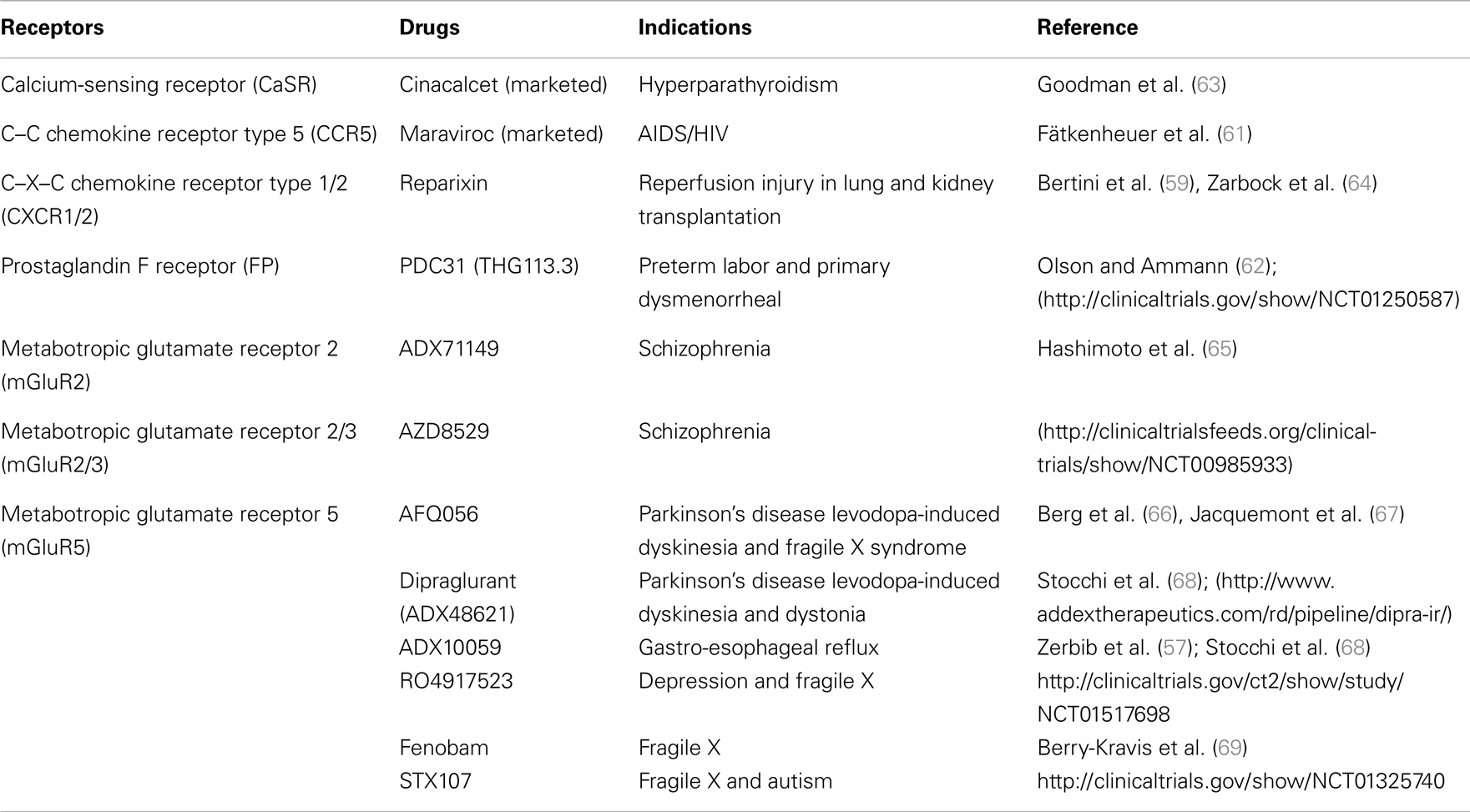
Table 2. Potential therapeutic usage of allosteric and biased GPCR signaling compounds/drugs.
