 Dorit Naot
Dorit Naot Jillian Cornish
Jillian Cornish- Department of Medicine, University of Auckland, Auckland, New Zealand
The positive association between body weight and bone density has been established in numerous laboratory and clinical studies. Apart from the direct effect of soft tissue mass on bone through skeletal loading, a number of cytokines and hormones contribute to the positive association between adipose and bone tissue, acting either locally in sites where cells of the two tissues are adjacent to each other or systemically through the circulation. The current review describes the effects of such local and systemic factors on bone physiology. One class of factors are the adipocyte-secreted peptides (adipokines), which affect bone turnover through a combination of direct effects in bone cells and indirect mechanisms mediated by the central nervous system. Another source of hormones that contribute to the coupling between fat and bone tissue are beta cells of the pancreas. Insulin, amylin, and preptin are co-secreted from pancreatic beta cells in response to increased glucose levels after feeding, and are also found in high circulating levels in obesity. A number of peptide hormones secreted from the gastrointestinal tract in response to feeding affect both fat and bone cells and thus can also act as mediators of the association between the two tissues. The current review focuses on results of laboratory studies investigating possible mechanism involved in the positive association between fat mass and bone mass.
Introduction
The skeleton provides structural support to soft tissues, attachment sites for muscles, and protection of vital organs. In addition, bone tissue acts as a reservoir for ions that can be sequestered from or released into the serum as required for the maintenance of calcium and phosphate homeostasis. Complex regulatory networks control bone physiology and optimize the response of bone to the changing mechanical and metabolic requirements (1, 2). The size and strength of the skeleton is adjusted to the mass of soft tissue to provide optimal support without being heavier than necessary and the rate of bone turnover has to be constantly adjusted to the availability of nutrients and the levels of circulating calcium and phosphate.
Body weight is positively correlated with bone mineral density (BMD) and negatively correlated with fracture risk (3). In contrast to the consistent finding that underweight is a major risk factor for fracture, the protective effect of increased weight against fracture appears to be more complex. Thus, when analyzing the contribution of the two components of body weight; fat mass and lean mass to the correlation with BMD, most studies identify fat mass as the major contributing factor while some studies find significant correlation with lean mass (4–6). Differential effects of visceral and subcutaneous fat have also been described, suggesting that subcutaneous fat is beneficial to bone whereas visceral fat is deleterious (7, 8). In addition, there is evidence that the relationship between weight and fracture risk is site-specific (9). A recently published study of the relationship of weight, height, and body mass index (BMI) with fracture risk at different sites in post-menopausal women found a significant inverse association between BMI and hip, spine, and wrist fractures and positive association between weight and ankle fractures (10). The effect of body weight on fracture risk also appears to be age-dependent, as obesity is a risk factor for fracture in children but is protective in adults (7).
Obesity is considered a global epidemic with serious co-morbidities that include type-2 diabetes, hypertension, and cancer (11). A number of clinical studies of the relationship between fat and bone mass and fracture risk in obese people demonstrated that the higher BMD in obesity is not as protective against fractures as it is in people with normal BMI (12). During the past two decades, the various procedures of bariatric surgery have increasingly become a common treatment for obesity (11). Both the weight gain in obesity and the rapid weight loss following bariatric surgery have major impacts on the skeleton, and many aspects of the relationship among nutritional status, fat tissue, and the skeleton have been studied in these conditions (11, 13–16).
In recent years, the traditional distinction between endocrine organs and target tissues has become blurred as it is now clear that many tissues secrete factors that circulate and act on target organs, thus creating complex communication networks where many tissues have endocrine functions. In the crosstalk between fat and bone, there is a major role for circulating factors secreted from adipose tissue; adipokines, as well as factors secreted from beta cells of the pancreas and from the gastrointestinal tract in response to feeding. The current review focuses on the bone activities of the local and circulating factors, which generate the positive association between fat and bone.
Adipokines
Adipokines are regulatory factors secreted from adipocytes that reside within adipose tissue and in the bone marrow. Several adipokines have been studied extensively and their effects in bone and other tissues are well-described, whereas the activities of additional novel adipokines are still largely unknown.
Leptin
The endocrine function of fat tissue was first discovered with the cloning of leptin and the finding that leptin is secreted from adipocytes and is part of a negative feedback loop that regulates fat tissue mass (17). Circulating levels of leptin positively correlate with body fat and its primary target in the regulation of fat mass was identified as the hypothalamus, where leptin binds to its receptor and regulates appetite and energy expenditure (18). Consequent studies showed a wide distribution of leptin receptors outside the central nervous system, and the expression of the signaling form of the leptin receptor in osteoblasts and chondrocytes suggested the skeleton as a potential target (19–21). The effects of leptin on bone mass are complex, with a combination of direct activity in bone cells, indirect effects through the central nervous systems as well as secondary effects through changes in body mass, and levels of other circulating hormones.
The direct effects of leptin in bone cells are anabolic. Leptin increases proliferation and differentiation of osteoblasts and chondrocytes (19–22) and reduces osteoclastogenesis through inhibiting the expression of receptor activator of nuclear factor κB ligand (RANKL) and induction of osteoprotegerin (19, 23). These results suggest that leptin has an overall positive effect on bone, and could therefore be a possible mediator of the positive correlation between fat and bone tissue.
A number of groups studied the phenotype of leptin-deficient mice (ob/ob) and leptin receptor-deficient mice (db/db), but the results of these studies are sometimes conflicting, possibly due to the different skeletal sites investigated and the specific parameters measured (1, 3, 24). The two animal models show profound obesity with reduced trabecular bone volume and BMD in femora and tibiae and an increase in the number and size of adipocytes in the marrow (25, 26). Systemic administration of leptin has an overall positive effect on bone; injection into ob/ob mice led to an increase in BMD (21, 25), while in wild-type mice leptin injection increased bone strength in males (26) and reduced ovariectomy-induced bone loss in females (27).
In contrast to these studies, Ducy et al. (28) found that circulating leptin inhibits bone formation via the hypothalamus and the sympathetic nervous system. They reported that both ob/ob and db/db mice have increased bone formation that results in high vertebral bone mass, and that intracerebroventricular (ICV) infusion of leptin causes bone loss in leptin-deficient and in wild-type mice through inhibition of bone formation and stimulation of bone resorption.
The main difference between the studies that show anabolic activity of leptin in bone and the one that demonstrate catabolic effects is the difference in the way leptin was administered: systemically in the former and centrally into the brain in the latter. However, Bartell et al. (29) directly compared the effect of leptin injected SC or through ICV infusion and found that regardless of the way of administration, leptin decreased body weight, food intake, and body fat and increased BMD. Thus, the exact nature of leptin activity in bone remains controversial and requires further investigations (30).
Clinical studies of the effect of leptin in bone are inconclusive, with some finding a positive correlation between leptin serum levels and BMD (31, 32) and others finding no correlation (33–35). Circulating leptin levels were not associated with fracture risk in a large prospective cohort study (36).
Adiponectin
Adiponectin, produced almost exclusively by adipocytes, is a 28 kDa protein that circulates in humans in trimers, hexamers, and high-molecular weight oligomers, in total concentrations of 0.5–30 μg/mL (37–39). Adiponectin regulates energy homeostasis, glucose and lipid metabolism, and inflammatory pathways (40). Plasma concentrations of adiponectin are inversely related to visceral fat mass and BMI, possibly due to the inhibition of adiponectin secretion by cytokines and hormones that are increased in obesity, by adipose tissue hypoxia, or by a negative effect of adiponectin on its own production (37). Clinical studies consistently show inverse relationships between circulating adiponectin concentrations and BMD, which persist after adjustment for potential confounding factors, including BMI, serum leptin, and central fat mass (33, 36, 41–43). There is a suggestion that an increased level of circulating adiponectin is a risk factor for fracture independent of body composition and BMD (36).
