 Andras Kern
Andras Kern Cristina Grande
Cristina Grande Roy G. Smith
Roy G. Smith- Department of Metabolism and Aging, Scripps Research Institute Florida, Jupiter, FL, USA
The orexigenic peptide hormone ghrelin is synthesized in the stomach and its receptor growth hormone secretagogue receptor (GHSR1a) is expressed mainly in the central nervous system (CNS). In this review, we confine our discussion to the physiological role of GHSR1a in the brain. Paradoxically, despite broad expression of GHSR1a in the CNS, other than trace amounts in the hypothalamus, ghrelin is undetectable in the brain. In our efforts to elucidate the function of the ligand-free ghrelin receptor (apo-GHSR1a), we identified subsets of neurons that co-express GHSR1a and dopamine receptors. In this review, we focus on interactions between apo-GHSR1a and dopamine-2 receptor (DRD2) and formation of GHSR1a:DRD2 heteromers in hypothalamic neurons that regulate appetite, and discuss implications for the treatment of Prader–Willi syndrome (PWS). GHSR1a antagonists of distinct chemical structures, a quinazolinone and a triazole, respectively, enhance and inhibit dopamine signaling through GHSR1a:DRD2 heteromers by an allosteric mechanism. This finding illustrates a potential strategy for designing the next generation of drugs for treating eating disorders as well as psychiatric disorders caused by abnormal dopamine signaling. Treatment with a GHSR1a antagonist that enhances dopamine/DRD2 activity in GHSR1a:DRD2 expressing hypothalamic neurons has the potential to inhibit the uncontrollable hyperphagia associated with PWS. DRD2 antagonists are prescribed for treating schizophrenia, but these block dopamine signaling in all DRD2 expressing neurons and are associated with adverse side effects, including enhanced appetite and excessive weight gain. A GHSR1a antagonist of structural class that allosterically blocks dopamine/DRD2 action in GHSR1a:DRD2 expressing neurons would have no effect on neurons expressing DRD2 alone; therefore, the side effects of DRD2 antagonists would potentially be reduced thereby enhancing patient compliance.
Introduction
The orexigenic hormone ghrelin is produced in stomach and influences feeding behavior, metabolism, pulsatile growth hormone (GH) release, and immune function (1–4). Ghrelin’s action is regulated by the GH secretagogue receptor (GHSR1a, aka the ghrelin receptor) that was originally identified by reverse pharmacology using a small molecule, MK-0677, developed to rejuvenate the GH/insulin-like growth factor axis in elderly subjects (5, 6). GHSR1a belongs to the Class A G-protein coupled receptor (GPCR) family. In isolation, under defined conditions, GHSR1a couples to Gαq resulting in activation of phospholipase C (PLC), inositol trisphosphate (IP3), and mobilization of [Ca2+]i (7).
The precise physiological function of ghrelin remains to be defined. Traditionally ghrelin was believed to control appetite and facilitate excessive weight gain in response to a high-fat diet, but recent findings question these conclusions. Studies in congenic C57BL/6J ghrelin knockout (KO) and ghsr KO mice showed food intake is independent of ghrelin signaling, and that the absence of ghrelin fails to protect mice from diet-induced obesity (8–10). Indeed, recent results in transgenic mice where ghrelin producing cells were selectively ablated confirm these findings (11). Acute stimulation of food intake in ghrelin-cell ablated mice requires doses of exogenous ghrelin that produce plasma ghrelin concentrations many-fold higher than the endogenous concentrations found in wildtype mice, suggesting endogenous ghrelin is not a critical regulator of food intake. With prolonged calorie restriction ghrelin-cell ablated mice exhibit profound hypoglycemia (11). Similarly, profound hypoglycemia was reported by the same group in calorie-restricted ghrelin-deficient mice generated by ablating medium chain fatty acid acyl-transferase that is essential for converting the inactive 28-aminoacid ghrelin peptide into its biologically active form (12). Injection of ghrelin or GH rescued the hypoglycemia. Based on the results from these two transgenic mouse models, the authors concluded that ghrelin’s major metabolic role is to regulate blood glucose under conditions of famine.
GHSR1a is expressed broadly in the brain and localized mainly in the hippocampal structures, hypothalamus, midbrain, cortex, and amygdala (13). These findings led us to investigate possible interactions of GHSR1a with dopamine receptors. By employing Ghsr-IRES-tau-GFP mice, we showed that subsets of neurons in the midbrain and hippocampus co-express GHSR1a and dopamine receptor-1 (DRD1) (14), and in the hypothalamus subsets co-express GHSR1a and dopamine receptor-2 (DRD2) (15). In vitro studies illustrated the formation of GHSR1a:DRD1 heteromers, and treatment with ghrelin and dopamine in combination augmented cAMP accumulation (14). Again, using Ghsr-IRES-tau-GFP mice, we identified subsets of hypothalamic neurons that co-express GHSR1a and dopamine-2 receptor (DRD2) (15). DRD2 signaling influences feeding frequency and volume, and mutations in DRD2 are associated with human obesity (16–20). In this review, we describe the co-expression of GHSR1a with dopamine receptors in neurons of the CNS, the dependence on GHSR1a for dopamine/DRD2 suppression of appetite and implications for the uncontrollable hyperphagia associated with Prader–Willi syndrome (PWS).
Ghrelin Activation of Hypothalamic Neurons Enhances Dopamine Release from Midbrain Dopaminergic Neurons
Pharmacological doses of ghrelin activate extra-hypothalamic neurons implicating a role for ghrelin in memory, addiction, depression, and neuroprotection (21–27). Indeed, ghsr−/−mice exhibit impaired contextual memory (28). In addition, improvements in spatial memory induced by exogenous ghrelin are inhibited by a dopamine-1 receptor (DRD1) antagonist (29). Ghrelin also augments cocaine-induced locomotor activity and the rewarding effects of alcohol, consistent with effects on dopamine signaling (30, 31). Although ghrelin administration induces dopamine release (30), there is no evidence that ghrelin has direct access to midbrain dopaminergic neurons (32). Exogenously applied ghrelin activates c-Fos expression in the arcuate nucleus, paraventricular nucleus (PVN), and lateral hypothalamus (LH), but not in GHSR1a expressing neurons of the midbrain or hippocampus (33, 34). Tracing studies with inactivated rabies virus show that neurons in the ventral tegmental area (VTA) and pars compacta of the substantia nigra (SNpc) receive projections from the PVN and LH (33–35). Furthermore, infusion of ghrelin into the LH, but not in the VTA, stimulates orexin release, which activates orexin receptors on VTA neurons causing release of dopamine (36). Neuroanatomy, neuropharmacology, and cell biology studies indicate VTA dopaminergic neurons innervate the hippocampus (37–41). These collective observations are consistent with exogenous ghrelin inducing hippocampal plasticity indirectly by first activating neurons in the LH and PVN that in turn enhance dopamine release from the VTA resulting in activation of hippocampal neurons. However, these results obtained using pharmacological doses of ghrelin cannot simply be extrapolated to predict the physiological role of endogenous ghrelin. A recent series of studies in mice and sheep indicate that other than trace amounts in the hypothalamus, endogenous ghrelin is not present in the CNS (42–45). This is remarkable given the broad distribution of GHSR1a in the brain and underlines a potential role for GHSR1a independent of ghrelin, which led us to speculate that apo-GHSR1a regulates neuronal function through protein–protein interactions.
