 Jonathan Janssens
Jonathan Janssens Harmonie Etienne
Harmonie Etienne Sherif Idriss1
Sherif Idriss1 Bronwen Martin
Bronwen Martin Stuart Maudsley
Stuart Maudsley- 1Translational Neurobiology Group, VIB Department of Molecular Genetics, University of Antwerp, Antwerp, Belgium
- 2Metabolism Unit, National Institute on Aging, National Institutes of Health, Bethesda, MD, USA
With our increasing appreciation of the true complexity of diseases and pathophysiologies, it is clear that this knowledge needs to inform the future development of pharmacotherapeutics. For many disorders, the disease mechanism itself is a complex process spanning multiple signaling networks, tissues, and organ systems. Identifying the precise nature and locations of the pathophysiology is crucial for the creation of systemically effective drugs. Diseases once considered constrained to a limited range of organ systems, e.g., central neurodegenerative disorders such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’ disease (HD), the role of multiple central and peripheral organ systems in the etiology of such diseases is now widely accepted. With this knowledge, it is increasingly clear that these seemingly distinct neurodegenerative disorders (AD, PD, and HD) possess multiple pathophysiological similarities thereby demonstrating an inter-related continuum of disease-related molecular alterations. With this systems-level appreciation of neurodegenerative diseases, it is now imperative to consider that pharmacotherapeutics should be developed specifically to address the systemic imbalances that create the disorders. Identification of potential systems-level signaling axes may facilitate the generation of therapeutic agents with synergistic remedial activity across multiple tissues, organ systems, and even diseases. Here, we discuss the potentially therapeutic systems-level interaction of the glucagon-like peptide 1 (GLP-1) ligand–receptor axis with multiple aspects of the AD, PD, and HD neurodegenerative continuum.
Introduction
Heptahelical G protein-coupled receptors (GPCRs) represent perhaps the most important single-protein class of pharmacotherapeutic targets. GPCR systems have adapted to perceive almost all forms of environmental and endogenous signaling entities, e.g., photons, odorants, lipids, carbohydrates, peptides, and nucleic acids. Currently, nearly 50% of existing licensed therapeutic compounds functionally interact with GPCRs (1, 2). Not only do GPCRs constitute one of the primary therapeutic targets but also their importance in systemic biology cannot be underestimated, as GPCRs constitute approximately 5% of the genome in early species such as the nematode worm and still approximately 1.5% of the genome in Homo sapiens (3–5). Therefore, it is likely that the molecular diversity is closely associated with changes in organismal complexity and compartmentalization. Rudimentary organisms such as nematode worms, with little tissue separation or definition, often require multiple ligands and receptors to specify particular signaling modalities. However, with the increased tissue complexity and physical separation present in highly complex multisystem organisms such as Homo sapiens, this ligand–receptor multiplicity has been refined to achieve the same degree of functional specificity. Therefore, instead of producing a single ligand–receptor system for each distinct function, ligand–receptor systems have been reproduced in distinct and often distant sites but are functionally differentiated from each other via cell-type specific scaffolding and transduction system interactions (6–13). Such a paradigm therefore creates a whole-organism mechanism to coordinate the activities of ligand–receptor systems at multiple tissue sites. While simple organisms essentially can constitute one physical interaction environment, more complex organisms require the subtly regulated behavior of multiple tissue/organ systems to be present for the homeostasis of such complex events as energy metabolism or reproduction. It is now clear from recent research that more complex communication systems, which overarch traditional organ axis [e.g., the hypothalamic–pituitary–gonadal (HPG) axis], potentially exist and that these “super-axes” are instead coordinated and generated around repeated and transposed ligand–receptor systems (14–20). The generation of these ligand–receptor super-axes therefore demonstrates an alternative method compared to more clinically based anatomical axes, in which to study and appreciate both physiological and pathophysiological processes. While normal homeostatic processes can be coordinated through these systems, it is likely that disease processes as well could be specifically present at this level of molecular complexity. While these added dimensions of physiological appreciation may seem daunting, they do, however, present the capacity to identify ligand–receptor super-axes that can be targeted for therapeutic situations in which a wide-scope of therapeutic activity is desired (6). It is in this context that our review is focused, i.e., is there potential ligand–receptor super-axis therapeutics that can ameliorate the multiple central and peripheral pathological processes that are present in neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’s disease (HD). In addition to the identification of potential super-axis therapeutics, we shall also discuss the potential strategies for engineered specificity of functional activity and signaling bias.
Systems-Level Expression of Neurodegenerative Disorders
In recent years, considerable evidence has accumulated that demonstrates that many of the classical central neurodegenerative disorders also coincide with significant and widespread peripheral pathophysiologies. In this respect, we must start to consider that diseases such as AD, PD, and HD may all share some common and coordinated pathological axes that are present in both central and peripheral sites. The presence of such a controlling system provides the possibility for a mechanism to systemically regulate and eventually remediate these diseases in a whole-body-wide approach. In the following sections, we will outline the multiple commonalities across these diseases and then allude to potential therapeutic systems that may facilitate such a multi-level therapy.
Alzheimer’s Disease
Alzheimer’s disease (AD) is the most common form of dementia that currently affects over 30 million people worldwide. With increasing life-span and lack of interventions to slow or stop the progression of AD, epidemiological studies indicate that this number is expected to reach 115.4 million in 2050 (21–25). Two different forms of AD have been described: the inherited form called “early-onset familial AD” (FAD) is caused by rare genetic variations in genes encoding amyloid precursor protein (APP), presenilin 1 (PSEN1), and 2 (PSEN2) involved in β-amyloid (Aβ) processing and which affect the intracellular trafficking of Aβ or APP (26, 27). Since <1% of AD cases are caused by genetic variations (27), the vast majority of AD cases are affected by the “sporadic late-onset” form of AD (SAD) (26, 27), which remains poorly understood. No genetic cause is known for SAD, but numerous risk factors exist such as aging, which is considered the most common risk factor for developing AD, head trauma, traumatic brain injury, metabolic dysfunctions, and many others. Furthermore, the ε4 allele of apolipoprotein E (APOE) represents another very important risk factor in both FAD and SAD (28, 29). The APOE protein is crucial for phospholipid and cholesterol transport and for neuronal damage repair. AD-related neurodegeneration causes progressive memory loss and a decline in cognitive abilities. These symptoms are associated with two neuropathological lesions: senile plaques that correspond to an extracellular deposition of Aβ peptides (27, 28), and neurofibrillary tangles (NFTs) due to the intracellular accumulation of the hyperphosphorylated microtubule protein Tau (30–32). AD-related behavioral abnormalities result from neuronal dysfunctions and death in different brain regions involved in cognition and mood such as the hippocampus, the amygdala, and the temporal, parietal, and frontal lobes of the cerebral cortex (25, 33, 34). AD pathology also includes coexisting metabolic and hormonal dysfunctions, inflammatory processes, excitotoxic damage, altered energy metabolism, and oxidative stress, not only in the brain but also throughout the body. These metabolic dysfunctions observed throughout the body are likely to contribute as well to neurological symptoms observed in AD (26, 27). Therefore, it is now clearly established that we need to take in consideration many other somatic and metabolic dysfunctions present in AD for a better understanding of the disease and to provide new targets for novel preventive or therapeutic efficacious treatments.
Metabolic Dysfunction in AD
Metabolic syndrome (MetS) is a disorder of energy utilization and storage and is characterized by abdominal obesity, elevated blood pressure, increased plasma glucose levels, high serum triglycerides, and low high-density cholesterol (HDL) levels (27, 28). Consequently, MetS has been considered as a risk factor for the development of cardiovascular diseases, type 2 diabetes mellitus (T2DM), and a number of dementias such as AD in patients over age 75 years (27). This is also reflected by the cellular and biochemical alterations observed in MetS since some of them have also been observed in AD patients (27, 28). In addition to MetS, insulin resistance in peripheral tissues corresponds to one of the main syndromes in T2DM and constitutes a high risk factor of developing AD (35). Insulin resistance results in the incapacity of insulin, generated in pancreatic β-cells, to mediate effective glucose uptake. Epidemiological studies have reported that people with diabetes mellitus have a 1.5- to 2.5-fold greater risk of developing cognitive impairment and dementia (36, 37). Moreover, hyperglycemia and insulin resistance are also likely to deteriorate the neuropathology of AD, especially in APOE4 carriers. Mechanistically, the increased risk of dementia with T2DM or obesity could be due to hyperglycemia, peripheral insulin resistance, oxidative stress, accumulation of advanced glycation end products, increased production of pro-inflammatory cytokines, and cerebral microvascular disease (38).
Inflammatory Processes in AD
Alzheimer’s disease is also closely associated with low-grade chronic inflammation (39) as highlighted by a number of increased systemic inflammation markers including C-reactive protein (CRP), fibrinogen, interleukin-6 (IL-6), and tumor necrosis factor α (TNF-α) (39, 40). It is well established that chronic inflammation in AD associated with MetS may arise from a deregulation of the endocrine homeostasis of adipose tissue, which is infiltrated by immune cells and produces pro-inflammatory molecules such as adipokines, cytokines, and chemokines (40). Pro-inflammatory molecules inhibit mitochondrial respiration in the brain, decrease the activities of the electron transport chain and mitochondrial membrane potential, increase mitochondrial membrane permeability, enhance reactive oxygen species (ROS) production, interfere with ATP production and cause mitochondrial shutdown, ultimately leading to neuronal degeneration (41). Increased levels of inflammatory molecules such as interleukin-1 (IL-1), IL-6, TNF-α, CRP, granulocyte macrophage colony-stimulating factor (GM-CSF), eotaxin, and macrophages inflammatory protein 1-α (MIP-1 α) have been reported in brain tissue from patients with AD (40, 42). In addition, 18 plasma biomarkers related to inflammation had been identified in patients affected with AD (23). Chronic inflammation is characterized by mononuclear cell infiltration of the affected tissue, angiogenesis, tissue clearance, fibrosis, and tissue remodeling that can interfere with the optimal tissue functions (43) and lead to some pathological disorders. Moreover, chronic inflammation may play a role in accelerated cognitive impairment by a direct effect on the brain or by influencing the development of vascular disease (36).
AD and Gastrointestinal Function
In addition to alterations of the central neuronal activity, it is now clear that a similar degradation of the enteric nervous system (ENS) in AD is apparent (44, 45). Gastrointestinal (GI) motility deficits are frequently occurring co-morbidities in dementia and represent a long-term debilitating problem. The structural integrity of gut muscle as well as the innervating ENS is both subject to amyloid-related cellular toxicity. Disruption of gut motility and the ability to absorb nutrients will significantly reduce metabolic support for stressed tissues across the whole body as nutrient absorption will be curtailed. With respect to GI energy uptake, alterations of multiple pancreatic factors are also involved in AD pathology, including amylin, leptin, and ghrelin. Amylin and leptin are both hormones that activate overlapping intracellular signaling pathways such as STAT3, Akt, and AMPK, which have a role in glucose and lipid metabolism and have an additive effect in signaling pathways. Amylin is co-localized and co-secreted with insulin from pancreatic β-cells in response to nutrient stimuli. As insulin secretion, the secretion of amylin is also impaired in T2DM. As for the deposition of Aβ in AD, amylin is also an amyloidogenic protein (islet amyloid peptide), which gets deposited in T2DM indicating that both AD and T2DM share many common disease features (46). In addition to disruption of the gut activity itself, there are also additional effects of AD pathology on the gut–brain axis communication. The orexigenic peptide ghrelin is primarily produced by the stomach, controls the central hunger sensation, and forms one of the major components of the gut–brain axis (26, 47). Ghrelin levels are often altered in AD patients suggesting that the gut–brain axis may contribute to defective cognition (48, 49). Ghrelin exerts an opposite effect to the anorexigenic leptin on food intake and energy homeostasis (26). Interestingly, it has been demonstrated that intracerebrovascular injection of ghrelin improved cognitive ability in streptozotocin-induced diabetic-rats by increasing the expression of cAMP response element-binding protein (CREB) and brain-derived neurotrophic factor (BDNF), and by attenuating the neuronal apoptosis in the hippocampus (27, 50). These findings suggested that ghrelin plays a pivotal role in metabolic control but also in regulating cognitive function and memory capacity.