The results of laboratory studies investigating the activity of adiponectin in bone are not entirely congruent, but there is ample evidence that adiponectin affects bone through a combination of direct and indirect mechanisms. The direct activity of adiponectin in bone cells is mediated through the AdipoR1 and AdipoR2 receptors, which are expressed in both osteoblasts and osteoclasts (44, 45). In vitro, most groups found that adiponectin stimulates the proliferation and differentiation of osteoblastic cells (46–48), whereas studies of the effect of adiponectin on osteoclastogenesis produced conflicting results. Inhibition of osteoclastogenesis was determined in bone marrow cultures (47, 48) while induction of osteoclastogenesis was found in co-cultures of osteoblasts and CD14 positive cells from peripheral blood (49). Two groups reported a direct inhibitory effect of adiponectin on osteoclast formation in CD14 positive cells and in RAW264.7 cells (47, 50), whereas two studies that used similar experimental systems showed no effects (48, 49). The inconsistency of some of the in vitro data could be explained by the finding that commercially available preparations of adiponectin are contaminated with lipopolysaccharide, and that this contaminant is responsible for some of the reported in vitro effects observed (51, 52).
A number of animal models were used to investigate adiponectin activity in bone. Adiponectin-deficient mice showed no significant abnormality in bone phenotype at 8 weeks of age (48, 53), but at 14 weeks, micro-computed tomography demonstrated increase in trabecular bone volume and trabecular number (48). Transgenic mice over-expressing adiponectin in the liver showed no bone abnormality (53), whereas in another study transient over-expression of adiponectin in mice increased trabecular bone mass and reduced osteoclast number and bone resorption (50). A recent in vivo study showed that the effects of adiponectin in bone are age-dependent (54). In young animals, the dominant effect of adiponectin is the attenuation of bone formation through inhibition of osteoblast proliferation and induction of their apoptosis, whereas in older animals, these local effects are masked by the activity of adiponectin to decrease the sympathetic tone, indirectly leading to an increase in bone mass. Thus, according to this study, adiponectin has both positive and negative effects on bone mass and adiponectin-deficient mice show increase in bone mass at a young age and an osteoporotic phenotype at 36 weeks (54). In another recent study, Wang et al. (55) compared the effect of ovariectomy on BMD and biomechanical strength in wild-type and adiponectin-deficient mice. The study showed that adiponectin-deficiency protects against ovariectomy-induced osteoporosis in mice, and suggested that a possible underlying mechanism for that effect is the enhanced osteogenic differentiation of mesenchymal stem cells identified in the adiponectin-deficient mice.
Other Adipokines
Resistin, fasting-induced adipose factor (FIAF), visfatin, vaspin, and apelin have been identified in recent years as additional factors secreted from adipocytes (56). Only a few clinical studies investigated the relationship between circulating levels of these factors and BMD, and studies of their activities in bone cells in vitro are still in preliminary stages. Most of the clinical studies that investigated the relationship between a number of the adipokines, including apelin, resistin, and visfatin and BMD found no significant correlations (34, 35, 57). A systematic review and meta-analysis found no association between resistin or visfatin and BMD and the authors concluded that inconsistent associations between these adipokines and BMD are probably confounded by body composition, in particular fat mass parameters (58).
The activities of FIAF, resistin, and visfatin in bone cells have been studied in vitro. FIAF is cleaved in vivo into an N-terminal coiled-coil domain [FIAF (CCD)] and a C-terminal fibrinogen-like domain [FIAF (FLD)] (59, 60). FIAF (CCD) inhibited osteoclast differentiation and function in mouse primary bone marrow and in RAW264.7 cell cultures, and decreased expression of genes encoding key osteoclastogenic factors such as M-CSF, connective tissue growth factor (CTGF), NFATc1, and DC-STAMP (61). FIAF (FLD) and intact FIAF were without effect in these experimental systems. In addition, FIAF (CCD) stimulated the proliferation of rat primary osteoblasts, but had no effect on osteoblasts differentiation. Thommesen et al. found that resistin is expressed in primary human bone marrow stem cells and in mature human osteoblasts (62). The expression of resistin mRNA in RAW 264.7 is increased during differentiation into mature osteoclasts, and treatment with recombinant resistin increased the number of differentiated osteoclasts in RAW 264.7 cells and enhanced the proliferation of MC3T3-E1 osteoblast-like cells. Studies of visfatin, an adipokine highly expressed in visceral fat and up-regulated in obesity and type-2 diabetes, showed that it has insulin-mimetic effects in various cell lines. In human primary osteoblasts, similar to insulin, visfatin enhanced glucose uptake, proliferation, and type I collagen synthesis (63). Further studies of the different adipokines and their effects in bone in vitro and in vivo are required to determine their possible role in the positive association between fat and bone.
Beta-Pancreatic Hormones
Insulin resistance in obesity leads to hypersecretion of insulin from β-pancreatic cells and two other hormones co-secreted with insulin, amylin, and preptin, also circulate at high levels in obesity. As these three β-pancreatic hormones directly affect bone cells they are likely to contribute to the fat mass/bone mass relationship.
Insulin
A large number of clinical studied in women and men determined that circulating insulin concentrations are related to BMD (64–68). Osteoblasts express insulin and IGF-1 receptors and in vitro, insulin directly stimulates osteoblast proliferation (69, 70). When administered locally over the calvarias of adult male mice, insulin produces two- to three-fold increases in histomorphometric indices of bone formation (71).
In recent years, insulin signaling in bone was the focus of a number of studies that proposed coupling between bone remodeling and glucose and energy metabolism mediated through an endocrine function of bone cells (72–74). In rodent models, insulin induces the expression of osteocalcin in osteoblasts, and inhibits the expression of osteoprotegerin, therefore stimulating osteoclast activity. Osteoclast-mediated resorption releases uncarboxylated osteocalcin into the circulation, where it ultimately enhances pancreatic insulin production (73, 75). Uncarboxylated osteocalcin is therefore a hormone secreted from bone, which acts as a positive regulator of insulin secretion, and was also shown to promote insulin sensitivity in peripheral tissues. However, the relevance of these novel findings to human physiology is uncertain. Antiresorptive therapies for osteoporosis reduce bone formation markers, including osteocalcin, and therefore patients treated with these medications would be expected to have increased risk for insulin resistance and diabetes. However, a post hoc analysis of large randomized placebo-controlled trials of antiresorptive therapies found that suppression of bone resorption, which significantly reduces the level of circulating osteocalcin, does not have a clinically important effect on fasting glucose or diabetes incidence (76).
In addition to its direct activity in bone cells, insulin also affects BMD indirectly, through increasing the levels of circulating sex hormones and saturated fatty acids (1). Clinical studies show that women with hyperinsulinemia have increased production estrogen and androgen in the ovary and reduced production of sex hormone binding globulin in the liver. These indirect mechanisms contribute to the high BMD in states of hyperinsulinemia, including obesity and generalized lipodystrophy (1).
Amylin
Amylin is a peptide hormone that belongs to the calcitonin family and is evolutionary related to insulin. In vitro, amylin directly stimulates osteoblast proliferation (77) and inhibits osteoclast differentiation and activity, although with lower potency than calcitonin (78). Systemic administration of amylin substantially increases bone volume in mice (79) and rats (80). In a study of the osteogenic effects of amylin in OVX rats, amylin protected the animals against OVX-induced bone loss at the distal metaphysis and total femora (81). Higher circulating levels of osteocalcin and lower levels of urinary deoxypyridinoline excretion in the amylin-treated animals in comparison to OVX untreated rats, suggests that amylin acts through the stimulation of osteoblast activity as well as inhibition of bone resorption. A recent study tested the osteogenic activity of amylin in normal rats, fructose-induced insulin-resistant and streptozotocin-induced type-2 diabetic rats (82). Infusion of amylin for 3 days had different outcomes in the three groups of animals. In normal rats, amylin induced an increase in bone formation rate and reduced osteoclast surface and erosive surface in the femur, in type-2 diabetic rats amylin normalized trabecular structure parameters and increased osteoblast number, whereas in insulin-resistant rats amylin appeared to have no osteogenic effects (82).