GHSR1a and DRD2 are Co-Expressed in Hypothalamic Neurons Resulting in Modification of Canonical Dopamine Signaling
Since endogenous ghrelin is undetectable in the brain, we addressed the question of what biological role un-liganded GHSR1a (apo-GHSR1a) might play by focusing on the function of hypothalamic neurons that co-express GHSR1a and DRD2. To determine the impact of expression of apo-GHSR1a on DRD2 signaling, we initially used HEK293 cells where we could control the relative expression levels of the two receptors and investigate signal transduction pathways. Dopamine activation of DRD2 in HEK293 cells results in canonical DRD2 coupling to Gαi/o and suppression of intracellular cAMP accumulation without mobilization of intracellular Ca2+ ([Ca2+]i) (46). However, when GHSR1a is expressed with DRD2, treatment with dopamine, or the DRD2 agonist quinpirole results in [Ca2+]i mobilization. When DRD2 is expressed with the motilin receptor, which is closely related to GHSR1a and is also coupled to Gαq, neither dopamine nor quinpirole treatment mobilizes [Ca2+]i, illustrating the significance of specific interactions between GHSR1a and DRD2. When GHSR1a is co-expressed with DRD2, dopamine-induced [Ca2+]i release is blocked by: pertussis toxin, Gβγ antagonists, PLC inhibitor, thapsigargin, and IP3 receptor blocker. We conclude that dopamine activation causes liberation of Gβγ subunits from Gαi/o, activation of PLC, IP3 mobilization, and release of [Ca2+]i (Figure 1). Importantly, this mechanism is activated in the complete absence of ghrelin. A mechanism involving down-stream cross-talk between GHSR1a and DRD2 caused by GHSR1a basal activity was ruled out. Blocking GHSR1a basal activity with Gαq siRNA or GHSR1a point mutants that lack constitutive activity failed to inhibit dopamine-induced [Ca2+]i release. The lack of cross-talk between the two receptors supports the hypothesis that GHSR1a and DRD2 co-expression results in non-canonical DRD2 signal transduction that is dependent on allosteric interactions between GHSR1a and DRD2 protomers. These results were recapitulated in DRD2 expressing SH-SY5Y neuroblastoma cells engineered to stably express GHSR1a, and in primary cultures of hypothalamic neurons (15), confirming that non-canonical DRD2 signaling is dependent upon interactions between GHSR1a and DRD2.
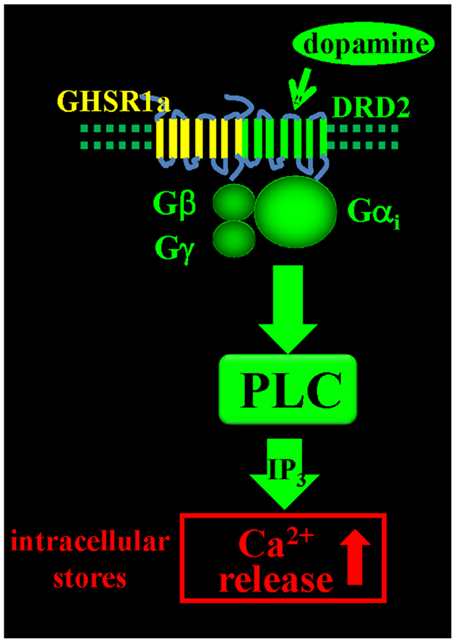
Figure 1. Dopamine-induced [Ca2+]i mobilization via GHSR1a:DRD2 heteromers. Dopamine activation of GHSR1a:DRD2 causes release of Ca2+ from intracellular stores by Gβγ dependent PLC activation and mobilization of IP3.
DRD2 Agonist Suppression of Feeding Behavior is Dependent on GHSR1a and Formation of GHSR1a:DRD2 Heteromers in Hypothalamic Neurons
The change in canonical to non-canonical DRD2 signal transduction when GHSR1a is co-expressed with DRD2 implicates allosteric interactions between GHSR1a and DRD2 as a result of GHSR1a:DRD2 heteromer formation. It is well documented, based mainly on in vitro experiments, that GPCRs form homomers and heteromers, and that in heteromers one protomer in the complex allosterically modifies signaling of the other (47, 48). To test if modification of signal transduction is a consequence of a physical interaction between GHSR1a and DRD2, time resolved fluorescence resonance energy transfer (Tr-FRET) experiments were conducted in HEK293 cells (49, 50). The high sensitivity of Tr-FRET where SNAP- or CLIP-tags are introduced at the N-terminus of GHSR1a and DRD2 allowed monitoring of GHSR1a:GHSR1a and GHSR1a:DRD2 heteromer formation at physiological concentrations on the surface of living cells (15).
When SNAP-GHSR1a is expressed in the presence of untagged DRD2 at a 1:1 ratio the Tr-FRET signal generated by GHSR1a homomers was reduced by ~50%, consistent with formation of GHSR1a:DRD2 heteromers. As further confirmation of direct interactions and heteromer formation, Tr-FRET assays were performed using SNAP-GHSR1a and CLIP-DRD2 in a receptor titration assay. High Tr-FRET signals were detected over a wide range of receptor expression, confirming GHSR1a:DRD2 heteromerization. To test if formation of heteromers was functionally relevant, Tr-FRET results were compared to the magnitude of dopamine-induced [Ca2+]i mobilization at various ratios of GHSR1a to DRD2. Dopamine-induced [Ca2+]i release correlated with the concentration of GHSR1a:DRD2 heteromers. Indeed, in support of an allosteric mechanism, the GHSR1a neutral antagonist, JMV2959, inhibited dopamine-induced [Ca2+]i mobilization without disrupting heteromer formation; similarly, a DRD2 antagonist attenuated ghrelin signaling (15). Collectively, these data show that in cell lines modification of canonical DRD2 signaling is dependent on formation of GHSR1a:DRD2 heteromers (15).