AD and Adipose Tissue
While insulin increases the production of leptin by adipose tissue, leptin exerts a negative feedback on both insulin secretion and insulin gene expression in pancreatic islet β-cells. These effects are mediated by both the autonomic nervous system and by direct actions via leptin receptors on pancreatic β-cells (51, 52). AD-related insulin resistance syndromes could be associated with the dysfunction of the adipoinsular axis leading to obesity and hyperinsulinemia (26). Leptin is directly neuroprotective in dopaminergic cells, potentially via APP- and tau-related mechanisms (26). Adiponectin is a protein hormone specifically released from adipose tissue and regulates the sensitivity of insulin, modulates fatty acid catabolism, glucose homeostasis, and anti-inflammatory systems. Adiponectin plays also a role in memory and cognitive impairment and contributes to a dysregulated glucose metabolism and mitochondrial dysfunction observed in patients with AD (27). Adiponectin modulates the expression of inflammatory molecules and protects neurons against β-amyloid toxicity (53), supporting the evidence of a link between the immune response and insulin resistance. Taken together, these findings support the role of adipokines and hormones related to the adipoinsular axis in the development of Alzheimer’s disease. Leptin and amylin can modulate the activity of key molecules involved in energy balance and neurogenesis. Ghrelin is also able to regulate not only energy metabolism but also cognitive function and active memory capacity. Adipokines are also capable to modulate the inflammatory response mediated by the glial cells. Thus, adipokines can play a relevant role as targets in the treatment of AD neurodegeneration processes and support to the relationship between AD and diabetes or MetS (26).
AD and Cardiovascular Disease
Multiple lines of evidence demonstrate a strong functional linkage between cardiovascular functionality and AD-related pathophysiology (54). Amyloid peptides have been demonstrated to be directly toxic to endothelial cells in the peripheral and cerebral circulation (55, 56). Due to AD-related vascular dysfunction, local hypoxic events in the central nervous system (CNS) may further elevate metabolic dysfunction and eventual amyloid plaque deposition and NFT generation. Initial vascular damage in the CNS can also cause a significant reduction in blood–brain-barrier (BBB) integrity, which can result in pathophysiological exchange of humoral agents, toxic proteins (e.g., amyloid), or abnormal hormone levels between the periphery and CNS (57). In addition to the detrimental effects of amyloid upon vascular structures, there is also considerable evidence of AD-related cellular toxicity induced by amyloid aggregation-mediated disruption of normal protein stability, folding, and proteolysis in cardiac tissues (58).
AD and Circadian Rhythms
Significant alterations in daily behavioral and sleep patterns are commonly described in AD and other neurodegenerative disorders (59). Notable disruptions of circadian rhythm in AD include the fragmentation of the sleep–wake cycle leading to increased nocturnal awakenings with increased daytime sleep bouts (60). Prospective human studies have indicated that circadian activity pattern disruption (decreased rhythm amplitude and phase-delays) are significant predictors of subsequent AD, suggesting that compromised rhythms might actually be a reliable preclinical phenomenon (61). Circadian rhythms are generated in the suprachiasmatic nucleus (SCN) as an output of the clock gene cycle, produced by a series of interlocking transcriptional feedback/feedforward loops of a panel of clock genes (e.g., PER1,2, CRY1,2, CLOCK, BMAL1) (62). Outside of the SCN, there are circadian oscillators throughout the body that control multiple autonomic functions. As the activity of many tissues is strongly regulated in a circadian manner, it is not surprising that disruption can negatively impact immune, metabolic, and cardiovascular systems (59).
AD and Sensory Modalities
Neurodegeneration associated with AD affects multiple central and peripheral systems, including several perceptive modalities including vision, taste, pain, and olfaction (63–65). Olfactory impairment has been shown to be predictive of conversion from mild cognitive impairment to AD with a higher than 80% sensitivity (66). With respect to it has also been demonstrated that both cognitively impaired and AD patients present a significant reduction of total taste scores and also individual taste scores on either side of the tongue (67). As sensory modalities are currently further investigated in AD, it appears that these systems may eventually serve as potential “therapeutic gateways” into the psychosocial and physiological alterations in AD (66).
Parkinson’s Disease
Parkinson’s disease (PD) is the most common movement disorder with an age-dependent prevalence, affecting about 1% of individuals over 60 years of age and increasing up to 4–5% at an age of 85 years (68). Studies have demonstrated a broad genetic etiology for several forms of PD and related Parkinsonian disorders (69). Pathomechanisms associated with mutations in PD-related genes involve mitochondrial dysfunction, abnormal protein degradation, and oxidative stress (70, 71). In about 95% of PD patients, however, no apparent genetic linkage (sporadic PD) has been described, indicating that PD is a multifactorial disease that arises owing to both genetic and environmental factors (69). The classical diagnosis of PD is based on the presence of bradykinesia (slowness of movement) together with at least one other cardinal motor feature such as resting tremor, rigidity, and postural instability (72, 73). Many PD patients experience also a wide range of non-motor (NM) deficiencies, including neuropsychiatric symptoms, dysautonomia (i.e., malfunction of the autonomic nervous system; ANS), hyposmia, sensory loss, sleep disturbances, and GI dysfunction. Many of these NM symptoms precede the onset of core motor dysfunctions and have a multisystem origin. Cognitive and neuropsychiatric symptoms of PD range from depression, anxiety, and apathy to frank dementia (68, 74). The occurrence of mild cognitive impairment and subsequent dementia is common in PD patients, but is more prevalent in later stages of the disease where it affects up to 80% of patients (74, 75). Two dementia types are associated with PD: Parkinson’s disease dementia (PDD) and dementia with Lewy bodies (DLB), which can be distinguished based on the onset of extrapyramidal (EP) signs. Depression is another important feature of PD and is reported in up to 45% of PD patients. Motor dysfunction in PD is closely linked to a striatonigral dopaminergic denervation and a progressive loss of dopaminergic neurons located in the substantia nigra pars compacta (SNPC) (76, 77). Lewy body (LB) pathology, characterized as cytoplasmic inclusions of α-synuclein in neuronal perikarya, and neurodegeneration in PD patients extends well beyond the dopaminergic striatonigral system. Additional extranigral pathology in the CNS involves noradrenergic, serotonergic, and cholinergic systems, as well as the cerebral cortex and olfactory bulb (78). Involvement of the ANS occurs at early stages of PD involving both the sympathetic and parasympathetic ganglia, and the ENS. While nigrostriatal degeneration is responsible for motor features of PD, the extranigral degeneration is thought to account for global NM symptoms of the disease (79–81). These observations have changed our traditional view of PD as being a predominant single-system disorder with selective involvement of nigrostriatal dopaminergic neurons toward a much broader multisystem LB disorder, also defined as Parkinson complex or Lewy-complex.
PD and Sensory Dysfunction
A number of NM features reported in PD can be attributed to cholinergic dysfunction, e.g., altered rapid eye movement (REM) sleep, mood, cognition, and olfaction. Olfactory impairment is highly prevalent in PD and reported in approximately 95% of early-stage PD patients. Loss of smell correlates well with neurodegeneration and concomitant α-synuclein pathology in the olfactory bulb and other secondary olfactory regions (82, 83). Hyposmia occurs early in the disease progress and may precede overt motor symptoms suggesting that PD might be a primary disorder of olfaction (84). Whereas cholinergic dysfunction is suggested as a molecular basis for hyposmia, decrements in other neurotransmitter systems involving noradrenaline and serotonin production are also reported (85–87).
PD and Gastrointestinal Dysfunction
Gastrointestinal dysfunction is seen as one of the earliest manifestations of PD preceding motor involvement. The GI symptoms in PD include weight loss, dysphagia, and concomitant excessive drooling, reduced salivation, nausea, constipation, and defecatory dysfunction, which reflect a deregulation of the GI motility along the entire length of the GI tract (88). Moreover, a number of the GI manifestations such as constipation are suggested to be proportional to the risk of developing the disease (89). Neuropathological studies have indicated early enteric pathology in PD in terms of LB and Lewy neurite (LN) accumulation in the dorsal motor nucleus of the vagus (DMV) and the ENS (80, 90). Importantly, the ENS shares a number of characteristics with the CNS, but is easier accessible and analyzable compared to the CNS through colonic biopsies. Therefore, the ENS offers reasonable potential for identifying novel disease markers in living patients with PD (80). In addition to the bowel dysfunctions (constipation) reported in PD, impairments in other pelvic organs including the urinary bladder and reproductive organs (also called the genitourinary system) are common NM disorders in PD (91).
PD and Sleep Pathologies
Since dopamine is known to have a role in the sleep–wake cycle (92), it is not surprising that PD patients present with sleep pathologies, e.g., sleep-maintenance insomnia, and REM sleep behavior disorder (RBD) (74). The occurrence of RBD is common in PD and manifests before the onset of clinical PD (93). In these patients, decreased cholinergic innervation was reported in sleep nuclei located in the lower brainstem. Furthermore, patients with RBD are more likely to have hyposmia, cardiac sympathetic denervation, and an increased risk for developing dementia in PD (94, 95).
Cardiovascular System Dysfunction and PD
Parkinson’s disease also involves considerable cardiovascular manifestations of dysautonomias (96). More specifically, sympathetic noradrenergic denervation of the heart is an integral part of the disease and is associated with LBs in sympathetic ganglia including those that innervate the heart. The sympathetic nerve fibers in the heart are decreased in PD. Reduced cardiac uptake and concomitant sympathetic denervation is already reported in the early disease process and increases with disease duration and severity, however, the heart also receives parasympathetic input from the dorsal vagal nucleus; there is less known about parasympathetic denervation in the heart of PD patients (97–101). Another determinant that underlies the cardiovascular dysautonomia is arterial baroreflex failure. The baroreflex is the most important vasoconstrictor mechanism to buffer acute fluctuations of arterial blood pressure (ABP) that occur during changes in posture, exercise, emotion, and other conditions (102). Together, these cardiovascular deficits result in orthostatic hypotension (OH), which is present in about 30–40% of PD patients (103).
Metabolic Dysfunction and PD
Recent epidemiological and molecular genetic studies have highlighted strong links between PD and T2DM. Patients with T2DM have an increased risk of developing AD (65%) and PD (35%) (104, 105). Overlap between PD and T2DM is further strengthened by the fact that more than 60% of PD patients have impaired insulin signaling and are glucose intolerant (106, 107). This is in line with neuropathological studies of PD patients, which have shown that insulin receptors are also densely packed on dopaminergic neurons of the SNPC (108). Furthermore, a number of animal and in vitro studies have indicated a role for insulin and glucose metabolism in the regulation of brain dopaminergic activity and firing (106). Chronic hyperglycemia is known to induce oxidative stress and concomitant production of ROS, factors implicated in T2DM and PD etiology. Moreover, energy starvation and metabolic impairment can induce the aggregation of α-synuclein in dopaminergic cells (109). Despite the marked clinical differences between PD and T2DM, an intriguing common pathogenesis is emerging and involves alterations in mitochondrial turnover, neuroinflammation, protein degradation, and glucose metabolism [for review, see Ref. (107)].
PD and Inflammation
Chronic inflammation is a prominent feature of multiple neurodegenerative disorders including PD (110, 111). Neuroinflammation is strongly associated with dopaminergic neuron degeneration and progression of PD. Leucine-rich repeat kinase 2 (LRRK2), a kinase mutated in both autosomal-dominantly inherited and sporadic PD cases, modulates inflammation in response to different pathological stimuli. PD-associated LRRK2 mutations may sensitize microglia cells toward a pro-inflammatory state, which in turn results in exacerbated inflammation with consequent neurodegeneration (112). Inflammatory pathways in PD appear to play a crucial role in the destruction of both pancreatic islet β-cells and dopaminergic neurons in the substantia nigra (113). Emerging evidence indicates that system-wide metabolic dysfunction in PD can induce metabolic inflammation, thus exacerbating the neurodegenerative activity in this disorder. The combined interaction between energy balance and inflammatory responses in PD therefore represent an important field for therapeutic study.
Huntington’s Disease
Huntington’s disease (HD) is a disabling neurodegenerative disorder characterized by a progressive impairment of motor and cognitive functions and is caused by a mutation that takes the form of a CAG trinucleotide repeat expansion in exon 1 of the huntingtin (htt) gene on chromosome 4 (114, 115). Presently, the precise nature of the molecular functionality of endogenous non-mutant huntingtin is not comprehensively appreciated. However, huntingtin appears to be associated with modulation of BDNF expression (116), cytoskeletal organization (117), vesicle trafficking (118), and clathrin-mediated endocytic pathways (119). The mutated form, with the polyglutamine expansion, possesses an altered protein structure leading to its aggregation in the CNS. These changes in protein function and aggregation then invariably lead to neuronal degeneration. HD manifests in a variety of symptoms, which can be behavioral, motoric, and cognitive. Behavioral changes commonly occur before motor symptoms and include mood changes, irritability, restlessness, psychosis, and hallucinations. Motor symptoms mainly occur as quick sudden movements in the arms, legs, and face (chorea). Tremors, unsteady gait and head turning, and shift eye position also occur as motor symptoms. As the disease prognosis worsens, a progressive dementia occurs in the form of memory loss, disorientation, confusion, and loss of judgment. Huntington’s disease is also associated with several other debilitating non-neuronal impairments that also occur in other neurodegenerative disorders, particularly PD. These include weight loss as one of the major symptoms, insulin resistance, changes in energy metabolism, and sleep disturbance due to the disturbance of the circadian rhythm (120–122).