The phenotype of amylin-deficient mice was described by Dacquin et al. (83). In this model, amylin deficiency did not alter the regulation of food intake, body weight, and glucose metabolism. At the age of 24 weeks, amylin-deficient mice had a typical osteoporotic phenotype; showing decreased BMD of long bones, low bone mass, and a 50% decrease in trabecular bone volume. The number of osteoblasts and bone formation rate were similar in amylin-deficient mice and wild-type controls, suggesting that the osteoporotic phenotype was not a result of a defect in bone formation. The amylin-deficient mice had an increased number of osteoclasts and an increase in degradation products of collagen in the urine, suggestive of accelerated bone resorption. Further studies of amylin-deficient mice demonstrated that the bone effects were sex-dependent. Amylin-deficient males showed increased trabecular thickness at 4 and 6 weeks of age and increased femoral length at 26 weeks, whereas female mice were no different than the wild type (84).
Only a small number of studies investigated the bone effects of amylin in humans. In a study of 16 women with anorexia nervosa and 15 healthy controls, Wojcik et al. found that amylin levels were positively associated with BMD and Z-scores at the femoral neck and total hip, and the association remained significant after controlling for weight or fat mass (85). In addition, the study found that women with anorexia nervosa had significantly lower levels of fasting amylin, suggesting that amylin could be one of the mechanisms underlying bone loss in anorexia nervosa (85). In another study, a group of patients with type 1 diabetes were treated with pramlintide (amylin analog) for 12 months (86). Although these patients were expected to benefit from the therapy due to the chronic amylin deficiency associated with diabetes, no change in BMD or biochemical markers of bone metabolism was observed following the treatment.
Preptin
Preptin, a peptide that corresponds to Asp69–Leu102 of pro-insulin-like growth factor-2 (pro-IGF-2), was identified as an additional molecule stored in secretory vesicles of pancreatic beta cells, and co-secreted with insulin and amylin (87). Preptin increases glucose-mediated insulin secretion (87). Circulating levels of preptin were found to be higher in diabetic patients compared to patients with impaired glucose tolerance and controls, suggesting a potential role for preptin in the pathogenesis of type-2 diabetes mellitus (88). Similar to the other beta-pancreatic hormones, preptin has anabolic activity in bone.
In vitro studies demonstrated that preptin stimulates the proliferation of primary rat osteoblasts and osteoblast-like cell lines and reduces osteoblast apoptosis induced by serum deprivation (89). Preptin-induced phosphorylation of p42/p44 MAP kinases in osteoblastic cells and its proliferative effects were blocked by MAP kinase inhibitors (89). In human primary osteoblasts, preptin promoted proliferation and alkaline phosphatase activity through induction of p42/p44 MAP kinase and CTGF (90). Preptin did not affect bone resorption in mouse bone marrow cultures (89). In contrast, a recent study that compared the effect of IGF-I, IGF-II, insulin, and preptin on human bone cells, suggested that preptin induces differentiation and activity of both osteoblasts and osteoclasts (91). In vivo, local administration of preptin increased bone formation and bone area in adult male mice (89).
Indications for the anabolic effect of preptin on bone in humans arise from a number of clinical observations. Increased circulating levels of a pro-IGF-2-(89-101) peptide in complex with IGF-binding protein-2 have been implicated in osteosclerosis observed in a number of patients with chronic hepatitis C infections (92). Patients with excess of other forms of pro-IGF-2, which do not contain the preptin sequence, do not develop osteosclerosis. A recent study measured preptin levels and bone metabolic markers in serum samples from male patients with osteoporosis, osteopenia, and normal bone mass (93). Serum preptin levels were lowest in the osteoporosis group, and were positively correlated with bone formation markers, whereas no correlation was observed with markers of bone resorption.
Gastrointestinal Hormones
Diet plays a critical role in mineral homeostasis as bone turnover is acutely responsive to food intake (94, 95). Feeding results in a decrease of up to 50% in markers of bone resorption and a more modest decrease in markers of bone formation (94, 96). Markers of bone resorption decrease 20 min after food intake and return to baseline after 6 h. In a study of controlled food intake over 5 days, a 70% energy restriction produced about 30% decrease in markers of bone formation and a similar increase in markers of bone resorption (97). A number of peptide hormones secreted from the gastrointestinal tract in response to feeding affect bone tissue, either directly or indirectly, and thus contribute to the coupling between feeding status and bone turnover.
Ghrelin
Ghrelin, a growth hormone (GH) secretagogue, is a peptide hormone primarily synthesized by cells in the stomach and released in response to fasting, such that circulating levels are maximal prior to meals and fall upon feeding (98, 99). In vivo, ghrelin is an orexigenic factor, increasing food intake and inducing release of GH from the pituitary via the hypothalamus.
The ghrelin receptor, GHS-R, is expressed in osteoblastic cells, but we found that ghrelin only weakly activated cell proliferation in cultures of primary rat and human osteoblasts (100). Other groups have shown that ghrelin increases proliferation and differentiation in cultures of animal and human osteoblastic cells (101–105). There is also evidence for ghrelin expression and secretion by osteoblast (101). The effect of ghrelin on bone was studied in a number of animal models. Ghrelin infusion increased BMD in rats, and a similar effect was observed in GH-deficient rats, suggesting the existence of a GH-independent effect of ghrelin in bone (102). In contrast, ghrelin knockout mice and GHS-R knockout mice had no changes in either bone mineral content or density in comparison to control mice (106, 107). Transgenic mice over-expressing ghrelin in the brain and stomach showed increased food intake and impaired glucose tolerance but otherwise had normal body size (108). No analysis of bone metabolism was carried out in this study.
In humans, 4 h of ghrelin infusion in healthy subjects and post-gastrostomy patients showed no acute effects on markers of bone turnover (109). Treatment with an oral ghrelin mimetic for 12 months was associated with a small increase in femoral neck BMD, whereas no changes were observed in other sites (110).
Glucose-Dependent Insulinotropic Peptide
Glucose-dependent insulinotropic polypeptide or gastric inhibitory polypeptide (GIP) is a 42 amino acid protein secreted from K-cells of the duodenum and from the small intestine in response to feeding, and induces insulin and glucagon secretion from the pancreas (111).
GIP receptor is expressed in osteoblasts, osteocytes, and osteoclasts (95) and in vitro, GIP increases osteoblast number and activity and inhibits osteoclast activity, suggestive of an overall direct anabolic effect in bone (112–114). Transgenic mice over-expressing GIP have increased bone formation, decreased bone resorption, and increased bone mass, and a long-term study also demonstrated reduced bone loss with aging (115, 116). In addition, administration of GIP reduces bone loss in ovariectomized mice (112). Mice lacking the GIP receptor, who have blunted insulin response to feeding, have low bone mass with reduced bone formation and increased numbers of mature osteoclasts (117, 118). In humans, 48 h after an intravenous bolus of GIP, there were no significant changes in serum levels of either markers of bone resorption or bone formation (119).
Glucagon-Like Peptides
Glucagon-like peptide-1 (GLP-1) and GLP-2 are two hormones derived from proglucagon precursor by proteolytic cleavage. These hormones are co-secreted from intestinal L-cells and their levels rise rapidly after food intake. Similar to GIP, GLP-1 stimulates insulin secretion from the pancreas, whereas GLP-2 acts in the intestine to stimulate mucosal growth and nutrient absorption (94, 95). A number of GLP-1 analogs are being developed as pharmaceutical agents for the treatment of type-2 diabetes, as GLP-1 increases postprandial insulin secretion, suppresses postprandial glucagon secretion, and delays gastric emptying (120). A small clinical study compared the postprandial plasma concentrations of GLP-1 and GLP-2 between a group that had previously consumed calcium phosphate-enriched diet for 3 weeks and a control group. The study showed that postprandial GLP-1 and GLP-2 levels were significantly increased in the group that had calcium phosphate-enriched diet, whereas levels of insulin and glucose showed no differences between the groups (121).
Although GLP-1 receptor is expressed in bone marrow stromal cells and in immature osteoblasts, it is not expressed in mature osteoblasts and therefore it is unlikely to have direct activity in these cells (113, 122, 123). Evidence from in vitro experiments suggests no direct effect of GLP-1 in either osteoblasts or osteoclasts (124). However, GLP-1 receptors are expressed in thyroid C cells, where GLP-1 induces postprandial calcitonin release and thus indirect inhibition of bone resorption (124). Mice deficient of the GLP-1 receptor have cortical osteopenia and increased osteoclast numbers and bone resorption activity (124).
