 Joe X. Xie1
Joe X. Xie1 Joseph Isaac Shapiro
Joseph Isaac Shapiro- 1Department of Medicine, University of Colorado School of Medicine, Aurora, CO, USA
- 2Department of Medicine, University of Toledo College of Medicine, Toledo, OH, USA
- 3Department of Medicine, Joan C. Edwards School of Medicine, Marshall University, Huntington, WV, USA
It has been postulated for some time that endogenous digitalis-like substances, also called cardiotonic steroids (CTS), exist, and that these substances are involved in sodium handling. Within the past 20 years, these substances have been unequivocally identified and measurements of circulating and tissue concentrations have been made. More recently, it has been identified that CTS also mediate signal transduction through the Na/K-ATPase, and consequently been implicated in profibrotic pathways. This review will discuss the mechanism of CTS in renal sodium handling and a potential “trade-off” effect from their role in inducing tissue fibrosis.
Introduction
Increased dietary sodium chloride (NaCl) intake has been implicated in cardiovascular and renal diseases for some time (1), and this implication has recently become fairly solid (2). This relationship between dietary sodium intake and cardiovascular disease is demonstrated in several large scale studies, such as the international study of salt and blood pressure (INTERSALT) (3) and the dietary approaches to stop hypertension (DASH) (4). With this relationship so demonstrated, understanding the specific mechanisms underlying the deleterious effects of NaCl becomes timely and relevant to clinical management.
This review will focus on one of the factors linking dietary NaCl to cardiovascular and renal disease. We will specifically discuss the role of digitalis-like factors, also known as endogenous cardiotonic steroids (CTS), which function as innate inhibitors of the Na/K-ATPase (5). Although the existence of these endogenous factors has been controversial (6–8), this is no longer the case. Some of these recent breakthroughs include the chemical identification of specific CTS in experimental animals and humans (9, 10), establishment of normal and pathological concentrations for these substances as well as defining possible roles for CTS in animal models of and human disease states (11–13). We would also stress that the discovery of the cell signaling functions of the Na/K-ATPase and its role in molecular cellular biology (14–16) has also been quite relevant to this field. Here, we will emphasize the role of trade-off with respect to CTS signaling and Na homeostasis.
Renal Salt Reabsorption and the Evidence for “Third Factor”
The microscopic architecture of the kidney involves the attachment of vascular filtering units called glomeruli with tubules that modulate the quantity, electrolytes, and acid-base content of tubular fluid, which ultimately becomes urine. Simplistically, the tubules can be roughly broken down into proximal, where 60–80% of all Na and water reabsorption occur and distal, the nephron segments responsible for the fine tuning of what is excreted as urine.
Clearly the renin–angiotensin–aldosterone system, vasopressin and the sympathetic nervous system are critically important in mammalian volume regulation as well as to the maintenance of blood pressure in the face of a hypovolemic insult (17). However, it is very clear that perturbations in these systems cannot explain natriuretic responses to acute or chronic expansion of blood volume (18). This point was first demonstrated in 1961 in a classic paper by de Wardener and colleagues (19). This study showed that natriuresis induced by saline infusion occurred even if renal perfusion pressure and glomerular filtration rate (GFR, factor 1) and aldosterone concentrations (factor 2) were prevented from changing. This so called “third factor,” which we now understand is (are) CTS, was a “hot” topic in the 1960s and 1970s, and was even incorporated into Guyton’s model for circulatory homeostasis (20). Cort and Lichardus observed that a circulating substance in animals subjected to carotid artery occlusion induced natriuresis in different mammals and inhibited sodium transport in frog skin (21). Buckalew showed that an ultrafiltrate of volume-expanded dogs inhibited sodium transport in toad bladders. They went on to propose that the active substance was an inhibitor of the Na/K-ATPase (22). Gonick and coworkers showed that volume expansion in rats, in fact, produced a chemical which did inhibit the ATPase activity of rat kidneys (11). In 1980, Gruber and Buckalew noted that elevated levels of circulating digoxin-like material was seen in volume-expanded dogs (23). Other important contributions were made in the laboratory of Schrier and de Wardener over the next decade (24–26). However, doubt as to the validity of Na/K-ATPase inhibitors developed during the 1980s and 1990s because of inconsistencies in the reported results. In particular, prevailing CTS assays were based on cross-reactivity of CTS with antibodies to digoxin. This cross-reactivity of the commercially employed anti-digoxin antibodies to CTS varied considerably (27–32). Probably, the most important inconsistency was that digitalis did not appear to be natriuretic in normal subjects (33). On this background, atrial (and brain) natriuretic peptide(s) were discovered, were obviously natriuretic, and their concentrations (which could be easily measured) were increased in volume-expanded states (34–38). Undoubtedly, these points deflected interest from the study of CTS. However, enthusiasm was renewed in the recent past for the following reasons. First, several CTS have been isolated from experimental animals and humans and chemically characterized. Specifically, marinobufagenin (MBG) as well as telecinobufagin (TCB) have been isolated from plasma and urine (9). Ouabain has also been identified although there is still some debate as to whether this is ouabain or something distinct, which also reacts to anti-ouabain antibodies (10, 39). The concentrations of ouabain (or ouabain like compound) and MBG appear to be in the range of 200–2700/min in humans, depending on whether disease is present (5, 40, 41). Plasma levels of TCB and bufalin are less well defined at present. Also, quite importantly, a signal cascade has been identified, which does not appear to involve enzymatic inhibition of the Na/K-ATPase. This signaling pathway involves CTS binding of the caveolar Na/K-ATPase in the company of Src and the EGFR and the elaboration of a signal cascade, which involves the generation of reactive oxygen species (ROS) (14, 16). Both of these concepts have been extensively reviewed (42–44).
“Trade-Off” Concept, a Historical Perspective
The concept of “trade-off” plays an extremely powerful role in physiology. This is perhaps best described by Neal Bricker who postulated that in renal disease, the hormonal forces driving nephrons to maintain fluid and electrolyte homeostasis would be complicated by the untoward consequences of these elevated hormones mediating other effects, essentially creating the signs, symptoms, and pathophysiologic changes associated with the uremic syndrome (45, 46). As sodium (Na) handling is so critical to volume balance, electrolyte homeostasis, and acid-base status, it is not surprising that Bricker formulated this hypothesis to involve the Na/K-ATPase.
Bricker speculated that an inhibitor of the Na/K-ATPase would circulate in increased concentration as a response to decreased GFR in order to maintain Na homeostasis (45). This inhibition would subsequently lead to decreased renal Na reabsorption, hence the maintenance of Na homeostasis (Figure 1). Unintended effects of higher concentrations of this Na/K-ATPase inhibitor would be responsible for some of the symptoms, signs, and abnormal laboratory results seen with chronic renal failure as well as potentially contribute to the progressive nature of chronic kidney disease (45, 47–50). As we will detail in this review, a potential consequence of increases in natriuretic hormone levels, specifically elevated CTS levels may be the profibrotic effects of these molecules (51). Before we address this, however, it may be useful to briefly discuss the evolution of our understanding of the Na/K-ATPase (45, 46), which had been described and characterized several decades before (52).