HD and Metabolic Dysfunction
In the pre-symptomatic phase and over the course of HD progression, several disruptive changes occur to the neuroendocrine system as a result of the htt mutation. Such changes can lead to detrimental effects to appetite, body weight, mood changes, and alterations of metabolism. One of the hubs of neuroendocrine metabolic disruption in neurodegenerative disease is the hypothalamus. Proteomic analysis of rat hypothalami expressing mutant htt has demonstrated alterations of heat shock protein-70 (Hsp-70), glutathione peroxidase (Gpx4) responsible for oxidative damage, glial fibrillary acidic protein (Gfap), and the enzyme acylglycerol-3-phosphate O-acyltransferase 1 (Agpat1) responsible for lipid synthesis (122). In addition to these hypothalamic findings, alterations in circulating endocrine factors are apparent in HD models, i.e., reduced insulin and leptin as well as reduced triglycerides and HDL. These changes were present presymptomatically and are concordant with other human studies. Aziz et al. (123) demonstrated that there are no significant differences in the characteristics of secretion of growth hormone (GH) and ghrelin of HD patients compared to controls. With disease progression, there are significant increases in secretion and irregularity of GH combined with an increased suppression of post-prandial ghrelin suppression. Such findings indicate that the impairment of regulation of GH and ghrelin secretion is positively correlated with worsening HD prognosis. HD patients as well as murine HD models demonstrate considerable diabetic-like pathologies, which create a severe metabolic stress and energetic dysregulation (121, 124–126). HD patients are more prone to T2DM than the general population (127). Such pathologies manifest in the form of hyperglycemia and abnormal lipid metabolism. HD is linked directly with a form of dysglycemia leading to a catabolic state characterized by weight loss and a lower body mass index than the healthy population. The lipid dysregulation involves high levels of triglycerides and LDL, which also creates a risk of microvascular and macrovascular pathologies on the long (124, 125). Insulin resistance is therefore thought to be one of the main factors of the pathogenesis of HD leading to severe neurobiological impairments. This hypothesis is supported by the InCHIANTI study where people with cognitive impairments were found to be more likely to suffer insulin resistance compared to controls (128). A clear relationship has been established between insulin secretion and HD using the R6/2 mouse model. These mice have shown glycosuria and glucose intolerance while above 70% of them develop diabetes at 14 weeks (126). The bacterial artificial chromosome-mediated transgenic HD (BACHD) mouse model has also shown impaired glucose metabolism and increased resistance to insulin and leptin (diabetic-like pathology). These effects were replicated by expressing a short fragment of mutant htt using an adenoviral vector, which causes hypothalamic inactivation (129).
HD and Gastrointestinal Functionality
Huntington’s disease patients have severe autonomic dysfunctions manifesting as disruptions of the GI tract, urinary and cardiovascular systems, as well as sexual dysfunction in men (114). Symptoms include difficulties of swallowing, dysphagia, early abdominal filling, defecation difficulties, urinary incontinence, and incomplete bladder emptying and postural hypotension. Such symptoms are very common in HD patients before the appearance of overt motor symptoms and also in carriers of the mutation but with less severity. Mutant htt is expressed along the GI tract and throughout the ENS. HD mouse models demonstrate a significant loss of functional neuropeptides in enteric nerves, decreased thickening of the GI mucosa, and villi length (130). Functionality of the GI tract is also significantly impaired in terms of gut motility and absorption of food. The degree of malabsorption inversely correlated with body weight indicating the importance of the GI tract dysfunction in weight loss and thus patient quality of life.
HD and Circadian Rhythm Alterations
Circadian studies have shown that HD patients suffer from abnormal night–day ratios and in addition the R6/2 HD mouse model also showed an abnormal night–day activity that was further disturbed as the disease progressed (131, 132). These disruptions were accompanied by an abnormal expression of clock genes in the SCN, striatum, and the motor cortex, correlating with the cognitive impairment and decline (132). In HD, it appears that functionality of the isolated SCN is specifically intact but that the pathophysiology is due to a dysfunction of the systemic circuitry rather than the SCN itself (133–135). Circadian changes in melatonin levels have been reported in HD patients (123). Synthesis of melatonin is regulated by the SCN and has a major role in the regulation of sleep as well as in peripheral circadian rhythms. Due to the ubiquitous expression of mutant htt and the presence of both central and peripheral circadian rhythms, HD-related pathology results in disruption of sleep as well as in uncoupling of peripheral and central circadian rhythms. For example, if liver metabolism is uncoupled from circadian rhythms, this leads to metabolic disruption and a detrimental effect on the disease progression (136). Levels of wakefulness-promoting factors such as orexin, ghrelin, adrenocorticotrophin hormone, and corticotrophin-releasing hormone have all been found to be abnormal in HD (136). Consequences of sleep deprivation including stress, depression, reduced immunity, memory and learning impairments, and metabolic and hormonal abnormalities are all likely to further exacerbate HD-related pathophysiology across the whole body (137–140).
HD and Cardiovascular Dysfunction
Consistent with a significant autonomic dysfunction, the cardiovascular system is also impaired in HD patients as well as in murine HD models (141, 142). It appears that both sympathetic and parasympathetic cardiovascular system-regulating systems as well as the baroreceptor reflexes are impaired along with their central regulatory pathways (143). HD patients are more than 10 times more likely to suffer from cardiovascular health issues compared to normal patients (144). While a significant component of cardiovascular pathology in HD may be due to autonomic nervous disruption, recent evidence has also demonstrated a direct pathological action of mutant htt upon cardiomyocytes as well (144, 145).
HD and Inflammatory Processes
Studies have provided evidence that the immune system is pathophysiologically active in HD even before overt disease manifestation (146). In addition, deficits of immune cell migration in response to chemoattractants have been obtained from human studies (147). Chemokine profiles are also significantly altered in HD patients as the disease progresses, with elevations of eotaxin and eotaxin-3, the chemokine (C–C motif) ligand 2 (CCL2), 3 (CCL3), and 4 (CCL4) (148). In addition to altered chemokine profiles, there is considerable evidence that demonstrates increased levels of circulating and CNS-borne pro-inflammatory cytokines, including IL-6, IL-8, and TNF-α (147). Reinforcing the potential importance of TNF-α in HD pathogenesis, it has recently been shown that therapeutic inhibition of TNF-α activity can significantly attenuate central and peripheral inflammation in the R6/2 HD model mice (149).
HD and Sensory Modalities
As with the other major neurodegenerative disorders discussed here, HD pathophysiology has been demonstrated to impact several sensory modalities. Hence, HD disease presentation and progression has been associated with disruption of visual perceptive systems (150), impairment of olfactory sensitivity (151), and alterations in gustatory responses to multiple stimulating tastants (152). With respect to gustation, it has been hypothesized that the specific disruption of this sensory modality may be a specific early indication of the loss of sufficient neurotrophic support in the CNS (153).
Signaling System “Super-Axes”
From multiple sources of information, both physiological and molecular biological, it now seems apparent that for many of the major CNS neurodegenerative disorders. i.e., AD, PD, and HD, their true pathophysiological spectrum is more widespread across the body than previously considered. In addition, it is clear that there are strong similarities between these neurodegenerative conditions, suggesting perhaps that a considerable proportion of these diseases is controlled by endogenous signaling systems that are merely perturbed by the initial disease locus, but then once activated stimulate a coherent series of pathophysiological activities. This potential system-wide disease–response axis clearly needs mechanisms to maintain its activity and also coordinate its functionality across diverse tissues in varied locations across the body. As we have previously discussed in the Section “Introduction,” the evolution of receptor signaling systems has had to deal with the challenges of intense somatic development from nematode worms to the hyper-complex Homo sapiens. The presence of the same receptor signaling system, in multiple specialized conformations, and in diverse tissues has been demonstrated for cholinergic ligands such as acetylcholine and peptidergic ligands such as gonadotropin-releasing hormone (GnRH: 12). With respect to a receptor signaling that may be preferentially involved in regulating the generic neurodegenerative “super-axis” system, we have identified a potential ligand–receptor system, the glucagon-like peptide 1 (GLP-1) system that may be critical for regulating pathophysiology, and therefore, also facilitating potential neurodegenerative remediation. Historically, GLP-1 has been considered primarily a gut incretin that is vitally involved, in concert with insulin, with glucose metabolism. GLP-1 is produced both in pancreatic α-cells as well as intestinal L-cells (154). Upon release into the circulation after food ingestion, GLP-1 facilitates glucose uptake by directly acting on pancreatic islet β-cells to enhance post-prandial insulin secretion (155). This process is mediated by GLP-1-mediated activation of a class B1 (secretin-like family) seven transmembrane spanning GPCRs (156, 157). The GLP-1 receptor (GLP-1R) has been shown to functionally interact with both heterotrimeric G proteins [Gαs, Gαq (158) as well as β-arrestin (159)]. This promiscuity of the GLP-1R therefore facilitates the ability to flexibly stimulate this receptor system to engender multiple downstream signaling cascades (1). Underpinning our assertion that the GLP-1 signaling system may represent an organism-wide functional “super-axis,” it has been demonstrated that components of the GLP-1 system are found in multiple tissues all the way across the body from the tongue, olfactory epithelia, CNS, heart, pancreas, intestine to reproductive tissues [Figure 1 (160–166)]. Considering the vital role of GLP-1 in energy metabolism and in maintaining the viability of multiple tissues, it is unsurprising that this receptor system has been transposed across the body. This super-axis therefore creates that possibility of multi-site, multi-tissue drug remediation of neurodegenerative disorders. Given the recent emergence of appreciation of the importance of metabolic support to diseases such as AD, PD, and HD (107, 114, 122), it is evident that advanced therapeutic control of the GLP-1 super-axis could therefore generate an excellent capacity to generate whole-organism systemic therapeutic actions. With respect to the connection between neuropathophysiology, it has been demonstrated that GLP-1 signaling is critically involved in metabolic regulation (165), controlling inflammatory processes (167), regulate gut–brain axis activity (168), control multiple sensory modalities (169, 170), modulate cardiovascular activity (171), coordinate sleep–wake cycles and circadian rhythms (172, 173). The GLP-1 system is likely to be only one of the several ligand–receptor systems that exerts a super-axis level of impact upon neurodegenerative mechanisms from multiple divergent initiator loci, however, due to considerable advances in therapeutic ligand design, it does represent an important target for the creation of a systems-level remedial agent. Concordant with the findings that the GLP-1 receptor system is intimately involved in multiple aspects of the neurodegenerative axes of AD, PD, and HD, it is unsurprising that ligands that can target this receptor system have been demonstrated to exert multiple remedial and effective actions (168, 174–178). The therapeutic regulation of such systems-level receptor systems clearly represents an excellent target for more nuanced therapeutic design as the efficacy of such “super-axis” compounds may be reinforced very strongly via multiple forms of tissue-to-tissue communication. Thus, it is likely that systems-level therapies may be far more efficacious than compared to receptor-modulating ligands that are only targeted to one specific component of the neurodegenerative axis (Figure 2). In this context of the potential “super-axis” therapies, a more advanced appreciation of the functional pharmacology of these receptor–ligand systems is vital. The generation of novel tissue and/or signal-selective GLP-1 modulating agents (1, 10, 179) is therefore perhaps one of the most important future fields of study for neurodegeneration.
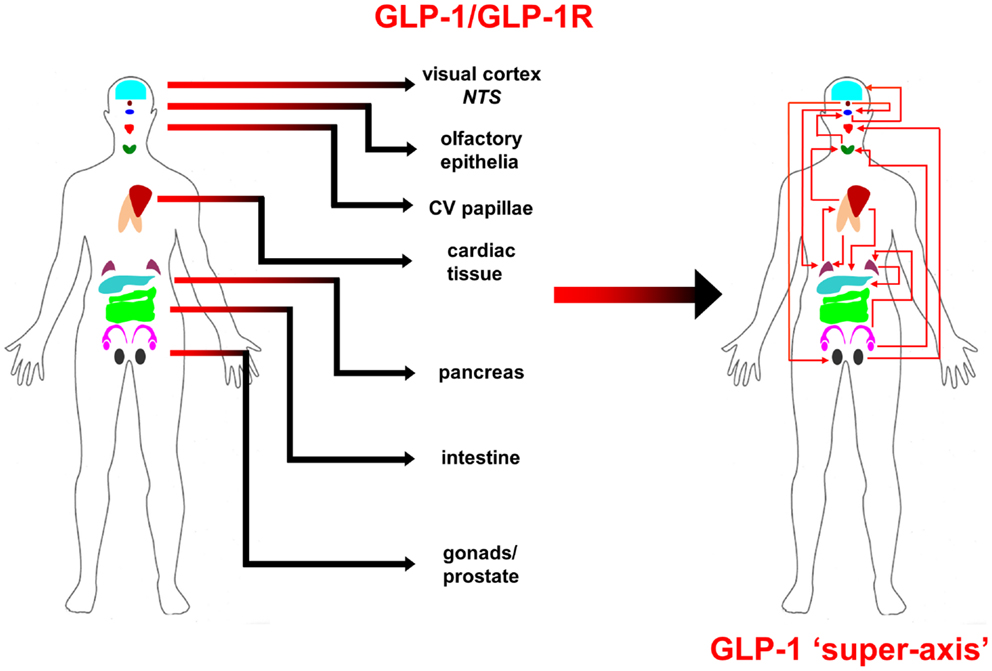
Figure 1. Glucagon-like peptide 1 (GLP-1) ligand and receptor system super-axis are shown. The expression of the ligand and receptor components of the GLP-1 system spans the whole human body. The repetitive expression of this GPCR system in multiple tissue types reinforces the importance of maintaining energy balance across the whole organism with an easily coordinated mechanism. The physical and hormonal connection between these multiple sites of GPCR functionality therefore can represent a “super-axis” of signaling connectivity that spreads across and over more classically defined tissue–tissue axes, such as the hypothalamic–pituitary– gonadotropic axis.