Conclusion and Perspectives
The possibility of directing GPCR signaling with either OL or AL has opened new opportunities for developing more selective/effective drugs; because, in principal, it would allow a better control of the signaling pathway(s) involved in the underlying pathophysiology, hence limiting side effects that may be engendered from the non-selective engagement of other GPCR subtypes and/or from unwanted downstream signaling effectors/responses. However, much more is needed to be understood about directed signaling in order to develop better therapeutics; especially about GPCR interaction with biased ligands (orthosteric and allosteric ones) and their ensuing effects on receptor conformation, and how such receptor–ligand complex can convey signaling information in a selective manner. Challenges also include a better understanding of the role played by GPCR domains such as the TMs, ECLs, and ICLs in the conformation of receptors and how different OL and AL ligands affect these domains to stabilize subsets of conformations for selective signaling. In particular, because allosteric compounds may represent a promising new class of drugs, predicting their sites of binding on GPCRs and modes of action – on the hormone binding, conformation, and “conduit” of signaling (e.g., ago-agonist, PAM, or NAM) – becomes fundamental. Moreover, because most GPCR can engage many downstream signaling pathways, which in many cases are cell-, tissue-, and/or context-specific, it is essential to better define the entire “signaling repertoire” for the hormones/drugs of reference and the putative-biased ligands to be studied, particularly, in normal vs. pathological conditions. Establishing such “signaling repertoire” becomes therefore essential in programs that aim at improving drug efficacy or at repurposing drugs; hence, future research efforts should be oriented toward the development of approaches for assessing the full spectrum of signaling of the ligands of interest, in different cell models, including those physiological and pathophysiological relevant ones, and integrative analytical methods for linking these “signaling signatures” to pre-clinical and/or existing clinical data.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Laporte laboratory for helpful discussions. Etienne Khoury and Stéphanie Clément, respectively, hold studentships from the McGill Division of Endocrinology and Metabolism, and from the Department of Pharmacology and Therapeutics. This work was supported by Canadian Institutes of Health Research (CIHR) Grants to Stéphane A. Laporte. Stéphane A. Laporte holds a “Chercheur Boursier Senior” from the “Fonds de recherche du Québec – Santé” (FRQS).
References
1. Lagerstrom MC, Schioth HB. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov (2008) 7(4):339–57. doi: 10.1038/nrd2518
2. Violin JD, Lefkowitz RJ. Beta-arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol Sci (2007) 28(8):416–22. doi:10.1016/j.tips.2007.06.006
3. Rajagopal S, Rajagopal K, Lefkowitz RJ. Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat Rev Drug Discov (2010) 9(5):373–86. doi:10.1038/nrd3024
4. Luttrell LM. Arrestin pathways as drug targets. Prog Mol Biol Transl Sci (2013) 118:469–97. doi:10.1016/B978-0-12-394440-5.00018-8
5. Monod J, Wyman J, Changeux JP. On the nature of allosteric transitions: a plausible model. J Mol Biol (1965) 12:88–118. doi:10.1016/S0022-2836(65)80285-6
6. May LT, Leach K, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol (2007) 47:1–51. doi:10.1146/annurev.pharmtox.47.120505.105159
7. Mishra RK, Makman MH, Costain WJ, Nair VD, Johnson RL. Modulation of agonist stimulated adenylyl cyclase and GTPase activity by l-pro-l-leu-glycinamide and its peptidomimetic analogue in rat striatal membranes. Neurosci Lett (1999) 269(1):21–4. doi:10.1016/S0304-3940(99)00413-9
8. Goupil E, Laporte SA, Hébert TE. Functional selectivity in GPCR signaling: understanding the full spectrum of receptor conformations. Mini Rev Med Chem (2012) 12(9):817–30. doi:10.2174/138955712800959143
9. Wootten D, Christopoulos A, Sexton PM. Emerging paradigms in GPCR allostery: implications for drug discovery. Nat Rev Drug Discov (2013) 12(8):630–44. doi:10.1038/nrd4052