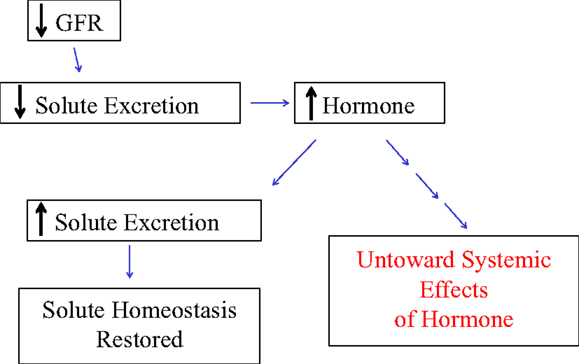
Figure 1. The schematic in figure demonstrates Bricker’s proposed trade-off mechanism by which physiologic changes such as reduced glomerular filtration rate (GFR) leading to the increased generation of a hormone can produce the desired effect in solute homeostasis, but with untoward effects in renal and other tissues.
Discovery of the Na/K-ATPase, Its Role in Signaling Cascades vs. Ion Transportation
The Na/K-ATPase was discovered by Skou in 1957 (53). This protein was demonstrated to be responsible for the electrogenic exchange of sodium and potassium (54). The Na/K-ATPase, also called the sodium pump, is present in all living cells (55). Although there has been some evolutionary modification of the sodium pump, in all multicellular animal cells, the sodium pump consists of (at least) a dimer of an alpha and beta subunit and is considered a member of P-type ATPases (43). Different isoforms of the alpha and beta subunits have been identified and are believed to have functional differences, a topic which has been extensively reviewed (56). Genes encoding the alpha-1 and alpha-2 isoforms reside on the chromosome 1 whereas alpha-3 appears to be coded for on chromosome 19 and alpha-4 (present only in sperm) is mapped to chromosome 13 in humans (57). The act of pumping sodium and potassium is accompanied by changes in conformation and phosphorylation state (43). It also requires energy provided by the hydrolysis of ATP as was initially identified also by Skou (58).The work of Skou was ultimately matured into the currently accepted Post-Albers model for Na/K-ATPase pumping function (43). The alpha 1 subunit of the Na/K-ATPase has 11 transmembrane domains as well as several well defined cytosolic regions referred to as the N, P, catalytic, and A domains (43). Interestingly, the development and maintenance on an evolutionary scale of caveolin and Src binding motifs, which are scattered throughout these cytosolic domains appeared to occur between single celled animal structures and slime mold (59).
In the late 1990s, the laboratory of Dr. Zijian Xie added a significant wrinkle to this understanding. While it is certainly possible that some signaling does occur through the chemical inhibition of the plasmalemmal Na/K-ATPase, it does appear that other mechanisms must be proposed to explain the signaling. In fact, it appears that the specific Na/K-ATPase molecules responsible for the greatest amount of signaling in response to the binding of CTS are actually not involved in pumping sodium or potassium (60). In the late 1990s, Dr. Xie and colleagues observed that in neonatal cardiac myocytes, ouabain caused increases in ROS measured with CMDCF (14). It was further noted that some of the downstream effects of ouabain were blocked by N-acetyl cysteine (NAC) or vitamin E. These increases in ROS could be demonstrated even when cytosolic calcium was maintained low by removal of extracellular calcium (16). It was further noted that Ras activation appeared to be necessary to see increases in ROS (16). Other studies determined that interactions between the Na/K-ATPase and Src appeared to initiate the signal cascade. The alpha 1 subunit of the Na/K-ATPase binds Src and appears to maintain it in an inactive state. However, binding a CTS appears to alter the Na/K-ATPase structure allowing Src to became activated which, in turn, trans-activates the EGFR, and begins the signal cascade which causes increases in ROS (61–64). The Na/K-ATPase–Src complex appears to function similar to a receptor tyrosine kinase. Downstream activation of PLC, PI(3)K, and PKC has also been established (15, 65–68) (Figure 2). The role of ROS in pump signaling has been extensively reviewed elsewhere (14, 16, 51, 69).
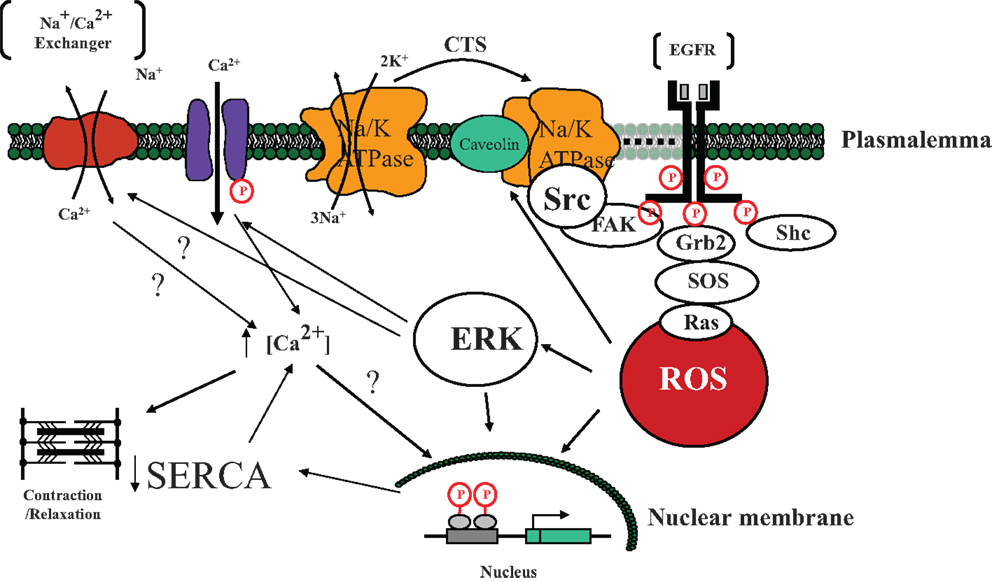
Figure 2. A schematic illustrating the involvement of cardiotonic steroid (CTS) – induced Na/K-ATPase signal cascade initiated by the Na/K-ATPase mediated activation of Src tyrosine kinase and subsequent downstream targets eventually leading to the development of reactive oxygen species (ROS). Specifically, we postulate that in the microdomain of caveolae, the Na/K-ATPase functions as a scaffolding protein, interacting with CTS and changing conformation so as to active Src. Src then trans-activates the EGFR which leads to a signal cascade involving FAK, Shc, Grb2, and SOS resulting in the generation of ROS which in turn activates additional Na/K-ATPase molecules as well as causes downstream activation of ERK as well as effects on the nuclear transcription (43). ERK activation has effects on both L-type channels and possibly the Na/Ca exchanger with net effect to increase cytosolic Ca in some tissues (15). Nuclear effects in myocardial tissue include downregulation of SERCA transcription and translation (70). Abbreviations: EGFR, epidermal growth factor receptor; FAK, focal adhesion kinase; Shc, Src homology-2 domain containing protein; Grb2, growth factor receptor-bound protein-2; SOS, son of sevenless protein; ERK, extracellular-signal-regulated kinase; SERCA, sarcoplasmic/endoplasmic reticulum calcium ATPase.