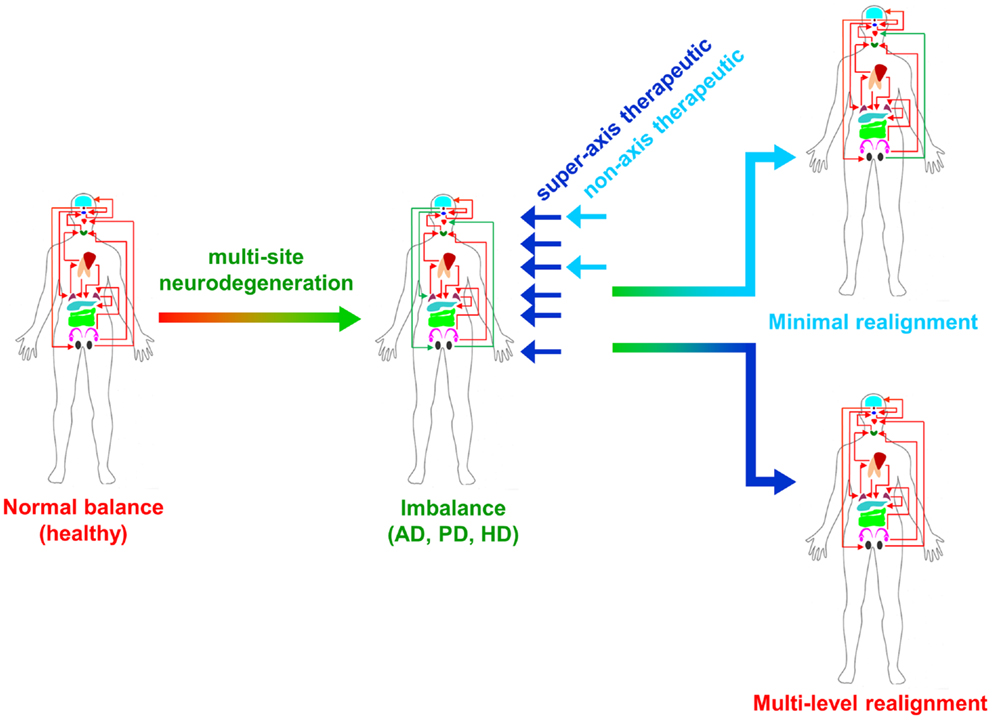
Figure 2. Super-axis remediation of complex systemic disorders is shown. Classical neurodegenerative diseases such as Alzheimer’s, Parkinson’s, and Huntington’s disease (AD, PD, and HD) represent intensely complex pathophysiological perturbations of normal systemic biology. These neurodegenerative disorders generate their full phenotypes through the disruption of multiple connected tissue–tissue signaling systems. Therapeutics that can interdict these perturbations at multiple sites in the disease process, i.e., “super-axis” therapeutics (dark blue) possess a much greater capacity to redress the systemic imbalances induced by disease than traditional non-axis therapeutics (light blue) that possess a limited functional repertoire.
GLP-1 Ligand Pharmacology
The GLP-1 glycoprotein hormone exists in two circulating molecular forms, GLP-1(7–37) and GLP-1(7–36) amide, both of which are full agonists at the GLP-1R (180). As stated before, the GLP-1R is expressed in multiple tissues including the central and peripheral nervous systems, heart, pancreas, kidney, lung, and GI tract. Once in the circulation, the GLP-1 peptide possesses a very short half-life (1–2 min) due to the proteolytic activity of dipeptidyl peptidase IV (DPP IV). Therefore, the creation of systemic therapeutics targeting the GLP-1R system first focused upon modification of the peptide backbone to prevent this degradation. Multiple strategies have been employed to improve the bioavailability of GLP-1 including, fatty acid acylation (181), addition of polyethylene glycol groups (182), d-amino acid substitutions (183), and transferrin hybridization (164). Perhaps, one of the most effective strategies for therapeutic generation was the creation of agents based on reptile forms of GLP-1 (exendin-4) that possess natural mutations that prevent DPP IV proteolysis (184). Exendin-4 has subsequently demonstrated multiple efficacious effects against a wide range of neurodegenerative disorders including AD, PD, and HD, reinforcing the importance of therapeutic targeting of this signaling super-axis. While the initial series of GLP-1 system-targeted therapies were peptidergically based, there is now considerable interest in the generation of novel, non-peptidergic ligands that can modify specific activities of the GLP-1 receptors (185–187). Non-peptidergic small-molecule ligands typically represent a more facile mechanism for drug production and administration. As the GLP-1R is a Class B secretin-like receptor, designing small-molecule agents that regulate the activity through an orthosteric interaction has proven difficult. However, the generation of small molecules that can allosterically interact to modulate GLP-1-mediated receptor activation has yielded interesting results, especially with regard to signal selectivity of functions. The search for small-molecule agonists/modulators of the GLP-1R has led to the identification of multiple compounds that can bind to and modulate GLP-1R function. Many of the newly developed small-molecule ligands for the GLP-1R (BETP, Novo Nordisk quinoxaline-based “Compound 2” [(6,7-dichloro2-methylsulfonyl-3-tert-butylaminoquinoxaline) (188), TT15, Boc5 (189–194)] possess intrinsic efficacy at the receptor, with respect to cAMP generation, and can augment insulin secretion either alone or in combination with peptide occupation of the GLP-1R orthosteric site. As the GLP-1R represents an interesting mechanism for controlling multiple levels of neurodegenerative behavior, it may be important for future therapeutic design to engineer the capacity for specific and beneficial tissue-based signaling bias to exploit the full therapeutic potential of this super-axis.
GLP-1R Signaling Bias
For ligand–receptor systems that may control whole-organism super-axes, it is clearly important that their regulation is tightly controlled in a highly nuanced manner. Thus, the molecular regulation networks of the GLP-1 receptor system are highly complex, with multiple endogenous and exogenous peptides [at least six: GLP-1(7–36)NH2, GLP-1(1–36)NH2, GLP-1(7–37), GLP-1(1–37), GLP-1(9–36)NH2, and oxyntomodulin] that interact with the receptor that results in the activation of numerous downstream signaling cascades (195). In the GLP-1R super-axis system, it is clearly crucial that selectively exploiting, via signal selectivity and bias, the full signaling repertoire of the GLP-1R could have tremendous benefit for multidimensional neurodegenerative research.
Surprisingly, for such an important emerging super-axis level therapeutic target, the current molecular appreciation of GLP-1R signaling and regulation is relatively limited compared to Class I rhodopsin-like receptors. Thus, the full gamut of GLP-1-modulated signaling paradigms is still a subject of intense research. In this respect, the GLP-1R has already demonstrated a considerable degree of signaling promiscuity: coupling has been demonstrated functional interactions with multiple heterotrimeric G proteins αs, αi/o, and αq/11 (196) as well as with β-arrestin-mediated pathways (159, 197). In addition to these classical signaling mechanisms, lateral signal transfer, i.e., “transactivation” (198), from the GLP-1R to epidermal growth factor receptor has also been reported (199). With most GPCRs studied to date, there is clearly a generic capacity for the activation of multiple and diverse signaling paradigms (1). For a receptor that can interact with multiple ligands, both orthosteric and allosteric, it is highly unlikely that the identical downstream signaling behavior, when studied at a high-dimensionality level (200), can be induced by chemically distinct ligands. Therefore, among both endogenous peptidergic agents and xenobiotics, there is a tremendous capacity to identify and therapeutically engineer ligand bias at such pleiotropic receptors. An additional layer of signaling complexity is also induced by the expression of the specific GPCRs in diverse tissues that contain differing types of GPCR-interacting proteins that again add further “texture” to the eventual signal.
In recent years using differential structure–activity-relationship analysis, effective signaling bias between cAMP-related pathways (MAPK signaling and calcium mobilization) for peptidergic ligands at the GLP-1R has been demonstrated (188, 192, 201). In addition to bias at the GLP-1R of peptidergic agents, small-molecule ligands that typically interact allosterically with the GLP-1R have also demonstrated an ability to exert selective signaling actions. These allosteric receptor interactions of the small molecules can be contemporaneous with the orthosteric ligand and can affect the conformational induction or selection of the receptor. Allosteric modulators, e.g., Novo Nordisk “Compound 2” (188, 201) can demonstrate their allosteric efficacy differentially between some of the endogenous stimulatory peptides as well as for peptidergic xenobiotics as well (188, 202). The allosteric interaction of Novo Nordisk “Compound 2” was also shown to significantly alter the qualitative nature of the transduced molecular signal as well from the orthosteric peptide ligands. Such allosteric activity at the GLP-1R can also be generated by naturally occurring medicinal agents such as quercetin and catechin, which can selectively augment specific signaling activities from specific subsets of peptidergic orthosteric agents (188, 203). Other modulators such as BETP, which still exerts qualitative signal conditioning effects on orthosteric ligand activity, have also shown an intrinsic efficacy at the GLP-1R in the absence of orthosteric engagement of the receptor (201). Not only are the activities of allosteric agents dependent on the qualitative nature of the resident agent in the orthosteric site but the resultant effects of the allosteric ligand can be further conditioned by changes in receptor transport/desensitization dynamics as well as receptor scaffolding and dimerization (196, 204).
It is clear that biased signaling is a significant pharmacological aspect of the GLP-1R and that this can include both G protein-dependent and G protein-independent pathways. This situations is then further complicated with the addition of orthosteric-selective allosteric receptor modulation. Rational and targeted exploitation of these pharmacological characteristics to engender tissue- and signaling-specific activity of the GLP-1 super-axis could yield tremendous therapeutic benefit for multiple neurodegenerative disorders. The functional and pathophysiological similarities of disorders such as AD, PD, and HD, potentially generated by a commonality of energy insufficiency in each disease (27, 205–209), presents a systemic level drug target whose exploitation may create remedial activities far beyond those induced by more monopharmacological agents. Drugs that target and remediate signaling super-axes in disease will likely be more tolerable to the patient as well, as endogenous tissue-to-tissue communication pipelines will not be significantly disrupted and thus the drug activity will be less opposed by homeostatic mechanisms. Our further enhanced understanding of how multiple pathophysiological processes in the body can be connected by functional signaling super-axes will hopefully allow the creation of a new series of more effective, tolerable, and ultimately beneficial anti-neurodegenerative therapies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Maudsley S, Martin B, Luttrell LM. The origins of diversity and specificity in G protein-coupled receptor signaling. J Pharmacol Exp Ther (2005) 314(2):485–94. doi: 10.1124/jpet.105.083121
2. Brink CB, Harvey BH, Bodenstein J, Venter DP, Oliver DW. Recent advances in drug action and therapeutics: relevance of novel concepts in G-protein-coupled receptor and signal transduction pharmacology. Br J Clin Pharmacol (2004) 57(4):373–87. doi:10.1111/j.1365-2125.2003.02046.x
3. Takeda S, Kadowaki S, Haga T, Takaesu H, Mitaku S. Identification of G protein-coupled receptor genes from the human genome sequence. FEBS Lett (2002) 520:97–101. doi:10.1016/S0014-5793(02)02775-8
4. Chadwick W, Maudsley S. The devil is in the dose: complexity of receptor systems and responses. In: Mattson MP, Calabrese EJ, editors. Hormesis: A Revolution in Biology, Toxicology and Medicine. Springer-Verlag (2010). p. 95–108.