To test if GHSR1a:DRD2 heteromer formation has physiological relevance, we sought evidence for the presence of the heteromers in native brain tissue. We applied confocal FRET microscopy on mouse hypothalamic neurons using a fluorescently labeled ghrelin analog to visualize GHSR1a, and a fluorescently tagged DRD2 monoclonal antibody to localize DRD2. In hypothalamic neurons from Ghsr+/+ mice, confocal FRET microscopy analysis shows GHSR1a and DRD2 in close proximity within 5 nm, consistent with heteromer formation (Figure 2). In striatal neurons of Ghsr+/+ mice, the confocal FRET signal is very weak indicating the absence of heteromers in the striatum. In hypothalamic neurons of Ghsr−/− mice, a confocal FRET signal is not detected confirming the specificity of imaging GHSR1a:DRD2 heteromers in Ghsr + + mice (15).
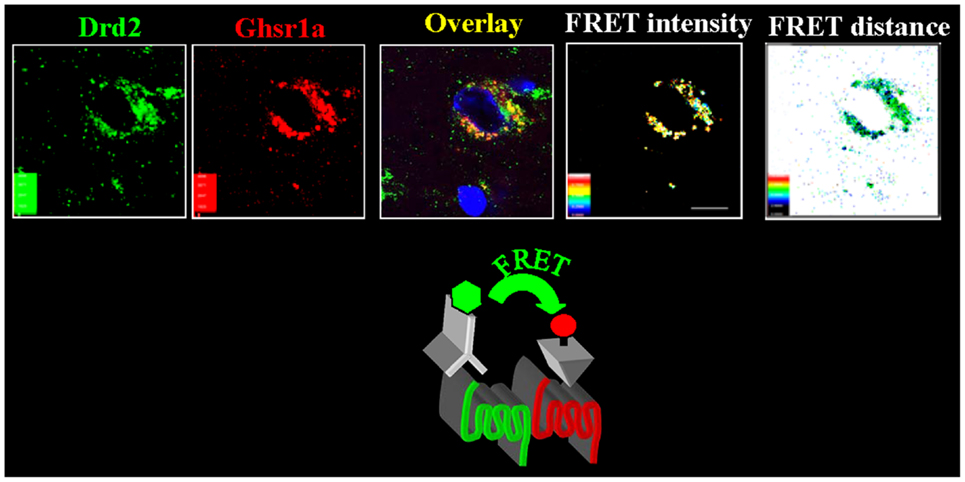
Figure 2. In hypothalamic neurons GHSR1a and DRD2 form heteromers. Hypothalamic brain slices from Ghsr+/+ mice were used to identify GHSR1a:DRD2 heteromers. Confocal FRET microscopy show that GHSR1a [identified using red fluorescent ghrelin (Cy5)] and DRD2 [in green, identified by immunofluorescent DRD2 monoclonal antibody (Cy3)] are in close proximity at a distance of 5–6 nm and FRET intensity 0.4–0.6 (15).
The physiological importance of GHSR1a:DRD2 interactions was tested by monitoring food intake of fasted Ghsr+/+ and Ghsr−/−mice following treatment with the selective DRD2 agonist cabergoline. In Ghsr+/+ mice, food intake was markedly inhibited by cabergoline compared to vehicle treated animals, but Ghsr−/−mice were refractory to cabergoline-induced anorexia; hence, the anorexigenic activity of DRD2 is dependent upon GHSR1a. To test if endogenous ghrelin played a role, food intake was compared in Ghrelin+/+ and Ghrelin−/−mice treated with cabergoline. Cabergoline inhibited food intake in both genotypes, showing that the anorexigenic activity of the DRD2 agonist is dependent on GHSR1a but not on ghrelin. Having shown in cells co-expressing GHSR1a and DRD2 that the GHSR1a neutral antagonist, JMV2959, inhibits dopamine-induced [Ca2+]i mobilization, mice were pretreated with JMV2959 before cabergoline injection. JMV2959 inhibited the anorexigenic effect of cabergoline in Ghsr+/+ mice, but not in Ghsr−/−mice; thus the antagonistic action of JMV2959 on DRD2 activity is dependent on GHSR1a. These data illustrate that allosteric interactions between GHSR1a and DRD2 as a consequence of heteromer formation are physiologically relevant mediators of feeding behavior (Figure 3) (15).
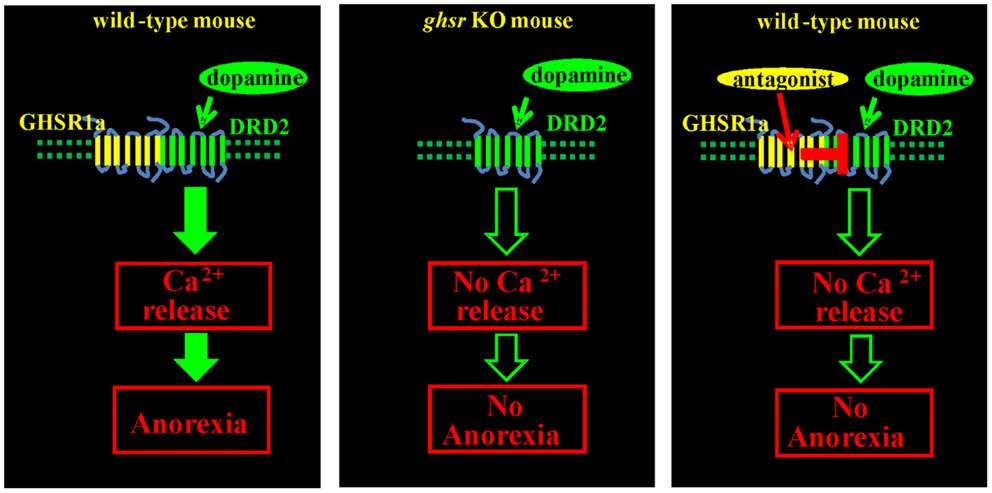
Figure 3. Anorexigenic effect of a DRD2 agonist is mediated by GHSR1a:DRD2 heteromers. Treatment of fasted Ghsr+/+ mice with the DRD2 agonist cabergoline inhibits food intake. Ghsr−/−mice, and Ghsr+/+ mice treated with the ghrelin receptor neutral antagonist, JMV2959, are refractory to the anorexigenic effects of cabergoline; hence, inhibition of food intake by cabergoline is dependent upon allosteric interactions between GHSR1a and DRD2.