Although inhibition of the Na/K-ATPase is certainly one possible mechanism by which digitalis and related molecules might “signal,” it is important to emphasize that even transporting epithelia typically have a redundancy of Na/K-ATPase pumping units given that cytosolic Na levels live within a range ideally suited to regulate Na/K-ATPase activity. While it is possible that certain compartments of the cell see higher local concentrations of Na with modest inhibition of Na/K-ATPase pump activity, we emphasize that physiological and even pharmacological concentrations of digitalis do not demonstrably increase cytosolic Na concentrations in physiologically relevant preparations (42). We would further point out that most studies, including those from our lab, which demonstrate inhibition of the Na/K-ATPase by circulating substances do so with strategies to control for the cytosolic Na concentration (71–74).
Approximately one decade ago, a further analogy of Na/K-ATPase signaling to the signaling of receptor tyrosine kinases was established with the observation that CTS binding to the Na/K-ATPase in renal tissues triggers endocytosis of the CTS-Na/K-ATPase complex (75). Subsequent studies have demonstrated that this internalization is associated with endosomal accumulation of the Na/K-ATPase and its caveolar signaling partners, and that the process requires both caveolin (and caveolar structure) and clathrin (76, 77). We have gone on to demonstrate that this process appears to also regulate the expression of the apical sodium transporter, NHE3, as well as impact renal salt excretion in vivo (78–80). Recent data from the laboratory of Dr. Lingrel utilizing novel genetic manipulations of the different alpha 1 isoforms in mice indicate that it is the alpha 1 subunit, which can be considered the functional receptor for these CTS. Interestingly, the amount of Na/K-ATPase alpha 1 subunit as well as it is affinity for CTS appear to both positively correlate with the magnitude of the signaling effect (81–84).
Recently, we have made several observations that bring the consideration of ROS in the context of Na pump signaling in a new light. First, we found that the Dahl salt-resistant (R) strain of rats had a natriuretic response to a high salt diet, which did not require substantial increases in blood pressure (hence the term “salt resistant”) and was accompanied by activation of Src and ERK as well as redistribution in the renal proximal tubule cells of the basolateral Na/K-ATPase and apical NHE3. This was previously observed with the wild type Sprague Dawley animals (which were used as a founder population to generate Dahl R and salt sensitive, S, rats). In contrast, the Dahl S rats did not have this redistribution. Isolated proximal tubules from young Dahl R and S rats maintained on a low salt diet demonstrated ouabain sensitivity and insensitivity, respectfully, in terms of Src and ERK activation as well as redistribution of the NaK-ATPase and NHE3 (85). Moving back to LLC-PK1 cells, we noted that the signaling observed with ouabain or other CTS could be duplicated by exposure to an ROS generation system (Glucose Oxidase + Glucose), blocked by anti-oxidants (e.g., N-acetyl cysteine) and was accompanied by specific carbonylation of two amino acids in the A domain portion of the alpha 1 subunit (86). Given that the proximal tubules of Dahl S rats demonstrate considerable carbonylation of plasma proteins including the Na/K-ATPase prior to exposure to high salt in vivo or ouabain in vitro (unpublished data), this suggests that chronic oxidation of the Na/K-ATPase may lead to impaired signal transduction in the proximal tubule and a form of oxidant “fatigue.” Perhaps of even greater importance, the protein oxidation seen with both ouabain and glucose oxidase/glucose was found to be reversible in a biochemical rather than a physiological sense since removing ouabain or glucose oxidase/glucose led to the return to non-carbonylated proteins regardless of whether new protein synthesis or protein degradation were inhibited. In addition, signaling through the Na/K-ATPase appeared to impact the amount and degree of protein carbonylation induced by glucose oxidase/glucose suggesting a role for the Na/K-ATPase as both a receptor and amplifier of ROS (86). We had seen in vivo data supporting this concept in earlier studies discussed below. Although a feed-forward system (which this appears to be) suggests ongoing amplification, it seems clear that endocytosis of this molecular machinery would be an effective termination mechanism (87). Whether the oxidatively modified Na/K-ATPase is a trigger for endocytosis is a topic we are actively investigating at present.
On this background, it is useful to consider whether a CTS is effectively natriuretic in vivo. This discussion began many years ago regarding the CTS pharmacological agent, digoxin, or digitalis, which was noted to effect natriuresis in patients with congestive heart failure but not normal subjects (88). Currently, there remains debate as to whether a CTS such as ouabain is, in fact, natriuretic (89). Although clearly this is important in understanding the physiological relevance of the molecular mechanisms described above, we would caution the reader that the answer to this question may be different depending on the physiological state of the experimental animal or subject at the time of the study (80, 85, 90). That said, we would certainly concede that a correlation between renal Na/K-ATPase signaling or inhibition and natriuresis may not always be present.
Role in Cardiac and Renal Fibrosis with Experimental Renal Failure
Concern that CTS signaling through the Na/K-ATPase might be profibrotic grew from several studies. First, we observed that experimental renal failure produced cardiac fibrosis in both rat and mouse (91). We would stress that human uremic cardiomyopathy is believed to also be complicated by fibrosis. When we performed active immunization prior to induction of experimental renal failure, the cardiac fibrosis was markedly attenuated. In a separate group of animals, infusion of MBG designed to achieve similar plasma levels of MBG as seen with experimental renal failure also caused cardiac fibrosis. Evidence for Na/K-ATPase signaling (e.g., Src and ERK activation) was see in both animals subjected to experimental renal failure or MBG infusion whereas active immunization against the MBG-Albumin conjugate attenuated this in the experimental renal failure group (51, 70, 91, 92). In addition, blockade of Na/K-ATPase signaling with active (or passive) immunization as well as pharmacologic blockade (see below) dramatically attenuated the oxidant stress in tissues seen with experimental renal failure (51, 91, 93, 94). Based on these animal studies, we next examined how CTS affected fibroblasts grown in culture. We noted that CTS (e.g., MBG, ouabain) induced increases in fibroblast collagen production as evidenced by either increased labeled proline incorporation or procollagen expression determined with Western blot. Evidence for Na/K-ATPase signaling (e.g., Src or ERK activation) could be observed as well. Moreover, ROS scavenging or pharmacological or molecular biological Src inhibition prevented increases in proline incorporation and collagen production seen with CTS. An increase in transcription was identified as we saw substantial increases in both mRNA for collagen as well as luciferase in cells transfected with a reporter construct following exposure to CTS. However, we did not see evidence for increased TGF beta signaling in these cells although pharmacological antagonism of the TGF beta system did block CTS stimulated collagen production (51). We next examined how CTS affected Fli-1 expression, stimulated by work performed by Watson and colleagues. Fli-1 is a negative regulator of collagen synthesis (95), and we noted that CTS induce decreases in Fli-1 expression in several types of fibroblasts (cardiac, renal, and dermal). We also observed that decreases in Fli-1 appear to be necessary for MBG to induce increases in collagen. Additional work showed that CTS induce translocation of PKCdelta from the cytosol to the nucleus in a PLC dependent manner. It appears that the translocation of PKCdelta causes Fli-1 phosphorylation and subsequent degradation (94).