In contrast to GLP-1, GLP-2 receptors are expressed on osteoclasts and GLP-2 has been shown to decrease bone resorption in vitro (125). In several human studies, administration of exogenous GLP-2 reduced bone resorption, as measured by reduction in circulating bone turnover markers whereas bone formation markers were unaffected (119, 126, 127).
Conclusion
The mechanisms of communication between different tissues in the body are very complex and the various in vitro and in vivo experimental systems designed to investigate these mechanisms do not always produce congruent results. In the study of the relationship between fat mass and bone mass, the most consistent clinical observation is that body weight is positively correlated with BMD and negatively associated with fracture risk. Of the numerous laboratory studies of the possible mediators of the fat–bone association, the current review focused on studies of the direct bone effects of adipokines (summarized in Figure 1A) and of hormones that respond to feeding (Figure 1B). Hormones secreted in response to feeding shift bone turnover balance to favor formation, whereas low levels of these hormones induce bone resorption. It is likely that these responses have evolved to promote bone mass and strength when nutrition is abundant and to maintain calcium homeostasis in times of food shortage. Other important physiological links between fat and bone, which were not discussed in detail here, include their indirect communication through the central nervous system, and the fact that osteoblasts and adipocytes differentiate from a common mesenchymal precursor cell. In vitro studies in combination with investigations of genetically modified animals substantially advanced our understanding of the relationship and crosstalk between fat and bone tissue. However, only clinical studies can ultimately determine whether hypotheses produced using experimental model systems have relevance to human physiology and pathology.
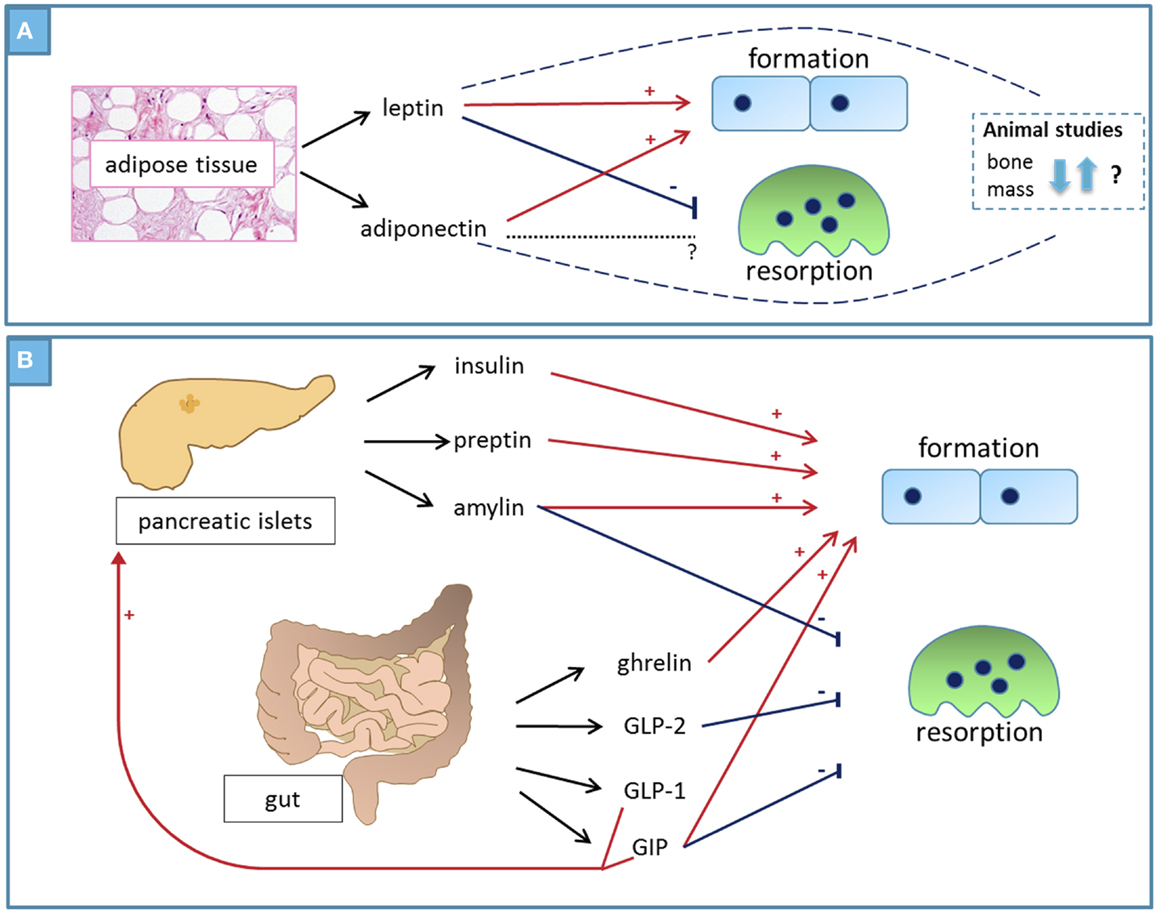
Figure 1. Summary – factors that mediate the positive association between fat and bone. (A) Adipokines: leptin directly increases osteoblast number and activity while inhibiting osteoclast activity, producing an overall anabolic bone effect. Animal studies produced conflicting results, some suggesting anabolic effect on bones and others showing negative bone effects through indirect mechanisms. Adiponectin – circulating levels of adiponectin correlate negatively with body fat and there is strong inverse relationship between circulating adiponectin and BMD. Adiponectin directly increases osteoblast number and activity, whereas its effects in osteoclasts vary in different experimental systems. The bone phenotype of adiponectin-deficient animals varies with age. (B) Peptide hormones secreted from pancreatic islets and from the gut in response to feeding have direct and indirect anabolic bone effects.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
2. Sadie-Van Gijsen H, Crowther NJ, Hough FS, Ferris WF. The interrelationship between bone and fat: from cellular see-saw to endocrine reciprocity. Cell Mol Life Sci (2013) 70(13):2331–49. doi:10.1007/s00018-012-1211-2
3. Reid IR. Relationships between fat and bone. Osteoporos Int (2008) 19(5):595–606. doi:10.1007/s00198-007-0492-z
4. Reid IR. Relationships among body mass, its components, and bone. Bone (2002) 31(5):547–55. doi:10.1016/S8756-3282(02)00864-5
5. Sornay-Rendu E, Boutroy S, Vilayphiou N, Claustrat B, Chapurlat RD. In obese postmenopausal women, bone microarchitecture and strength are not commensurate to greater body weight: the Os des Femmes de Lyon (OFELY) study. J Bone Miner Res (2013) 28(7):1679–87. doi:10.1002/jbmr.1880
6. Wang MC, Bachrach LK, Van Loan M, Hudes M, Flegal KM, Crawford PB. The relative contributions of lean tissue mass and fat mass to bone density in young women. Bone (2005) 37(4):474–81. doi:10.1016/j.bone.2005.04.038
7. Dimitri P, Bishop N, Walsh JS, Eastell R. Obesity is a risk factor for fracture in children but is protective against fracture in adults: a paradox. Bone (2012) 50(2):457–66. doi:10.1016/j.bone.2011.05.011
8. Gilsanz V, Chalfant J, Mo AO, Lee DC, Dorey FJ, Mittelman SD. Reciprocal relations of subcutaneous and visceral fat to bone structure and strength. J Clin Endocrinol Metab (2009) 94(9):3387–93. doi:10.1210/jc.2008-2422
9. Compston J. Obesity and bone. Curr Osteoporos Rep (2013) 11(1):30–5. doi:10.1007/s11914-012-0127-y
10. Compston JE, Flahive J, Hosmer DW, Watts NB, Siris ES, Silverman S, et al. Relationship of weight, height, and body mass index with fracture risk at different sites in postmenopausal women: the global longitudinal study of osteoporosis in women (GLOW). J Bone Miner Res (2014) 29(2):487–93. doi:10.1002/jbmr.2051
11. Folli F, Sabowitz BN, Schwesinger W, Fanti P, Guardado-Mendoza R, Muscogiuri G. Bariatric surgery and bone disease: from clinical perspective to molecular insights. Int J Obes (Lond) (2012) 36(11):1373–9. doi:10.1038/ijo.2012.115
12. Ong T, Sahota O, Tan W, Marshall L. A United Kingdom perspective on the relationship between body mass index (BMI) and bone health: a cross sectional analysis of data from the Nottingham Fracture Liaison Service. Bone (2013) 59:207–10. doi:10.1016/j.bone.2013.11.024
13. Brzozowska MM, Sainsbury A, Eisman JA, Baldock PA, Center JR. Bariatric surgery, bone loss, obesity and possible mechanisms. Obes Rev (2013) 14(1):52–67. doi:10.1111/j.1467-789X.2012.01050.x
14. Leslie WD, Rubin MR, Schwartz AV, Kanis JA. Type 2 diabetes and bone. J Bone Miner Res (2012) 27(11):2231–7. doi:10.1002/jbmr.1759