Our results support the concept that heteromerization of GPCRs is an important mechanism for regulating GPCR function. Many GPCRs function as oligomeric complexes, where receptors physically interact to form homo- or heteromers. These protein–protein interactions stabilize specific conformations of GPCRs to activate specific down-stream effectors and signaling pathways (51–54). For example, ghrelin inhibition of insulin release from pancreatic beta-cells is mediated by GHSR1a coupling to Gαi rather than canonical coupling through Gαq (55); this modification in G-protein coupling is a consequence of formation of heteromers formed between GHSR1a and somatostatin receptor-5 (56). Heterodimers formed between GHSR1a and melanocortin-3 receptor has also been reported, and a combination of in situ hypridization and immunohistochemistry indicated co-localization of the receptors in hypothalamic neurons (57).
Examples of dimerization of GPCRs in vitro are well documented, but the physiological relevance is repeatedly questioned because of the paucity of data illustrating dimerization in native tissues. However, we show by applying FRET confocal microscopy the presence of GHSR1a:DRD2 heteromers in native hypothalamic neurons and further that inhibition of food intake by a DRD2 agonist is dependent on GHSR1a. Formation of the heteromeric complex results in non-canonical signaling dependent on Gβγ activation of PLC and mobilization of [Ca2+]i. This occurs in the absence of ghrelin, showing apo-GHSR1a is capable of allosterically modifying DRD2 signaling. The allosteric interaction within the GHSR1a:DRD2 heteromer is altered by the presence of a neutral GHSR1a antagonist (15); hence, the conformation of one protomer influences the signaling properties of the other. These findings demonstrate an important role for apo-GHSR1a in the brain and resolve the paradox that GHSR1a is expressed in brain areas not accessible to peripherally produced ghrelin, and where there is no evidence of ghrelin production.
Implications of GHSR1:DRD2 Heteromers in Obsessive Eating Associated with Prader–Willi Syndrome
Prader–Willi syndrome is a genetic disorder occurring in approximately 1 in 10,000 births and is associated with parent of origin imprinting. In normal subjects, maternal 15q11.2 is imprinted, but the paternal chromosome has a working copy of 15q11.2. Patients with PWS lack a working copy because of paternal deletion (70% of cases), maternal uniparental disomy (mUPD, 25% of cases), or imprinting errors in the imprinting center (IC, 5% of cases) (58). The deletion or lack of expression of 15q11.2 on the paternal chromosome results in a complex multi-systemic disorder. The phenotype includes short stature, low muscle tone, uncontrollable appetite, incomplete sexual development, and impaired cognition. The behavioral characteristics fall into the spectrum of autism spectrum disorders. In narrowing down the DNA region responsible for PWS, it was found that individuals with paternal deletion of the SNORD116 gene cluster had all the major characteristic features of PWS (59). This locus encompasses a long non-coding RNA transcript that is processed into multiple small nuclear RNAs, as well as spliced exons of host gene 116G (60). The expression of Snord116 in mice becomes progressively more prominent in the arcuate nucleus, PVN, and ventromedial nucleus of the hypothalamus as the mice mature (61); these particular areas express GHSR1a and are involved in regulation of appetite mediated by DRD2.
While GH replacement is frequently used to treat impaired growth and hypotonia associated with PWS, the most distinguishing feature, for which there is no medical therapy, is the involuntary and uncontrollable chronic feeling of hunger, combined with a slow metabolism that leads to excessive eating and life-threatening obesity (62–64). The major theory proposed for explaining the insatiable appetite and excessive weight gain associated with PWS is elevated circulating levels of the orexigenic hormone ghrelin in the blood (65–67). Despite this, the direct approach of treating PWS patients with a GHSR1a antagonist, to our knowledge, has not been tested; this is likely because early clinical studies where GHSR1a antagonists were tested as antiobesity agents in normal subjects proved disappointing. The age-dependent (7 months to 5 years) transition to hyperphagia in PWS does not correlate with a change in ghrelin levels (68). However, as PWS children progress into adulthood hyperphagia becomes more severe; notably, the postprandial decrease in ghrelin observed in normal subjects is absent (69). Indeed, this is a most important distinction between normal and PWS adults. The postprandial nadir in endogenous ghrelin observed in normal subjects provides a mechanism for GHSR1a re-sensitization, and a satiety signal to tell the brain – stop eating! Indeed, the overall phenotype of PWS can be interpreted as impaired GHSR1a re-sensitization and as consequence low levels of active GHSR1a.
Prader–Willi syndrome patients present with high ghrelin and low GH levels; however, the anterior pituitary gland appears structurally normal and produces GH, suggesting impaired hypothalamic-pituitary signaling. Since GHSR1a agonists, including ghrelin, stimulate GH release, a possible explanation, other than GHSR1a desensitization, is that GHSR expression is attenuated. Expression of GHSR is positively regulated by thyroid hormone and estradiol, and negatively by cortisol (70). Indeed, hypothyroidism, hypogonadism, and elevated cortisol are all associated with PWS (71). Although not assessed quantitatively, GHSR1a expression appears normal in brains from deceased PWS patients (72). Nevertheless, compared to normal subjects, PWS patients elicit attenuated GH release in response to a GHSR1a agonist (73). High plasma concentrations of ghrelin should result in hyperglycemia and insulin resistance, but these metabolic changes are not consistently observed in PWS (73, 74). Lowering ghrelin levels by administering somatostatin or octreotide had no effect on appetite, weight gain, or behavior in PWS subjects (75–77). However, this does not preclude a role for ghrelin, because these compounds also suppress secretion of gastrointestinal anorexigenic hormones (76).
A recent report on the Snord116± mouse model of PWS showed that different inhibitors of GHSR1a failed to inhibit food intake (78) and that the involvement of some new pathway is linked to changes in feeding and psychiatric behavior (79). The authors concluded that ghrelin signaling is not involved in PWS and perhaps the elevated plasma ghrelin concentration is playing a compensatory role in PWS subjects. These findings support our suggestion that elevated endogenous ghrelin results in GHSR1a desensitization and lower levels of active GHSR1a. Low GHSR1a levels on the plasma membrane of hypothalamic neurons would predictably cause hyperphagia because as described above the appetite suppressing effect of DRD2 agonists is dependent on molecular interactions with apo-GHSR1a and DRD2 (15). Indeed, ghsr−/− mice are completely resistant to the anorexigenic effect of the DRD2 agonist cabergoline. Down-stream, cabergoline increases expression and signaling of brain-derived neurotrophic factor (BDNF). BDNF is one of the most important suppressors of food intake, and BDNF is low in PWS (80, 81).