These studies next led to work examining the effects of mineralocorticoid antagonists. We should first say that Finotti and colleagues reported 30 years ago that spironolactone and canrenone were antagonists of ouabain binding to the Na/K-ATPase (96). We looked at whether this observation was applicable to our system. In vitro, we saw that both spironolactone and canrenone could attenuate MBG-induced increases in collagen production in cardiac fibroblasts. Interestingly, we could not see a substantial effect of aldosterone on cardiac collagen production. Our in vitro observations were extended to in vivo studies where we saw that administration of spironolactone to rats with experimental renal failure markedly attenuated the observed cardiac fibrosis (94). This suggests the Na/K-ATPase signaling cascade may be a useful target for therapeutic drug development.
Further studies have demonstrated that the effects of MBG (and other CTS) are not specific for cardiac fibroblasts. We have noted that renal fibroblasts have a very similar response as cardiac fibroblasts, suggesting a potential pathological role for MBG in producing renal fibrosis and progressive renal failure. Using MBG infusion in the rat, we saw that such infusion was associated with the induction of Snail, a transcription factor known to be involved in epithelial–mesenchymal transformation (EMT). In LLC-PK1 cells grown in culture, MBG induces EMT in a dose and time dependent way (97).
Trade-Off with Respect to CTS
With the aforementioned data, we would suggest that the CTS signal cascade through the Na/K-ATPase fits the concept of “trade-off.” Specifically, CTS concentrations increase in response to volume expansion and/or salt loading. These CTS mediate increases in urinary Na excretion, maintaining Na homeostasis, but the endocytosis machinery may fatigue with ongoing stimulation. Moreover, there are other consequences of the elevated CTS concentrations, namely vasoconstriction and hypertension along with fibrosis, which was described above (Figure 3). The fibrosis may lead to further renal insensitivity in terms of natriuresis, and the combination of events cascading to produce progressive cardiovascular disease.
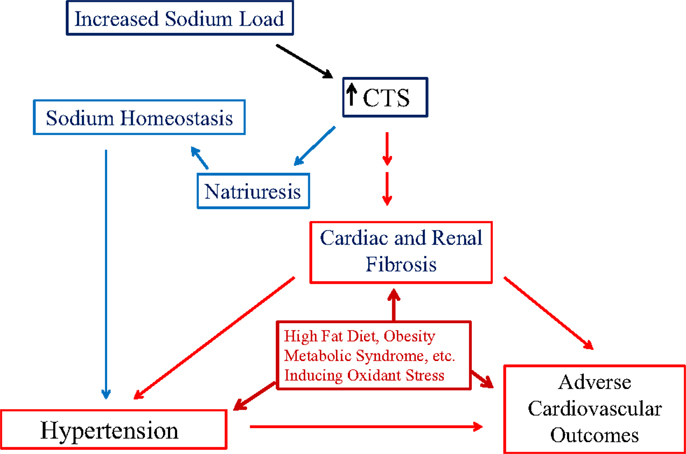
Figure 3. The schematic shown in figure illustrates the balance between the natriuretic effect of cardiotonic steroids (CTS) and the trade-off of inducing Na/K-ATPase-mediated signal transduction leading to cardiac and renal fibrosis, eventually contributing to the development of hypertension and adverse cardiovascular outcomes. In addition, chronic metabolic states resulting in the production of reactive oxygen species (ROS) creating oxidative stress may exacerbate the progression of cardiac and renal disease.
Future Directions
As we better understand the role of CTS signaling through the Na/K-ATPase, several therapeutic targets come to mind, which may provide novel and effective therapy for different chronic diseases. First, there is the interaction of the CTS with the Na/K-ATPase. This has been addressed experimentally in our laboratory with both active and passive immunization (51, 91, 93, 98) as well as pharmacologically with several different approaches (94, 99). Other groups have developed different substances which can loosely describe as “ouabain antagonists” which we have recently reviewed (5). Rostafuroxin has been very well characterized and appears to have potential for the treatment of hypertension (100, 101). Recently, our laboratory has begun to develop strategies to alter the interaction between the Na/K-ATPase alpha 1 subunit and Src (102). However, it is clear that the aforementioned signaling cascade affords a number of possible sites for intervention including but not limited to the generation of ROS (69), activation of Src and activation of ERK. Unfortunately, these molecular targets will also fit under the general rubric of “trade-off.” Although some aspects of CTS and signaling through the Na/K-ATPase may be maladaptive as we have discussed in this review, it is almost certain that that inhibition of this CTS-Na/K-ATPase pathway may have deleterious effects which need to be navigated.
Conflict of Interest Statement
Neither Dr. Joe Xie nor Ms. Anna Pearl Shapiro has any conflicts to report. Dr. Joseph Isaac Shapiro currently receives grant support from the NIH concerning this review topic (HL109015 as principal investigator, HL071556 and HL105649 as Co-investigator). Dr. Joseph Isaac Shapiro also holds some awarded patents related to this work (US Patent 8,283,441, Canadian Patents 2641303, 2667251, 2774486, 2360383).
Abbreviations
Ca, calcium; Cl, chloride; CTS, cardiotonic steroid; EGFR, epidermal growth factor receptor; EMT, epithelial-mesenchymal transformation; ERK, extracellular signal-regulated kinase; FAK, focal adhesion kinase; Fli-1, friend leukemia integration 1 transcription factor; GFR, glomerular filtration rate; Grb2, growth factor receptor-bound protein-2; MBG, marinobufagenin; Na, sodium; Na/K-ATPase, sodium potassium ATPase; NAC, N-acetyl cysteine; NHE3, sodium-hydrogen exchanger 3; PI(3)K, phosphoinositide 3-kinase; PKC, protein kinase C; PLC, phospholipase C; ROS, reactive oxygen species; SERCA, sarcoplasmic endoplasmic reticulum calcium ATPase; Shc, Src homology-2 domain containing protein; SOS, Son of Sevenless protein; TCB, telecinobufagin; TGF, transforming growth factor.