5. Fredriksson R, Schiöth HB. The repertoire of G-protein-coupled receptors in fully sequenced genomes. Mol Pharmacol (2005) 67(5):1414–25. doi:10.1124/mol.104.009001
6. Maudsley S, Martin B, Luttrell LM. G protein-coupled receptor signaling complexity in neuronal tissue: implications for novel therapeutics. Curr Alzheimer Res (2007) 4(1):3–19. doi:10.2174/156720507779939850
7. Maurice P, Guillaume JL, Benleulmi-Chaachoua A, Daulat AM, Kamal M, Jockers R. GPCR-interacting proteins, major players of GPCR function. Adv Pharmacol (2011) 62:349–80. doi:10.1016/B978-0-12-385952-5.00001-4
8. Maudsley S, Siddiqui S, Martin B. Systems analysis of arrestin pathway functions. Prog Mol Biol Transl Sci (2013) 118:431–67. doi:10.1016/B978-0-12-394440-5.00017-6
9. Gesty-Palmer D, Yuan L, Martin B, Wood WH III, Lee MH, Janech MG, et al. β-arrestin-selective G protein-coupled receptor agonists engender unique biological efficacy in vivo. Mol Endocrinol (2013) 27(2):296–314. doi:10.1210/me.2012-1091
10. Maudsley S, Patel SA, Park SS, Luttrell LM, Martin B. Functional signaling biases in G protein-coupled receptors: game theory and receptor dynamics. Mini Rev Med Chem (2012) 12(9):831–40. doi:10.2174/138955712800959071
11. Gesty-Palmer D, Luttrell LM. Refining efficacy: exploiting functional selectivity for drug discovery. Adv Pharmacol (2011) 62:79–107. doi:10.1016/B978-0-12-385952-5.00009-9
12. Maudsley S, Davidson L, Pawson AJ, Chan R, López de Maturana R, Millar RP. Gonadotropin-releasing hormone (GnRH) antagonists promote proapoptotic signaling in peripheral reproductive tumor cells by activating a Galphai-coupling state of the type I GnRH receptor. Cancer Res (2004) 64(20):7533–44. doi:10.1158/0008-5472.CAN-04-1360
13. Gesty-Palmer D, Luttrell LM. Heptahelical terpsichory. Who calls the tune? J Recept Signal Transduct Res (2008) 28(1–2):39–58. doi:10.1080/10799890801941921
14. Wang L, Chadwick W, Park SS, Zhou Y, Silver N, Martin B, et al. Gonadotropin-releasing hormone receptor system: modulatory role in aging and neurodegeneration. CNS Neurol Disord Drug Targets (2010) 9(5):651–60. doi:10.2174/187152710793361559
15. Tiwary BK. Correlated evolution of gonadotropin-releasing hormone and gonadotropin-inhibitory hormone and their receptors in mammals. Neuroendocrinology (2013) 97(3):242–51. doi:10.1159/000342694
16. Kim DK, Cho EB, Moon MJ, Park S, Hwang JI, Kah O, et al. Revisiting the evolution of gonadotropin-releasing hormones and their receptors in vertebrates: secrets hidden in genomes. Gen Comp Endocrinol (2011) 170(1):68–78. doi:10.1016/j.ygcen.2010.10.018
17. Barreiro ML, Tena-Sempere M. Ghrelin and reproduction: a novel signal linking energy status and fertility? Mol Cell Endocrinol (2004) 226(1–2):1–9. doi:10.1016/j.mce.2004.07.015
18. O’Carroll AM, Lolait SJ, Harris LE, Pope GR. The apelin receptor APJ: journey from an orphan to a multifaceted regulator of homeostasis. J Endocrinol (2013) 219(1):R13–35. doi:10.1530/JOE-13-0227
19. Pi M, Quarles LD. Multiligand specificity and wide tissue expression of GPRC6A reveals new endocrine networks. Endocrinology (2012) 153(5):2062–9. doi:10.1210/en.2011-2117
20. Ferreira AJ, Murça TM, Fraga-Silva RA, Castro CH, Raizada MK, Santos RA. New cardiovascular and pulmonary therapeutic strategies based on the angiotensin-converting enzyme 2/angiotensin-(1-7)/mas receptor axis. Int J Hypertens (2012) 2012:147825. doi:10.1155/2012/147825
21. Zurbig P, Jahn H. Use of proteomic methods in the analysis of human body fluids in Alzheimer research. Electrophoresis (2012) 33(24):3617–30. doi:10.1002/elps.201200360
22. Sinha RN. Make dementia a public health priority in India. Indian J Public Health (2011) 55(2):67–9. doi:10.4103/0019-557X.85234
23. Mapstone M, Cheema AK, Fiandaca MS, Zhong X, Mhyre TR, MacArthur LH, et al. Plasma phospholipids identify antecedent memory impairment in older adults. Nat Med (2014) 20(4):415–8. doi:10.1038/nm.3466
24. Ferri CP, Sousa R, Albanese E, Ribeiro WS, Honyashiki M. World Alzheimer’s Report 2009. In: Prince M, Jackson J, editors. London: Alzheimer’s Disease International (2009).
25. Di Domenico F, Sultana R, Barone E, Perluigi M, Cini C, Mancuso C, et al. Quantitative proteomics analysis of phosphorylated proteins in the hippocampus of Alzheimer’s disease subjects. J Proteomics (2011) 74(7):1091–103. doi:10.1016/j.jprot.2011.03.033
26. Folch J, Pedrós I, Patraca I, Martínez N, Sureda F, Camins A. Metabolic basis of sporadic Alzheimer’s disease. Role of hormones related to energy metabolism. Curr Pharm Des (2013) 19(38):6739–48. doi:10.2174/13816128113199990612
27. Cai H, Cong WN, Ji S, Rothman S, Maudsley S, Martin B. Metabolic dysfunction in Alzheimer’s disease and related neurodegenerative disorders. Curr Alzheimer Res (2012) 9(1):5–17. doi:10.2174/156720512799015064
28. Zabel C, Nguyen HP, Hin SC, Hartl D, Mao L, Klose J. Proteasome and oxidative phoshorylation changes may explain why aging is a risk factor for neurodegenerative disorders. J Proteomics (2010) 73:2230–8. doi:10.1016/j.jprot.2010.08.008
29. Grela A, Turek A, Piekoszewski W. Application of matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) in Alzheimer’s disease. Clin Chem Lab Med (2012) 50(8):1297–304. doi:10.1515/cclm-2011-0550
30. Adlard PA, Cummings BJ. Alzheimer’s disease – a sum greater than its parts? Neurobiol Aging (2004) 25(6):725–33. doi:10.1016/j.neurobiolaging.2003.12.016
31. Holtzman DM, Morris JC, Goate AM. Alzheimer’s disease: the challenge of the second century. Sci Transl Med (2011) 3(77):77sr1. doi:10.1126/scitranslmed.3002369
32. Moro ML, Giaccone G, Lombardi R, Indaco A, Uggetti A. APP mutations in the Abeta coding region are associated with abundant cerebral deposition of Abeta38. Acta Neuropathol (2012) 124(6):809–21. doi:10.1007/s00401-012-1061-x
33. Chadwick W, Brenneman R, Martin B, Maudsley S. Complex and multidimensional lipid raft alterations in a murine model of Alzheimer’s disease. Int J Alzheimers Dis (2010) 2010:604792. doi:10.4061/2010/604792
34. Sutcliffe JG, Hedlund PB, Thomas EA, Bloom FE, Hilbush BS. Peripheral reduction of beta-amyloid is sufficient to reduce brain beta-amyloid: implications for Alzheimer’s disease. J Neurosci Res (2011) 89(6):808–14. doi:10.1002/jnr.22603
35. Watts AS, Loskutova N, Burns JM, Johnson DK. Metabolic syndrome and cognitive decline in early Alzheimer’s disease and healthy older adults. J Alzheimers Dis (2013) 35(2):253–65. doi:10.3233/JAD-121168
36. Ninomiya T. Diabetes mellitus and dementia. Curr Diab Rep (2014) 14(5):487. doi:10.1007/s11892-014-0487-z
37. Pasquier F, Boulogne A, Leys D, Fontaine P. Diabetes mellitus and dementia. Diabetes Metab (2006) 32(5 Pt 1):403–14. doi:10.1016/S1262-3636(07)70298-7
38. de la Monte SM, Wands JR. Alzheimer’s disease is type 3 diabetes-evidence reviewed. J Diabetes Sci Technol (2008) 2(6):1101–13. doi:10.1177/193229680800200619
39. Nakagawa T, Hu H, Zharikov S, Tuttle KR, Short RA, Glushakova O, et al. A causal role for uric acid in fructose-induced metabolic syndrome. Am J Physiol Renal Physiol (2006) 290(3):F625–31.
40. Grunblatt E, Bartl J, Riederer P. The link between iron, metabolic syndrome, and Alzheimer’s disease. J Neural Transm (2011) 118(3):371–9. doi:10.1007/s00702-010-0426-3
41. Misiak B, Leszek J, Kiejna A. Metabolic syndrome, mild cognitive impairment and Alzheimer’s disease – the emerging role of systemic low-grade inflammation and adiposity. Brain Res Bull (2012) 89(3–4):144–9. doi:10.1016/j.brainresbull.2012.08.003
42. Guix FX, Wahle T, Vennekens K, Snellinx A, Chávez-Gutiérrez L, Ill-Raga G, et al. Modification of g-secretase by nitrosative stress links neuronal ageing to sporadic Alzheimer’s disease. EMBO Mol Med (2012) 4:660–73. doi:10.1002/emmm.201200243
43. Manabe I. Chronic inflammation links cardiovascular, metabolic and renal diseases. Circ J (2011) 75(12):2739–48. doi:10.1253/circj.CJ-11-1184
44. Van Ginneken C, Schäfer KH, Van Dam D, Huygelen V, De Deyn PP. Morphological changes in the enteric nervous system of aging and APP23 transgenic mice. Brain Res (2011) 1378:43–53. doi:10.1016/j.brainres.2011.01.030
45. Semar S, Klotz M, Letiembre M, Van Ginneken C, Braun A, Jost V, et al. Changes of the enteric nervous system in amyloid-β protein precursor transgenic mice correlate with disease progression. J Alzheimers Dis (2013) 36(1):7–20. doi:10.3233/JAD-120511
46. Fu W, Ruangkittisakul A, MacTavish D, Shi JY, Ballanyi K, Jhamandas JH. Amyloid beta (Abeta) peptide directly activates amylin-3 receptor subtype by triggering multiple intracellular signaling pathways. J Biol Chem (2012) 287(22):18820–30. doi:10.1074/jbc.M111.331181
47. De Vriese C, Delporte C. Influence of ghrelin on food intake and energy homeostasis. Curr Opin Clin Nutr Metab Care (2007) 10(5):615–9. doi:10.1097/MCO.0b013e32829fb37c
48. Theodoropoulou A, Metallinos IC, Psyrogiannis A, Vagenakis GA, Kyriazopoulou V. Ghrelin and leptin secretion in patients with moderate Alzheimer’s disease. J Nutr Health Aging (2012) 16(5):472–7. doi:10.1007/s12603-012-0058-4
49. Gahete MD, Rubio A, Córdoba-Chacón J, Gracia-Navarro F, Kineman RD, Avila J, et al. Expression of the ghrelin and neurotensin systems is altered in the temporal lobe of Alzheimer’s disease patients. J Alzheimers Dis (2010) 22(3):819–28. doi:10.3233/JAD-2010-100873
50. Ma LY, Zhang DM, Tang Y, Lu Y, Zhang Y, Gao Y, et al. Ghrelin-attenuated cognitive dysfunction in streptozotocin-induced diabetic rats. Alzheimer Dis Assoc Disord (2011) 25(4):352–63. doi:10.1097/WAD.0b013e31820ce536
51. Kieffer TJ, Habener JF. The adipoinsular axis: effects of leptin on pancreatic beta-cells. Am J Physiol Endocrinol Metab (2000) 278(1):E1–14.
52. Kim JB, Sarraf P, Wright M, Yao KM, Mueller E. Nutritional and insulin regulation of fatty acid synthetase and leptin gene expression through ADD1/SREBP1. J Clin Invest (1998) 101(1):1–9. doi:10.1172/JCI1411
53. Giordano V, Peluso G, Iannuccelli M, Benatti P, Nicolai R, Calvani M. Systemic and brain metabolic dysfunction as a new paradigm for approaching Alzheimer’s dementia. Neurochem Res (2007) 32(4–5):555–67. doi:10.1007/s11064-006-9125-8
54. Kelleher RJ, Soiza RL. Evidence of endothelial dysfunction in the development of Alzheimer’s disease: is Alzheimer’s a vascular disorder? Am J Cardiovasc Dis (2013) 3(4):197–226.