Endogenous dopamine signaling via DRD2 inhibits excessive food intake and is dependent upon apo-GHSR1a and formation of GHSR1a:DRD2 heteromers. Formation of heteromers results in non-canonical DRD2 signal transduction and suppression of food intake. We suggest that persistently elevated circulating ghrelin in PWS reduces accumulation of GHSR1a on the plasma membrane of hypothalamic neurons. Low concentrations of GHSR1a contribute to hyperphagia by lowering the concentration of GHSR1a:DRD2, thereby attenuating satiety signals regulated by dopamine. The feasibility of targeting heterodimers formed between GHSR1a and DRD2 to modify non-canonical dopamine signaling was illustrated by us previously (15). This finding provides an approach to suppress or augment dopamine signaling selectively in neurons that express GHSR1a:DRD2 through an allosteric mechanism. Indeed, we have identified GHSR1a antagonists belonging to distinct structural classes, quinazolinones and triazoles that respectively enhance and inhibit DRD2 signaling through GHSR1a:DRD2 heteromers. Since these heteromers are found in a hypothalamic regions involved in development of PWS, GHSR1a antagonists that enhance DRD2 signaling are potential candidates for therapeutic intervention to suppress appetite. Targeting dopamine signaling in GHSR1a:DRD2 expressing neurons with GHSR1a antagonists provides the opportunity to design drugs that selectively target neurons co-expressing GHSR1a and DRD2 without affecting neurons expressing DRD2 alone.
Summary
In subsets of hypothalamic neurons that co-express GHSR1a and DRD2, GHSR1a:DRD2 heteromers are formed. Formation of these heteromers results in allosteric modification of the conformation of DRD2, thereby causing non-canonical signal transduction in response to dopamine. In contrast to canonical DRD2 signal transduction that involves suppression of cAMP accumulation, the non-canonical pathway results in Gβγγ subunit-dependent activation of PLC and mobilization of [Ca2+]i. The physiological relevance of GHSR1a:DRD2 heteromers is illustrated by experiments in Ghsr+/+, Ghsr−/−, and Ghrelin−/− mice. DRD2 agonism produces anorexia in Ghsr+/+ and Ghrelin−/− mice, but not in Ghsr−/−mice. Hence, the anorexigenic effects of DRD2 agonists are dependent on GHSR1a, but not ghrelin. Further, Ghsr+/+ mice treated with a neutral GHSR1a antagonist (the triazole, JMV2959) are resistant to DRD2 agonist-induced anorexia. Hence, pharmacological intervention with a GHSR1a antagonist according to structure, triazole vs. quinazolinone will respectively allosterically block or enhance dopamine signaling in neurons expressing GHSR1a:DRD2 heteromers without affecting signaling in neurons expressing DRD2 alone. These results show the potential of developing drugs that selectively act on subsets of neurons that express GHSR1a:DRD2 heteromers for treating obsessive eating disorders such as in PWS and for psychiatric disorders associated with irregularities in dopamine signaling.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by a grant from the US National Institutes of Health (R01AG019230 to Roy G. Smith). We gratefully also thank the Josilyn Faith Levine and Harold Wilkinson IV and Leta Lindley Prader–Willi Research Fellowship awarded to Dr. Cristina Grande.
References
1. Dixit VD, Yang H, Sun Y, Weeraratna AT, Youm YH, Smith RG, et al. Ghrelin promotes thymopoiesis during aging. J Clin Invest (2007) 117:2778–90. doi: 10.1172/JCI30248
2. Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature (1999) 402:656–60. doi:10.1038/45230
3. Sun Y, Wang P, Zheng H, Smith RG. Ghrelin stimulation of growth hormone release and appetite is mediated through the growth hormone secretagogue receptor. Proc Natl Acad Sci U S A (2004) 101:4679–84. doi:10.1073/pnas.0305930101
4. Wren AM, Seal LJ, Cohen MA, Brynes AE, Frost GS, Murphy KG, et al. Ghrelin enhances appetite and increases food intake in humans. J Clin Endocrinol Metab (2001) 86:5992. doi:10.1210/jcem.86.12.8111
5. Howard AD, Feighner SD, Cully DF, Arena JP, Liberator PA, Rosenblum CI, et al. A receptor in pituitary and hypothalamus that functions in growth hormone release. Science (1996) 273:974–7. doi:10.1126/science.273.5277.974
6. Smith RG. Development of growth hormone secretagogues. Endocr Rev (2005) 26:346–60. doi:10.1210/er.2004-0019
7. Smith RG, Van der Ploeg LH, Howard AD, Feighner SD, Cheng K, Hickey GJ, et al. Peptidomimetic regulation of growth hormone secretion. Endocr Rev (1997) 18:621–45. doi:10.1210/edrv.18.5.0316
8. Sun Y, Ahmed S, Smith RG. Deletion of ghrelin impairs neither growth nor appetite. Mol Cell Biol (2003) 23:7973–81. doi:10.1128/MCB.23.22.7973-7981.2003
9. Sun Y, Asnicar M, Saha PK, Chan L, Smith RG. Ablation of ghrelin improves the diabetic but not obese phenotype of ob/ob mice. Cell Metab (2006) 3:379–86. doi:10.1016/j.cmet.2006.04.004
10. Sun Y, Butte NF, Garcia JM, Smith RG. Characterization of adult ghrelin and ghrelin receptor knockout mice under positive and negative energy balance. Endocrinology (2008) 149:843–50. doi:10.1210/en.2007-0271
11. McFarlane MR, Brown MS, Goldstein JL, Zhao TJ. Induced ablation of ghrelin cells in adult mice does not decrease food intake, body weight, or response to high-fat diet. Cell Metab (2014) 20:54–60. doi:10.1016/j.cmet.2014.04.007
12. Zhao TJ, Liang G, Li RL, Xie X, Sleeman MW, Murphy AJ, et al. Ghrelin O-acyltransferase (GOAT) is essential for growth hormone-mediated survival of calorie-restricted mice. Proc Natl Acad Sci U S A (2010) 107:7467–72. doi:10.1073/pnas.1002271107
13. Guan XM, Yu H, Palyha OC, McKee KK, Feighner SD, Sirinathsinghji DJ, et al. Distribution of mRNA encoding the growth hormone secretagogue receptor in brain and peripheral tissues. Brain Res Mol Brain Res (1997) 48:23–9. doi:10.1016/S0169-328X(97)00071-5
14. Jiang H, Betancourt L, Smith RG. Ghrelin amplifies dopamine signaling by crosstalk involving formation of GHS-R/D1R heterodimers. Mol Endocrinol (2006) 20:1772–85. doi:10.1210/me.2005-0084
15. Kern A, Albarran-Zeckler R, Walsh HE, Smith RG. Apo-ghrelin receptor forms heteromers with DRD2 in hypothalamic neurons and is essential for anorexigenic effects of DRD2 agonism. Neuron (2012) 73:317–32. doi:10.1016/j.neuron.2011.10.038
16. Fetissov SO, Meguid MM, Sato T, Zhang LH. Expression of dopaminergic receptors in the hypothalamus of lean and obese Zucker rats and food intake. Am J Physiol Regul Integr Comp Physiol (2002) 283:R905–10.