References
1. Luft FC, Weinberger MH. Sodium intake and essential hypertension. Hypertension (1982) 4:III14–9. doi: 10.1161/01.HYP.4.5_Pt_2.III14
2. Ritz E. Salt and hypertension. Nephrology (2010) 15(Suppl 2):49–52. doi:10.1111/j.1440-1797.2010.01311.x
3. Elliott P. The INTERSALT study: an addition to the evidence on salt and blood pressure, and some implications. J Hum Hypertens (1989) 3:289–98.
4. Appel LJ, Moore TJ, Obarzanek E, Vollmer WM, Svetkey LP, Sacks FM, et al. A clinical trial of the effects of dietary patterns on blood pressure. DASH Collaborative Research Group. N Engl J Med (1997) 336:1117–24. doi:10.1056/NEJM199704173361601
5. Bagrov AY, Shapiro JI, Fedorova OV. Endogenous cardiotonic steroids: physiology, pharmacology, and novel therapeutic targets. Pharmacol Rev (2009) 61:9–38. doi:10.1124/pr.108.000711
6. Kelly RA. Excretion of artifactual endogenous digitalis-like factors. Am J Physiol (1986) 251:H205–9.
7. Kelly RA, O’Hara DS, Canessa ML, Mitch WE, Smith TW. Characterization of digitalis-like factors in human plasma. Interactions with NaK-ATPase and cross-reactivity with cardiac glycoside-specific antibodies. J Biol Chem (1985) 260:11396–405.
8. Hansen O. Do putative endogenous digitalis-like factors have a physiological role? Hypertension (1994) 24:640–4. doi:10.1161/01.HYP.24.5.640
9. Komiyama Y, Dong XH, Nishimura N, Masaki H, Yoshika M, Masuda M, et al. A novel endogenous digitalis, telocinobufagin, exhibits elevated plasma levels in patients with terminal renal failure. Clin Biochem (2005) 38:36–45. doi:10.1016/j.clinbiochem.2004.08.005
10. Hamlyn JM, Blaustein MP, Bova S, DuCharme DW, Harris DW, Mandel F, et al. Identification and characterization of a ouabain-like compound from human plasma. Proc Natl Acad Sci U S A (1991) 88:6259–63. doi:10.1073/pnas.88.21.9907-d
11. Gonick HC, Kramer HJ, Paul W, Lu E. Circulating inhibitor of sodium-potassium-activated adenosine triphosphatase after expansion of extracellular fluid volume in rats. Clin Sci Mol Med (1977) 53:329–34.
12. Schoner W, Scheiner-Bobis G. Endogenous and exogenous cardiac glycosides and their mechanisms of action. Am J Cardiovasc Drugs (2007) 7:173–89. doi:10.2165/00129784-200707030-00004
13. Kolmakova EV, Haller ST, Kennedy DJ, Isachkina AN, Budny GV, Frolova EV, et al. Endogenous cardiotonic steroids in chronic renal failure. Nephrol Dial Transplant (2011) 26:2912–9. doi:10.1093/ndt/gfq772
14. Xie Z, Kometiani P, Liu J, Li J, Shapiro JI, Askari A. Intracellular reactive oxygen species mediate the linkage of Na+/K+-ATPase to hypertrophy and its marker genes in cardiac myocytes. J Biol Chem (1999) 274:19323–8. doi:10.1074/jbc.274.27.19323
15. Tian J, Liu J, Garlid KD, Shapiro JI, Xie Z. Involvement of mitogen-activated protein kinases and reactive oxygen species in the inotropic action of ouabain on cardiac myocytes. A potential role for mitochondrial K(ATP) channels. Mol Cell Biochem (2003) 242:181–7. doi:10.1023/A:1021114501561
16. Liu J, Tian J, Haas M, Shapiro JI, Askari A, Xie Z. Ouabain interaction with cardiac Na+/K+-ATPase initiates signal cascades independent of changes in intracellular Na+ and Ca2+ concentrations. J Biol Chem (2000) 275:27838–44. doi:10.1074/jbc.M002950200
17. Zhou HZ, Shapiro JI, Chan L, Schrier RW. Atrial natriuretic peptide protects against cold ischemic injury in the isolated and in situ rat kidney. J Am Soc Nephrol (1990) 1:927–8.
18. Abraham WT, Schrier RW. Body fluid volume regulation in health and disease. Adv Intern Med (1994) 39:23–47.
19. de Wardener H, Mills IH, Clapham WF, Hayter CJ. Studies on the efferent mechanism of the sodium diuresis which follows the administration of intravenous saline in the dog. Clin Sci (1961) 21:249–58.
20. Guyton AC, Coleman TG, Granger HJ. Circulation: overall regulation. Annu Rev Physiol (1972) 34:13–46. doi:10.1146/annurev.ph.34.030172.000305
21. Cort JH, Rudinger J, Lichardus B, Hagemann I. Effects of oxytocin antagonists on the saluresis accompanying carotid occlusion. Am J Physiol (1966) 210:162–8.
22. Buckalew VM Jr, Martinez FJ, Green WE. The effect of dialysates and ultrafiltrates of plasma of saline-loaded dogs on toad bladder sodium transport. J Clin Invest (1970) 49:926–35. doi:10.1172/JCI106312
23. Gruber KA, Whitaker JM, Buckalew VM Jr. Endogenous digitalis-like substance in plasma of volume-expanded dogs. Nature (1980) 287:743–5. doi:10.1038/287743a0
24. Schrier RW, McDonald KM, Marshall RA, Lauler DP. Absence of natriuretic response to acute hypotonic intravascular volume expansion in dogs. Clin Sci (1968) 34:57–72.
25. De Wardener HE, Fabian M, Jones JJ, Lee J, Schrier RW, Verroust PJ. The effect of acute extracellular fluid volume expansion and acute haemorrhage on plasma antidiuretic hormone and oxytocin levels in dogs. J Physiol (1968) 196:122.
26. de Wardener HE, Clarkson EM, Nutbourne DM, Schrier RW, Talner LB, Ventom MG, et al. Evidence for a hormone other than aldosterone which controls urinary sodium excretion. Adv Nephrol Necker Hosp (1971) 1:97–111.