10. Hemstapat K, Da Costa H, Nong Y, Brady AE, Luo Q, Niswender CM, et al. A novel family of potent negative allosteric modulators of group II metabotropic glutamate receptors. J Pharmacol Exp Ther (2007) 322(1):254–64. doi:10.1124/jpet.106.117093
11. Lee T, Schwandner R, Swaminath G, Weiszmann J, Cardozo M, Greenberg J, et al. Identification and functional characterization of allosteric agonists for the G protein-coupled receptor FFA2. Mol Pharmacol (2008) 74(6):1599–609. doi:10.1124/mol.108.049536
12. Goupil E, Tassy D, Bourguet C, Quiniou C, Wisehart V, Pétrin D, et al. A novel biased allosteric compound inhibitor of parturition selectively impedes the prostaglandin F2alpha-mediated Rho/ROCK signaling pathway. J Biol Chem (2010) 285(33):25624–36. doi:10.1074/jbc.M110.115196
13. Makita N, Sato J, Manaka K, Shoji Y, Oishi A, Hashimoto M, et al. An acquired hypocalciuric hypercalcemia autoantibody induces allosteric transition among active human Ca-sensing receptor conformations. Proc Natl Acad Sci U S A (2007) 104(13):5443–8. doi:10.1073/pnas.0701290104
14. Tateyama M, Kubo Y. Dual signaling is differentially activated by different active states of the metabotropic glutamate receptor 1alpha. Proc Natl Acad Sci U S A (2006) 103(4):1124–8. doi:10.1073/pnas.0505925103
15. Quoyer J, Janz JM, Luo J, Ren Y, Armando S, Lukashova V, et al. Pepducin targeting the C-X-C chemokine receptor type 4 acts as a biased agonist favoring activation of the inhibitory G protein. Proc Natl Acad Sci U S A (2013) 110(52):E5088–97. doi:10.1073/pnas.1312515110
16. Ahn KH, Mahmoud MM, Kendall DA. Allosteric modulator ORG27569 induces CB1 cannabinoid receptor high affinity agonist binding state, receptor internalization, and Gi protein-independent ERK1/2 kinase activation. J Biol Chem (2012) 287(15):12070–82. doi:10.1074/jbc.M111.316463
17. Ahn KH, Mahmoud MM, Shim JY, Kendall DA. Distinct roles of beta-arrestin 1 and beta-arrestin 2 in ORG27569-induced biased signaling and internalization of the cannabinoid receptor 1 (CB1). J Biol Chem (2013) 288(14):9790–800. doi:10.1074/jbc.M112.438804
18. Percherancier Y, Berchiche YA, Slight I, Volkmer-Engert R, Tamamura H, Fujii N, et al. Bioluminescence resonance energy transfer reveals ligand-induced conformational changes in CXCR4 homo- and heterodimers. J Biol Chem (2005) 280(11):9895–903. doi:10.1074/jbc.M411151200
19. Décaillot FM, Kazmi MA, Lin Y, Ray-Saha S, Sakmar TP, Sachdev P. CXCR7/CXCR4 heterodimer constitutively recruits beta-arrestin to enhance cell migration. J Biol Chem (2011) 286(37):32188–97. doi:10.1074/jbc.M111.277038
20. Smith NJ, Milligan G. Allostery at G protein-coupled receptor homo- and heteromers: uncharted pharmacological landscapes. Pharmacol Rev (2010) 62(4):701–25. doi:10.1124/pr.110.002667
21. Goupil E, Laporte SA, Hébert TE. GPCR heterodimers: asymmetries in ligand binding and signalling output offer new targets for drug discovery. Br J Pharmacol (2013) 168(5):1101–3. doi:10.1111/bph.12040
22. Satake H, Matsubara S, Aoyama M, Kawada T, Sakai T. GPCR Heterodimerization in the Reproductive System: functional Regulation and Implication for Biodiversity. Front Endocrinol (Lausanne) (2013) 4:100. doi:10.3389/fendo.2013.00100
23. Hebert TE, Moffett S, Morello JP, Loisel TP, Bichet DG, Barret C, et al. A peptide derived from a beta2-adrenergic receptor transmembrane domain inhibits both receptor dimerization and activation. J Biol Chem (1996) 271(27):16384–92. doi:10.1074/jbc.271.27.16384
24. Tarasova NI, Rice WG, Michejda CJ. Inhibition of G-protein-coupled receptor function by disruption of transmembrane domain interactions. J Biol Chem (1999) 274(49):34911–5. doi:10.1074/jbc.274.49.34911
25. Hawes BE, Luttrell LM, Exum ST, Lefkowitz RJ. Inhibition of G protein-coupled receptor signaling by expression of cytoplasmic domains of the receptor. J Biol Chem (1994) 269(22):15776–85.
26. Hayashida W, Horiuchi M, Dzau VJ. Intracellular third loop domain of angiotensin II type-2 receptor. Role in mediating signal transduction and cellular function. J Biol Chem (1996) 271(36):21985–92. doi:10.1074/jbc.271.36.21985
27. Krupnick JG, Gurevich VV, Schepers T, Hamm HE, Benovic JL. Arrestin-rhodopsin interaction. Multi-site binding delineated by peptide inhibition. J Biol Chem (1994) 269(5):3226–32.