17. Johnson PM, Kenny PJ. Dopamine D2 receptors in addiction-like reward dysfunction and compulsive eating in obese rats. Nat Neurosci (2010) 13:635–41. doi:10.1038/nn.2519
18. Palmiter RD. Is dopamine a physiologically relevant mediator of feeding behavior? Trends Neurosci (2007) 30:375–81. doi:10.1016/j.tins.2007.06.004
19. Pijl H. Reduced dopaminergic tone in hypothalamic neural circuits: expression of a “thrifty” genotype underlying the metabolic syndrome? Eur J Pharmacol (2003) 480:125–31. doi:10.1016/j.ejphar.2003.08.100
20. Volkow ND, Wang G-J, Baler RD. Reward, dopamine and the control of food intake: implications for obesity. Trends Cogn Sci (2011) 15:37–46. doi:10.1016/j.tics.2010.11.001
21. Andrews ZB. The extra-hypothalamic actions of ghrelin on neuronal function. Trends Neurosci (2011) 34:31–40. doi:10.1016/j.tins.2010.10.001
22. Atcha Z, Chen WS, Ong AB, Wong FK, Neo A, Browne ER, et al. Cognitive enhancing effects of ghrelin receptor agonists. Psychopharmacology (Berl) (2009) 206:415–27. doi:10.1007/s00213-009-1620-6
23. Carlini VP, Varas MM, Cragnolini AB, Schiöth HB, Scimonelli TN, de Barioglio SR. Differential role of the hippocampus, amygdala, and dorsal raphe nucleus in regulating feeding, memory, and anxiety-like behavioral responses to ghrelin. Biochem Biophys Res Commun (2004) 313:635–41. doi:10.1016/j.bbrc.2003.11.150
24. Chen L, Xing T, Wang M, Miao Y, Tang M, Chen J, et al. Local infusion of ghrelin enhanced hippocampal synaptic plasticity and spatial memory through activation of phosphoinositide 3-kinase in the dentate gyrus of adult rats. Eur J Neurosci (2011) 33:266–75. doi:10.1111/j.1460-9568.2010.07491.x
25. Davis JF, Choi DL, Clegg DJ, Benoit SC. Signaling through the ghrelin receptor modulates hippocampal function and meal anticipation in mice. Physiol Behav (2011) 103:39–43. doi:10.1016/j.physbeh.2010.10.017
26. Diano S, Farr SA, Benoit SC, McNay EC, da Silva I, Horvath B, et al. Ghrelin controls hippocampal spine synapse density and memory performance. Nat Neurosci (2006) 9:381–8. doi:10.1038/nn1656
27. Jerlhag E, Egecioglu E, Landgren S, Salomé N, Heilig M, Moechars D, et al. Requirement of central ghrelin signaling for alcohol reward. Proc Natl Acad Sci U S A (2009) 106:11318–23. doi:10.1073/pnas.0812809106
28. Albarran-Zeckler RG, Brantley AF, Smith RG. Growth hormone secretagogue receptor (GHS-R1a) knockout mice exhibit improved spatial memory and deficits in contextual memory. Behav Brain Res (2012) 232:13–9. doi:10.1016/j.bbr.2012.03.012
29. Jacoby SM, Currie PJ. SKF 83566 attenuates the effects of ghrelin on performance in the object location memory task. Neurosci Lett (2011) 504:316–20. doi:10.1016/j.neulet.2011.09.056
30. Jerlhag E, Egecioglu E, Dickson SL, Engel JA. Ghrelin receptor antagonism attenuates cocaine- and amphetamine-induced locomotor stimulation, accumbal dopamine release, and conditioned place preference. Psychopharmacology (Berl) (2010) 211:415–22. doi:10.1007/s00213-010-1907-7
31. Wellman PJ, Davis KW, Nation JR. Augmentation of cocaine hyperactivity in rats by systemic ghrelin. Regul Pept (2005) 125:151–4. doi:10.1016/j.regpep.2004.08.013
32. Schaeffer M, Langlet F, Lafont C, Molino F, Hodson DJ, Roux T, et al. Rapid sensing of circulating ghrelin by hypothalamic appetite-modifying neurons. Proc Natl Acad Sci U S A (2013) 110:1512–7. doi:10.1073/pnas.1212137110
33. Lawrence CB, Snape AC, Baudoin FM, Luckman SM. Acute central ghrelin and GH secretagogues induce feeding and activate brain appetite centers. Endocrinology (2002) 143:155–62. doi:10.1210/endo.143.1.8561
34. Pirnik Z, Bundziková J, Holubová M, Pýchová M, Fehrentz JA, Martinez J, et al. Ghrelin agonists impact on Fos protein expression in brain areas related to food intake regulation in male C57BL/6 mice. Neurochem Int (2011) 59:889–95. doi:10.1016/j.neuint.2011.08.001
35. Watabe-Uchida M, Zhu L, Ogawa SK, Vamanrao A, Uchida N. Whole-brain mapping of direct inputs to midbrain dopamine neurons. Neuron (2012) 74:858–73. doi:10.1016/j.neuron.2012.03.017
36. Cone JJ, McCutcheon JE, Roitman MF. Ghrelin acts as an interface between physiological state and phasic dopamine signaling. J Neurosci (2014) 34:4905–13. doi:10.1523/JNEUROSCI.4404-13.2014
37. Gasbarri A, Sulli A, Packard MG. The dopaminergic mesencephalic projections to the hippocampal formation in the rat. Prog Neuropsychopharmacol Biol Psychiatry (1997) 21:1–22. doi:10.1016/S0278-5846(96)00157-1
38. Gasbarri A, Verney C, Innocenzi R, Campana E, Pacitti C. Mesolimbic dopaminergic neurons innervating the hippocampal formation in the rat: a combined retrograde tracing and immunohistochemical study. Brain Res (1994) 668:71–9. doi:10.1016/0006-8993(94)90512-6
39. Swanson LW. The projections of the ventral tegmental area and adjacent regions: a combined fluorescent retrograde tracer and immunofluorescence study in the rat. Brain Res Bull (1982) 9:321–53. doi:10.1016/0361-9230(82)90145-9
40. Lisman JE, Grace AA. The hippocampal-VTA loop: controlling the entry of information into long-term memory. Neuron (2005) 46:703–13. doi:10.1016/j.neuron.2005.05.002