27. Bergdahl B, Dahlstrom G, Molin L, Bertler A. Inter and intra laboratory variation of digoxin radioimmunoassay in Sweden. Acta Pharmacol Toxicol (Copenh) (1979) 45:66–72. doi:10.1111/j.1600-0773.1979.tb02362.x
28. Bergdahl B, Molin L. Precision of digoxin radioimmunoassays and matrix effects: four kits compared. Clin Biochem (1981) 14:67–71. doi:10.1016/S0009-9120(81)90704-9
29. Gusdon JP Jr, Buckalew VM Jr, Hennessy JF. A digoxin-like immunoreactive substance in preeclampsia. Am J Obstet Gynecol (1984) 150:83–5. doi:10.1016/S0002-9378(84)80114-3
30. Pleasants RA, Gadsden RH Sr, McCormack JP, Piveral K, Sawyer WT. Interference of digoxin-like immunoreactive substances with three digoxin immunoassays in patients with various degrees of renal function. Clin Pharm (1986) 5:810–6.
31. Miyashita H, Sato T, Tamura T, Tamura O, Tazawa H. The problems of digitalis therapy from the viewpoint of serum concentration with special reference to the sampling time, to the overlapping range of serum concentration where intoxicated and non-intoxicated patients are located and to atrial fibrillation. Jpn Circ J (1986) 50:628–35. doi:10.1253/jcj.50.628
32. Ebara H, Suzuki S, Nagashima K, Koizumi T, Nishida A, Kanbe Y, et al. Digoxin- and digitoxin-like immunoreactive substances in amniotic fluid, cord blood, and serum of neonates. Pediatr Res (1986) 20:28–31. doi:10.1203/00006450-198601000-00007
34. de Bold AJ, Borenstein HB, Veress AT, Sonnenberg H. A rapid and potent natriuretic response to intravenous injection of atrial myocardial extract in rats. Life Sci (1981) 28:89–94. doi:10.1016/0024-3205(81)90370-2
35. de Bold AJ, Flynn TG. Cardionatrin I – a novel heart peptide with potent diuretic and natriuretic properties. Life Sci (1983) 33:297–302. doi:10.1016/0024-3205(83)90390-9
36. Pamnani MB, Clough DL, Chen JS, Link WT, Haddy FJ. Effects of rat atrial extract on sodium transport and blood pressure in the rat. Proc Soc Exp Biol Med (1984) 176:123–31. doi:10.3181/00379727-176-41851
37. Nakamoto M, Shapiro JI, Shanley PF, Chan L, Schrier RW. In vitro and in vivo protective effect of atriopeptin III on ischemic acute renal failure. J Clin Invest (1987) 80:698–705. doi:10.1172/JCI113124
39. Baecher S, Kroiss M, Fassnacht M, Vogeser M. No endogenous ouabain is detectable in human plasma by ultra-sensitive UPLC-MS/MS. Clin Chim Acta (2014) 431:87–92. doi:10.1016/j.cca.2014.01.038
40. Bagrov AY, Shapiro JI. Endogenous digitalis: pathophysiologic roles and therapeutic applications. Nat Clin Pract Nephrol (2008) 4:378–92. doi:10.1038/ncpneph0848
41. Fedorova OV, Shapiro JI, Bagrov AY. Endogenous cardiotonic steroids and salt-sensitive hypertension. Biochim Biophys Acta (2010) 1802:1230–6. doi:10.1016/j.bbadis.2010.03.011
42. Xie Z, Askari A. Na(+)/K(+)-ATPase as a signal transducer. Eur J Biochem (2002) 269:2434–9. doi:10.1046/j.1432-1033.2002.02910.x
43. Pierre SV, Xie Z. The Na, K-ATPase receptor complex: its organization and membership. Cell Biochem Biophys (2006) 46:303–16. doi:10.1385/CBB:46:3:303
44. Tian J, Xie ZJ. The Na-K-ATPase and calcium-signaling microdomains. Physiology (2008) 23:205–11. doi:10.1152/physiol.00008.2008
45. Bricker NS. On the pathogenesis of the uremic state. An exposition of the “trade-off hypothesis”. N Engl J Med (1972) 286:1093–9. doi:10.1056/NEJM197205182862009
46. Bricker NS, Fine LG. Uremia: formulations and expectations. The trade-off hypothesis: current status. Kidney Int Suppl (1978) 8:S5–8.
47. Bricker NS, Schmidt RW, Favre H, Fine L, Bourgoignie JJ. On the biology of sodium excretion: the search for a natriuretic hormone. Yale J Biol Med (1975) 48:293–303.
48. Bourgoignie JJ, Hwang KH, Ipakchi E, Bricker NS. The presence of a natriuretic factor in urine of patients with chronic uremia. The absence of the factor in nephrotic uremic patients. J Clin Invest (1974) 53:1559–67. doi:10.1172/JCI107706
49. Licht A, Stein S, Bricker NS. Hormonal changes and transport adaptation in chronic renal failure: the possible role of a natriuretic hormone. Biochem Soc Trans (1978) 6:837–9.
50. Licht A, Stein S, McGregor CW, Bourgoignie JJ, Bricker NS. Progress in isolation and purification of an inhibitor of sodium transport obtained from dog urine. Kidney Int (1982) 21:339–44. doi:10.1038/ki.1982.27
51. Elkareh J, Kennedy DJ, Yashaswi B, Vetteth S, Shidyak A, Kim EG, et al. Marinobufagenin stimulates fibroblast collagen production and causes fibrosis in experimental uremic cardiomyopathy. Hypertension (2007) 49:215–24. doi:10.1161/01.HYP.0000252409.36927.05
52. Skou JC, Zerahn K. Investigations on the effect of some local anaesthetics and other amines on the active transport of sodium through the isolated short-circuited frog skin. Biochim Biophys Acta (1959) 35:324–33. doi:10.1016/0006-3002(59)90381-6
53. Skou JC. The influence of some cations on an adenosine triphosphatase from peripheral nerves. Biochim Biophys Acta (1957) 23:394–401. doi:10.1016/0006-3002(57)90343-8
54. Skou JC. Nobel Lecture. The identification of the sodium pump. Biosci Rep (1998) 18:155–69. doi:10.1023/A:1020196612909
56. McDonough AA, Azuma KK, Lescale-Matys L, Tang MJ, Nakhoul F, Hensley CB, et al. Physiologic rationale for multiple sodium pump isoforms. Differential regulation of alpha 1 vs alpha 2 by ionic stimuli. Ann N Y Acad Sci (1992) 671:156–68. doi:10.1111/j.1749-6632.1992.tb43793.x