28. Mukherjee S, Palczewski K, Gurevich VV, Hunzicker-Dunn M. Beta-arrestin-dependent desensitization of luteinizing hormone/choriogonadotropin receptor is prevented by a synthetic peptide corresponding to the third intracellular loop of the receptor. J Biol Chem (1999) 274(19):12984–9. doi:10.1074/jbc.274.19.12984
29. Staus DP, Wingler LM, Strachan RT, Rasmussen SG, Pardon E, Ahn S, et al. Regulation of beta2-adrenergic receptor function by conformationally selective single-domain intrabodies. Mol Pharmacol (2014) 85(3):472–81. doi:10.1124/mol.113.089516
30. Auger GA, Pease JE, Shen X, Xanthou G, Barker MD. Alanine scanning mutagenesis of CCR3 reveals that the three intracellular loops are essential for functional receptor expression. Eur J Immunol (2002) 32(4):1052–8. doi:10.1002/1521-4141(200204)32:4<1052::AID-IMMU1052>3.3.CO;2-C
31. Lan H, Teeter MM, Gurevich VV, Neve KA. An intracellular loop 2 amino acid residue determines differential binding of arrestin to the dopamine D2 and D3 receptors. Mol Pharmacol (2009) 75(1):19–26. doi:10.1124/mol.108.050542
32. Small KM, Forbes SL, Rahman FF, Bridges KM, Liggett SB. A four amino acid deletion polymorphism in the third intracellular loop of the human alpha 2C-adrenergic receptor confers impaired coupling to multiple effectors. J Biol Chem (2000) 275(30):23059–64. doi:10.1074/jbc.M000796200
33. Conner M, Hawtin SR, Simms J, Wootten D, Lawson Z, Conner AC, et al. Systematic analysis of the entire second extracellular loop of the V(1a) vasopressin receptor: key residues, conserved throughout a G-protein-coupled receptor family, identified. J Biol Chem (2007) 282(24):17405–12. doi:10.1074/jbc.M702151200
34. Rihakova L, Quiniou C, Hamdan FF, Kaul R, Brault S, Hou X, et al. VRQ397 (CRAVKY): a novel noncompetitive V2 receptor antagonist. Am J Physiol Regul Integr Comp Physiol (2009) 297(4):R1009–18. doi:10.1152/ajpregu.90766.2008
35. Scarselli M, Li B, Kim SK, Wess J. Multiple residues in the second extracellular loop are critical for M3 muscarinic acetylcholine receptor activation. J Biol Chem (2007) 282(10):7385–96. doi:10.1074/jbc.M610394200
36. Leu FP, Nandi M. GPCR somatostatin receptor extracellular loop 2 is a key ectodomain for making subtype-selective antibodies with agonist-like activities in the pancreatic neuroendocrine tumor BON cell line. Pancreas (2010) 39(8):1155–66. doi:10.1097/MPA.0b013e3181de8c05
37. Peri KG, Quiniou C, Hou X, Abran D, Varma DR, Lubell WD, et al. THG113: a novel selective FP antagonist that delays preterm labor. Semin Perinatol (2002) 26(6):389–97. doi:10.1053/sper.2002.37307
38. Blanpain C, Vanderwinden JM, Cihak J, Wittamer V, Le Poul E, Issafras H, et al. Multiple active states and oligomerization of CCR5 revealed by functional properties of monoclonal antibodies. Mol Biol Cell (2002) 13(2):723–37. doi:10.1091/mbc.01-03-0129
39. Dogo-Isonagie C, Lam S, Gustchina E, Acharya P, Yang Y, Shahzad-ul-Hussan S, et al. Peptides from second extracellular loop of C-C chemokine receptor type 5 (CCR5) inhibit diverse strains of HIV-1. J Biol Chem (2012) 287(18):15076–86. doi:10.1074/jbc.M111.332361
40. Thathiah A, Horré K, Snellinx A, Vandewyer E, Huang Y, Ciesielska M, et al. Beta-arrestin 2 regulates Abeta generation and gamma-secretase activity in Alzheimer’s disease. Nat Med (2013) 19(1):43–9. doi:10.1038/nm.3023
41. Lee C, Luck MD, Jüppner H, Potts JT Jr, Kronenberg HM, Gardella TJ. Homolog-scanning mutagenesis of the parathyroid hormone (PTH) receptor reveals PTH-(1-34) binding determinants in the third extracellular loop. Mol Endocrinol (1995) 9(10):1269–78. doi:10.1210/me.9.10.1269