41. Rossato JI, Bevilaqua LR, Izquierdo I, Medina JH, Cammarota M. Dopamine controls persistence of long-term memory storage. Science (2009) 325:1017–20. doi:10.1126/science.1172545
42. Banks WA, Tschop M, Robinson SM, Heiman ML. Extent and direction of ghrelin transport across the blood-brain barrier is determined by its unique primary structure. J Pharmacol Exp Ther (2002) 302:822–7. doi:10.1124/jpet.102.034827
43. Furness JB, Hunne B, Matsuda N, Yin L, Russo D, Kato I, et al. Investigation of the presence of ghrelin in the central nervous system of the rat and mouse. Neuroscience (2011) 193:1–9. doi:10.1016/j.neuroscience.2011.07.063
44. Grouselle D, Chaillou E, Caraty A, Bluet-Pajot MT, Zizzari P, Tillet Y, et al. Pulsatile cerebrospinal fluid and plasma ghrelin in relation to growth hormone secretion and food intake in the sheep. J Neuroendocrinol (2008) 20:1138–46. doi:10.1111/j.1365-2826.2008.01770.x
45. Sakata I, Nakano Y, Osborne-Lawrence S, Rovinsky SA, Lee CE, Perello M, et al. Characterization of a novel ghrelin cell reporter mouse. Regul Pept (2009) 155:91–8. doi:10.1016/j.regpep.2009.04.001
46. Missale C, Nash S, Robinson S, Jaber M, Caron M. Dopamine receptors: from structure to function. Physiol Rev (1998) 78:189–225.
47. Milligan G. G protein-coupled receptor hetero-dimerization: contribution to pharmacology and function. Br J Pharmacol (2009) 158:5–14. doi:10.1111/j.1476-5381.2009.00169.x
48. Smith NJ, Milligan G. Allostery at G protein-coupled receptor homo- and heteromers: uncharted pharmacological landscapes. Pharmacol Rev (2010) 62:701–25. doi:10.1124/pr.110.002667
49. Albizu L, Cottet M, Kralikova M, Stoev S, Seyer R, Brabet I, et al. Time-resolved FRET between GPCR ligands reveals oligomers in native tissues. Nat Chem Biol (2010) 6:587–94. doi:10.1038/nchembio.396
50. Maurel D, Comps-Agrar L, Brock C, Rives ML, Bourrier E, Ayoub MA, et al. Cell-surface protein-protein interaction analysis with time-resolved FRET and snap-tag technologies: application to GPCR oligomerization. Nat Methods (2008) 5:561–7. doi:10.1038/nmeth.1213
51. González S, Moreno-Delgado D, Moreno E, Pérez-Capote K, Franco R, Mallol J, et al. Circadian-related heteromerization of adrenergic and dopamine D4 receptors modulates melatonin synthesis and release in the pineal gland. PLoS Biol (2012) 10:e1001347. doi:10.1371/journal.pbio.1001347
52. Hasbi A, Fan T, Alijaniaram M, Nguyen T, Perreault ML, O’Dowd BF, et al. Calcium signaling cascade links dopamine D1-D2 receptor heteromer to striatal BDNF production and neuronal growth. Proc Natl Acad Sci U S A (2009) 106:21377–82. doi:10.1073/pnas.0903676106
53. Jones KA, Borowsky B, Tamm JA, Craig DA, Durkin MM, Dai M, et al. GABA(B) receptors function as a heteromeric assembly of the subunits GABA(B)R1 and GABA(B)R2. Nature (1998) 396:674–9. doi:10.1038/25460
54. Rashid AJ, So CH, Kong MM, Furtak T, El-Ghundi M, Cheng R, et al. D1-D2 dopamine receptor heterooligomers with unique pharmacology are coupled to rapid activation of Gq/11 in the striatum. Proc Natl Acad Sci U S A (2007) 104:654–9. doi:10.1073/pnas.0604049104
55. Dezaki K, Kakei M, Yada T. Ghrelin uses Galphai2 and activates voltage-dependent K+ channels to attenuate glucose-induced Ca2+ signaling and insulin release in islet beta-cells: novel signal transduction of ghrelin. Diabetes (2007) 56:2319–27. doi:10.2337/db07-0345
56. Park S, Jiang H, Zhang H, Smith RG. Modification of ghrelin receptor signaling by somatostatin receptor-5 regulates insulin release. Proc Natl Acad Sci (2012) 109:19003–8. doi:10.1073/pnas.1209590109
57. Rediger A, Piechowski CL, Yi CX, Tarnow P, Strotmann R, Gruters A, et al. Mutually opposite signal modulation by hypothalamic heterodimerization of ghrelin and melanocortin-3 receptors. J Biol Chem (2011) 286:39623–31. doi:10.1074/jbc.M111.287607
58. Whittington JE, Butler JV, Holland AJ. Changing rates of genetic subtypes of Prader-Willi syndrome in the UK. Eur J Hum Genet (2007) 15:127–30. doi:10.1038/sj.ejhg.5201716
59. Duker AL, Ballif BC, Bawle EV, Person RE, Mahadevan S, Alliman S, et al. Paternally inherited microdeletion at 15q11.2 confirms a significant role for the SNORD116 C/D box snoRNA cluster in Prader-Willi syndrome. Eur J Hum Genet (2010) 18:1196–201. doi:10.1038/ejhg.2010.102
60. Powell WT, Coulson RL, Crary FK, Wong SS, Ach RA, Tsang P, et al. A Prader-Willi locus lncRNA cloud modulates diurnal genes and energy expenditure. Hum Mol Genet (2013) 22:4318–28. doi:10.1093/hmg/ddt281
61. Zhang Q, Bouma GJ, McClellan K, Tobet S. Hypothalamic expression of snoRNA Snord116 is consistent with a link to the hyperphagia and obesity symptoms of Prader-Willi syndrome. Int J Dev Neurosci (2012) 30:479–85. doi:10.1016/j.ijdevneu.2012.05.005
62. Driscoll DJ, Miller JL, Schwartz S, Cassidy SB. Prader-Willi Syndrome. In: Pagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR, Fong CT, et al. editors. GeneReviews® [Internet]. Seattle WA: University of Washington; 1993–2014 (1993). Available from http://www.ncbi.nlm.nih.gov/books/NBK1330/
63. Schulze AH, Petersen MB, Blichfeldt SS, Kastrup KW, Brondum-Nielsen K. [Prader-Willi syndrome – clinical picture and genetics]. Ugeskr Laeger (1995) 157:1513–9.