55. Thomas T, Thomas G, McLendon C, Sutton T, Mullan M. Beta-amyloid-mediated vasoactivity and vascular endothelial damage. Nature (1996) 380(6570):168–71. doi:10.1038/380168a0
56. Sutton ET, Hellermann GR, Thomas T. Beta-amyloid-induced endothelial necrosis and inhibition of nitric oxide production. Exp Cell Res (1997) 230(2):368–76. doi:10.1006/excr.1996.3440
57. Erickson MA, Banks WA. Blood-brain barrier dysfunction as a cause and consequence of Alzheimer’s disease. J Cereb Blood Flow Metab (2013) 33(10):1500–13. doi:10.1038/jcbfm.2013.135
58. Willis MS, Patterson C. Proteotoxicity and cardiac dysfunction – Alzheimer’s disease of the heart? N Engl J Med (2013) 368(5):455–64. doi:10.1056/NEJMra1106180
59. Coogan AN, Schutová B, Husung S, Furczyk K, Baune BT, Kropp P, et al. The circadian system in Alzheimer’s disease: disturbances, mechanisms, and opportunities. Biol Psychiatry (2013) 74(5):333–9. doi:10.1016/j.biopsych.2012.11.021
60. McClung CA. Circadian rhythms and mood regulation: insights from pre-clinical models. Eur Neuropsychopharmacol (2011) 21:S683–93. doi:10.1016/j.euroneuro.2011.07.008
61. Tranah GJ, Blackwell T, Stone KL, Ancoli-Israel S, Paudel ML, Ensrud KE, et al. Circadian activity rhythms and risk of incident dementia and mild cognitive impairment in older women. Ann Neurol (2011) 70(5):722–32. doi:10.1002/ana.22468
62. Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature (2002) 418:935–41. doi:10.1038/nature00965
63. Aliani M, Udenigwe CC, Girgih AT, Pownall TL, Bugera JL, Eskin MN. Aroma and taste perceptions with Alzheimer disease and stroke. Crit Rev Food Sci Nutr (2013) 53(7):760–9. doi:10.1080/10408398.2011.559557
64. Cole LJ, Gavrilescu M, Johnston LA, Gibson SJ, Farrell MJ, Egan GF. The impact of Alzheimer’s disease on the functional connectivity between brain regions underlying pain perception. Eur J Pain (2011) 15(6):e1–11. doi:10.1016/j.ejpain.2010.10.010
65. Stover KR, Brown RE. Age-related changes in visual acuity, learning and memory in the APPswe/PS1dE9 mouse model of Alzheimer’s disease. Behav Brain Res (2012) 231(1):75–85. doi:10.1016/j.bbr.2012.02.044
66. Behrman S, Chouliaras L, Ebmeier KP. Considering the senses in the diagnosis and management of dementia. Maturitas (2014) 77(4):305–10. doi:10.1016/j.maturitas.2014.01.003
67. Steinbach S, Hundt W, Vaitl A, Heinrich P, Förster S, Bürger K, et al. Taste in mild cognitive impairment and Alzheimer’s disease. J Neurol (2010) 257(2):238–46. doi:10.1007/s00415-009-5300-6
68. Aarsland D, Larsen JP, Lim NG, Janvin C, Karlsen K, Tandberg E, et al. Range of neuropsychiatric disturbances in patients with Parkinson’s disease. J Neurol Neurosurg Psychiatry (1999) 67:492–6. doi:10.1136/jnnp.67.4.492
69. Trinh J, Farrer M. Advances in the genetics of Parkinson disease. Nat Rev Neurol (2013) 9:445–54. doi:10.1038/nrneurol.2013.132
70. Antony PM, Diederich NJ, Kruger R, Balling R. The hallmarks of Parkinson’s disease. FEBS J (2013) 280:5981–93. doi:10.1111/febs.12335
71. Licker V, Kovari E, Hochstrasser DF, Burkhard PR. Proteomics in human Parkinson’s disease research. J Proteomics (2009) 73:10–29. doi:10.1016/j.jprot.2009.07.007
72. McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology (2005) 65:1863–72. doi:10.1212/01.wnl.0000187889.17253.b1
73. Savitt JM, Dawson VL, Dawson TM. Diagnosis and treatment of Parkinson disease: molecules to medicine. J Clin Invest (2006) 116:1744–54. doi:10.1172/JCI29178
74. Chaudhuri KR, Schapira AH. Non-motor symptoms of Parkinson’s disease: dopaminergic pathophysiology and treatment. Lancet Neurol (2009) 8:464–74. doi:10.1016/S1474-4422(09)70068-7
75. Jellinger KA. Synuclein deposition and non-motor symptoms in Parkinson disease. J Neurol Sci (2011) 310:107–11. doi:10.1016/j.jns.2011.04.012
76. Kovari E, Horvath J, Bouras C. Neuropathology of Lewy body disorders. Brain Res Bull (2009) 80:203–10. doi:10.1016/j.brainresbull.2009.06.018
77. Tsuboi Y, Uchikado H, Dickson DW. Neuropathology of Parkinson’s disease dementia and dementia with Lewy bodies with reference to striatal pathology. Parkinsonism Relat Disord (2007) 13(S3):S221–4. doi:10.1016/S1353-8020(08)70005-1
78. Dickson DW, Fujishiro H, Orr C, DelleDonne A, Josephs KA. Neuropathology of non-motor features of Parkinson disease. Parkinsonism Relat Disord (2009) 15(S3):S1–5. doi:10.1016/S1353-8020(09)70769-2
79. Beach TG, Adler CH, Sue LI, Vedders L, Lue L, White Iii CL, et al. Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol (2010) 119:689–702. doi:10.1007/s00401-010-0664-3
80. Derkinderen P, Rouaud T, Lebouvier T, Bruley des Varannes S, Neunlist M. Parkinson disease: the enteric nervous system spills its guts. Neurology (2011) 77:1761–7. doi:10.1212/WNL.0b013e318236ef60
81. Wakabayashi K, Takahashi H. Neuropathology of autonomic nervous system in Parkinson’s disease. Eur Neurol (1997) 38(S2):2–7. doi:10.1159/000113469
82. Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging (2003) 24:197–211. doi:10.1016/S0197-4580(02)00065-9
83. Doty RL. Olfactory dysfunction in Parkinson disease. Nat Rev Neurol (2012) 8:329–39. doi:10.1038/nrneurol.2012.80
84. Ponsen MM, Stoffers D, Booij J, van Eck-Smit BL, Wolters ECH, Berendse HW. Idiopathic hyposmia as a preclinical sign of Parkinson’s disease. Ann Neurol (2004) 56:173–81. doi:10.1002/ana.20160
85. Arendt T, Bigl V, Arendt A, Tennstedt A. Loss of neurons in the nucleus basalis of Meynert in Alzheimer’s disease, paralysis agitans and Korsakoff’s Disease. Acta Neuropathol (1983) 61:101–8. doi:10.1007/BF00697388
86. Petzold GC, Hagiwara A, Murthy VN. Serotonergic modulation of odor input to the mammalian olfactory bulb. Nat Neurosci (2009) 12:784–91. doi:10.1038/nn.2335
87. Zarow C, Lyness SA, Mortimer JA, Chui HC. Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol (2003) 60:337–41. doi:10.1001/archneur.60.3.337
88. Cloud LJ, Greene JG. Gastrointestinal features of Parkinson’s disease. Curr Neurol Neurosci Rep (2011) 11:379–84. doi:10.1007/s11910-011-0204-0
89. Savica R, Carlin JM, Grossardt BR, Bower JH, Ahlskog JE, Maraganore DM, et al. Medical records documentation of constipation preceding Parkinson disease: a case-control study. Neurology (2009) 73:1752–8. doi:10.1212/WNL.0b013e3181c34af5
90. Cersosimo MG, Benarroch EE. Pathological correlates of gastrointestinal dysfunction in Parkinson’s disease. Neurobiol Dis (2012) 46:559–64. doi:10.1016/j.nbd.2011.10.014
91. Sakakibara R, Kishi M, Ogawa E, Tateno F, Uchiyama T, Yamamoto T, et al. Bladder, bowel, and sexual dysfunction in Parkinson’s disease. Parkinsons Dis (2011) 2011:924605. doi:10.4061/2011/924605
92. Gulyani S, Earley CJ, Camandola S, Maudsley S, Ferré S, Mughal MR, et al. Diminished iron concentrations increase adenosine A(2A) receptor levels in mouse striatum and cultured human neuroblastoma cells. Exp Neurol (2009) 215(2):236–42. doi:10.1016/j.expneurol.2008.10.007
93. Boeve BF, Silber MH, Ferman TJ, Kokmen E, Smith GE. REM sleep behavior disorder and degenerative dementia: an association likely reflecting Lewy body disease. Neurology (1998) 51:363–70. doi:10.1212/WNL.51.2.363
94. Muller ML, Bohnen NI. Cholinergic dysfunction in Parkinson’s disease. Curr Neurol Neurosci Rep (2013) 13:377. doi:10.1007/s11910-013-0377-9
95. Vendette M, Gagnon JF, Décary A, Massicotte-Marquez J, Postuma RB, Doyon J, et al. REM sleep behavior disorder predicts cognitive impairment in Parkinson disease without dementia. Neurology (2007) 69:1843–9. doi:10.1212/01.wnl.0000278114.14096.74
96. Goldstein DS. Dysautonomia in Parkinson disease. Compr Physiol (2014) 4:805–26. doi:10.1002/cphy.c130026
97. Jain S, Goldstein DS. Cardiovascular dysautonomia in Parkinson disease: from pathophysiology to pathogenesis. Neurobiol Dis (2012) 46:572–80. doi:10.1016/j.nbd.2011.10.025
98. Orimo S, Takahashi A, Uchihara T, Mori F, Kakita A, Wakabayashi K, et al. Degeneration of cardiac sympathetic nerve begins in the early disease process of Parkinson’s disease. Brain Pathol (2007) 17:24–30. doi:10.1111/j.1750-3639.2006.00032.x
99. Orimo S, Uchihara T, Nakamura A, Mori F, Kakita A, Wakabayashi K, et al. Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain (2008) 131:642–50. doi:10.1093/brain/awm302
100. Barrio JR, Huang SC, Melega WP, Yu DC, Hoffman JM, Schneider JS, et al. 6-[18F]fluoro-l-dopa probes dopamine turnover rates in central dopaminergic structures. J Neurosci Res (1990) 27:487–93. doi:10.1002/jnr.490270408
101. Chung EJ, Lee WY, Yoon WT, Kim BJ, Lee GH. MIBG scintigraphy for differentiating Parkinson’s disease with autonomic dysfunction from Parkinsonism-predominant multiple system atrophy. Mov Disord (2009) 24:1650–5. doi:10.1002/mds.22649
102. Benarroch EE. The arterial baroreflex: functional organization and involvement in neurologic disease. Neurology (2008) 71:1733–8. doi:10.1212/01.wnl.0000335246.93495.92
103. Ha AD, Brown CH, York MK, Jankovic J. The prevalence of symptomatic orthostatic hypotension in patients with Parkinson’s disease and atypical parkinsonism. Parkinsonism Relat Disord (2011) 17:625–8. doi:10.1016/j.parkreldis.2011.05.020
104. Arvanitakis Z, Wilson RS, Bienias JL, Evans DA, Bennett DA. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol (2004) 61:661–6. doi:10.1001/archneur.61.5.661
105. Xu Q, Park Y, Huang X, Hollenbeck A, Blair A. Diabetes and risk of Parkinson’s disease. Diabetes Care (2011) 34:910–5. doi:10.2337/dc10-1922
106. Aviles-Olmos I, Limousin P, Lees A, Foltynie T. Parkinson’s disease, insulin resistance and novel agents of neuroprotection. Brain (2013) 136:374–84. doi:10.1093/brain/aws009
107. Santiago JA, Potashkin JA. Shared dysregulated pathways lead to Parkinson’s disease and diabetes. Trends Mol Med (2013) 19:176–86. doi:10.1016/j.molmed.2013.01.002
108. Unger JW, Livingston JN, Moss AM. Insulin receptors in the central nervous system: localization, signalling mechanisms and functional aspects. Prog Neurobiol (1991) 36:343–62. doi:10.1016/0301-0082(91)90015-S
109. Bellucci A, Collo G, Sarnico I, Battistin L, Missale C, Spano P. Alpha-synuclein aggregation and cell death triggered by energy deprivation and dopamine overload are counteracted by D2/D3 receptor activation. J Neurochem (2008) 106:560–77. doi:10.1111/j.1471-4159.2008.05406.x
110. Russo I, Barlati S, Bosetti F. Effects of neuroinflammation on the regenerative capacity of brain stem cells. J Neurochem (2011) 116(6):947–56. doi:10.1111/j.1471-4159.2010.07168.x
111. Pradhan S, Andreasson K. Commentary: progressive inflammation as a contributing factor to early development of Parkinson’s disease. Exp Neurol (2013) 241:148–55. doi:10.1016/j.expneurol.2012.12.008
112. Russo I, Bubacco L, Greggio E. LRRK2 and neuroinflammation: partners in crime in Parkinson’s disease? J Neuroinflammation (2014) 11:52. doi:10.1186/1742-2094-11-52
113. Lu M, Hu G. Targeting metabolic inflammation in Parkinson’s disease: implications for prospective therapeutic strategies. Clin Exp Pharmacol Physiol (2012) 39(6):577–85. doi:10.1111/j.1440-1681.2011.05650.x
114. Martin B, Golden E, Keselman A, Stone M, Mattson MP, Egan JM, et al. Therapeutic perspectives for the treatment of Huntington’s disease: treating the whole body. Histol Histopathol (2008) 23(2):237–50.
115. Collaborative Group HD. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell (1993) 72(6):971–83. doi:10.1016/0092-8674(93)90585-E
116. Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science (2001) 293(5529):493–8. doi:10.1126/science.1059581
117. Hoffner G, Kahlem P, Djian P. Perinuclear localization of huntingtin as a consequence of its binding to microtubules through an interaction with beta-tubulin: relevance to Huntington’s disease. J Cell Sci (2002) 115(5):941–8.