57. Yang-Feng TL, Schneider JW, Lindgren V, Shull MM, Benz EJ Jr, Lingrel JB, et al. Chromosomal localization of human Na+, K+-ATPase alpha- and beta-subunit genes. Genomics (1988) 2:128–38. doi:10.1016/0888-7543(88)90094-8
58. Skou JC. The identification of the sodium pump. Biosci Rep (2004) 24:436–51. doi:10.1007/s10540-005-2740-9
59. Xie Z, Xie J. The Na/K-ATPase-mediated signal transduction as a target for new drug development. Front Biosci (2005) 10:3100–9. doi:10.2741/1766
60. Liang M, Tian J, Liu L, Pierre S, Liu J, Shapiro J, et al. Identification of a pool of non-pumping Na/K-ATPase. J Biol Chem (2007) 282:10585–93. doi:10.1074/jbc.M609181200
61. Haas M, Askari A, Xie Z. Involvement of Src and epidermal growth factor receptor in the signal-transducing function of Na+/K+-ATPase. J Biol Chem (2000) 275:27832–7. doi:10.1074/jbc.M002951200
62. Haas M, Wang H, Tian J, Xie Z. Src-mediated inter-receptor cross-talk between the Na+/K+-ATPase and the epidermal growth factor receptor relays the signal from ouabain to mitogen-activated protein kinases. J Biol Chem (2002) 277:18694–702. doi:10.1074/jbc.M111357200
63. Wang H, Haas M, Liang M, Cai T, Tian J, Li S, et al. Ouabain assembles signaling cascades through the caveolar Na+/K+-ATPase. J Biol Chem (2004) 279:17250–9. doi:10.1074/jbc.M313239200
64. Tian J, Cai T, Yuan Z, Wang H, Liu L, Haas M, et al. Binding of Src to Na+/K+-ATPase forms a functional signaling complex. Mol Biol Cell (2006) 17:317–26. doi:10.1091/mbc.E05-08-0735
65. Elkareh J, Periyasamy SM, Shidyak A, Vetteth S, Schroeder J, Raju V, et al. Marinobufagenin induces increases in procollagen expression in a process involving protein kinase C and Fli-1: implications for uremic cardiomyopathy. Am J Physiol Renal Physiol (2009) 296:F1219–26. doi:10.1152/ajprenal.90710.2008
66. Chen Y, Cai T, Yang C, Turner DA, Giovannucci DR, Xie Z. Regulation of inositol 1,4,5-trisphosphate receptor-mediated calcium release by the Na/K-ATPase in cultured renal epithelial cells. J Biol Chem (2008) 283:1128–36. doi:10.1074/jbc.M708025200
67. Pierre SV, Yang C, Yuan Z, Seminerio J, Mouas C, Garlid KD, et al. Ouabain triggers preconditioning through activation of the Na+, K+-ATPase signaling cascade in rat hearts. Cardiovasc Res (2007) 73:488–96. doi:10.1016/j.cardiores.2006.11.003
68. Yuan Z, Cai T, Tian J, Ivanov AV, Giovannucci DR, Xie Z. Na/K-ATPase tethers phospholipase C and IP3 receptor into a calcium-regulatory complex. Mol Biol Cell (2005) 16:4034–45. doi:10.1091/mbc.E05-04-0295
69. Priyadarshi S, Valentine B, Han C, Fedorova OV, Bagrov AY, Liu J, et al. Effect of green tea extract on cardiac hypertrophy following 5/6 nephrectomy in the rat. Kidney Int (2003) 63:1785–90. doi:10.1046/j.1523-1755.2003.00914.x
70. Kennedy DJ, Malhotra D, Shapiro JI. Molecular insights into uremic cardiomyopathy: cardiotonic steroids and Na/K ATPase signaling. Cell Mol Biol (2006) 52:3–14.
71. Xie ZJ, Wang YH, Ganjeizadeh M, McGee R Jr, Askari A. Determination of total (Na+ + K+)-ATPase activity of isolated or cultured cells. Anal Biochem (1989) 183:215–9. doi:10.1016/0003-2697(89)90470-3
72. Jack-Hays MG, Xie Z, Wang Y, Huang WH, Askari A. Activation of Na+/K(+)-ATPase by fatty acids, acylglycerols, and related amphiphiles: structure-activity relationship. Biochim Biophys Acta (1996) 1279:43–8. doi:10.1016/0005-2736(95)00245-6
73. Periyasamy SM, Chen J, Cooney D, Carter P, Omran E, Tian J, et al. Effects of uremic serum on isolated cardiac myocyte calcium cycling and contractile function. Kidney Int (2001) 60:2367–76. doi:10.1046/j.1523-1755.2001.00053.x
74. Xie Z. Ouabain interaction with cardiac Na/K-ATPase reveals that the enzyme can act as a pump and as a signal transducer. Cell Mol Biol (2001) 47:383–90.
75. Liu J, Periyasamy SM, Gunning W, Fedorova OV, Bagrov AY, Malhotra D, et al. Effects of cardiac glycosides on sodium pump expression and function in LLC-PK1 and MDCK cells. Kidney Int (2002) 62:2118–25. doi:10.1046/j.1523-1755.2002.00672.x
76. Liu J, Liang M, Liu L, Malhotra D, Xie Z, Shapiro JI. Ouabain-induced endocytosis of the plasmalemmal Na/K-ATPase in LLC-PK1 cells requires caveolin-1. Kidney Int (2005) 67:1844–54. doi:10.1111/j.1523-1755.2005.00283.x
77. Liu J, Kesiry R, Periyasamy SM, Malhotra D, Xie Z, Shapiro JI. Ouabain induces endocytosis of plasmalemmal Na/K-ATPase in LLC-PK1 cells by a clathrin-dependent mechanism. Kidney Int (2004) 66:227–41. doi:10.1111/j.1523-1755.2004.00723.x
78. Cai H, Wu L, Qu W, Malhotra D, Xie Z, Shapiro JI, et al. Regulation of apical NHE3 trafficking by ouabain-induced activation of the basolateral Na+-K+-ATPase receptor complex. Am J Physiol Cell Physiol (2008) 294:C555–63. doi:10.1152/ajpcell.00475.2007
79. Oweis S, Wu L, Kiela PR, Zhao H, Malhotra D, Ghishan FK, et al. Cardiac glycoside downregulates NHE3 activity and expression in LLC-PK1 cells. Am J Physiol Renal Physiol (2006) 290:F997–1008. doi:10.1152/ajprenal.00322.2005
80. Periyasamy SM, Liu J, Tanta F, Kabak B, Wakefield B, Malhotra D, et al. Salt loading induces redistribution of the plasmalemmal Na/K-ATPase in proximal tubule cells. Kidney Int (2005) 67:1868–77. doi:10.1111/j.1523-1755.2005.00285.x
81. Wansapura AN, Lasko V, Xie Z, Fedorova OV, Bagrov AY, Lingrel JB, et al. Marinobufagenin enhances cardiac contractility in mice with ouabain-sensitive alpha1 Na+-K+-ATPase. Am J Physiol Heart Circ Physiol (2009) 296:H1833–9. doi:10.1152/ajpheart.00285.2009
82. Loreaux EL, Kaul B, Lorenz JN, Lingrel JB. Ouabain-sensitive alpha1 Na, K-ATPase enhances natriuretic response to saline load. J Am Soc Nephrol (2008) 19:1947–54. doi:10.1681/ASN.2008020174