64. Whittington J, Holland A. Neurobehavioral phenotype in Prader-Willi syndrome. Am J Med Genet C Semin Med Genet (2010) 154C:438–47. doi:10.1002/ajmg.c.30283
65. Cummings DE, Clement K, Purnell JQ, Vaisse C, Foster KE, Frayo RS, et al. Elevated plasma ghrelin levels in Prader-Willi syndrome. Nat Med (2002) 8:643–4. doi:10.1038/nm0702-643
66. Haqq AM, Farooqi IS, O’Rahilly S, Stadler DD, Rosenfeld RG, Pratt KL, et al. Serum ghrelin levels are inversely correlated with body mass index, age, and insulin concentrations in normal children and are markedly increased in Prader-Willi syndrome. J Clin Endocrinol Metab (2003) 88:174–8. doi:10.1210/jc.2002-021052
67. Yi CX, Heppner K, Tschop MH. Ghrelin in eating disorders. Mol Cell Endocrinol (2011) 340:29–34. doi:10.1016/j.mce.2011.03.001
68. Goldstone AP, Holland AJ, Butler JV, Whittington JE. Appetite hormones and the transition to hyperphagia in children with Prader-Willi syndrome. Int J Obes (Lond) (2012) 36:1564–70. doi:10.1038/ijo.2011.274
69. DelParigi A, Tschop M, Heiman ML, Salbe AD, Vozarova B, Sell SM, et al. High circulating ghrelin: a potential cause for hyperphagia and obesity in Prader-Willi syndrome. J Clin Endocrinol Metab (2002) 87:5461–4. doi:10.1210/jc.2002-020871
70. Petersenn S, Rasch AC, Penshorn M, Beil FU, Schulte HM. Genomic structure and transcriptional regulation of the human growth hormone secretagogue receptor. Endocrinology (2001) 142:2649–59. doi:10.1210/en.142.6.2649
71. Vaiani E, Herzovich V, Chaler E, Chertkoff L, Rivarola MA, Torrado M, et al. Thyroid axis dysfunction in patients with Prader-Willi syndrome during the first 2 years of life. Clin Endocrinol (2010) 73:546–50. doi:10.1111/j.1365-2265.2010.03840.x
72. Talebizadeh Z, Kibiryeva N, Bittel DC, Butler MG. Ghrelin, peptide YY and their receptors: gene expression in brain from subjects with and without Prader-Willi syndrome. Int J Mol Med (2005) 15:707–11.
73. Grugni G, Guzzaloni G, Morabito F. Impairment of GH responsiveness to GH-releasing hexapeptide (GHRP-6) in Prader-Willi syndrome. J Endocrinol Invest (2001) 24:340–8. doi:10.1007/BF03343871
74. Choe YH, Jin DK, Kim SE, Song SY, Paik KH, Park HY, et al. Hyperghrelinemia does not accelerate gastric emptying in Prader-Willi syndrome patients. J Clin Endocrinol Metab (2005) 90:3367–70. doi:10.1210/jc.2004-1651
75. De Waele K, Ishkanian SL, Bogarin R, Miranda CA, Ghatei MA, Bloom SR, et al. Long-acting octreotide treatment causes a sustained decrease in ghrelin concentrations but does not affect weight, behaviour and appetite in subjects with Prader-Willi syndrome. Eur J Endocrinol (2008) 159:381–8. doi:10.1530/EJE-08-0462
76. Haqq AM, Stadler DD, Rosenfeld RG, Pratt KL, Weigle DS, Frayo RS, et al. Circulating ghrelin levels are suppressed by meals and octreotide therapy in children with Prader-Willi syndrome. J Clin Endocrinol Metab (2003) 88:3573–6. doi:10.1210/jc.2003-030205
77. Tan TM, Vanderpump M, Khoo B, Patterson M, Ghatei MA, Goldstone AP. Somatostatin infusion lowers plasma ghrelin without reducing appetite in adults with Prader-Willi syndrome. J Clin Endocrinol Metab (2004) 89:4162–5. doi:10.1210/jc.2004-0835
78. Lin D, Wang Q, Ran H, Liu K, Wang Y, Wang J, et al. Abnormal response to the anorexic effect of GHS-R inhibitors and exenatide in male Snord116 deletion mouse model for Prader-Willi syndrome. Endocrinology (2014) 155:2355–62. doi:10.1210/en.2013-2083
79. Tauber M, Diene G, Mimoun E, Cabal-Berthoumieu S, Mantoulan C, Molinas C, et al. Prader-Willi syndrome as a model of human hyperphagia. Front Horm Res (2014) 42:93–106. doi:10.1159/000358317
80. Chiba S, Numakawa T, Ninomiya M, Yoon HS, Kunugi H. Cabergoline, a dopamine receptor agonist, has an antidepressant-like property and enhances brain-derived neurotrophic factor signaling. Psychopharmacology (Berl) (2010) 211:291–301. doi:10.1007/s00213-010-1894-8
Keywords: ghrelin, growth hormone secretagogue receptor, dopamine, heterodimers, hypothalamus
Citation: Kern A, Grande C and Smith RG (2014) Apo-ghrelin receptor (apo-GHSR1a) regulates dopamine signaling in the brain. Front. Endocrinol. 5:129. doi: 10.3389/fendo.2014.00129
Received: 20 May 2014; Paper pending published: 23 June 2014;
Accepted: 18 July 2014; Published online: 18 August 2014.
Edited by:
Jacques Epelbaum, Université Paris Descartes, FranceReviewed by:
Justo P Castaño, University of Cordoba and Maimonides Biomedical Research Institute, SpainJacques Pantel, INSERM, France
Copyright: © 2014 Kern, Grande and Smith. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roy G. Smith, Department of Metabolism and Aging, Scripps Research Institute Florida, 130 Scripps Way #3B3, Jupiter, FL 33458, USA e-mail:cmdzbWl0aEBzY3JpcHBzLmVkdQ==
†Joint first authors