118. DiFiglia M, Sapp E, Chase K, Schwarz C, Meloni A, Young C, et al. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron (1995) 14(5):1075–81. doi:10.1016/0896-6273(95)90346-1
119. Velier J, Kim M, Schwarz C, Kim TW, Sapp E, Chase K, et al. Wild-type and mutant huntingtins function in vesicle trafficking in the secretory and endocytic pathways. Exp Neurol (1998) 152(1):34–40. doi:10.1006/exnr.1998.6832
120. Morton AJ. Circadian and sleep disorder in Huntington’s disease. Exp Neurol (2013) 243:34–44. doi:10.1016/j.expneurol.2012.10.014
121. Aziz NA, Pijl H, Frölich M, Snel M, Streefland TC, Roelfsema F, et al. Systemic energy homeostasis in Huntington’s disease patients. J Neurol Neurosurg Psychiatry (2010) 81(11):1233–7. doi:10.1136/jnnp.2009.191833
122. Cong WN, Cai H, Wang R, Daimon CM, Maudsley S, Raber K, et al. Altered hypothalamic protein expression in a rat model of Huntington’s disease. PLoS One (2012) 7(10):e47240. doi:10.1371/journal.pone.0047240
123. Aziz NA, Pijl H, Frölich M, Schröder-van der Elst JP, van der Bent C, Roelfsema F, et al. Delayed onset of the diurnal melatonin rise in patients with Huntington’s disease. J Neurol (2009) 256(12):1961–5. doi:10.1007/s00415-009-5196-1
124. Farrer LA. Diabetes mellitus in Huntington disease. Clin Genet (1985) 27:62–7. doi:10.1111/j.1399-0004.1985.tb00185.x
125. Dorsey ER, Beck CA, Brenna JT, Shoulson I. Altered cholesterol and fatty acid metabolism in Huntington disease. J Clin Lipidol (2010) 4(1):17–23. doi:10.1016/j.jacl.2009.11.003
126. Hunt MJ, Morton AJ. Atypical diabetes associated with inclusion formation in the R6/2 mouse model of Huntington’s disease is not improved by treatment with hypoglycaemic agents. Exp Brain Res (2005) 166:220–9. doi:10.1007/s00221-005-2357-z
127. Lalic NM, Maric J, Svetel M, Jotic A, Stefanova E, Lalic K, et al. Glucose homeostasis in Huntington disease: abnormalities in insulin sensitivity and early-phase insulin secretion. Arch Neurol (2008) 65(4):476–80. doi:10.1001/archneur.65.4.476
128. Geroldi C, Frisoni GB, Paolisso G, Bandinelli S, Lamponi M, Abbatecola AM, et al. Insulin resistance in cognitive impairment: the InCHIANTI study. Arch Neurol (2005) 62(7):1067–72. doi:10.1001/archneur.62.7.1067
129. Hult S, Soylu R, Björklund T, Belgardt BF, Mauer J, Brüning JC, et al. Mutant huntingtin causes metabolic imbalance by disruption of hypothalamic neurocircuits. Cell Metab (2011) 13(4):428–39. doi:10.1016/j.cmet.2011.02.013
130. van der Burg JM, Winqvist A, Aziz NA, Maat-Schieman ML, Roos RA, Bates GP, et al. Gastrointestinal dysfunction contributes to weight loss in Huntington’s disease mice. Neurobiol Dis (2011) 44(1):1–8. doi:10.1016/j.nbd.2011.05.006
131. Kudo T, Schroeder A, Loh DH, Kuljis D, Jordan MC, Roos KP, et al. Dysfunctions in circadian behavior and physiology in mouse models of Huntington’s disease. Exp Neurol (2011) 228(1):80–90. doi:10.1016/j.expneurol.2010.12.011
132. Morton AJ, Wood NI, Hastings MH, Hurelbrink C, Barker RA, Maywood ES. Disintegration of the sleep-wake cycle and circadian timing in Huntington’s disease. J Neurosci (2005) 25(1):157–63. doi:10.1523/JNEUROSCI.3842-04.2005
133. Pallier PN, Maywood ES, Zheng Z, Chesham JE, Inyushkin AN, Dyball R, et al. Pharmacological imposition of sleep slows cognitive decline and reverses dysregulation of circadian gene expression in a transgenic mouse model of Huntington’s disease. J Neurosci (2007) 27(29):7869–78. doi:10.1523/JNEUROSCI.0649-07.2007
134. Pallier PN, Morton AJ. Management of sleep/wake cycles improves cognitive function in a transgenic mouse model of Huntington’s disease. Brain Res (2009) 1279:90–8. doi:10.1016/j.brainres.2009.03.072
135. Maywood ES, Fraenkel E, McAllister CJ, Wood N, Reddy AB, Hastings MH, et al. Disruption of peripheral circadian timekeeping in a mouse model of Huntington’s disease and its restoration by temporally scheduled feeding. J Neurosci (2010) 30(30):10199–204. doi:10.1523/JNEUROSCI.1694-10.2010
136. Bates GP, Harper PS, Jones L, editors. Huntington’s Disease. Third ed. Oxford: Oxford University Press (2002).
137. Fava M. Daytime sleepiness and insomnia as correlates of depression. J Clin Psychiatry (2004) 65(Suppl 16):27–32.
138. Sephton S, Spiegel D. Circadian disruption in cancer: a neuroendocrine immune pathway from stress to disease? Brain Behav Immun (2003) 17:321–8. doi:10.1016/S0889-1591(03)00078-3
139. Spiegel K, Tasali E, Penev P, Van Cauter E. Brief communication: sleep curtailment in healthy young men is associated with decreased leptin levels, elevated ghrelin levels, and increased hunger and appetite. Ann Intern Med (2004) 7:846–50.
140. Eismann EA, Lush E, Sephton SE. Circadian effects in cancer relevant psychoneuroendocrine and immune pathways. Psychoneuroendocrinology (2010) 35:963–76. doi:10.1016/j.psyneuen.2009.12.011
141. Kiriazis H, Jennings NL, Davern P, Lambert G, Su Y, Pang T, et al. Neurocardiac dysregulation and neurogenic arrhythmias in a transgenic mouse model of Huntington’s disease. J Physiol (2012) 590(Pt 22):5845–60. doi:10.1113/jphysiol.2012.238113
142. Buonincontri G, Wood NI, Puttick SG, Ward AO, Carpenter TA, Sawiak SJ, et al. Right ventricular dysfunction in the R6/2 transgenic mouse model of Huntington’s disease is unmasked by dobutamine. J Huntingtons Dis (2014) 3(1):25–32. doi:10.3233/JHD-130083
143. Schroeder AM, Loh DH, Jordan MC, Roos KP, Colwell CS. Baroreceptor reflex dysfunction in the BACHD mouse model of Huntington’s disease. PLoS Curr (2011) 3:RRN1266. doi:10.1371/currents.RRN1266
144. van der Burg JMM, Björkqvist M, Brundin P. Beyond the brain: widespread pathology in Huntington’s disease. Lancet Neurol (2009) 8:765–74. doi:10.1016/S1474-4422(09)70178-4
145. Pattison JS, Sanbe A, Maloyan A, Osinska H, Klevitsky R, Robbins J. Cardiomyocyte expression of a polyglutamine preamyloid oligomer causes heart failure. Circulation (2008) 117:2743–51. doi:10.1161/CIRCULATIONAHA.107.750232
146. Björkqvist M, Wild EJ, Thiele J, Silvestroni A, Andre R, Lahiri N, et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J Exp Med (2008) 205(8):1869–77. doi:10.1084/jem.20080178
147. Kwan W, Träger U, Davalos D, Chou A, Bouchard J, Andre R, et al. Mutant huntingtin impairs immune cell migration in Huntington disease. J Clin Invest (2012) 122(12):4737–47. doi:10.1172/JCI64484
148. Wild E, Magnusson A, Lahiri N, Krus U, Orth M, Tabrizi SJ, et al. Abnormal peripheral chemokine profile in Huntington’s disease. PLoS Curr (2011) 3:RRN1231. doi:10.1371/currents.RRN1231
149. Hsiao HY, Chiu FL, Chen CM, Wu YR, Chen HM, Chen YC, et al. Inhibition of soluble tumor necrosis factor is therapeutic in Huntington’s disease. Hum Mol Genet (2014) 23(16):4328–44. doi:10.1093/hmg/ddu151
150. O’Donnell BF, Blekher TM, Weaver M, White KM, Marshall J, Beristain X, et al. Visual perception in prediagnostic and early stage Huntington’s disease. J Int Neuropsychol Soc (2008) 14(3):446–53. doi:10.1017/S1355617708080405
151. Scahill RI, Hobbs NZ, Say MJ, Bechtel N, Henley SM, Hyare H, et al. Clinical impairment in premanifest and early Huntington’s disease is associated with regionally specific atrophy. Hum Brain Mapp (2013) 34(3):519–29. doi:10.1002/hbm.21449
152. Mitchell IJ, Heims H, Neville EA, Rickards H. Huntington’s disease patients show impaired perception of disgust in the gustatory and olfactory modalities. J Neuropsychiatry Clin Neurosci (2005) 17(1):119–21.