15. Nakamura KM, Haglind EG, Clowes JA, Achenbach SJ, Atkinson EJ, Melton LJ III, et al. Fracture risk following bariatric surgery: a population-based study. Osteoporos Int (2014) 25(1):151–8. doi:10.1007/s00198-013-2463-x
16. Nielson CM, Srikanth P, Orwoll ES. Obesity and fracture in men and women: an epidemiologic perspective. J Bone Miner Res (2012) 27(1):1–10. doi:10.1002/jbmr.1486
17. Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature (1998) 395(6704):763–70. doi:10.1038/27376
18. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature (1994) 372(6505):425–32. doi:10.1038/372425a0
19. Cornish J, Callon KE, Bava U, Lin C, Naot D, Hill BL, et al. Leptin directly regulates bone cell function in vitro and reduces bone fragility in vivo. J Endocrinol (2002) 175(2):405–15. doi:10.1677/joe.0.1750405
20. Maor G, Rochwerger M, Segev Y, Phillip M. Leptin acts as a growth factor on the chondrocytes of skeletal growth centers. J Bone Miner Res (2002) 17(6):1034–43. doi:10.1359/jbmr.2002.17.6.1034
21. Steppan CM, Crawford DT, Chidsey-Frink KL, Ke H, Swick AG. Leptin is a potent stimulator of bone growth in ob/ob mice. Regul Pept (2000) 92(1–3):73–8. doi:10.1016/S0167-0115(00)00152-X
22. Thomas T, Gori F, Khosla S, Jensen MD, Burguera B, Riggs BL. Leptin acts on human marrow stromal cells to enhance differentiation to osteoblasts and to inhibit differentiation to adipocytes. Endocrinology (1999) 140(4):1630–8. doi:10.1210/en.140.4.1630
23. Holloway WR, Collier FM, Aitken CJ, Myers DE, Hodge JM, Malakellis M, et al. Leptin inhibits osteoclast generation. J Bone Miner Res (2002) 17(2):200–9. doi:10.1359/jbmr.2002.17.2.200
24. Cornish J, Naot D, Costa JL. The bone-fat mass relationship: laboratory studies. IBMS BoneKey (2009) 6:311–22. doi:10.1138/20090395
25. Hamrick MW, Della-Fera MA, Choi YH, Pennington C, Hartzell D, Baile CA. Leptin treatment induces loss of bone marrow adipocytes and increases bone formation in leptin-deficient ob/ob mice. J Bone Miner Res (2005) 20(6):994–1001. doi:10.1359/JBMR.050103
26. Williams GA, Callon KE, Watson M, Costa JL, Ding Y, Dickinson M, et al. Skeletal phenotype of the leptin receptor-deficient db/db mouse. J Bone Miner Res (2011) 26(8):1698–709. doi:10.1002/jbmr.367
27. Burguera B, Hofbauer LC, Thomas T, Gori F, Evans GL, Khosla S, et al. Leptin reduces ovariectomy-induced bone loss in rats. Endocrinology (2001) 142(8):3546–53. doi:10.1210/en.142.8.3546
28. Ducy P, Amling M, Takeda S, Priemel M, Schilling AF, Beil FT, et al. Leptin inhibits bone formation through a hypothalamic relay: a central control of bone mass. Cell (2000) 100(2):197–207. doi:10.1016/S0092-8674(00)81558-5
29. Bartell SM, Rayalam S, Ambati S, Gaddam DR, Hartzell DL, Hamrick M, et al. Central (ICV) leptin injection increases bone formation, bone mineral density, muscle mass, serum IGF-1, and the expression of osteogenic genes in leptin-deficient ob/ob mice. J Bone Miner Res (2011) 26(8):1710–20. doi:10.1002/jbmr.406
30. Gimble JM. Leptin’s balancing act between bone and fat. J Bone Miner Res (2011) 26(8):1694–7. doi:10.1002/jbmr.445
31. Blain H, Vuillemin A, Guillemin F, Durant R, Hanesse B, de Talance N, et al. Serum leptin level is a predictor of bone mineral density in postmenopausal women. J Clin Endocrinol Metab (2002) 87(3):1030–5. doi:10.1210/jc.87.3.1030
32. Thomas T, Burguera B, Melton LJ III, Atkinson EJ, O’Fallon WM, Riggs BL, et al. Role of serum leptin, insulin, and estrogen levels as potential mediators of the relationship between fat mass and bone mineral density in men versus women. Bone (2001) 29(2):114–20. doi:10.1016/S8756-3282(01)00487-2
33. Mohiti-Ardekani J, Soleymani-Salehabadi H, Owlia MB, Mohiti A. Relationships between serum adipocyte hormones (adiponectin, leptin, resistin), bone mineral density and bone metabolic markers in osteoporosis patients. J Bone Miner Metab (2013). doi:10.1007/s00774-013-0511-4
34. Peng XD, Xie H, Zhao Q, Wu XP, Sun ZQ, Liao EY. Relationships between serum adiponectin, leptin, resistin, visfatin levels and bone mineral density, and bone biochemical markers in Chinese men. Clin Chim Acta (2008) 387(1–2):31–5. doi:10.1016/j.cca.2007.08.012
35. Zhang H, Xie H, Zhao Q, Xie GQ, Wu XP, Liao EY, et al. Relationships between serum adiponectin, apelin, leptin, resistin, visfatin levels and bone mineral density, and bone biochemical markers in post-menopausal Chinese women. J Endocrinol Invest (2010) 33(10):707–11. doi:10.3275/6886
36. Barbour KE, Zmuda JM, Boudreau R, Strotmeyer ES, Horwitz MJ, Evans RW, et al. Adipokines and the risk of fracture in older adults. J Bone Miner Res (2011) 26(7):1568–76. doi:10.1002/jbmr.361
37. Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun (1999) 257(1):79–83. doi:10.1006/bbrc.1999.0255
38. Nakano Y, Tobe T, Choi-Miura NH, Mazda T, Tomita M. Isolation and characterization of GBP28, a novel gelatin-binding protein purified from human plasma. J Biochem (1996) 120(4):803–12. doi:10.1093/oxfordjournals.jbchem.a021483
39. Wang Y, Lam KS, Chan L, Chan KW, Lam JB, Lam MC, et al. Post-translational modifications of the four conserved lysine residues within the collagenous domain of adiponectin are required for the formation of its high molecular weight oligomeric complex. J Biol Chem (2006) 281(24):16391–400. doi:10.1074/jbc.M513907200
40. Ye R, Scherer PE. Adiponectin, driver or passenger on the road to insulin sensitivity? Mol Metab (2013) 2(3):133–41. doi:10.1016/j.molmet.2013.04.001
41. Jurimae J, Jurimae T, Ring-Dimitriou S, LeMura LM, Arciero PJ, von Duvillard SP. Plasma adiponectin and insulin sensitivity in overweight and normal-weight middle-aged premenopausal women. Metabolism (2009) 58(5):638–43. doi:10.1016/j.metabol.2008.12.009