42. Peeters MC, van Westen GJ, Guo D, Wisse LE, Müller CE, Beukers MW, et al. GPCR structure and activation: an essential role for the first extracellular loop in activating the adenosine A2B receptor. FASEB J (2011) 25(2):632–43. doi:10.1096/fj.10-164319
43. Samson M, LaRosa G, Libert F, Paindavoine P, Detheux M, Vassart G, et al. The second extracellular loop of CCR5 is the major determinant of ligand specificity. J Biol Chem (1997) 272(40):24934–41. doi:10.1074/jbc.272.40.24934
44. Shi L, Javitch JA. The second extracellular loop of the dopamine D2 receptor lines the binding-site crevice. Proc Natl Acad Sci U S A (2004) 101(2):440–5. doi:10.1073/pnas.2237265100
45. Klco JM, Wiegand CB, Narzinski K, Baranski TJ. Essential role for the second extracellular loop in C5a receptor activation. Nat Struct Mol Biol (2005) 12(4):320–6. doi:10.1038/nsmb913
46. Ahuja S, Hornak V, Yan EC, Syrett N, Goncalves JA, Hirshfeld A, et al. Helix movement is coupled to displacement of the second extracellular loop in rhodopsin activation. Nat Struct Mol Biol (2009) 16(2):168–75. doi:10.1038/nsmb.1549
47. Xiao J, Huang Z, Chen CZ, Agoulnik IU, Southall N, Hu X, et al. Identification and optimization of small-molecule agonists of the human relaxin hormone receptor RXFP1. Nat Commun (2013) 4:1953. doi:10.1038/ncomms2953
48. Smith NJ, Ward RJ, Stoddart LA, Hudson BD, Kostenis E, Ulven T, et al. Extracellular loop 2 of the free fatty acid receptor 2 mediates allosterism of a phenylacetamide ago-allosteric modulator. Mol Pharmacol (2011) 80(1):163–73. doi:10.1124/mol.110.070789
49. Wang C, Wu H, Katritch V, Han GW, Huang XP, Liu W, et al. Structure of the human smoothened receptor bound to an antitumour agent. Nature (2013) 497(7449):338–43. doi:10.1038/nature12167
50. Valant C, Felder CC, Sexton PM, Christopoulos A. Probe dependence in the allosteric modulation of a G protein-coupled receptor: implications for detection and validation of allosteric ligand effects. Mol Pharmacol (2012) 81(1):41–52. doi:10.1124/mol.111.074872
51. Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature (2013) 504(7478):101–6. doi:10.1038/nature12735
52. Fisher A, Heldman E, Gurwitz D, Haring R, Barak D, Meshulam H, et al. Selective signaling via unique M1 muscarinic agonists. Ann N Y Acad Sci (1993) 695:300–3. doi:10.1111/j.1749-6632.1993.tb23070.x
53. Gurwitz D, Haring R, Heldman E, Fraser CM, Manor D, Fisher A. Discrete activation of transduction pathways associated with acetylcholine m1 receptor by several muscarinic ligands. Eur J Pharmacol (1994) 267(1):21–31. doi:10.1016/0922-4106(94)90220-8
54. Violin JD, DeWire SM, Yamashita D, Rominger DH, Nguyen L, Schiller K, et al. Selectively engaging beta-arrestins at the angiotensin II type 1 receptor reduces blood pressure and increases cardiac performance. J Pharmacol Exp Ther (2010) 335(3):572–9. doi:10.1124/jpet.110.173005
55. Boerrigter G, Soergel DG, Violin JD, Lark MW, Burnett JC Jr. TRV120027, a novel beta-arrestin biased ligand at the angiotensin II type I receptor, unloads the heart and maintains renal function when added to furosemide in experimental heart failure. Circ Heart Fail (2012) 5(5):627–34. doi:10.1161/CIRCHEARTFAILURE.112.969220
56. DeWire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM, et al. A G protein-biased ligand at the mu-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J Pharmacol Exp Ther (2013) 344(3):708–17. doi:10.1124/jpet.112.201616
57. Zerbib F, Keywood C, Strabach G. Efficacy, tolerability and pharmacokinetics of a modified release formulation of ADX10059, a negative allosteric modulator of metabotropic glutamate receptor 5: an esophageal pH-impedance study in healthy subjects. Neurogastroenterol Motil (2010) 22(8):859–65. doi:10.1111/j.1365-2982.2010.01484.x