153. Gardiner J, Barton D, Vanslambrouck JM, Braet F, Hall D, Marc J, et al. Defects in tongue papillae and taste sensation indicate a problem with neurotrophic support in various neurological diseases. Neuroscientist (2008) 14(3):240–50. doi:10.1177/1073858407312382
154. Zhou J, Livak MF, Bernier M, Muller DC, Carlson OD, Elahi D, et al. Ubiquitination is involved in glucose-mediated downregulation of GIP receptors in islets. Am J Physiol Endocrinol Metab (2007) 293(2):E538–47. doi:10.1152/ajpendo.00070.2007
155. Elliott RM, Morgan LM, Tredger JA, Deacon S, Wright J, Marks V. Glucagon-like peptide-1(7–36)amide and glucose-dependent insulinotropic polypeptide secretion in response to nutrient ingestion in man: acute post-prandial and 24-h secretion patterns. J Endocrionol (1993) 138:159–66. doi:10.1677/joe.0.1380159
156. Thorens B, Porret A, Bühler L, Deng SP, Morel P, Widmann C. Cloning and functional expression of the human islet GLP-1 receptor: demonstration that exendin-4 is an agonist and exendin-(9–39) an antagonist of the receptor. Diabetes (1993) 42:1678–82. doi:10.2337/diabetes.42.11.1678
157. Chapter MC, White CM, DeRidder A, Chadwick W, Martin B, Maudsley S. Chemical modification of class II G protein-coupled receptor ligands: frontiers in the development of peptide analogs as neuroendocrine pharmacological therapies. Pharmacol Ther (2010) 125:39–54. doi:10.1016/j.pharmthera.2009.07.006
158. Martin B, Lopez de Maturana R, Brenneman R, Walent T, Mattson MP, Maudsley S. Class II G protein-coupled receptors and their ligands in neuronal function and protection. Neuromolecular Med (2005) 7(1–2):3–36. doi:10.1385/NMM:7:1-2:003
159. Sonoda N, Imamura T, Yoshizaki T, Babendure JL, Lu JC, Olefsky JM. Beta-arrestin-1 mediates glucagon-like peptide-1 signaling to insulin secretion in cultured pancreatic beta cells. Proc Natl Acad Sci USA (2008) 105(18):6614–9. doi:10.1073/pnas.0710402105
160. Ladenheim EE. Gastrointestinal regulatory peptides and central nervous system mechanisms of weight control. Curr Opin Endocrinol Diabetes Obes (2012) 19(1):13–8. doi:10.1097/MED.0b013e32834ea723
161. Cai H, Maudsley S, Martin B. What is the role of metabolic hormones in taste buds of the tongue. Front Horm Res (2014) 42:134–46. doi:10.1159/000358322
162. Banks WA, During MJ, Niehoff ML. Brain uptake of the glucagon-like peptide-1 antagonist exendin(9-39) after intranasal administration. J Pharmacol Exp Ther (2004) 309(2):469–75. doi:10.1124/jpet.103.063222
163. van Bloemendaal L, Ten Kulve JS, la Fleur SE, Ijzerman RG, Diamant M. Effects of glucagon-like peptide 1 on appetite and body weight: focus on the CNS. J Endocrinol (2014) 221(1):T1–16. doi:10.1530/JOE-13-0414
164. Kim BJ, Zhou J, Martin B, Carlson OD, Maudsley S, Greig NH, et al. Transferrin fusion technology: a novel approach to prolonging biological half-life of insulinotropic peptides. J Pharmacol Exp Ther (2010) 334(3):682–92. doi:10.1124/jpet.110.166470
165. Phillips LK, Prins JB. Update on incretin hormones. Ann N Y Acad Sci (2011) 1243:E55–74. doi:10.1111/j.1749-6632.2012.06491.x
166. Körner M, Stöckli M, Waser B, Reubi JC. GLP-1 receptor expression in human tumors and human normal tissues: potential for in vivo targeting. J Nucl Med (2007) 48(5):736–43. doi:10.2967/jnumed.106.038679
167. Hölscher C. Potential role of glucagon-like peptide-1 (GLP-1) in neuroprotection. CNS Drugs (2012) 26(10):871–82. doi:10.2165/11635890-000000000-00000
168. Martin B, Golden E, Carlson OD, Pistell P, Zhou J, Kim W, et al. Exendin-4 improves glycemic control, ameliorates brain and pancreatic pathologies, and extends survival in a mouse model of Huntington’s disease. Diabetes (2009) 58(2):318–28. doi:10.2337/db08-0799
169. Ahrén B. GLP-1 and extra-islet effects. Horm Metab Res (2004) 36(11–12):842–5. doi:10.1055/s-2004-826173
170. Cai H, Daimon CM, Cong WN, Wang R, Chirdon P, de Cabo R, et al. Longitudinal analysis of calorie restriction on rat taste bud morphology and expression of sweet taste modulators. J Gerontol A Biol Sci Med Sci (2014) 69(5):532–44. doi:10.1093/gerona/glt129
171. Avogaro A, Vigili de Kreutzenberg S, Fadini GP. Cardiovascular actions of GLP-1 and incretin-based pharmacotherapy. Curr Diab Rep (2014) 14(5):483. doi:10.1007/s11892-014-0483-3
172. Gil-Lozano M, Romaní-Pérez M, Outeiriño-Iglesias V, Vigo E, Brubaker PL, González-Matías LC, et al. Effects of prolonged exendin-4 administration on hypothalamic-pituitary-adrenal axis activity and water balance. Am J Physiol Endocrinol Metab (2013) 304(10):E1105–17. doi:10.1152/ajpendo.00529.2012
173. Brunner JI, Gotter AL, Millstein J, Garson S, Binns J, Fox SV, et al. Pharmacological validation of candidate causal sleep genes identified in an N2 cross. J Neurogenet (2011) 25(4):167–81. doi:10.3109/01677063.2011.628426
174. Martin B, Chadwick W, Cong WN, Pantaleo N, Daimon CM, Golden EJ, et al. Euglycemic agent-mediated hypothalamic transcriptomic manipulation in the N171-82Q model of Huntington disease is related to their physiological efficacy. J Biol Chem (2012) 287(38):31766–82. doi:10.1074/jbc.M112.387316
175. Duarte AI, Candeias E, Correia SC, Santos RX, Carvalho C, Cardoso S, et al. Crosstalk between diabetes and brain: glucagon-like peptide-1 mimetics as a promising therapy against neurodegeneration. Biochim Biophys Acta (2013) 1832(4):527–41. doi:10.1016/j.bbadis.2013.01.008
176. Aviles-Olmos I, Dickson J, Kefalopoulou Z, Djamshidian A, Ell P, Soderlund T, et al. Exenatide and the treatment of patients with Parkinson’s disease. J Clin Invest (2013) 123(6):2730–6. doi:10.1172/JCI68295
177. Chen S, An FM, Yin L, Liu AR, Yin DK, Yao WB, et al. Glucagon-like peptide-1 protects hippocampal neurons against advanced glycation end product-induced tau hyperphosphorylation. Neuroscience (2014) 256:137–46. doi:10.1016/j.neuroscience.2013.10.038
178. Hölscher C. The incretin hormones glucagon like peptide 1 and glucose-dependent insulinotropic polypeptide are neuroprotective in mouse models of Alzheimer’s disease. Alzheimers Dement (2014) 10(1S):S47–54. doi:10.1016/j.jalz.2013.12.009
179. Maudsley S, Davidson L, Pawson AJ, Chan R, López de Maturana R, et al. Gonadotropin-releasing hormone (GnRH) antagonists promote proapoptotic signaling in peripheral reproductive tumor cells by activating a Galphai-coupling state of the type I GnRH receptor. Cancer Res (2004) 64(20):7533–44. doi:10.1158/0008-5472.CAN-04-1360
180. Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet (2006) 368(9548):1696–705. doi:10.1016/S0140-6736(06)69705-5
181. Knudsen LB, Nielsen PF, Huusfeldt PO, Johansen NL, Madsen K, Pedersen FZ, et al. Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration. J Med Chem (2000) 43:1664–9. doi:10.1021/jm9909645
182. Lee SH, Lee S, Youn YS, Na DH, Chae SY, Byun Y, et al. Synthesis, characterization, and pharmacokinetic studies of PEGylated glucagon-like peptide-1. Bioconjug Chem (2005) 16:377–82. doi:10.1021/bc049735+
183. Siegel EG, Scharf G, Gallwitz B, Mentlein R, Morys-Wortmann C, Fölsch UR, et al. Comparison of the effect of native glucagon-like peptide 1 and dipeptidyl peptidase IV-resistant analogues on insulin release from rat pancreatic islets. Eur J Clin Invest (1999) 29:610–4. doi:10.1046/j.1365-2362.1999.00440.x
184. Nielsen LL, Young AA, Parkes DG. Pharmacology of exenatide (synthetic exendin-4): a potential therapeutic for improved glycemic control of type 2 diabetes. Regul Pept (2004) 117(2):77–88. doi:10.1016/j.regpep.2003.10.028
185. Moon HS, Kim MK, Son MH. The development of non-peptide glucagon-like peptide-1 receptor agonist for the treatment of type 2 diabetes. Arch Pharm Res (2011) 34(7):1041–3. doi:10.1007/s12272-011-0721-z
186. Morris LC, Days EL, Turney M, Mi D, Lindsley CW, Weaver CD, et al. A duplexed high-throughput screen to identify allosteric modulators of the Glucagon-Like Peptide 1 and Glucagon receptors. J Biomol Screen (2014) 19(6):847–58. doi:10.1177/1087057114520971
187. Zhang YJ, Shen LL, Cheon HG, Xu YN, Jeong JH. Synthesis and biological evaluation of glucagon-like peptide-1 receptor agonists. Arch Pharm Res (2014) 37(5):588–99. doi:10.1007/s12272-013-0253-9
188. Koole C, Wootten D, Simms J. Allosteric ligands of the glucagon-like peptide 1 receptor (GLP-1R) differentially modulate endogenous and exogenous peptide responses in a pathway-selective manner: implications for drug screening. Mol Pharmacol (2010) 78:456–65. doi:10.1124/mol.110.065664
189. Chen D, Liao J, Li N. A nonpeptidic agonist of glucagon-like peptide 1 receptors with efficacy in diabetic db/db mice. Proc Natl Acad Sci USA (2007) 104:943–8. doi:10.1073/pnas.0610173104
190. Sloop KW, Willard FS, Brenner MB. Novel small molecule glucagon-like peptide-1 receptor agonist stimulates insulin secretion in rodents and from human islets. Diabetes (2010) 59:3099–107. doi:10.2337/db10-0689
191. Knudsen LB, Kiel D, Teng M. Small-molecule agonists for the glucagon-like peptide 1 receptor. Proc Natl Acad Sci USA (2007) 104:937–42. doi:10.1073/pnas.0605701104
192. Willard FS, Wootten D, Showalter AD. Small molecule allosteric modulation of the glucagon-like peptide-1 receptor enhances the insulinotropic effect of oxyntomodulin. Mol Pharmacol (2012) 82:1066–73. doi:10.1124/mol.112.080432
193. Wootten D, Savage EE, Valant C, May LT. Allosteric modulation of endogenous metabolites as an avenue for drug discovery. Mol Pharmacol (2012) 82:281–90. doi:10.1124/mol.112.079319
194. Mapelli C, Natarajan SI, Meyer JP. Eleven amino acid glucagon-like peptide-1 receptor agonists with antidiabetic activity. J Med Chem (2009) 52:7788–99. doi:10.1021/jm900752a
195. Koole C, Savage EE, Christopoulos A, Miller LJ, Sexton PM, Wootten D. Minireview: signal bias, allosterism, and polymorphic variation at the GLP-1R: implications for drug discovery. Mol Endocrinol (2013) 27(8):1234–44. doi:10.1210/me.2013-1116
196. Montrose-Rafizadeh C, Avdonin P, Garant MJ. Pancreatic glucagon-like peptide-1 receptor couples to multiple G proteins and activates mitogen-activated protein kinase pathways in Chinese hamster ovary cells. Endocrinology (1999) 140:1132–40. doi:10.1210/endo.140.3.6550
197. Quoyer J, Longuet C, Broca C. GLP-1 mediates antiapoptotic effect by phosphorylating Bad through a β-arrestin 1-mediated ERK1/2 activation in pancreatic β-cells. J Biol Chem (2010) 285:1989–2002. doi:10.1074/jbc.M109.067207
198. Maudsley S, Pierce KL, Zamah AM, Miller WE, Ahn S, Daaka Y, et al. The beta(2)-adrenergic receptor mediates extracellular signal-regulated kinase activation via assembly of a multi-receptor complex with the epidermal growth factor receptor. J Biol Chem (2000) 275(13):9572–80. doi:10.1074/jbc.275.13.9572
199. Buteau J, Foisy S, Joly E, Prentki M. Glucagon-like peptide 1 induces pancreatic β-cell proliferation via transactivation of the epidermal growth factor receptor. Diabetes (2003) 52:124–32. doi:10.2337/diabetes.52.1.124
200. Martin B, Chen H, Daimon CM, Chadwick W, Siddiqui S, Maudsley S. Plurigon: three dimensional visualization and classification of high-dimensionality data. Front Physiol (2013) 4:190. doi:10.3389/fphys.2013.00190
201. Wootten D, Savage EE, Willard FS, Bueno AB, Sloop KW, Christopoulos A, et al. Differential activation and modulation of the glucagon-like peptide-1 receptor by small molecule ligands. Mol Pharmacol (2013) 83(4):822–34. doi:10.1124/mol.112.084525
202. Coopman K, Huang Y, Johnston N, Bradley SJ, Wilkinson GF, Willars GB. Comparative effects of the endogenous agonist glucagon-like peptide-1 (GLP-1)-(7–36) amide and the small-molecule ago-allosteric agent “compound 2” at the GLP-1 receptor. J Pharmacol Exp Ther (2010) 334:795–808. doi:10.1124/jpet.110.166009
203. Wootten D, Simms J, Koole C. Modulation of the glucagon-like peptide-1 receptor signaling by naturally occurring and synthetic flavonoids. J Pharmacol Exp Ther (2011) 336:540–50. doi:10.1124/jpet.110.176362
204. Harikumar KG, Wootten D, Pinon DI, Koole C, Ball AM, Furness SG, et al. Glucagon-like peptide-1 receptor dimerization differentially regulates agonist signaling but does not affect small molecule allostery. Proc Natl Acad Sci USA (2012) 109(45):18607–12. doi:10.1073/pnas.1205227109
205. de la Monte SM, Tong M. Brain metabolic dysfunction at the core of Alzheimer’s disease. Biochem Pharmacol (2014) 88(4):548–59. doi:10.1016/j.bcp.2013.12.012
206. Amadoro G, Corsetti V, Florenzano F, Atlante A, Bobba A, Nicolin V, et al. Morphological and bioenergetic demands underlying the mitophagy in post-mitotic neurons: the pink-parkin pathway. Front Aging Neurosci (2014) 6:18. doi:10.3389/fnagi.2014.00018
207. Morais VA, Haddad D, Craessaerts K, De Bock PJ, Swerts J, Vilain S, et al. PINK1 loss-of-function mutations affect mitochondrial complex I activity via NdufA10 ubiquinone uncoupling. Science (2014) 344(6180):203–7. doi:10.1126/science.1249161
208. Bassil F, Fernagut PO, Bezard E, Meissner WG. Insulin, IGF-1 and GLP-1 signaling in neurodegenerative disorders: targets for disease modification? Prog Neurobiol (2014) 118C:1–18. doi:10.1016/j.pneurobio.2014.02.005
Keywords: transcriptomics and proteomics, pharmacotherapeutics, pathophysiology, heptahelical G protein-coupled receptor
Citation: Janssens J, Etienne H, Idriss S, Azmi A, Martin B and Maudsley S (2014) Systems-level G protein-coupled receptor therapy across a neurodegenerative continuum by the GLP-1 receptor system. Front. Endocrinol. 5:142. doi: 10.3389/fendo.2014.00142
Received: 28 May 2014; Accepted: 14 August 2014;
Published online: 01 September 2014.
Edited by:
Briony Forbes, The University of Adelaide, AustraliaCopyright: © 2014 Janssens, Etienne, Idriss, Azmi, Martin and Maudsley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stuart Maudsley, Translational Neurobiology Group, VIB Department of Molecular Genetics, University of Antwerp, Universiteitsplein 1, Building V, Antwerpen B2610, Belgium e-mail:c3R1YXJ0Lm1hdWRzbGV5QG1vbGdlbi52aWItdWEuYmU=