42. Lenchik L, Register TC, Hsu FC, Lohman K, Nicklas BJ, Freedman BI, et al. Adiponectin as a novel determinant of bone mineral density and visceral fat. Bone (2003) 33(4):646–51. doi:10.1016/S8756-3282(03)00237-0
43. Richards JB, Valdes AM, Burling K, Perks UC, Spector TD. Serum adiponectin and bone mineral density in women. J Clin Endocrinol Metab (2007) 92(4):1517–23. doi:10.1210/jc.2006-2097
44. Berner HS, Lyngstadaas SP, Spahr A, Monjo M, Thommesen L, Drevon CA, et al. Adiponectin and its receptors are expressed in bone-forming cells. Bone (2004) 35(4):842–9. doi:10.1016/j.bone.2004.06.008
45. Shinoda Y, Yamaguchi M, Ogata N, Akune T, Kubota N, Yamauchi T, et al. Regulation of bone formation by adiponectin through autocrine/paracrine and endocrine pathways. J Cell Biochem (2006) 99(1):196–208. doi:10.1002/jcb.20890
46. Luo XH, Guo LJ, Yuan LQ, Xie H, Zhou HD, Wu XP, et al. Adiponectin stimulates human osteoblasts proliferation and differentiation via the MAPK signaling pathway. Exp Cell Res (2005) 309(1):99–109. doi:10.1016/j.yexcr.2005.05.021
47. Oshima K, Nampei A, Matsuda M, Iwaki M, Fukuhara A, Hashimoto J, et al. Adiponectin increases bone mass by suppressing osteoclast and activating osteoblast. Biochem Biophys Res Commun (2005) 331(2):520–6. doi:10.1016/j.bbrc.2005.03.210
48. Williams GA, Wang Y, Callon KE, Watson M, Lin JM, Lam JB, et al. In vitro and in vivo effects of adiponectin on bone. Endocrinology (2009) 150(8):3603–10. doi:10.1210/en.2008-1639
49. Luo XH, Guo LJ, Xie H, Yuan LQ, Wu XP, Zhou HD, et al. Adiponectin stimulates RANKL and inhibits OPG expression in human osteoblasts through the MAPK signaling pathway. J Bone Miner Res (2006) 21(10):1648–56. doi:10.1359/jbmr.060707
50. Yamaguchi N, Kukita T, Li YJ, Kamio N, Fukumoto S, Nonaka K, et al. Adiponectin inhibits induction of TNF-alpha/RANKL-stimulated NFATc1 via the AMPK signaling. FEBS Lett (2008) 582(3):451–6. doi:10.1016/j.febslet.2007.12.037
51. Naot D, Williams GA, Lin JM, Cornish J, Grey A. Evidence that contamination by lipopolysaccharide confounds in vitro studies of adiponectin activity in bone. Endocrinology (2012) 153(5):2076–81. doi:10.1210/en.2011-2004
52. Turner JJ, Smolinska MJ, Sacre SM, Foxwell BM. Induction of TLR tolerance in human macrophages by adiponectin: does LPS play a role? Scand J Immunol (2009) 69(4):329–36. doi:10.1111/j.1365-3083.2008.02224.x
53. Yamaguchi N, Kukita T, Li YJ, Martinez Argueta JG, Saito T, Hanazawa S, et al. Adiponectin inhibits osteoclast formation stimulated by lipopolysaccharide from Actinobacillus actinomycetemcomitans. FEMS Immunol Med Microbiol (2007) 49(1):28–34. doi:10.1111/j.1574-695X.2006.00164.x
54. Kajimura D, Lee HW, Riley KJ, Arteaga-Solis E, Ferron M, Zhou B, et al. Adiponectin regulates bone mass via opposite central and peripheral mechanisms through FoxO1. Cell Metab (2013) 17(6):901–15. doi:10.1016/j.cmet.2013.04.009
55. Wang F, Wang PX, Wu XL, Dang SY, Chen Y, Ni YY, et al. Deficiency of adiponectin protects against ovariectomy-induced osteoporosis in mice. PLoS One (2013) 8(7):e68497. doi:10.1371/journal.pone.0068497
56. Magni P, Dozio E, Galliera E, Ruscica M, Corsi MM. Molecular aspects of adipokine-bone interactions. Curr Mol Med (2010) 10(6):522–32. doi:10.2174/156652410792231295
57. Sucunza N, Barahona MJ, Resmini E, Fernandez-Real JM, Ricart W, Farrerons J, et al. A link between bone mineral density and serum adiponectin and visfatin levels in acromegaly. J Clin Endocrinol Metab (2009) 94(10):3889–96. doi:10.1210/jc.2009-0474
58. Biver E, Salliot C, Combescure C, Gossec L, Hardouin P, Legroux-Gerot I, et al. Influence of adipokines and ghrelin on bone mineral density and fracture risk: a systematic review and meta-analysis. J Clin Endocrinol Metab (2011) 96(9):2703–13. doi:10.1210/jc.2011-0047
59. Ge H, Yang G, Huang L, Motola DL, Pourbahrami T, Li C. Oligomerization and regulated proteolytic processing of angiopoietin-like protein 4. J Biol Chem (2004) 279(3):2038–45. doi:10.1074/jbc.M307583200
60. Kersten S, Mandard S, Tan NS, Escher P, Metzger D, Chambon P, et al. Characterization of the fasting-induced adipose factor FIAF, a novel peroxisome proliferator-activated receptor target gene. J Biol Chem (2000) 275(37):28488–93. doi:10.1074/jbc.M004029200
61. Lin JM, Naot D, Watson M, Costa JL, Reid IR, Cornish J, et al. Skeletal actions of fasting-induced adipose factor (FIAF). Endocrinology (2013) 154(12):4685–94. doi:10.1210/en.2013-1238
62. Thommesen L, Stunes AK, Monjo M, Grosvik K, Tamburstuen MV, Kjobli E, et al. Expression and regulation of resistin in osteoblasts and osteoclasts indicate a role in bone metabolism. J Cell Biochem (2006) 99(3):824–34. doi:10.1002/jcb.20915
63. Xie H, Tang SY, Luo XH, Huang J, Cui RR, Yuan LQ, et al. Insulin-like effects of visfatin on human osteoblasts. Calcif Tissue Int (2007) 80(3):201–10. doi:10.1007/s00223-006-0155-7
64. Abrahamsen B, Rohold A, Henriksen JE, Beck-Nielsen H. Correlations between insulin sensitivity and bone mineral density in non-diabetic men. Diabet Med (2000) 17(2):124–9. doi:10.1046/j.1464-5491.2000.00234.x
65. Ahmed LA, Schirmer H, Berntsen GK, Fonnebo V, Joakimsen RM. Features of the metabolic syndrome and the risk of non-vertebral fractures: the Tromso study. Osteoporos Int (2006) 17(3):426–32. doi:10.1007/s00198-005-0003-z
66. Reid IR, Ames R, Evans MC, Sharpe S, Gamble G, France JT, et al. Determinants of total body and regional bone mineral density in normal postmenopausal women – a key role for fat mass. J Clin Endocrinol Metab (1992) 75(1):45–51. doi:10.1210/jc.75.3.779
67. Reid IR, Evans MC, Cooper GJ, Ames RW, Stapleton J. Circulating insulin levels are related to bone density in normal postmenopausal women. Am J Physiol (1993) 265(4 Pt 1):E655–9.