83. Dostanic-Larson I, Lorenz JN, Van Huysse JW, Neumann JC, Moseley AE, Lingrel JB. Physiological role of the alpha1- and alpha2-isoforms of the Na+-K+-ATPase and biological significance of their cardiac glycoside binding site. Am J Physiol Regul Integr Comp Physiol (2006) 290:R524–8. doi:10.1152/ajpregu.00838.2005
84. Dostanic I, Schultz Jel J, Lorenz JN, Lingrel JB. The alpha 1 isoform of Na, K-ATPase regulates cardiac contractility and functionally interacts and co-localizes with the Na/Ca exchanger in heart. J Biol Chem (2004) 279:54053–61. doi:10.1074/jbc.M410737200
85. Liu J, Yan Y, Liu L, Xie Z, Malhotra D, Joe B, et al. Impairment of Na/K-ATPase signaling in renal proximal tubule contributes to Dahl salt-sensitive hypertension. J Biol Chem (2011) 286:22806–13. doi:10.1074/jbc.M111.246249
86. Yan Y, Shapiro AP, Haller S, Katragadda V, Liu L, Tian J, et al. Involvement of reactive oxygen species in a feed-forward mechanism of Na/K-ATPase-mediated signaling transduction. J Biol Chem (2013) 288:34249–58. doi:10.1074/jbc.M113.461020
87. Liu J, Shapiro JI. Regulation of sodium pump endocytosis by cardiotonic steroids: molecular mechanisms and physiological implications. Pathophysiology (2007) 14:171–81. doi:10.1016/j.pathophys.2007.09.008
88. Eichna LW, Farber SJ, Berger AR, Earle DP, Rader B, Pellegrino E, et al. The interrelationships of the cardiovascular, renal and electrolyte effects of intravenous digoxin in congestive heart failure. J Clin Invest (1951) 30:1250–61. doi:10.1172/JCI102545
89. Buckalew V. Is endogenous ouabain a physiological regulator of cardiovascular and renal function? Am J Physiol Heart Circ Physiol (2009) 297:H1972–3. doi:10.1152/ajpheart.01002.2009
90. Nesher M, Dvela M, Igbokwe VU, Rosen H, Lichtstein D. Physiological roles of endogenous ouabain in normal rats. Am J Physiol Heart Circ Physiol (2009) 297:H2026–34. doi:10.1152/ajpheart.00734.2009
91. Kennedy DJ, Vetteth S, Periyasamy SM, Kanj M, Fedorova L, Khouri S, et al. Central role for the cardiotonic steroid marinobufagenin in the pathogenesis of experimental uremic cardiomyopathy. Hypertension (2006) 47:488–95. doi:10.1161/01.HYP.0000202594.82271.92
92. Kennedy DJ, Elkareh J, Shidyak A, Shapiro AP, Smaili S, Mutgi K, et al. Partial nephrectomy as a model for uremic cardiomyopathy in the mouse. Am J Physiol Renal Physiol (2008) 294:F450–4. doi:10.1152/ajprenal.00472.2007
93. Haller ST, Kennedy DJ, Shidyak A, Budny GV, Malhotra D, Fedorova OV, et al. Monoclonal antibody against marinobufagenin reverses cardiac fibrosis in rats with chronic renal failure. Am J Hypertens (2012) 25:690–6. doi:10.1038/ajh.2012.17
94. Tian J, Shidyak A, Periyasamy SM, Haller S, Taleb M, El-Okdi N, et al. Spironolactone attenuates experimental uremic cardiomyopathy by antagonizing marinobufagenin. Hypertension (2009) 54:1313–20. doi:10.1161/HYPERTENSIONAHA.109.140038
95. Czuwara-Ladykowska J, Shirasaki F, Jackers P, Watson DK, Trojanowska M. Fli-1 inhibits collagen type I production in dermal fibroblasts via an Sp1-dependent pathway. J Biol Chem (2001) 276:20839–48. doi:10.1074/jbc.M010133200
96. Finotti P, Palatini P. Canrenone as a partial agonist at the digitalis receptor site of sodium-potassium-activated adenosine triphosphatase. J Pharmacol Exp Ther (1981) 217:784–90.
97. Fedorova LV, Raju V, El-Okdi N, Shidyak A, Kennedy DJ, Vetteth S, et al. The cardiotonic steroid hormone marinobufagenin induces renal fibrosis: implication of epithelial-to-mesenchymal transition. Am J Physiol Renal Physiol (2009) 296:F922–34. doi:10.1152/ajprenal.90605.2008
98. Haller ST, Drummond CA, Yan Y, Liu J, Tian J, Malhotra D, et al. Passive immunization against marinobufagenin attenuates renal fibrosis and improves renal function in experimental renal disease. Am J Hypertens (2013) 27(4):603–9. doi:10.1093/ajh/hpt169
99. Zhang Z, Li Z, Tian J, Jiang W, Wang Y, Zhang X, et al. Identification of hydroxyxanthones as Na/K-ATPase ligands. Mol Pharmacol (2010) 77:961–7. doi:10.1124/mol.110.063974
100. Ferrari P, Ferrandi M, Valentini G, Bianchi G. Rostafuroxin: an ouabain antagonist that corrects renal and vascular Na+-K+- ATPase alterations in ouabain and adducin-dependent hypertension. Am J Physiol Regul Integr Comp Physiol (2006) 290:R529–35. doi:10.1152/ajpregu.00518.2005
101. Ferrari P. Rostafuroxin: an ouabain-inhibitor counteracting specific forms of hypertension. Biochim Biophys Acta (2010) 1802:1254–8. doi:10.1016/j.bbadis.2010.01.009
Keywords: cardiotonic steroids, digitalis-like factors, fibrosis, sodium pump, signaling, renal failure, hypertension
Citation: Xie JX, Shapiro AP and Shapiro JI (2014) The trade-off between dietary salt and cardiovascular disease; a role for Na/K-ATPase signaling? Front. Endocrinol. 5:97. doi: 10.3389/fendo.2014.00097
Received: 11 April 2014; Accepted: 07 June 2014;
Published online: 17 July 2014.
Edited by:
Harvey Craig Gonick, University of California Berkeley, USAReviewed by:
Gert Jansen, Erasmus MC, NetherlandsHarvey Craig Gonick, University of California Berkeley, USA
Neal S. Bricker, University of California Los Angeles School of Medicine, USA
Copyright: © 2014 Xie, Shapiro and Shapiro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joseph Isaac Shapiro, Department of Medicine, Joan C. Edwards School of Medicine, Marshall University, 1600 Medical Center Drive Suite 3408, Huntington, WV 25701, USA e-mail:c2hhcGlyb2pAbWFyc2hhbGwuZWR1