58. Zerbib F, Bruley des Varannes S, Roman S, Tutuian R, Galmiche JP, Mion F, et al. Randomised clinical trial: effects of monotherapy with ADX10059, a mGluR5 inhibitor, on symptoms and reflux events in patients with gastro-oesophageal reflux disease. Aliment Pharmacol Ther (2011) 33(8):911–21. doi:10.1111/j.1365-2036.2011.04596.x
59. Bertini R, Allegretti M, Bizzarri C, Moriconi A, Locati M, Zampella G, et al. Noncompetitive allosteric inhibitors of the inflammatory chemokine receptors CXCR1 and CXCR2: prevention of reperfusion injury. Proc Natl Acad Sci U S A (2004) 101(32):11791–6. doi:10.1073/pnas.0402090101
60. Ginestier C, Liu S, Diebel ME, Korkaya H, Luo M, Brown M, et al. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J Clin Invest (2010) 120(2):485–97. doi:10.1172/JCI39397
61. Fätkenheuer G, Pozniak AL, Johnson MA, Plettenberg A, Staszewski S, Hoepelman AI, et al. Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1. Nat Med (2005) 11(11):1170–2. doi:10.1038/nm1319
62. Olson DM, Ammann C. Role of the prostaglandins in labour and prostaglandin receptor inhibitors in the prevention of preterm labour. Front Biosci (2007) 12:1329–43. doi:10.2741/2151
63. Goodman WG, Frazao JM, Goodkin DA, Turner SA, Liu W, Coburn JW. A calcimimetic agent lowers plasma parathyroid hormone levels in patients with secondary hyperparathyroidism. Kidney Int (2000) 58:436–45. doi:10.1046/j.1523-1755.2000.00183.x
64. Zarbock A, Allegretti M, Ley K. Therapeutic inhibition of CXCR2 by Reparixin attenuates acute lung injury in mice. Br J Pharmacol (2008) 155(3):357–64. doi:10.1038/bjp.2008.270
65. Hashimoto K, Malchow B, Falkai P, Schmitt A. Glutamate modulators as potential therapeutic drugs in schizophrenia and affective disorders. Eur Arch Psychiatry Clin Neurosci (2013) 263(5):367–77. doi:10.1007/s00406-013-0399-y
66. Berg D, Godau J, Trenkwalder C, Eggert K, Csoti I, Storch A, et al. AFQ056 treatment of levodopa-induced dyskinesias: results of 2 randomized controlled trials. Mov Disord (2011) 26(7):1243–50. doi:10.1002/mds.23616
67. Jacquemont S, Curie A, des Portes V, Torrioli MG, Berry-Kravis E, Hagerman RJ, et al. Epigenetic modification of the FMR1 gene in fragile X syndrome is associated with differential response to the mGluR5 antagonist AFQ056. Sci Transl Med (2011) 3(64):64ra1. doi:10.1126/scitranslmed.3001708
68. Stocchi F, Rascol O, Destee A, Hattori N, Hauser RA, Lang AE, et al. 15th International Congress of Parkinson’s Disease and Movement Disorders. Toronto (2011).
Keywords: G protein-coupled receptors, allosterism, biased signaling, functional selectivity, receptor domains
Citation: Khoury E, Clément S and Laporte SA (2014) Allosteric and biased G protein-coupled receptor signaling regulation: potentials for new therapeutics. Front. Endocrinol. 5:68. doi: 10.3389/fendo.2014.00068
Received: 13 March 2014; Paper pending published: 01 April 2014;
Accepted: 22 April 2014; Published online: 08 May 2014.
Edited by:
Stuart Maudsley, Vlaams Instituut voor Biotechnologie, BelgiumReviewed by:
Nicola J. Smith, Victor Chang Cardiac Research Institute, AustraliaEmma June Petrie, The University of Melbourne, Australia
Copyright: © 2014 Khoury, Clément and Laporte. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stéphane A. Laporte, Department of Medicine, Polypeptide Lab, McGill University, Strathcona Anatomy and Dentistry Building, 3640 University Street, Room W315, Montreal, QC H3A 2B2, Canada e-mail:c3RlcGhhbmUubGFwb3J0ZUBtY2dpbGwuY2E=
†Etienne Khoury and Stéphanie Clément have contributed equally to this work.