68. Stolk RP, Van Daele PL, Pols HA, Burger H, Hofman A, Birkenhager JC, et al. Hyperinsulinemia and bone mineral density in an elderly population: the Rotterdam study. Bone (1996) 18(6):545–9. doi:10.1016/8756-3282(96)00079-8
69. Hickman J, McElduff A. Insulin promotes growth of the cultured rat osteosarcoma cell line UMR-106-01: an osteoblast-like cell. Endocrinology (1989) 124(2):701–6. doi:10.1210/endo-124-2-701
70. Pun KK, Lau P, Ho PW. The characterization, regulation, and function of insulin receptors on osteoblast-like clonal osteosarcoma cell line. J Bone Miner Res (1989) 4(6):853–62. doi:10.1002/jbmr.5650040610
71. Cornish J, Callon KE, Reid IR. Insulin increases histomorphometric indices of bone formation in vivo. Calcif Tissue Int (1996) 59(6):492–5. doi:10.1007/BF00369216
72. Ferron M, Hinoi E, Karsenty G, Ducy P. Osteocalcin differentially regulates beta cell and adipocyte gene expression and affects the development of metabolic diseases in wild-type mice. Proc Natl Acad Sci USA (2008) 105(13):5266–70. doi:10.1073/pnas.0711119105
73. Fulzele K, Riddle RC, DiGirolamo DJ, Cao X, Wan C, Chen D, et al. Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell (2010) 142(2):309–19. doi:10.1016/j.cell.2010.06.002
74. Lee NK, Sowa H, Hinoi E, Ferron M, Ahn JD, Confavreux C, et al. Endocrine regulation of energy metabolism by the skeleton. Cell (2007) 130(3):456–69. doi:10.1016/j.cell.2007.05.047
75. Lee GS, Simpson C, Sun BH, Yao C, Foer D, Sullivan B, et al. Measurement of plasma, serum, and platelet serotonin in individuals with high bone mass and mutations in LRP5. J Bone Miner Res (2014) 29(4):976–81. doi:10.1002/jbmr.2086
76. Schwartz AV, Schafer AL, Grey A, Vittinghoff E, Palermo L, Lui LY, et al. Effects of antiresorptive therapies on glucose metabolism: results from the FIT, HORIZON-PFT, and FREEDOM trials. J Bone Miner Res (2013) 28(6):1348–54. doi:10.1002/jbmr.1865
77. Cornish J, Callon KE, Cooper GJ, Reid IR. Amylin stimulates osteoblast proliferation and increases mineralized bone volume in adult mice. Biochem Biophys Res Commun (1995) 207(1):133–9. doi:10.1006/bbrc.1995.1163
78. Cornish J, Callon KE, Bava U, Kamona SA, Cooper GJ, Reid IR. Effects of calcitonin, amylin, and calcitonin gene-related peptide on osteoclast development. Bone (2001) 29(2):162–8. doi:10.1016/S8756-3282(01)00494-X
79. Cornish J, Callon KE, King AR, Cooper GJ, Reid IR. Systemic administration of amylin increases bone mass, linear growth, and adiposity in adult male mice. Am J Physiol (1998) 275(4 Pt 1):E694–9.
80. Horcajada-Molteni MN, Chanteranne B, Lebecque P, Davicco MJ, Coxam V, Young A, et al. Amylin and bone metabolism in streptozotocin-induced diabetic rats. J Bone Miner Res (2001) 16(5):958–65. doi:10.1359/jbmr.2001.16.5.958
81. Horcajada-Molteni MN, Davicco MJ, Lebecque P, Coxam V, Young AA, Barlet JP. Amylin inhibits ovariectomy-induced bone loss in rats. J Endocrinol (2000) 165(3):663–8. doi:10.1677/joe.0.1650663
82. Gutierrez-Rojas I, Lozano D, Nuche-Berenguer B, Moreno P, Acitores A, Ramos-Alvarez I, et al. Amylin exerts osteogenic actions with different efficacy depending on the diabetic status. Mol Cell Endocrinol (2013) 365(2):309–15. doi:10.1016/j.mce.2012.11.013
83. Dacquin R, Davey RA, Laplace C, Levasseur R, Morris HA, Goldring SR, et al. Amylin inhibits bone resorption while the calcitonin receptor controls bone formation in vivo. J Cell Biol (2004) 164(4):509–14. doi:10.1083/jcb.200312135
84. Davey RA, Moore AJ, Chiu MW, Notini AJ, Morris HA, Zajac JD. Effects of amylin deficiency on trabecular bone in young mice are sex-dependent. Calcif Tissue Int (2006) 78(6):398–403. doi:10.1007/s00223-005-0286-2
85. Wojcik MH, Meenaghan E, Lawson EA, Misra M, Klibanski A, Miller KK. Reduced amylin levels are associated with low bone mineral density in women with anorexia nervosa. Bone (2010) 46(3):796–800. doi:10.1016/j.bone.2009.11.014
86. Borm AK, Klevesath MS, Borcea V, Kasperk C, Seibel MJ, Wahl P, et al. The effect of pramlintide (amylin analogue) treatment on bone metabolism and bone density in patients with type 1 diabetes mellitus. Horm Metab Res (1999) 31(8):472–5. doi:10.1055/s-2007-978777
87. Buchanan CM, Phillips AR, Cooper GJ. Preptin derived from proinsulin-like growth factor II (proIGF-II) is secreted from pancreatic islet beta-cells and enhances insulin secretion. Biochem J (2001) 360(Pt 2):431–9. doi:10.1042/0264-6021:3600431
88. Yang G, Li L, Chen W, Liu H, Boden G, Li K. Circulating preptin levels in normal, impaired glucose tolerance, and type 2 diabetic subjects. Ann Med (2009) 41(1):52–6. doi:10.1080/07853890802244142
89. Cornish J, Callon KE, Bava U, Watson M, Xu X, Lin JM, et al. Preptin, another peptide product of the pancreatic beta-cell, is osteogenic in vitro and in vivo. Am J Physiol (2007) 292(1):E117–22. doi:10.1152/ajpendo.00642.2005
90. Liu YS, Lu Y, Liu W, Xie H, Luo XH, Wu XP, et al. Connective tissue growth factor is a downstream mediator for preptin-induced proliferation and differentiation in human osteoblasts. Amino Acids (2010) 38(3):763–9. doi:10.1007/s00726-009-0281-4
91. Bosetti M, Sabbatini M, Nicoli E, Fusaro L, Cannas M. Effects and differentiation activity of IGF-I, IGF-II, insulin and preptin on human primary bone cells. Growth Factors (2013) 31(2):57–65. doi:10.3109/08977194.2013.770392
92. Khosla S, Ballard FJ, Conover CA. Use of site-specific antibodies to characterize the circulating form of big insulin-like growth factor II in patients with hepatitis C-associated osteosclerosis. J Clin Endocrinol Metab (2002) 87(8):3867–70. doi:10.1210/jcem.87.8.8783
93. Li N, Zheng YB, Han J, Liang W, Wang JY, Zhou JR, et al. Lower circulating preptin levels in male patients with osteoporosis are correlated with bone mineral density and bone formation. BMC Musculoskelet Disord (2013) 14:49. doi:10.1186/1471-2474-14-49
94. Walsh JS, Henriksen DB. Feeding and bone. Arch Biochem Biophys (2010) 503(1):11–9. doi:10.1016/j.abb.2010.06.020
95. Wong IP, Baldock PA, Herzog H. Gastrointestinal peptides and bone health. Curr Opin Endocrinol Diabetes Obes (2010) 17(1):44–50. doi:10.1097/MED.0b013e3283344a05
96. Clowes JA, Hannon RA, Yap TS, Hoyle NR, Blumsohn A, Eastell R. Effect of feeding on bone turnover markers and its impact on biological variability of measurements. Bone (2002) 30(6):886–90. doi:10.1016/S8756-3282(02)00728-7
97. Ihle R, Loucks AB. Dose-response relationships between energy availability and bone turnover in young exercising women. J Bone Miner Res (2004) 19(8):1231–40. doi:10.1359/JBMR.040410
98. Dornonville de la Cour C, Bjorkqvist M, Sandvik AK, Bakke I, Zhao CM, Chen D, et al. A-like cells in the rat stomach contain ghrelin and do not operate under gastrin control. Regul Pept (2001) 99(2–3):141–50. doi:10.1016/S0167-0115(01)00243-9
99. Tschop M, Smiley DL, Heiman ML. Ghrelin induces adiposity in rodents. Nature (2000) 407(6806):908–13. doi:10.1038/35038090
100. Costa JL, Naot D, Lin JM, Watson M, Callon KE, Reid IR, et al. Ghrelin is an osteoblast mitogen and increases osteoclastic bone resorption in vitro. Int J Pept (2011) 2011:605193. doi:10.1155/2011/605193
101. Delhanty PJ, van der Eerden BC, van der Velde M, Gauna C, Pols HA, Jahr H, et al. Ghrelin and unacylated ghrelin stimulate human osteoblast growth via mitogen-activated protein kinase (MAPK)/phosphoinositide 3-kinase (PI3K) pathways in the absence of GHS-R1a. J Endocrinol (2006) 188(1):37–47. doi:10.1677/joe.1.06404
102. Fukushima N, Hanada R, Teranishi H, Fukue Y, Tachibana T, Ishikawa H, et al. Ghrelin directly regulates bone formation. J Bone Miner Res (2005) 20(5):790–8. doi:10.1359/JBMR.041237
103. Kim SW, Her SJ, Park SJ, Kim D, Park KS, Lee HK, et al. Ghrelin stimulates proliferation and differentiation and inhibits apoptosis in osteoblastic MC3T3-E1 cells. Bone (2005) 37(3):359–69. doi:10.1016/j.bone.2005.04.020
104. MacCarinelli G, Sibilia V, Torsello A, Raimondo F, Pitto M, Giustina A, et al. Ghrelin regulates proliferation and differentiation of osteoblastic cells. J Endocrinol (2005) 184(1):249–56. doi:10.1677/joe.1.05837
105. Wang DH, Hu YS, Du JJ, Hu YY, Zhong WD, Qin WJ. Ghrelin stimulates proliferation of human osteoblastic TE85 cells via NO/cGMP signaling pathway. Endocrine (2009) 35(1):112–7. doi:10.1007/s12020-008-9117-3
106. Sun Y, Ahmed S, Smith RG. Deletion of ghrelin impairs neither growth nor appetite. Mol Cell Biol (2003) 23(22):7973–81. doi:10.1128/MCB.23.22.7973-7981.2003
107. Sun Y, Wang P, Zheng H, Smith RG. Ghrelin stimulation of growth hormone release and appetite is mediated through the growth hormone secretagogue receptor. Proc Natl Acad Sci USA (2004) 101(13):4679–84. doi:10.1073/pnas.0305930101
108. Bewick GA, Kent A, Campbell D, Patterson M, Ghatei MA, Bloom SR, et al. Mice with hyperghrelinemia are hyperphagic and glucose intolerant and have reduced leptin sensitivity. Diabetes (2009) 58(4):840–6. doi:10.2337/db08-1428
109. Huda MS, Durham BH, Wong SP, Dovey TM, McCulloch P, Kerrigan D, et al. Lack of an acute effect of ghrelin on markers of bone turnover in healthy controls and post-gastrectomy subjects. Bone (2007) 41(3):406–13. doi:10.1016/j.bone.2007.05.006
110. Nass R, Pezzoli SS, Oliveri MC, Patrie JT, Harrell FE Jr, Clasey JL, et al. Effects of an oral ghrelin mimetic on body composition and clinical outcomes in healthy older adults: a randomized trial. Ann Intern Med (2008) 149(9):601–11. doi:10.7326/0003-4819-149-9-200811040-00003
111. Meier JJ, Nauck MA, Schmidt WE, Gallwitz B. Gastric inhibitory polypeptide: the neglected incretin revisited. Regul Pept (2002) 107(1–3):1–13. doi:10.1016/S0167-0115(02)00039-3
112. Bollag RJ, Zhong Q, Ding KH, Phillips P, Zhong L, Qin F, et al. Glucose-dependent insulinotropic peptide is an integrative hormone with osteotropic effects. Mol Cell Endocrinol (2001) 177(1–2):35–41. doi:10.1016/S0303-7207(01)00405-1
113. Bollag RJ, Zhong Q, Phillips P, Min L, Zhong L, Cameron R, et al. Osteoblast-derived cells express functional glucose-dependent insulinotropic peptide receptors. Endocrinology (2000) 141(3):1228–35. doi:10.1210/endo.141.3.7366
114. Zhong Q, Itokawa T, Sridhar S, Ding KH, Xie D, Kang B, et al. Effects of glucose-dependent insulinotropic peptide on osteoclast function. Am J Physiol Metab (2007) 292(2):E543–8. doi:10.1152/ajpendo.00364.2006
115. Ding KH, Shi XM, Zhong Q, Kang B, Xie D, Bollag WB, et al. Impact of glucose-dependent insulinotropic peptide on age-induced bone loss. J Bone Miner Res (2008) 23(4):536–43. doi:10.1359/jbmr.071202
116. Xie D, Zhong Q, Ding KH, Cheng H, Williams S, Correa D, et al. Glucose-dependent insulinotropic peptide-overexpressing transgenic mice have increased bone mass. Bone (2007) 40(5):1352–60. doi:10.1016/j.bone.2007.01.007
117. Tsukiyama K, Yamada Y, Yamada C, Harada N, Kawasaki Y, Ogura M, et al. Gastric inhibitory polypeptide as an endogenous factor promoting new bone formation after food ingestion. Mol Endocrinol (2006) 20(7):1644–51. doi:10.1210/me.2005-0187
118. Xie D, Cheng H, Hamrick M, Zhong Q, Ding KH, Correa D, et al. Glucose-dependent insulinotropic polypeptide receptor knockout mice have altered bone turnover. Bone (2005) 37(6):759–69. doi:10.1016/j.bone.2005.06.021
119. Henriksen DB, Alexandersen P, Bjarnason NH, Vilsboll T, Hartmann B, Henriksen EE, et al. Role of gastrointestinal hormones in postprandial reduction of bone resorption. J Bone Miner Res (2003) 18(12):2180–9. doi:10.1359/jbmr.2003.18.12.2180
120. Cho YM, Wideman RD, Kieffer TJ. Clinical application of glucagon-like peptide 1 receptor agonists for the treatment of type 2 diabetes mellitus. Endocrinol Metab (Seoul) (2013) 28(4):262–74. doi:10.3803/EnM.2013.28.4.262
121. Trautvetter U, Jahreis G. Effect of supplementary calcium phosphate on plasma gastrointestinal hormones in a double-blind, placebo-controlled, cross-over human study. Br J Nutr (2014) 111(2):287–93. doi:10.1017/S0007114513002341
122. Pacheco-Pantoja EL, Ranganath LR, Gallagher JA, Wilson PJ, Fraser WD. Receptors and effects of gut hormones in three osteoblastic cell lines. BMC Physiol (2011) 11:12. doi:10.1186/1472-6793-11-12
123. Sanz C, Vazquez P, Blazquez C, Barrio PA, Alvarez Mdel M, Blazquez E. Signaling and biological effects of glucagon-like peptide 1 on the differentiation of mesenchymal stem cells from human bone marrow. Am J Physiol (2010) 298(3):E634–43. doi:10.1152/ajpendo.00460.2009
124. Yamada C, Yamada Y, Tsukiyama K, Yamada K, Udagawa N, Takahashi N, et al. The murine glucagon-like peptide-1 receptor is essential for control of bone resorption. Endocrinology (2008) 149(2):574–9. doi:10.1210/en.2007-1292
125. Karsdal MA, Holst JJ, Henriksen D. GLP-2 reduces bone resorption in vitro via the osteoclast GLP-2 receptor. J Bone Miner Res (2004) 19(S1):S416. doi:10.1002/jbmr.5650191306
126. Henriksen DB, Alexandersen P, Hartmann B, Adrian CL, Byrjalsen I, Bone HG, et al. Disassociation of bone resorption and formation by GLP-2: a 14-day study in healthy postmenopausal women. Bone (2007) 40(3):723–9. doi:10.1016/j.bone.2006.09.025
Keywords: adipokines, adipose tissue, fat–bone association, pancreatic hormones, gastrointestinal peptides
Citation: Naot D and Cornish J (2014) Cytokines and hormones that contribute to the positive association between fat and bone. Front. Endocrinol. 5:70. doi: 10.3389/fendo.2014.00070
Received: 14 February 2014; Accepted: 27 April 2014;
Published online: 09 May 2014.
Edited by:
Gustavo Duque, The University of Sydney, AustraliaReviewed by:
Jennifer Tickner, University of Western Australia, AustraliaBasem M. Abdallah, University of Southern Denmark, Denmark
Copyright: © 2014 Naot and Cornish. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dorit Naot, Department of Medicine, University of Auckland, Private Bag 92019, Auckland 1142, New Zealand e-mail:ZC5uYW90QGF1Y2tsYW5kLmFjLm56